

Abstract
Parkinson’s disease (PD) is a chronic, progressive movement disorder of adults and the second most common neurodegenerative disease after Alzheimer’s disease. Neuropathologic diagnosis of PD requires moderate-to-marked neuronal loss in the ventrolateral substantia nigra pars compacta and α-synuclein (αS) Lewy body pathology. Nigrostriatal dopaminergic neurodegeneration correlates with the Parkinsonian motor features, but involvement of other peripheral and central nervous system regions leads to a wide range of non-motor features. Nigrostriatal dopaminergic neurodegeneration is shared with other parkinsonian disorders, including some genetic forms of parkinsonism, but many of these disorders do not have Lewy bodies. An ideal animal model for PD, therefore, should exhibit age-dependent and progressive dopaminergic neurodegeneration, motor dysfunction and abnormal αS pathology. Rodent models of PD using genetic or toxin based strategies have been widely used in the past several decades to investigate the pathogenesis and therapeutics of PD, but few recapitulate all the major clinical and pathologic features of PD. It is likely that new strategies or better understanding of fundamental disease processes may facilitate development of better animal models. In this review, we highlight progress in generating rodent models of PD based on impairments of four major cellular functions: mitochondrial oxidative phosphorylation, autophagy-lysosomal metabolism, ubiquitin-proteasome protein degradation, and endoplasmic reticulum (ER) stress/unfolded protein response (UPR). We attempt to evaluate how impairment of these major cellular systems contribute to PD and how they can be exploited in rodent models. In addition, we review recent cell biological studies suggesting a link between αS aggregation and impairment of nuclear membrane integrity, as observed during cellular models of apoptosis. We also briefly discuss the role of incompetent phagocytic clearance and how this may be a factor to consider in developing new rodent models of PD.
Introduction
Parkinson’s disease (PD) is a chronic, progressive movement disorder and the second most common neurodegenerative disease after Alzheimer’s disease [143]. Pathologically, PD is characterized by selective and progressive loss of dopaminergic neurons projecting from the substantia nigra (SN) pars compacta to the corpus striatum, leading to an extrapyramidal motor disorder with bradykinesia, resting tremor, rigidity, and postural instability. Involvement of other regions of the central and peripheral nervous system contributes to a range of non-motor symptoms such as autonomic dysfunction, depression and cognitive deficits [103,155]. The characteristic pathological hallmarks of PD are cytoplasmic and neuritic aggregates of the presynaptic protein α-synuclein (α-Syn) in Lewy bodies and Lewy neurites, collectively referred to as Lewy-related pathology [45]. In addition to α-Syn, these structures also contain other proteins, whose specificity and precise role in the disease process are less certain [157]. The distribution of Lewy-related pathology in PD follows a fairly predictable pattern depending upon selective vulnerability or connectivity, which has led Braak and coworkers to propose a staging scheme for PD [23]. Other systems for classification Lewy-related pathology have also been proposed [9,3,196], but the Braak scheme has been groundbreaking in drawing attention to the multisystem pathology in PD and anatomical “predictability” of disease progression as it relates to evolving signs and symptoms of PD [21]. In the Braak staging scheme there is early involvement of olfactory bulb and central parasympathetic nuclei in the medulla, suggesting that the pathology may actually originate outside of the central nervous system [22]. Indeed, there is evidence for α-Syn pathology in peripheral tissues, albeit little evidence that it precedes brain pathology [85,11,10].
Although it has been two centuries since the description of shaking palsy by James Parkinson [144], the etiology of this disease remains enigmatic. A clue to possible role of environmental factors was the report of a PD-like syndrome in humans exposed to meperidine analogues such as MPTP [104]; however, Lewy-related pathology was not observed at autopsy [59]. Other neurotoxins implicated in PD include pesticides, organic solvents and air pollutants [68]. The discovery of mutations in the gene for α-synuclein (SNCA) in familial PD [158] and subsequent demonstration that Lewy-related pathology contained α-Syn [187] solidified the etiologic importance of αS to PD. Genetic studies of kindreds with PD in multiple affected family members led to identification of several other genetic loci implicated in autosomal dominant or recessive forms of PD [98]. Genetic factors implicated in sporadic PD can be ascertained from genome wide association studies (GWAS). A meta-analysis of currently available GWAS by Nalls et al. [140] implicated 28 loci, and an additional locus was identified in an autopsy-confirmed cohort of PD [13]. A curated on-line database of genetic loci implicated in PD is PDGene [109]. The top genetic loci are associated with genes that implicate distinct cellular processes affecting select cellular compartments, notably cytoplasm, cytoskeletal components, mitochondria, endosomes, and lysosomes (Table 1). Perturbations in proteostasis and mitochondrial quality control, and other basic cellular processes, converge in producing PD [40]. It is increasingly recognized that impairment of several major cellular functions associated with energy metabolism, protein degradation and stress response play important roles in αS deposition and programmed cell death in dopaminergic neurons of PD [138,202,117,44,110]. In this review, we highlight recent progress in generating rodent models of PD based on impairments of four major cellular functions: mitochondrial oxidative phosphorylation, autophagy-lysosomal metabolism, ubiquitin-proteasome protein degradation, and endoplasmic reticulum (ER) stress/unfolded protein response (UPR). The goal of this review is to provide insights into possible pathogenic mechanisms of PD at the cellular level, with a particular focus on rodent animal models.
Table 1.
middle PD genetic loci and their cellular compartments
| Gene | O.R | P-value | Function | EC | PM | Nucl | Cyto | Skel | Mito | Lyso | Endo | Golgi | ER |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNCA | 1.3 | 1.8 E-82 | synaptic vesicle release | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 3 | 3 | |
| MAPT | 0.8 | 6.1 E-49 | microtubule associated protein | 5 | 5 | 5 | 5 | ||||||
| TMEM175 | 1.3 | 6.0 E-41 | endosome-lysosome potassium channel | 3 | 5 | 5 | |||||||
| ASH1L | 0.5 | 6.9 E-28 | histone lysine N-methyltransferase | 5 | 5 | 5 | |||||||
| MCCC1 | 0.8 | 5.5 E-22 | mitochondrial enzyme | 5 | 5 | ||||||||
| STK39 | 1.2 | 1.7 E-20 | kinase/cell stress response | 3 | 5 | 5 | |||||||
| LRRK2 | 1.2 | 4.9 E-14 | kinase/regulation of aumiddlehagy/lysosomes, Golgi & synaptic vesicle trafficking | 5 | 4 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | |
| HLA-DQB1 | 0.8 | 5.8 E-13 | class II major histocompatibility complex | 4\ | |||||||||
| INPP5F | 1.8 | 1.29 E-11 | inositol polyphosphate-5-phosphatase | 3 | 5 |
Select middle genetic loci with largest effect sizes from PDGene (showing their odds ratio for risk/protection for PD and their p-values) as well as the cellular compartments with which putative genes reside. O.R. = Odds ratio; EC = extracellular; PM = plasma membrane; Nucl = nucleus; Cyto = cymiddlelasm; Skel = cytoskeleton; Mito = mitochondrion; Lyso = lysosome; Endo = endosome; Golgi – Golgi apparatus; ER = endoplasmic reticulum. Color code = darker color indicates strength of evidence (5, 4 & 3) from GeneCards® Compartments, which integrates data from literature curation, high-throughput microscopy-based screens, predictions from primary sequence, and automatic text mining
Impairment of mitochondrial oxidative phosphorylation in PD and in animal models
Mitochondria are critical components of the eukaryotic cell due to their unique function – oxidative phosphorylation [154]. Oxidative phosphorylation is based upon two tightly connected components: the electron transport chain and chemiosmosis, which are orchestrated by five multi-subunit protein complexes localized on the inner mitochondrial membrane. The electron transport chain (complex I, II, III and IV) pass electrons from one subunit to another, and energy released in these electron transfers is used to form an electrochemical gradient (proton gradient). The chemiosmosis is the last step of oxidative phosphorylation, where ATP synthase (complex V) utilizes the proton gradient to produce ATP [154]. This is normal process of physiological respiration. However, the electron transport chain contains several redox centers that may leak electrons to molecular oxygen, leading to the production of superoxide. It has been reported that approximately 1% to 2% of the molecular oxygen consumed during normal physiological respiration is converted into reactive oxygen species (ROS) [31]. Production of ROS is markedly increased in a variety of pathologic conditions, including defects in one of the electron transport chain complexes [31]. Therefore, the mitochondrial oxidative phosphorylation system produces not only ATP essential for cell metabolism and other normal activities, but also incidental ROS that can lead to cell injury and death.
The role of mitochondrial dysfunction in PD includes evidence for reduced complex I activity and oxidation in various tissues of PD patients [173,136,99,151], and notable increases in complex I related oxidative damage in postmortem PD brain [93]. Mitochondrial deficits and subsequent oxidative stress are a leading hypothesis for dopaminergic neuron death in PD [60]. A number of PD-related genes are tightly associated with mitochondrial dysfunction [198,188,178], and there is increasing evidence of mutations or deletions in mitochondrial DNA in non-familial PD [75,81,166]. Given the wealth of this supporting evidence, it is not surprising that a number of well-established PD animal models are based upon “mitochondrial toxins,” such as MPTP and rotenone [124]. Meanwhile, molecular strategies aimed at producing mitochondrial deficits have also been employed in PD animal models (Table 2).
Table 2.
Mitochondrial oxidative phosphorylation system in PD models
| Drug or gene modification | αS pathology | Neuronal loss | Striatal DA reduction | Motor behavior | Remarks | ||
|---|---|---|---|---|---|---|---|
| SN | Other region | ||||||
| Toxin | MPTP | No | Yes | No | Yes | Yes | Systematic administration |
| 6-OHDA | No | Yes | No | Yes | Yes | SN injection | |
| Paraquat/Maneb | Yes | Yes | No | Yes | Yes | Systematic administration | |
| Rotenone | Yes | Yes | No | Yes | Yes | Systematic administration | |
| Trichloroethylene | Yes | Yes | No | Yes | Yes | Systematic administration | |
| Homocysteine | Yes | Yes | No | Yes | Yes | SN injection | |
| Genetic | PINK1 (KO) | No | Yes/No | No | Yes/No | Yes/No | Results vary in different animal models |
| TFAM (KO) | No | Yes | No | Yes | Yes | Conditional knockout (DA only) | |
| Ndufs4 (KO) | No | No | No | Yes | No | ||
| Hspd1(KO) | ND | ND | ND | ND | Yes | Heterozygous knock-out | |
| Aldh1a1/Aldh2 (KO) | ND | Yes | No | Yes | Yes | Knock-out of both isoforms | |
| HtrA2 (TM and TW) | ND | Yes (For TW only) | Yes | ND | Yes | ||
| Twinkle (TM) | ND | Yes | No | ND | Yes | ||
| dOTC (TM) | ND | Yes | No | Yes | Yes | ||
ND: not determined; KO: knockout; KD: knockdown; TM: Transgenic mutant gene; TW: Transgenic wildtype gene.
In addition to MPTP other neurotoxins have been used to model mitochondrial dysfunction in PD, including pesticides such as rotenone [8], paraquat [147], maneb [46] and trichloroethylene [61], as well as endogenous neurotoxins such as 6-hydroxy-dopamine (6-OHDA) [87,83] and homocysteine [101,17].
MPTP and rotenone, two potent inhibitors of complex I, are the most popular mitochondrial toxins for modeling PD in rodents [124]. MPTP neurotoxicity is relatively limited to the nigrostriatal dopaminergic pathway and is associated with a characteristic and quantifiable motor phenotype [134]. This model, as well as most other neurotoxin models, lacks αS pathology typical of PD [134], although isolated reports have shown ubiquitin and αS immunoreactive inclusions in mice treated with chronic low dose of MPTP [58]. In contrast, a number of rodent models with chronic rotenone treatment have αS-positive intracellular inclusions in parallel with degeneration of nigrostriatal dopaminergic neurons and motor deficits. Phenotypic variability, high mortality rate and other non-PD related features are drawbacks of the rotenone model [34]. Paraquat is a weak inhibitor of mitochondrial complex I [165]. Some studies showed that it can induce motor dysfunction and loss of dopaminergic neurons in SN [24,127], and some studies have reported Lewy body-like structures in rodents treated with paraquat [120,54]. Interestingly, a combination of paraquat and an inhibitor of complex III – maneb [57] – significantly enhances SN neuronal loss in this model, indicative of synergistic effects of deficits in multiple subunits of the mitochondrial electron transport chain on neurodegeneration [193]. The relevance of paraquat to PD is questionable [135]. More recently, trichloroethylene, a complex I inhibitor [61], which has been used in PD animal models, has been epidemiologically linked to PD in humans who have been exposed to trichloroethylene-related products in the workplace [61,69,76]. Administration of trichloroethylene to rats induces SN neuron loss, dopamine depletion in the striatum, and behavioral deficits [112]. More importantly, accumulation of intraneuronal αS can be observed in this model.
In addition to environmental neurotoxins, mitochondrial deficits can also be induced by endogenous molecules, such as 6-hydroxydopamine (6-OHDA) [87,83] and homocysteine [101,17], which have been employed in animal models of PD. 6-OHDA is a hydroxylated analog of dopamine that can be detected in trace amounts in human brain and urine samples [87]. In fact, 6-OHDA was one of the earliest [197], and still widely used, neurotoxin models for PD, valued for its predictable degeneration of dopaminergic neurons and consistent behavioral phenotype [160,170]. The exact mechanism underlying its toxic effects is elusive, but recently, it was demonstrated that mitochondrial dysfunction is the primary cause of 6-OHDA-induced dopaminergic neuronal death [102]. Similar to MPTP, 6-OHDA rodent models lack Lewy-related pathology. Moreover, administration of 6-OHDA must be done by stereotaxic injection or direct infusion into the brain since it is impermeability to the blood brain barrier [18]. Homocysteine is another endogenous molecule that is toxic to dopaminergic neurons. It became a promising reagent for modeling PD in rodents due to its elevated level in plasma of PD patients and neurotoxicity to dopaminergic neurons in cell-based experiments [17]. Rats with unilateral intranigral infusion of homocysteine have inhibition of mitochondrial complex-I activity, reduced striatal dopamine, neurodegeneration of SN dopaminergic neurons, and impaired motor activity [17]. When applied to mice through other routes of administration homocysteine was not toxic; however, it was shown to enhance neurotoxicity of MPTP [47]. Therefore, sensitivity of rats or mice to homocysteine might account for the above discrepancy. It is worth noting that no αS pathology has been reported in homocysteine-induced rodent models.
Besides pharmacological approaches, genetic manipulations represent another strategy to generate mitochondrial dysfunction models. PTEN-induced putative kinase 1 (PINK1) is a mitochondrial serine/threonine-protein kinase, intimately involved in mitochondrial quality control. Mutations in PINK1, which are mostly loss of function mutations [198] or partial dominant negative mutations [161], are linked to autosomal recessive PD [199]. Several PINK1 knockout rodent models have been developed. One model established by Dave and coworkers has been well-characterized in a series of studies [39]. This model has significant mitochondrial proteomic alterations in the absence of mitochondrial functional changes at postnatal day 10; starts to show functional abnormalities at 4 months of age; and develops rapidly progressive neurodegeneration between 4 and 8 months of age. The mice have a progressive movement disorder accompanied by loss of midbrain dopaminergic neurons, with 25% reduction at 6 months and 50% at 8 months of age [206,205]. An unexpected finding is 2 to 3 fold increase in both DA and 5-HT striatal content as late as 8 months of age [39].
Another PINK1 deficient mouse model has defects of key mitochondrial functions that correlate with an age-dependent and moderate decrease of striatal dopamine content. They have impaired locomotor activity; however, no dopaminergic neuronal loss is observed even at 18 months of age [64]. Other reported PINK1 knockout mice do not show observable gross motor dysfunction or alterations in the dopaminergic system, except for gait changes and olfactory dysfunction [65]. It is worth noting that none of the PINK1 models has αS pathology. A limited number of autopsies on PD with PINK1 mutations has shown mild Lewy-related pathology [159]. Given these observations, it has been suggested that PINK1 knockout mice may be used as models of prodromal PD [183].
Mitochondrial transcription factor A (TFAM) is a key activator of mitochondrial transcription that plays an essential role in maintaining mitochondrial integrity. A mouse model with significant mitochondrial dysfunction – MitoPark – was generated by disruption of TFAM in dopaminergic neurons [49]. MitoPark mice have reduced mtDNA expression and respiratory chain deficiency in midbrain dopaminergic neurons, associated with a motor phenotype, slowly progressive SN neurodegeneration, and formation of intra-neuronal inclusions. Further investigation in this model revealed that altered nigrostriatal function precedes the onset of motor dysfunciton [73]. Loss of DA neurons also was associated with impairment in the circadian control of rest and activity [56]. Although MitoPark mice successfully recapitulate several characteristic features of PD, they do not have αS pathology.
There are several other genes that have been less frequently used to induce mitochondrial dysfunction. For example, mouse models with deficiency or abnormal expression of mitochondria function-related genes Ndufs4 [185,95], Hspd1 [118], Aldh1a1/Aldh2 [209], HtrA2 [26,152], Twinkle [186], and dOTC [137] have been generated. These mice show PD-related features to a varying degree. Interestingly, a mitochondrial targeted restriction endonuclease (Mito-PstI) has been used to develop a PD mouse model in which mitochondrial DNA double-strand breaks in dopaminergic neurons is induced by the expression of Mito-Pstl conditionally controlled under dopamine transporter (DAT) promoter-driven tetracycline transactivator protein [156]. This model has mitochondrial dysfunction in dopaminergic neurons and many features of PD, including an L-DOPA reversible motor phenotype, progressive neurodegeneration of DA neurons, and striatal DA depletion [156]. While these models strongly implicate mitochondrial dysfunciton in PD, none of them has αS pathology, which is true for both mitochondrial neurotoxin and genetic models. This raises the question of whether mitochondrial dysfunction would ever be sufficient for induction of αS pathology.
Impairment of autophagy-lysosomal system in PD and in animal models
The autophagy-lysosomal system is responsible for degrading and recycling unnecessary or damaged cytoplasmic molecules or organelles, an essential role in maintaining cellular energy and metabolic homeostasis [149]. Autophagy-lysosomal degradation is a delicately balanced and multistep process that involves orchestrated action of a large number of cellular factors. Autophagy includes three subtypes (macroautophagy, microautophagy and chaperone-mediated autophagy) that are distinguished by distinct routes for delivering targets to lysosomes [149]. Defects in any step or essential factor, such as formation of autophagosome or autophagolysosome, lead to accumulation of unwanted proteins (e.g., αS [208]) in neurons and possible neuron death. It has been suggested that this mechanism might be critical for Lewy-related pathology and neuronal loss in PD.
Support for this hypothesis is the mounting evidence of changes in autophagic vacuoles or autophagy-related markers in postmortem PD brains [5,212,43], as well as discoveries of autophagy-lysosomal related genetic abnormalities in familial PD [180,1,163]. Given the evidence that dysfunction of autophagy-lysosome system is crucial to PD, strategies aimed at compromising autophagy-lysosome system via genetic or pharmacological manipulations have been explored to develop PD animal models (Table 3).
Table 3.
Aumiddlehagy-lysosomal system in PD models
| Drug or gene modification | αS pathology | Neuronal loss | Striatal DA reduction | Motor behavior | Remarks | ||
|---|---|---|---|---|---|---|---|
| SN | Other region | ||||||
| Toxin | Conduritol-β-epoxide | Yes | No | Yes (Cortex) | ND | ND | Systematic administration |
| Genetics | Glucocerebrosidase (KO) | Yes | ND | Yes | ND | Yes | Conditional knockout |
| Cathepsin D (KO) | Yes | ND | ND | ND | ND | ||
| ATP13A2 (KO) | No | No | No | ND | Yes | ||
| Atg7 (KO) | Yes | Yes | No | Yes | Yes | Conditional knockout | |
| Lrrk2 (MT) | No | Yes | No | Yes | Yes | ||
| Interferon-β (KO) | Yes | Yes | Yes | No | Yes | ||
ND: not determined; KO: knockout; KD: knockdown; TM: Transgenic mutant gene; TW: Transgenic wildtype gene.
As the final common pathway in the autophagy-lysosomal system, lysosomes have been the most frequent target for disruption in PD models. Genetic variants in certain lysosomal enzymes, such as glucocerebrosidase (GBA) and ATP13A2, are associated with familial PD [171,150]. Moreover, some lysosomal enzymes have been implicated in α-Syn degradation [177]. These enzymes have been targets for modeling PD in rodents. A mouse model with lysosomal function inhibition by administration of a potent, selective and irreversible competitive inhibitor of glucocerebrosidase – conduritol-β-epoxide – was associated with insoluble αS aggregates in the SN, as well as widespread neuroinflammation, as well as abnormalities in synapses, axonal transport, and cytoskeletal proteins [167]. Another mouse model with reduction in glucocerebrosidase activity in all tissues except the skin with mutations in GBA had rapid motor dysfunction associated with severe neurodegeneration and apoptotic cell death in the brain [51]. Further studies of this model revealed defective autophagic and proteasomal machinery and accumulation of p62/SQSTM1, ubiquitinated proteins and insoluble αS [145].
A mouse model of lysosomal enzyme cathepsin D deficiency showed impaired macroautophagy and proteasome activity, as well as extensive accumulation of endogenous αS as oligomeric and insoluble αS species [37,162]. In contrast to glucocerebrosidase and cathepsin D, ATP13A2 is a late endosomal/lysosomal P5-type transport ATPase that has no direct role in protein degradation. A mouse model lacking ATP13A2 shows accumulation of ubiquitinated protein aggregates, lipofuscinosis, and endolysosomal abnormalities, as well as age-related motor dysfunction [94]. Surprisingly, these phenotypes were independent of αS [94].
Another strategy for disruption of autophagy-lysosome degradation pathway is to impair autophagy. It has been reported that a mouse model with impairment of autophagy caused by conditional deletion of Atg7, an E1-like activating enzyme that is essential for autophagy and cytoplasmic to vacuole formation, can recapitulate many pathological features of PD, including age-related loss of dopaminergic neurons, striatal dopamine reduction, αS accumulation and accumulation of ubiquitinated protein aggregates [2].
The most common genetic cause of late-onset PD, LRRK2 [214], is associated with defects in autophagy [121,71]. LRRK2 has been the target in a number of PD rodent models. Besides the changes of autophagic activity, some PD features, such as motor dysfunction, neuronal loss, dopamine depletion and axonal degeneration have been observed in different LRRK2 models [164,148,108,191]; however, none has αS pathology.
It is worth noting that a recent study shows that mice lacking IFN-beta-IFNAR function demonstrate motor and cognitive learning impairments, accompanied by reduction of dopaminergic neurons and defects of dopamine signaling in the nigrostriatal region, and formation of αS containing inclusions as well [48]. In-depth investigation of mouse brain revealed that the lack of IFN-β signaling caused defects in neuronal autophagy prior to synucleinopathy, which may be responsible for the observed PD features in this novel mouse model [48].
It is interesting to note that many rodent models with impaired autophagy-lysosomal system develop αS pathology without motor dysfunction. In contrast, models with genetic manipulation of ATP13A2 or LRRK2 show impaired motor function without αS pathology. This indicates that αS pathology and motor dysfunction may be independent processes in PD.
Impairment of ubiquitin proteasome system in PD and in animal models
The ubiquitin-proteasome system (UPS) is an essential and highly regulated mechanism for protein catabolism in the cytosol and nucleus that plays an important role in a variety of basic cellular processes [131]. The UPS degradation pathway starts with polyubiquitination of the target protein, which includes a series of ATP-dependent enzymatic steps and a series of necessary enzymes, ubiquitin-activating (E1), ubiquitin-conjugating (E2) and ubiquitin-ligase (E3) enzymes. The ubiquitinated protein is subsequently recognized by the 26S proteasome, and subjected to digestion into peptide fragments in its subunits (20S) after passing through the 19S subunit for the cleavage of polyubiquitin chain. Although 26S proteasome as an intact functional unit is the most common destination for protein degradation, its 20S subunit can also exist freely in mammalian cells to digest natively unfolded or damaged proteins in an ATP-independent manner. Under certain circumstances, the 20S proteasome can disassemble from the 26S and become a dominant degradation pathway [207]. Therefore, the 20S subunit is considered the final pathway of the UPS.
Since the UPS is a major route for αS degradation [208], it is conceivable that the blockage of this pathway could contribute to accumulation of αS and eventual Lewy-related pathology. Support for this hypothesis stems from a number of studies. Ubiquitinated αS coexists with proteasome subunits in Lewy bodies in PD [15], and both structural and functional defects in the 26/20S proteasome have been reported in PD brains [25,129]. In addition, several components of the UPS, including proteasomal subunits, proteasomal activators, heat shock proteins, ubiquitin and ubiquitinated proteins have been identified as constituents of Lewy bodies [130]. Consequently, impairment in the UPS has been considered important PD pathogenesis.
In order to investigate proteasome dysfunction as a model for PD, the UPS can be altered with toxins or genetic manipulations (Table 4). Several proteasome inhibitors (e.g., lactacystin, PSI, MG and EXPO) have been administered to rodents. Lactacystin is the most often used proteasome inhibitor that is applied by stereotaxic infusion into medial forebrain bundle targeting the nigrostriatal pathology or directly into the SN. Almost all animals show varying degrees of neuronal loss at the injection site, there is variability related to dose used. Most also have striatal DA reduction, αS accumulation in SN, and motor impairment [16,204]. A major criticism of these models is the lack of specificity of this toxin. Surprisingly, a study from Lorenc-Koci and coworkers showed that lactacystin infusion into the SN caused reduction of αS expression locally, as well as neuronal loss and reduced DA, but no motor impairment [113]. Inhibitors of UPS suitable for systemic administration include Z-lle-Glu(OtBu)-Ala-Leu-al [132,172], MG-132 and epoxomicin [132,189]. Results from these inhibitors have been inconsistent; some models recapitulate some features of PD pathology, while others have no effect at all [20]. These discrepancies are likely related to a number of experimental design issues, such dose and strain differences.
Table 4.
Ubiquitin-proteasome system in PD models
| Drug or gene modification | αS pathology | Neuronal loss | Striatal DA reduction | Motor behavior | Remarks | ||
|---|---|---|---|---|---|---|---|
| SN | Other region | ||||||
| Toxin | Lactacystin | Yes | Yes | No | Yes | Yes | SN injection |
| PSI | Yes | Yes | ND | ND | Yes | Systematic administration | |
| MG-132 | ND | Yes | No | Yes | No | SN injection | |
| Epoxomicin | ND | Yes | ND | Yes | Yes | Systematic administration | |
| Genetics | PSMC1 (KO) | Yes | Yes | Yes | Yes | Yes | Conditional knockout |
| Parkin (KO) | No | No | No | Yes | No | ||
| Parkin (TM) | Yes | Yes | No | Yes | Yes | Q311X, T240R mutant | |
| UCH-L1 (TM or KO) | No | Yes | No | Yes | Yes | ||
ND: not determined; KO: knockout; KD: knockdown; TM: Transgenic mutant gene; TW: Transgenic wildtype gene; PSI: Z-lle-Glu(OtBu)-Ala-Leu-al.
Genetic manipulations that perturb the UPS have also been developed for rodent models. Bedford and coworkers used a Cre/loxP system to establish a mouse model with conditional knockout of the proteasomal Psmc1 ATPase in neurons in different regions of the brain [12]. In this model, the 26S proteasome is disrupted due to failure of assembly of the 19S subunit, while 20S degradation remains functional. Mice in this model have significant loss of DA neurons and reduction of striatal dopamine [12,146]. More interestingly, the neurons that survive effects of 26S proteasome-depletion in both the forebrain and SN develop intraneuronal inclusions containing αS, ubiquitin and mitochondria, resembling pre-Lewy bodies [146,12].
Other rodent genetic models of UPS deficiency have targeted Parkin or UCH-L1. Parkin is an E3 ubiquitin ligase that plays an important role in UPS as well mitochondrial quality control [41]. Homozygous or compound heterozygous mutations in Parkin are associated with autosomal recessive juvenile onset Parkinsonism (ARJP), while heterozygous mutations may play a role in sporadic PD, as well [41]. Mutations in Parkin impair UPS protein degradation, leading to abnormal accumulation of proteins. Although it is well-established that parkin dysfunction disrupts mitophagy [141], this may also be an indirect consequence of impairment of the UPS because parkin-mediated proteasomal degradation of mitochondrial outer membrane proteins is an upstream determinant of mitophagy [28]. A number of parkin knockout mice have been reported; however, none of them has SN dopamine-related motor deficits, but mild reduction in DA without neuron loss has been observed in some models [66,123,213,153]. In contrast, a bacterial artificial chromosome transgenic mouse model expressing mutant parkin (Parkin-Q311X) in dopaminergic neurons demonstrated age-dependent neurodegeneration in the SN, significant reduction of striatal DA and motor deficits [114]. Moreover, this model had progressive accumulation of proteinase K-resistant αS in the SN [114]. Another interesting parkin PD model is a rat model with adeno-associated virus (AAV)-mediated overexpression of p.T240R Parkin mutant and wild type human parkin. Overexpression of both forms of parkin were associated with progressive and dose-dependent dopaminergic neuronal loss and mild motor deficits [201].
UCHL1 is included in the discussion of models of UPS dysfunciton because it is a member of a gene family whose products hydrolyze small C-terminal adducts of ubiquitin to generate ubiquitin monomers [122]. There are few rodent models for UCHL1 in part because disease-causing mutations (e.g. UCHL1 p.I93M) are extremely rare [107]. Transgenic mice with p.I93M UCHL1 mutation had significant reduction in SN neurons and decreased DA in the striatum, as well as decreased spontaneous, voluntary movements [176,210]. Another mouse model with UCHL1 knockout had early onset motor deficits, short lifespan and forebrain astrogliosis [179]. In these models, UCHL1 deficiency did not lead to changes in αS expression [176,210,179].
Of the various models for UPS dysfunciton, lactacystin may be better than other proteasome inhibitors with respect to modeling the most important clinical and pathological features of PD. While genetic manipulation of Parkin or UCHL1 may induce SN neuronal degeneration and consequent motor deficits, αS pathology is not consistently observed in these models.
Impairment of endoplasmic reticulum (ER) stress and unfolded protein response (UPR) in PD and in animal models
The ER plays a key role in properly folding proteins and in protein quality control [133]. Under normal conditions, ER homeostasis is a tightly regulated process. ER homeostasis can be disrupted by a number of factors that lead to protein misfolding, abnormal protein maturation or ER calcium release, a process referred to as “ER stress.” ER stress triggers a physiologic protective response termed the unfolded protein response (UPR) [133]. The UPR is regulated by three major ER stress sensor proteins – activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and protein kinase RNA-like ER kinase (PERK). With activation of the UPR, cells restore ER balance by reducing ER stress and recovering ER homeostasis [133]. If the UPR becomes dysregulated, it can lead to defective ER proteostasis (e.g., accumulation of toxic misfolded proteins) associated with cell dysfunction and even cell death.
Since the accumulation of misfolded αS is a fundamental pathogenic process in PD [11], it is not surprising that ER stress and UPR can be observed in PD brains. There is compelling evidence that UPR activation markers (phosphorylated PERK and eIF2a) are increased in the SN in PD [80]. In addition, ER stress-associated proteins, such as HERP, BIP and pPDI, are upregulated in SN of PD and some also co-localize with Lewy bodies [35,175,182]. Therefore, ER stress and UPR are likely involved in pathogenesis of PD; however, it is unclear if accumulation of aggregated αS is the cause or result of ER stress and disrupted UPR. Studies have showed that αS aggregation can induce ER stress and UPR [14] and vice versa [89].
Most studies of ER stress and the UPR in PD rodent models have focused on rescuing neuronal loss via alleviation of ER stress and regulation of UPR [133]. Only a few studies have produced ER stress via toxins or genetic manipulation to model PD in rodents (Table 5). An example is deletion of ATF6a gene in MPTP mice, which was associated with significantly increased neuronal loss and formation of ubiquitin-positive neuronal inclusions [79]. In a rat model overexpressing αS in the SN with AAV gene transfer, knockdown of GRP78 was shown to aggravate αS neurotoxicity, leading to significantly more neuronal loss and greater reduction of striatal DA; however, GRP78 knockdown had no influence on rat without αS overexpression [168]. As to PERK pathway in UPR, a recent study demonstrated that sustained up-regulation of ATF4 in the SN of rats via AAV mediated gene transfer produced motor dysfunction, neuronal loss and striatal DA reduction [77]. It is worth noting that none of the above models with perturbed UPR pathway had αS pathology. In contrast, a recently reported rat model using intranigral injection of a ER stress inducer – tunicamycin –not only had locomotor impairment and dopaminergic neuronal loss death, but also extensive αS oligomerization, activation of astroglia and increased expression of ER stress markers [36]. Missing from this report was description of whether they also had intra-neuronal αS inclusions.
Table 5.
ER stress and UPR system in PD models
| Drug or gene modification | αS pathology | Neuronal loss | Striatal DA reduction | Motor behavior | Remarks | ||
|---|---|---|---|---|---|---|---|
| SN | Other region | ||||||
| Toxin | Tunicamycin | Yes | Yes | No | ND | Yes | SN injection |
| Genetics | ATF6a (KO) | No | Worse | No | ND | No | MPTP & probenecid injection |
| GRP78 (KD) | Worse | Worse | No | Worse | No | αS expression | |
| ATF4 (TW) | No | Yes | No | Yes | Yes | rAAV expression in SN | |
ND: not determined; KO: knockout; KD: knockdown; TM: Transgenic mutant gene; TW: Transgenic wildtype gene.
It is clear that αS pathology is not a significant feature of rodent models with genetically induced ER stress or impairment of UPR, although they are associated with neuronal pathology and motor deficits. In contrast, administration of tunicamycin is a more robust method to model PD features, including αS pathology, by producing ER stress and perturbing the UPR.
Expression or deletion of PD-associated genes in PD models
Animal models based upon expression of PD genetic risk variants are useful for understanding the functional consequences of the variants and their role in the pathogenesis of PD. Some of these PD-related genes, such as Parkin and ATP13A2, have a clearly defined function in specific organelles or functional systems. Other genes do not fit well in this classification scheme. These include αS and DJ-1, neither of which seems to be directly related to mitochondrial stress, autophagy-lysosomal degradation, UPS or ER stress/UPR.
Alpha-synuclein in animal models of PD
αS is a major component of Lewy body, the pathological hallmark of PD [203,187]. Genetic coding region mutations, as well as duplication or triplication of the gene for αS (SNCA), are linked to autosomal dominant PD, suggesting that not only mutations, but also higher than normal levels of αS play a role in PD [203,53,181].
Transgenic rodent models overexpressing high level of wild type, mutant or truncated αS demonstrate regional expression of αS driven in part by mode of delivery and promoter, and most of them manifest a variety of pathologies and behaviors deficits. Several models have worth mentioning due to their robust phenotypes and development of features similar to those found in PD. Masliah et al. generated a wild type (WT) αS transgenic mouse model that had motor deficits and loss of dopaminergic terminals in the basal ganglia, as well as αS- and ubiquitin-immunoreactive neuronal inclusions in the neocortex, hippocampus, and SN [125]. Another mouse model expressing WT αS had progressive PD-like motor deficits, progressive nigrostriatal pathology, and regionally specific proteinase K-resistant αS aggregates; however, no neuron loss was observed in this model [32]. Transgenic mice with bacterial artificial chromosome expressing wild-type αS from the complete human SNCA locus at disease-relevant levels had age-dependent loss of nigrostriatal dopamine neurons and motor dysfunction, but they had no observable αS pathology [86].
Several rodent models are based upon overexpression of mutant αS. Three transgenic models with A53T αS overexpression driven by mouse prion protein promoter have been generated. All three models had motor deficits [63,62,106], but only two of them had robust αS pathology and neuronal loss [62,106]. The other one had no αS pathology at all [63]. Another robust model is a mouse that conditionally expresses A53T in midbrain dopaminergic neurons. This model had profound motor disabilities, significant dopaminergic neuronal loss, reduction of dopamine release and αS pathology in both neuronal soma and axons of dopaminergic neurons [111]. Compared to the number of A53T models, there are only a few mice generated with overexpression of A30P. One notable model is one with high expression of A30P driven by hamster prion protein promoter. This mouse had progressive degeneration with gliosis and motor deficits, but without lesions in nigrostriatal dopaminergic neurons or αS pathology [70]. Double transgenic mouse models overexpressing both A53T and A30P have also been generated. One had not only accumulation of sarkosyl-insoluble αS and αS with pathologic posttranslational modifications (e.g., phosphorylation, ubiquitination and nitration), but also motor dysfunction and neuronal loss [84]; the other one had progressive decline in locomotor activity and loss of tyrosine hydroxylase-positive neurons, but no αS pathology [194]. Transgenic mouse models overexpressing other αS mutants have also been reported. For example, overexpression of E46K in mice is associated with age-dependent motor impairments and age-dependent neuronal inclusions that resemble Lewy bodies. Interestingly, this model also had abundant neuronal tau inclusions that resemble neurofibrillary tangles [50].
Transgenic mouse models have also been developed that express truncated forms of αS. Mouse overexpressing αS (1–119) driven by ROSA26 promoter had marked reduction of striatal dopamine yet no dopaminergic neuronal loss in SN or other PD features [38]. Transgenic mice with overexpression of αS (1–120) driven by rat tyrosine hydroxylase promoter had progressive reduction in spontaneous locomotion accompanied by reduction in striatal dopamine levels and formation of pathological inclusions in SN and olfactory bulb neurons [195]. In contrast, transgenic mice generated with the CamKIIα promoter had expression in forebrain and formation of small granular neuronal cytoplasmic αS inclusions as well as memory deficits [78]; perhaps a better model for Lewy body dementia than for PD.
Although most transgenic αS rodent models have αS aggregates, there are some models with very subtle or no pathology throughout their life-span [52,126]. Moreover, marked differences can be also found in αS transgenic models generated in animals with similar genetic backgrounds and using identical promoters [63,62,106]. Therefore, unpredictable effects of genomic integration site, epigenetic modifications and transgene copy numbers might play important roles in variability in transgenic rodent models.
In addition to transgenic technology, lentivirus or adeno-associated virus (AAV) infection-mediated overexpression of WT or mutant αS have been used to produce expression in particular brain regions through stereotaxic injection. Rats tend to be more often used with this type of model than mice, likely due to surgical technical challenges. It is interesting that in most viral models there is marked localized αS pathology, but variable neuronal loss and motor deficits [42,105,97]. This is probably a function of the high virus-mediated expression over a relatively short term, which does not allow neurons to adapt and to effectively degrade the overexpressed αS or to develop functional deficits from neurodegeneration.
It is worth noting that in order to test Braak staging hypothesis that posits that αS pathology progressively spreads by cells between interconnected brain regions, there are many rodent models which have been developed to demonstrate prion-like propagation of αS [116]. Some rodent models successfully recapitulate the distribution of αS pathology in brain regions vulnerable in PD, accompanied by neurodegeneration and motor impairment [115], which seems to fit with the hypothesis of pathologic αS propagation. On the other hand, a problem in these models is that the initial transmissible αS aggregates are supplied exogenously through inoculations that would be associated with disruption of tissue integrity at least at the site of implantation, rather than endogenously generated pathologic αS [90].
DJ-1 in PD models
Genetic studies have showed that missense, truncation and mutations in DJ-1 can cause autosomal recessive, early-onset familial PD [19]. Although the exact role of DJ-1 in PD pathogenesis is still unknown, studies on this gene suggest that it is mostly known as a redox-sensitive chaperone and as a sensor for oxidative stress, but associated with various cellular processes, including regulation of mitochondrial complex I activity, 20S proteasome function and autophagy [4,6,139].
Several mouse models with loss of DJ-1 function due to gene deficiency have been generated. The first mouse model generated by Goldberg et al. had altered dopaminergic neurotransmission, reduced sensitivity of nigral neurons to dopamine, and pronounced defects in locomotor activity [67]. Another DJ-1 knockout mouse line from Chandran et al. had progressive behavioral deficits, but no nigrostriatal dopaminergic neuronal loss [29]. A mouse model from Chen et al. had age-dependent and task-dependent impairment in motor behaviors with nigrostriatal dopaminergic dysfunction, but no dopaminergic neuronal loss [30]. Kim et al. and Manning-Boğ et al., respectively, generated mouse models with DJ-1 function loss via different strategies. While both models had minimal PD-like changes, they both had enhanced vulnerability to PD-like pathology in response to MPTP [96,119]. Although dopaminergic neurodegeneration and motor impairment markedly vary in DJ-1 deficient mouse models, lack of αS pathology is consistently observed. Therefore, DJ-1 related PD may be independent of Lewy-related pathology. Unfortunately, autopsy findings in DJ-1-related PD have yet to be reported.
Other genes in PD models
Given the important role of nigrostriatal pathology in clinical manifestations of PD, several genes involved in development and maintenance of dopaminergic neurons have been targeted in PD rodent models. Conditional knockout of SHH in mouse DA neurons causes progressive loss of DA neurons, reduction of striatal dopamine and motor deficits [72]. Mice with heterozygous Nurr1 knockout, or conditional knockout, in DA neurons display the age-dependent decline in DA neurons and decreased DA signaling, but motor dysfunction was only apparent in heterozygous Nurr1 knockout (Nurr1+/−) mice [91,211]. Pitx3-deficient aphakia mice display marked reduction in spontaneous locomotor activity and very low striatal DA levels due to progressive loss of mesencephalic dopaminergic neurons during fetal and postnatal development [82,200]. Mouse lacking one En1 allele have decreased mitochondrial complex I activity and progressive SN dopamine neuron degeneration in adulthood, but no observable motor deficits [142]. It is worth noting that none of models targeting dopaminergic neurons have αS pathology. In contrast, mice deficient of c-Rel factor or VMAT2 show not only progressive SN neurodegeneration, but also accumulated of αS [192,7]. It is unclear why some of these models (e.g., c-Rel, VMAT2) can induce αS aggregates in rodent models, while others do not. One possible reason is that the molecular interaction network of these genes might interface with pathways of αS degradation or expression.
Concluding comments
In order to completely recapitulate the major clinical and neuropathological characteristics of PD, an ideal animal model should exhibit age-dependent and progressive neurodegeneration, motor dysfunction and abnormal αS pathology. Neurodegeneration should include dopaminergic neuronal loss, striatal dopamine reduction and αS pathology in the form of neuronal and axonal inclusions (Lewy bodies and Lewy neurites). Motor dysfunction should include slowing (i.e. bradykinesia) that is responsive to dopaminergic therapy. With increasing recognition of the importance of non-motor symptoms of PD [174,155], ideal models would have involvement of central and peripheral autonomic nervous system and the olfactory bulb [128]. Although there are many rodent models generated over the past few decades, which have provided important insights into mechanisms of PD pathogenesis, none of the current models recapitulates all of the major features of PD.
In this review, inspired by advances in our understanding of the pathogenesis of PD at the cellular level, we attempted to evaluate how impairment of major cellular functional systems contributes to PD and how it can be exploited in rodent models. We note that neurotoxin models tend to be more efficient than genetic models in producing dopaminergic neurodegeneration; however, genetic models have the advantage of specifically disrupting cellular functions that are affected in PD. An obvious limitation of neurotoxin models is the unwanted side effects of a toxin [27], while genetic models have their own limitations related to whether they are based upon cDNA or genomic DNA, as well as effects of genetic background of the particular animal, as well as the integration site and copy number of the transgene.
In general, rodent models with impairment of mitochondrial oxidative phosphorylation system, ER stress and UPR system are best for modeling nigrostriatal degeneration accompanied by motor dysfunction, but they rarely have αS pathology. In contrast, models with impairment of autophagy-lysosomal system or ubiquitin-proteasome system more often have αS pathology (e.g., cathepsin D KO mice [37,162]), or αS pathology in addition to neuronal loss and motor deficits (e.g., PSMC1 KO [12] and Parkin-Q311X [114]). Genetic models with expression or deletion of PD-associated genes often have neurodegeneration with varying motor deficits, but only models based upon αS (with few exceptions - VMAT2 [192], c-Rel [7]) have αS pathology. Therefore, αS pathology seems closely associated with impairment of protein degradation systems or overexpression of αS, but independent of mitochondrial dysfunction, ER stress and impaired UPR. Overexpression of αS is expected to lead to aggregation if it is a concentration-dependent process [100]; however, it is less clear how cell models based upon mitochondrial dysfunction or perturbation of ER stress lead to αS pathology [89,92].
Our recent cell biological studies may provide some important clues. We found that αS aggregates can be rapidly formed in cultured neurons undergoing apoptosis by interaction between αS and proaggregant nuclear factors (e.g., histones) associated with disruption of nuclear envelope integrity. The role of nuclear factors in cellular models of PD draws attention to the number of genetic risk factors that are associated proteins that are in the nuclear compartment (Table 1). In our cellular model, high molecular weight aggregates were observed at later stages of apoptosis [90], and eventually, the aggregates spread to adjacent cells. We postulate that this may be a mechanism for formation of abnormal αS seeds that eventually contributes to cell-to-cell propagation [90] (Figure 1). It is possible that the αS aggregates observed in cell models cause by mitochondrial dysfunction could be secondary to loss of nuclear membrane integrity associated with apoptosis [90]. In cell culture experiments, apoptotic neurons can be observed with live cell imaging, and the process can be followed over time, permitting detection of a range of intracellular αS aggregates at different stages of apoptosis. In the brain, apoptotic neurons are rapidly cleared by the innate immune system, which includes intrinsic tissue phagocytes. It is conceivable that in normal individuals, toxin-associated neuronal apoptosis and initial αS aggregates are rapidly cleared by phagocytosis, while in PD aggregates are inefficiently clearly and become seeds for subsequent propagation of αS pathology. There is evidence to suggest that phagocytic activity is significantly reduced in PD [169,74,184]. Moreover, clearance of apoptotic cells can be impaired under chronic inflammatory conditions [190], such as those reported in PD [55]. If this hypothesis can be proven, phagocytic deficits may contribute to pathogenesis of PD. Augmenting innate immunity may be a novel therapeutic target to limit initiation and spread of αS pathology. The animal models described in this review have not used this approach. It is intriguing to postulate that apoptosis-associated αS seeds might be more readily detected in rodent models if they also had deficiency in phagocytosis. We schematically illustrate in Figure 2 mechanisms that might contribute to such a hypothetical model of PD pathogenesis.
Figure 1. Schema for role of proaggregant nuclear factors in α-synuclein aggregation.
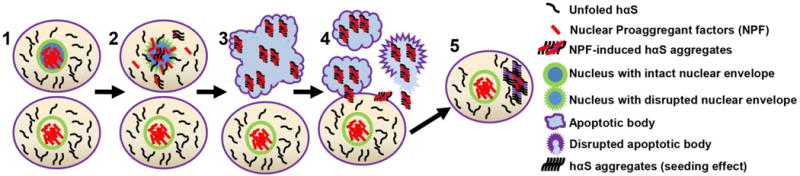
Filamentous αS aggregates can be rapidly formed in apoptotic neurons by interaction between αS and nuclear proaggregant factors associated with disruption of nuclear envelope. Such aggregates can be released from apoptotic bodies and taken up by surrounding healthy neurons as a potential seed for subsequent propagation.
Figure 2. Summary of cellular systems pathobiology in PD.

Schematic hypothesis that PD rodent models with impairment of the four cell functional systems in neurons may form filamentous αS aggregates as potential seeds for propagation of αS pathology. Neurons with mitochondrial impairment (pathway 1) and ER stress/UPR (pathway 2) do not induce αS aggregation if apoptosis is not triggered (I.a), while neurons with impairment of autophagy-lysosomal (pathway 3) or ubiquitin proteasome system (pathway 4) normally induce αS aggregation (I.b) even without triggering apoptosis. If the impairment persists, apoptosis will be eventually triggered. In normal subjects with properly functioning immune system, apoptotic neurons are rapidly cleared by phagocytes (II); however, in PD apoptotic neurons may remain to form filamentous αS aggregates (III). Subsequently, they are released and taken up by surrounding healthy neurons as a potential seed for subsequent propagation (IV).
The αS pathology observed in αS transgenic rodents varies in different models, and is also distinct from Lewy-related pathology in PD in terms of morphology, composition and biochemical properties. In addition to the models included in this review, there are a number of other αS transgenic rodent models that do not show observable PD features throughout their lives [126,52,88]. The explanation for differences between various models remains unclear, but could be related to unpredictable effects related to genomic integration site, epigenetic modifications and transgene copy numbers. Future transgenic models will likely increasingly exploit ROSA26 locus targeted strategies to avoid those potential pitfalls [33].
In summary, many rodent models have been developed to study PD using genetic or toxin based strategies, but all have significant limitations. None of the available experimental rodent models recapitulate all the major pathologic and behavioral phenotypes of PD. Considering the role that aging plays in etiology and pathogenesis of PD, models that take these factors into consideration may address some of these deficiencies. As one suggestion, strategies that combine available genetic or toxin based strategies combined with induction of phagocytosis deficits may be worth exploring in future models of PD in rodents.
Acknowledgments
This study was supported by the National Institute of Health (P50-NS072187 and R21-NS099757) and the Mangurian Foundation Lewy Body Dementia Program at Mayo Clinic (Dickson, Jiang).
Footnotes
Neither author has actual or potential conflicts of interest.
References
- 1.Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. New Engl J Med. 2004;351:1972–1977. doi: 10.1056/Nejmoa033277. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, Savitt JM. Development and characterization of a new Parkinson’s disease model resulting from impaired autophagy. J Neurosci. 2012;32:16503–16509. doi: 10.1523/JNEUROSCI.0209-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alafuzoff I, Ince PG, Arzberger T, Al-Sarraj S, Bell J, Bodi I, Bogdanovic N, Bugiani O, Ferrer I, Gelpi E, Gentleman S, Giaccone G, Ironside JW, Kavantzas N, King A, Korkolopoulou P, Kovacs GG, Meyronet D, Monoranu C, Parchi P, Parkkinen L, Patsouris E, Roggendorf W, Rozemuller A, Stadelmann-Nessler C, Streichenberger N, Thal DR, Kretzschmar H. Staging/typing of Lewy body related alpha-synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol. 2009;117:635–652. doi: 10.1007/s00401-009-0523-2. [DOI] [PubMed] [Google Scholar]
- 4.Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807–14812. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anglade P, Vyas S, JavoyAgid F, Herrero MT, Michel PP, Marquez J, MouattPrigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 6.Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev. 2013;2013:683920. doi: 10.1155/2013/683920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baiguera C, Alghisi M, Pinna A, Bellucci A, De Luca MA, Frau L, Morelli M, Ingrassia R, Benarese M, Porrini V, Pellitteri M, Bertini G, Fabene PF, Sigala S, Spillantini MG, Liou HC, Spano PF, Pizzi M. Late-onset Parkinsonism in NFkappaB/c-Rel-deficient mice. Brain. 2012;135:2750–2765. doi: 10.1093/brain/aws193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem. 1999;274:16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- 9.Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL, 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009;117:613–634. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beach TG, Adler CH, Serrano G, Sue LI, Walker DG, Dugger BN, Shill HA, Driver-Dunckley E, Caviness JN, Intorcia A, Filon J, Scott S, Garcia A, Hoffman B, Belden CM, Davis KJ, Sabbagh MN. Prevalence of Submandibular Gland Synucleinopathy in Parkinson’s Disease, Dementia with Lewy Bodies and other Lewy Body Disorders. J Parkinsons Dis. 2016;6:153–163. doi: 10.3233/JPD-150680. doi:10.3233/JPD-150680 JPD150680 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White CL, Iii, Akiyama H, Caviness JN, Shill HA, Sabbagh MN, Walker DG. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, Mee M, Soane T, Layfield R, Sheppard PW, Ebendal T, Usoskin D, Lowe J, Mayer RJ. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci. 2008;28:8189–8198. doi: 10.1523/JNEUROSCI.2218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beecham GW, Dickson DW, Scott WK, Martin ER, Schellenberg G, Nuytemans K, Larson EB, Buxbaum JD, Trojanowski JQ, Van Deerlin VM, Hurtig HI, Mash DC, Beach TG, Troncoso JC, Pletnikova O, Frosch MP, Ghetti B, Foroud TM, Honig LS, Marder K, Vonsattel JP, Goldman SM, Vinters HV, Ross OA, Wszolek ZK, Wang L, Dykxhoorn DM, Pericak-Vance MA, Montine TJ, Leverenz JB, Dawson TM, Vance JM. PARK10 is a major locus for sporadic neuropathologically confirmed Parkinson disease. Neurology. 2015;84:972–980. doi: 10.1212/WNL.0000000000001332. doi:10.1212/WNL.0000000000001332WNL.0000000000001332 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belal C, Ameli NJ, El Kommos A, Bezalel S, Al’Khafaji AM, Mughal MR, Mattson MP, Kyriazis GA, Tyrberg B, Chan SL. The homocysteine-inducible endoplasmic reticulum (ER) stress protein Herp counteracts mutant alpha-synuclein-induced ER stress via the homeostatic regulation of ER-resident calcium release channel proteins. Hum Mol Genet. 2012;21:963–977. doi: 10.1093/hmg/ddr502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of alpha-synuclein by proteasome. J Biol Chem. 1999;274:33855–33858. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- 16.Bentea E, Van der Perren A, Van Liefferinge J, El Arfani A, Albertini G, Demuyser T, Merckx E, Michotte Y, Smolders I, Baekelandt V, Massie A. Nigral proteasome inhibition in mice leads to motor and non-motor deficits and increased expression of Ser129 phosphorylated alpha-synuclein. Front Behav Neurosci. 2015;9:68. doi: 10.3389/fnbeh.2015.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharjee N, Borah A. Oxidative stress and mitochondrial dysfunction are the underlying events of dopaminergic neurodegeneration in homocysteine rat model of Parkinson’s disease. Neurochem Int. 2016;101:48–55. doi: 10.1016/j.neuint.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Blandini F, Armentero MT, Martignoni E. The 6-hydroxydopamine model: news from the past. Parkinsonism Relat Disord. 2008;14(Suppl 2):S124–129. doi: 10.1016/j.parkreldis.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 20.Bove J, Zhou C, Jackson-Lewis V, Taylor J, Chu Y, Rideout HJ, Wu DC, Kordower JH, Petrucelli L, Przedborski S. Proteasome inhibition and Parkinson’s disease modeling. Ann Neurol. 2006;60:260–264. doi: 10.1002/ana.20937. [DOI] [PubMed] [Google Scholar]
- 21.Braak H, Bohl JR, Muller CM, Rub U, de Vos RA, Del Tredici K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Del Tredici K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J Parkinsons Dis. 2017;7:S73–S87. doi: 10.3233/JPD-179001. doi:10.3233/JPD-179001 JPD179001 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. doi:S0197458002000659 [pii] [DOI] [PubMed] [Google Scholar]
- 24.Brooks AI, Chadwick CA, Gelbard HA, Cory-Slechta DA, Federoff HJ. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res. 1999;823:1–10. doi: 10.1016/s0006-8993(98)01192-5. [DOI] [PubMed] [Google Scholar]
- 25.Bukhatwa S, Zeng BY, Rose S, Jenner P. A comparison of changes in proteasomal subunit expression in the substantia nigra in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy. Brain Res. 2010;1326:174–183. doi: 10.1016/j.brainres.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 26.Casadei N, Sood P, Ulrich T, Fallier-Becker P, Kieper N, Helling S, May C, Glaab E, Chen J, Nuber S, Wolburg H, Marcus K, Rapaport D, Ott T, Riess O, Kruger R, Fitzgerald JC. Mitochondrial defects and neurodegeneration in mice overexpressing wild-type or G399S mutant HtrA2. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw353. [DOI] [PubMed] [Google Scholar]
- 27.Casey DE. The relationship of pharmacology to side effects. J Clin Psychiat. 1997;58:55–62. [PubMed] [Google Scholar]
- 28.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandran JS, Lin X, Zapata A, Hoke A, Shimoji M, Moore SO, Galloway MP, Laird FM, Wong PC, Price DL, Bailey KR, Crawley JN, Shippenberg T, Cai H. Progressive behavioral deficits in DJ-1-deficient mice are associated with normal nigrostriatal function. Neurobiol Dis. 2008;29:505–514. doi: 10.1016/j.nbd.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Cagniard B, Mathews T, Jones S, Koh HC, Ding Y, Carvey PM, Ling Z, Kang UJ, Zhuang X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280:21418–21426. doi: 10.1074/jbc.M413955200. [DOI] [PubMed] [Google Scholar]
- 31.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 32.Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR. A progressive mouse model of Parkinson’s disease: the Thy1-aSyn (“Line 61”) mice. Neurotherapeutics. 2012;9:297–314. doi: 10.1007/s13311-012-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu VT, Weber T, Graf R, Sommermann T, Petsch K, Sack U, Volchkov P, Rajewsky K, Kuhn R. Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol. 2016;16:4. doi: 10.1186/s12896-016-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cicchetti F, Drouin-Ouellet J, Gross RE. Environmental toxins and Parkinson’s disease: what have we learned from pesticide-induced animal models? Trends Pharmacol Sci. 2009;30:475–483. doi: 10.1016/j.tips.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Conn KJ, Gao W, McKee A, Lan MS, Ullman MD, Eisenhauer PB, Fine RE, Wells JM. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004;1022:164–172. doi: 10.1016/j.brainres.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 36.Coppola-Segovia V, Cavarsan C, Maia FG, Ferraz AC, Nakao LS, Lima MM, Zanata SM. ER Stress Induced by Tunicamycin Triggers alpha-Synuclein Oligomerization, Dopaminergic Neurons Death and Locomotor Impairment: a New Model of Parkinson’s Disease. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-0114-x. [DOI] [PubMed] [Google Scholar]
- 37.Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, Boston H, Saftig P, Woulfe J, Feany MB, Myllykangas L, Schlossmacher MG, Tyynela J. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2009;2:5. doi: 10.1186/1756-6606-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daher JP, Ying M, Banerjee R, McDonald RS, Hahn MD, Yang L, Flint Beal M, Thomas B, Dawson VL, Dawson TM, Moore DJ. Conditional transgenic mice expressing C-terminally truncated human alpha-synuclein (alphaSyn119) exhibit reduced striatal dopamine without loss of nigrostriatal pathway dopaminergic neurons. Mol Neurodegener. 2009;4:34. doi: 10.1186/1750-1326-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dave KD, De Silva S, Sheth NP, Ramboz S, Beck MJ, Quang C, Switzer RC, 3rd, Ahmad SO, Sunkin SM, Walker D, Cui X, Fisher DA, McCoy AM, Gamber K, Ding X, Goldberg MS, Benkovic SA, Haupt M, Baptista MA, Fiske BK, Sherer TB, Frasier MA. Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol Dis. 2014;70:190–203. doi: 10.1016/j.nbd.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 40.Dawson TM, Dawson VL. Rare genetic mutations shed light on the pathogenesis of Parkinson disease. J Clin Invest. 2003;111:145–151. doi: 10.1172/JCI17575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dawson TM, Dawson VL. The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S32–39. doi: 10.1002/mds.22798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Decressac M, Mattsson B, Lundblad M, Weikop P, Bjorklund A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol Dis. 2012;45:939–953. doi: 10.1016/j.nbd.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic Lysosomal Depletion in Parkinson’s Disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/Jneurosci.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, Bezard E. Lysosomal impairment in Parkinson’s disease. Mov Disord. 2013;28:725–732. doi: 10.1002/mds.25462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dickson DW. Neuropathology of Parkinson disease. Parkinsonism Relat Disord. 2017 doi: 10.1016/j.parkreldis.2017.07.033. doi:S1353-8020(17)30280-8 [pii] 10.1016/j.parkreldis.2017.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Domico LM, Zeevalk GD, Bernard LP, Cooper KR. Acute neurotoxic effects of mancozeb and maneb in mesencephalic neuronal cultures are associated with mitochondrial dysfunction. Neurotoxicology. 2006;27:816–825. doi: 10.1016/j.neuro.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 47.Duan W, Ladenheim B, Cutler RG, Kruman II, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J Neurochem. 2002;80:101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- 48.Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjorn M, Kuss M, Liu Y, Porcu G, Kolkova K, Friis Rundsten C, Ruscher K, Pakkenberg B, Goldmann T, Loreth D, Prinz M, Rubinsztein DC, Issazadeh-Navikas S. Lack of Neuronal IFN-beta-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia. Cell. 2015;163:324–339. doi: 10.1016/j.cell.2015.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emmer KL, Waxman EA, Covy JP, Giasson BI. E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem. 2011;286:35104–35118. doi: 10.1074/jbc.M111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Enquist IB, Lo Bianco C, Ooka A, Nilsson E, Mansson JE, Ehinger M, Richter J, Brady RO, Kirik D, Karlsson S. Murine models of acute neuronopathic Gaucher disease. Proc Natl Acad Sci U S A. 2007;104:17483–17488. doi: 10.1073/pnas.0708086104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Escobar VD, Kuo YM, Orrison BM, Giasson BI, Nussbaum RL. Transgenic mice expressing S129 phosphorylation mutations in alpha-synuclein. Neurosci Lett. 2014;563:96–100. doi: 10.1016/j.neulet.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 54.Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse. 2007;61:991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferrari CC, Tarelli R. Parkinson’s disease and systemic inflammation. Parkinsons Dis. 2011;2011:436813. doi: 10.4061/2011/436813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fifel K, Cooper HM. Loss of dopamine disrupts circadian rhythms in a mouse model of Parkinson’s disease. Neurobiol Dis. 2014;71:359–369. doi: 10.1016/j.nbd.2014.08.024. [DOI] [PubMed] [Google Scholar]
- 57.Fitsanakis VA, Amarnath V, Moore JT, Montine KS, Zhang J, Montine TJ. Catalysis of catechol oxidation by metal-dithiocarbamate complexes in pesticides. Free Radic Biol Med. 2002;33:1714–1723. doi: 10.1016/s0891-5849(02)01169-3. [DOI] [PubMed] [Google Scholar]
- 58.Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Sudhof TC. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Forno LS, DeLanney LE, Irwin I, Langston JW. Similarities and differences between MPTP-induced parkinsonsim and Parkinson’s disease. Neuropathologic considerations. Adv Neurol. 1993;60:600–608. [PubMed] [Google Scholar]
- 60.Franco-Iborra S, Vila M, Perier C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We at? Neuroscientist. 2016;22:266–277. doi: 10.1177/1073858415574600. [DOI] [PubMed] [Google Scholar]
- 61.Gash DM, Rutland K, Hudson NL, Sullivan PG, Bing G, Cass WA, Pandya JD, Liu M, Choi DY, Hunter RL, Gerhardt GA, Smith CD, Slevin JT, Prince TS. Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann Neurol. 2008;63:184–192. doi: 10.1002/ana.21288. [DOI] [PubMed] [Google Scholar]
- 62.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 63.Gispert S, Del Turco D, Garrett L, Chen A, Bernard DJ, Hamm-Clement J, Korf HW, Deller T, Braak H, Auburger G, Nussbaum RL. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci. 2003;24:419–429. doi: 10.1016/s1044-7431(03)00198-2. [DOI] [PubMed] [Google Scholar]
- 64.Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, Voos W, Leuner K, Muller WE, Kudin AP, Kunz WS, Zimmermann A, Roeper J, Wenzel D, Jendrach M, Garcia-Arencibia M, Fernandez-Ruiz J, Huber L, Rohrer H, Barrera M, Reichert AS, Rub U, Chen A, Nussbaum RL, Auburger G. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One. 2009;4:e5777. doi: 10.1371/journal.pone.0005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glasl L, Kloos K, Giesert F, Roethig A, Di Benedetto B, Kuhn R, Zhang J, Hafen U, Zerle J, Hofmann A, de Angelis MH, Winklhofer KF, Holter SM, Vogt Weisenhorn DM, Wurst W. Pink1-deficiency in mice impairs gait, olfaction and serotonergic innervation of the olfactory bulb. Exp Neurol. 2012;235:214–227. doi: 10.1016/j.expneurol.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 66.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 67.Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 68.Goldman SM. Environmental toxins and Parkinson’s disease. Annu Rev Pharmacol Toxicol. 2014;54:141–164. doi: 10.1146/annurev-pharmtox-011613-135937. [DOI] [PubMed] [Google Scholar]
- 69.Goldman SM, Quinlan PJ, Ross GW, Marras C, Meng C, Bhudhikanok GS, Comyns K, Korell M, Chade AR, Kasten M, Priestley B, Chou KL, Fernandez HH, Cambi F, Langston JW, Tanner CM. Solvent exposures and Parkinson disease risk in twins. Ann Neurol. 2012;71:776–784. doi: 10.1002/ana.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gomez-Isla T, Irizarry MC, Mariash A, Cheung B, Soto O, Schrump S, Sondel J, Kotilinek L, Day J, Schwarzschild MA, Cha JH, Newell K, Miller DW, Ueda K, Young AB, Hyman BT, Ashe KH. Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2003;24:245–258. doi: 10.1016/s0197-4580(02)00091-x. [DOI] [PubMed] [Google Scholar]
- 71.Gomez-Suaga P, Fdez E, Blanca Ramirez M, Hilfiker S. A Link between Autophagy and the Pathophysiology of LRRK2 in Parkinson’s Disease. Parkinsons Dis. 2012;2012:324521. doi: 10.1155/2012/324521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gonzalez-Reyes LE, Verbitsky M, Blesa J, Jackson-Lewis V, Paredes D, Tillack K, Phani S, Kramer ER, Przedborski S, Kottmann AH. Sonic hedgehog maintains cellular and neurochemical homeostasis in the adult nigrostriatal circuit. Neuron. 2012;75:306–319. doi: 10.1016/j.neuron.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Good CH, Hoffman AF, Hoffer BJ, Chefer VI, Shippenberg TS, Backman CM, Larsson NG, Olson L, Gellhaar S, Galter D, Lupica CR. Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson’s disease. Faseb J. 2011;25:1333–1344. doi: 10.1096/fj.10-173625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grozdanov V, Bliederhaeuser C, Ruf WP, Roth V, Fundel-Clemens K, Zondler L, Brenner D, Martin-Villalba A, Hengerer B, Kassubek J, Ludolph AC, Weishaupt JH, Danzer KM. Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol. 2014;128:651–663. doi: 10.1007/s00401-014-1345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gu G, Reyes PE, Golden GT, Woltjer RL, Hulette C, Montine TJ, Zhang J. Mitochondrial DNA deletions/rearrangements in parkinson disease and related neurodegenerative disorders. J Neuropathol Exp Neurol. 2002;61:634–639. doi: 10.1093/jnen/61.7.634. [DOI] [PubMed] [Google Scholar]
- 76.Guehl D, Bezard E, Dovero S, Boraud T, Bioulac B, Gross C. Trichloroethylene and parkinsonism: a human and experimental observation. Eur J Neurol. 1999;6:609–611. doi: 10.1046/j.1468-1331.1999.650609.x. doi:NE060513 [pii] [DOI] [PubMed] [Google Scholar]
- 77.Gully JC, Sergeyev VG, Bhootada Y, Mendez-Gomez H, Meyers CA, Zolotukhin S, Gorbatyuk MS, Gorbatyuk OS. Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci Lett. 2016;627:36–41. doi: 10.1016/j.neulet.2016.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hall K, Yang S, Sauchanka O, Spillantini MG, Anichtchik O. Behavioural deficits in transgenic mice expressing human truncated (1-120 amino acid) alpha-synuclein. Exp Neurol. 2015;264:8–13. doi: 10.1016/j.expneurol.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 79.Hashida K, Kitao Y, Sudo H, Awa Y, Maeda S, Mori K, Takahashi R, Iinuma M, Hori O. ATF6alpha promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson’s disease. PLoS One. 2012;7:e47950. doi: 10.1371/journal.pone.0047950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 81.Howell N, Elson JL, Chinnery PF, Turnbull DM. mtDNA mutations and common neurodegenerative disorders. Trends Genet. 2005;21:583–586. doi: 10.1016/j.tig.2005.08.012. doi:S0168-9525(05)00255-6 [pii] 10.1016/j.tig.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 82.Hwang DY, Ardayfio P, Kang UJ, Semina EV, Kim KS. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Brain Res Mol Brain Res. 2003;114:123–131. doi: 10.1016/s0169-328x(03)00162-1. [DOI] [PubMed] [Google Scholar]
- 83.Iglesias-Gonzalez J, Sanchez-Iglesias S, Mendez-Alvarez E, Rose S, Hikima A, Jenner P, Soto-Otero R. Differential Toxicity of 6-Hydroxydopamine in SH-SY5Y Human Neuroblastoma Cells and Rat Brain Mitochondria: Protective Role of Catalase and Superoxide Dismutase. Neurochem Res. 2012;37:2150–2160. doi: 10.1007/s11064-012-0838-6. [DOI] [PubMed] [Google Scholar]
- 84.Ikeda M, Kawarabayashi T, Harigaya Y, Sasaki A, Yamada S, Matsubara E, Murakami T, Tanaka Y, Kurata T, Wuhua X, Ueda K, Kuribara H, Ikarashi Y, Nakazato Y, Okamoto K, Abe K, Shoji M. Motor impairment and aberrant production of neurochemicals in human alpha-synuclein A30P+A53T transgenic mice with alpha-synuclein pathology. Brain Res. 2009;1250:232–241. doi: 10.1016/j.brainres.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 85.Ikemura M, Saito Y, Sengoku R, Sakiyama Y, Hatsuta H, Kanemaru K, Sawabe M, Arai T, Ito G, Iwatsubo T, Fukayama M, Murayama S. Lewy body pathology involves cutaneous nerves. J Neuropathol Exp Neurol. 2008;67:945–953. doi: 10.1097/NEN.0b013e318186de48. [DOI] [PubMed] [Google Scholar]
- 86.Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B, Deltheil T, Kosillo P, Cioroch M, Wagner K, Ansorge O, Bannerman DM, Bolam JP, Magill PJ, Cragg SJ, Wade-Martins R. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A. 2013;110:E4016–4025. doi: 10.1073/pnas.1309143110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jellinger K, Linert L, Kienzl E, Herlinger E, Youdim MB. Chemical evidence for 6-hydroxydopamine to be an endogenous toxic factor in the pathogenesis of Parkinson’s disease. J Neural Transm Suppl. 1995;46:297–314. [PubMed] [Google Scholar]
- 88.Jiang P, Gan M, Ebrahim AS, Lin WL, Melrose HL, Yen SH. ER stress response plays an important role in aggregation of alpha-synuclein. Mol Neurodegener. 2010;5:56. doi: 10.1186/1750-1326-5-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jiang P, Gan M, Lin WL, Yen SH. Nutrient deprivation induces alpha-synuclein aggregation through endoplasmic reticulum stress response and SREBP2 pathway. Front Aging Neurosci. 2014;6:268. doi: 10.3389/fnagi.2014.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang P, Gan M, Yen SH, Moussaud S, McLean PJ, Dickson DW. Proaggregant nuclear factor(s) trigger rapid formation of alpha-synuclein aggregates in apoptotic neurons. Acta Neuropathol. 2016;132:77–91. doi: 10.1007/s00401-016-1542-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kadkhodaei B, Ito T, Joodmardi E, Mattsson B, Rouillard C, Carta M, Muramatsu S, Sumi-Ichinose C, Nomura T, Metzger D, Chambon P, Lindqvist E, Larsson NG, Olson L, Bjorklund A, Ichinose H, Perlmann T. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci. 2009;29:15923–15932. doi: 10.1523/JNEUROSCI.3910-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kalivendi SV, Cunningham S, Kotamraju S, Joseph J, Hillard CJ, Kalyanaraman B. Alpha-synuclein up-regulation and aggregation during MPP+-induced apoptosis in neuroblastoma cells: intermediacy of transferrin receptor iron and hydrogen peroxide. J Biol Chem. 2004;279:15240–15247. doi: 10.1074/jbc.M312497200. [DOI] [PubMed] [Google Scholar]
- 93.Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kett LR, Stiller B, Bernath MM, Tasset I, Blesa J, Jackson-Lewis V, Chan RB, Zhou B, Di Paolo G, Przedborski S, Cuervo AM, Dauer WT. alpha-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci. 2015;35:5724–5742. doi: 10.1523/JNEUROSCI.0632-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim HW, Choi WS, Sorscher N, Park HJ, Tronche F, Palmiter RD, Xia Z. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiol Aging. 2015;36:2617–2627. doi: 10.1016/j.neurobiolaging.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Bjorklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. doi:20026246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Krige D, Carroll MT, Cooper JM, Marsden CD, Schapira AH. Platelet mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann Neurol. 1992;32:782–788. doi: 10.1002/ana.410320612. [DOI] [PubMed] [Google Scholar]
- 100.Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J, Randolph TW, Narhi LO, Biere AL, Citron M, Carpenter JF. Oxidative dimer formation is the critical rate-limiting step for Parkinson’s disease alpha-synuclein fibrillogenesis. Biochemistry. 2003;42:829–837. doi: 10.1021/bi026528t. [DOI] [PubMed] [Google Scholar]
- 101.Kuhn W, Roebroek R, Blom H, van Oppenraaij D, Przuntek H, Kretschmer A, Buttner T, Woitalla D, Muller T. Elevated plasma levels of homocysteine in Parkinson’s disease. Eur Neurol. 1998;40:225–227. doi: 10.1159/000007984. [DOI] [PubMed] [Google Scholar]
- 102.Kupsch A, Schmidt W, Gizatullina Z, Debska-Vielhaber G, Voges J, Striggow F, Panther P, Schwegler H, Heinze HJ, Vielhaber S, Gellerich FN. 6-Hydroxydopamine impairs mitochondrial function in the rat model of Parkinson’s disease: respirometric, histological, and behavioral analyses. J Neural Transm (Vienna) 2014;121:1245–1257. doi: 10.1007/s00702-014-1185-3. [DOI] [PubMed] [Google Scholar]
- 103.Langston JW. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann Neurol. 2006;59:591–596. doi: 10.1002/ana.20834. [DOI] [PubMed] [Google Scholar]
- 104.Langston JW, Langston EB, Irwin I. MPTP-induced parkinsonism in human and non-human primates–clinical and experimental aspects. Acta Neurol Scand Suppl. 1984;100:49–54. [PubMed] [Google Scholar]
- 105.Lauwers E, Beque D, Van Laere K, Nuyts J, Bormans G, Mortelmans L, Casteels C, Vercammen L, Bockstael O, Nuttin B, Debyser Z, Baekelandt V. Non-invasive imaging of neuropathology in a rat model of alpha-synuclein overexpression. Neurobiol Aging. 2007;28:248–257. doi: 10.1016/j.neurobiolaging.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 106.Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 –> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 108.Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, Schjeide LM, Meissner E, Zauft U, Allen NC, Liu T, Schilling M, Anderson KJ, Beecham G, Berg D, Biernacka JM, Brice A, DeStefano AL, Do CB, Eriksson N, Factor SA, Farrer MJ, Foroud T, Gasser T, Hamza T, Hardy JA, Heutink P, Hill-Burns EM, Klein C, Latourelle JC, Maraganore DM, Martin ER, Martinez M, Myers RH, Nalls MA, Pankratz N, Payami H, Satake W, Scott WK, Sharma M, Singleton AB, Stefansson K, Toda T, Tung JY, Vance J, Wood NW, Zabetian CP, Young P, Tanzi RE, Khoury MJ, Zipp F, Lehrach H, Ioannidis JP, Bertram L. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012;8:e1002548. doi: 10.1371/journal.pgen.1002548. doi:10.1371/journal.pgen.1002548 PGENETICS-D-11-02212 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lim KL. Ubiquitin-proteasome system dysfunction in Parkinson’s disease: current evidence and controversies. Expert Rev Proteomics. 2007;4:769–781. doi: 10.1586/14789450.4.6.769. [DOI] [PubMed] [Google Scholar]
- 111.Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, Ding J, Mateo Y, Sullivan PH, Wu LG, Goldstein DS, Lovinger D, Cai H. Conditional expression of Parkinson’s disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32:9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu M, Choi DY, Hunter RL, Pandya JD, Cass WA, Sullivan PG, Kim HC, Gash DM, Bing G. Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats. J Neurochem. 2010;112:773–783. doi: 10.1111/j.1471-4159.2009.06497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lorenc-Koci E, Lenda T, Antkiewicz-Michaluk L, Wardas J, Domin H, Smialowska M, Konieczny J. Different effects of intranigral and intrastriatal administration of the proteasome inhibitor lactacystin on typical neurochemical and histological markers of Parkinson’s disease in rats. Neurochem Int. 2011;58:839–849. doi: 10.1016/j.neuint.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 114.Lu XH, Fleming SM, Meurers B, Ackerson LC, Mortazavi F, Lo V, Hernandez D, Sulzer D, Jackson GR, Maidment NT, Chesselet MF, Yang XW. Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J Neurosci. 2009;29:1962–1976. doi: 10.1523/JNEUROSCI.5351-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Luk KC, Lee VM. Modeling Lewy pathology propagation in Parkinson’s disease. Parkinsonism Related Disord. 2014;20(Suppl 1):S85–87. doi: 10.1016/S1353-8020(13)70022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009357. doi: 10.1101/cshperspect.a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Magnoni R, Palmfeldt J, Christensen JH, Sand M, Maltecca F, Corydon TJ, West M, Casari G, Bross P. Late onset motoneuron disorder caused by mitochondrial Hsp60 chaperone deficiency in mice. Neurobiol Dis. 2013;54:12–23. doi: 10.1016/j.nbd.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 119.Manning-Bog AB, Caudle WM, Perez XA, Reaney SH, Paletzki R, Isla MZ, Chou VP, McCormack AL, Miller GW, Langston JW, Gerfen CR, Dimonte DA. Increased vulnerability of nigrostriatal terminals in DJ-1-deficient mice is mediated by the dopamine transporter. Neurobiol Dis. 2007;27:141–150. doi: 10.1016/j.nbd.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 120.Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- 121.Manzoni C. LRRK2 and autophagy: a common pathway for disease. Biochem Soc Trans. 2012;40:1147–1151. doi: 10.1042/BST20120126. [DOI] [PubMed] [Google Scholar]
- 122.Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Kruger R, Hattori N, Mellick GD, Quattrone A, Satoh J, Toda T, Wang J, Ioannidis JP, de Andrade M, Rocca WA. UCHL1 is a Parkinson’s disease susceptibility gene. Ann Neurol. 2004;55:512–521. doi: 10.1002/ana.20017. [DOI] [PubMed] [Google Scholar]
- 123.Martella G, Platania P, Vita D, Sciamanna G, Cuomo D, Tassone A, Tscherter A, Kitada T, Bonsi P, Shen J, Pisani A. Enhanced sensitivity to group II mGlu receptor activation at corticostriatal synapses in mice lacking the familial parkinsonism-linked genes PINK1 or Parkin. Exp Neurol. 2009;215:388–396. doi: 10.1016/j.expneurol.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Martinez TN, Greenamyre JT. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid Redox Signal. 2012;16:920–934. doi: 10.1089/ars.2011.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 126.Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- 127.McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 128.McDowell K, Chesselet MF. Animal models of the non-motor features of Parkinson’s disease. Neurobiol Dis. 2012;46:597–606. doi: 10.1016/j.nbd.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McNaught KS, Belizaire R, Jenner P, Olanow CW, Isacson O. Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci Lett. 2002;326:155–158. doi: 10.1016/s0304-3940(02)00296-3. [DOI] [PubMed] [Google Scholar]
- 130.McNaught KS, Jackson T, JnoBaptiste R, Kapustin A, Olanow CW. Proteasomal dysfunction in sporadic Parkinson’s disease. Neurology. 2006;66:S37–49. doi: 10.1212/wnl.66.10_suppl_4.s37. [DOI] [PubMed] [Google Scholar]
- 131.McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- 132.McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol. 2004;56:149–162. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- 133.Mercado G, Valdes P, Hetz C. An ERcentric view of Parkinson’s disease. Trends Mol Med. 2013;19:165–175. doi: 10.1016/j.molmed.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 134.Meredith GE, Rademacher DJ. MPTP mouse models of Parkinson’s disease: an update. J Parkinsons Dis. 2011;1:19–33. doi: 10.3233/JPD-2011-11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Miller GW. Paraquat: the red herring of Parkinson’s disease research. Toxicol Sci. 2007;100:1–2. doi: 10.1093/toxsci/kfm223. [DOI] [PubMed] [Google Scholar]
- 136.Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun. 1989;163:1450–1455. doi: 10.1016/0006-291x(89)91141-8. [DOI] [PubMed] [Google Scholar]
- 137.Moisoi N, Fedele V, Edwards J, Martins LM. Loss of PINK1 enhances neurodegeneration in a mouse model of Parkinson’s disease triggered by mitochondrial stress. Neuropharmacology. 2014;77:350–357. doi: 10.1016/j.neuropharm.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moon HE, Paek SH. Mitochondrial Dysfunction in Parkinson’s Disease. Exp Neurobiol. 2015;24:103–116. doi: 10.5607/en.2015.24.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Moscovitz O, Ben-Nissan G, Fainer I, Pollack D, Mizrachi L, Sharon M. The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nat Commun. 2015;6:6609. doi: 10.1038/ncomms7609. [DOI] [PubMed] [Google Scholar]
- 140.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–993. doi: 10.1038/ng.3043. doi:10.1038/ng.3043 ng.3043 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nordstrom U, Beauvais G, Ghosh A, Pulikkaparambil Sasidharan BC, Lundblad M, Fuchs J, Joshi RL, Lipton JW, Roholt A, Medicetty S, Feinstein TN, Steiner JA, Escobar Galvis ML, Prochiantz A, Brundin P. Progressive nigrostriatal terminal dysfunction and degeneration in the engrailed1 heterozygous mouse model of Parkinson’s disease. Neurobiol Dis. 2015;73:70–82. doi: 10.1016/j.nbd.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. New Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. doi:10.1056/NEJM2003ra020003 348/14/1356 [pii] [DOI] [PubMed] [Google Scholar]
- 144.Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, Burn D, Halliday GM, Bezard E, Przedborski S, Lehericy S, Brooks DJ, Rothwell JC, Hallett M, DeLong MR, Marras C, Tanner CM, Ross GW, Langston JW, Klein C, Bonifati V, Jankovic J, Lozano AM, Deuschl G, Bergman H, Tolosa E, Rodriguez-Violante M, Fahn S, Postuma RB, Berg D, Marek K, Standaert DG, Surmeier DJ, Olanow CW, Kordower JH, Calabresi P, Schapira AHV, Stoessl AJ. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 2017;32:1264–1310. doi: 10.1002/mds.27115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, Waddington SN, Schapira AH, Duchen MR. Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson’s disease. Cell Metab. 2013;17:941–953. doi: 10.1016/j.cmet.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Paine SM, Anderson G, Bedford K, Lawler K, Mayer RJ, Lowe J, Bedford L. Pale body-like inclusion formation and neurodegeneration following depletion of 26S proteasomes in mouse brain neurones are independent of alpha-synuclein. PLoS One. 2013;8:e54711. doi: 10.1371/journal.pone.0054711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Palmeira CM, Moreno AJ, Madeira VMC. Mitochondrial Bioenergetics Is Affected by the Herbicide Paraquat. Bba-Bioenergetics. 1995;1229:187–192. doi: 10.1016/0005-2728(94)00202-G. [DOI] [PubMed] [Google Scholar]
- 148.Palomo-Garo C, Gomez-Galvez Y, Garcia C, Fernandez-Ruiz J. Targeting the cannabinoid CB2 receptor to attenuate the progression of motor deficits in LRRK2-transgenic mice. Pharmacol Res. 2016;110:181–192. doi: 10.1016/j.phrs.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 149.Pan TH, Kondo S, Le WD, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- 150.Park JS, Blair NF, Sue CM. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov Disord. 2015;30:770–779. doi: 10.1002/mds.26243. [DOI] [PubMed] [Google Scholar]
- 151.Parker WD, Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 152.Patterson VL, Zullo AJ, Koenig C, Stoessel S, Jo H, Liu X, Han J, Choi M, DeWan AT, Thomas JL, Kuan CY, Hoh J. Neural-specific deletion of Htra2 causes cerebellar neurodegeneration and defective processing of mitochondrial OPA1. PLoS One. 2014;9:e115789. doi: 10.1371/journal.pone.0115789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Perier C, Vila M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009332. doi: 10.1101/cshperspect.a009332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pfeiffer RF. Non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord. 2016;22(Suppl 1):S119–122. doi: 10.1016/j.parkreldis.2015.09.004. doi:10.1016/j.parkreldis.2015.09.004 S1353-8020(15)00379-X [pii] [DOI] [PubMed] [Google Scholar]
- 156.Pickrell AM, Pinto M, Hida A, Moraes CT. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci. 2011;31:17649–17658. doi: 10.1523/JNEUROSCI.4871-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52:183–191. doi: 10.1097/00005072-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 158.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 159.Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson’s disease. Mov Disord. 2012;27:831–842. doi: 10.1002/mds.24962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Przedborski S, Levivier M, Jiang H, Ferreira M, Jackson-Lewis V, Donaldson D, Togasaki DM. Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience. 1995;67:631–647. doi: 10.1016/0306-4522(95)00066-r. [DOI] [PubMed] [Google Scholar]
- 161.Puschmann A, Fiesel FC, Caulfield TR, Hudec R, Ando M, Truban D, Hou X, Ogaki K, Heckman MG, James ED, Swanberg M, Jimenez-Ferrer I, Hansson O, Opala G, Siuda J, Boczarska-Jedynak M, Friedman A, Koziorowski D, Aasly JO, Lynch T, Mellick GD, Mohan M, Silburn PA, Sanotsky Y, Vilarino-Guell C, Farrer MJ, Chen L, Dawson VL, Dawson TM, Wszolek ZK, Ross OA, Springer W. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain. 2017;140:98–117. doi: 10.1093/brain/aww261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, Xie ZL, Speake LD, Parks R, Crabtree D, Liang Q, Crimmins S, Schneider L, Uchiyama Y, Iwatsubo T, Zhou Y, Peng L, Lu Y, Standaert DG, Walls KC, Shacka JJ, Roth KA, Zhang J. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 164.Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL, Moore DJ. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Richardson JR, Quan Y, Sherer TB, Greenamyre JT, Miller GW. Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci. 2005;88:193–201. doi: 10.1093/toxsci/kfi304. [DOI] [PubMed] [Google Scholar]
- 166.Richter G, Sonnenschein A, Grunewald T, Reichmann H, Janetzky B. Novel mitochondrial DNA mutations in Parkinson’s disease. J Neural Transm (Vienna) 2002;109:721–729. doi: 10.1007/s007020200060. [DOI] [PubMed] [Google Scholar]
- 167.Rocha EM, Smith GA, Park E, Cao H, Graham AR, Brown E, McLean JR, Hayes MA, Beagan J, Izen SC, Perez-Torres E, Hallett PJ, Isacson O. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain alpha-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid Redox Signal. 2015;23:550–564. doi: 10.1089/ars.2015.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Salganik M, Sergeyev VG, Shinde V, Meyers CA, Gorbatyuk MS, Lin JH, Zolotukhin S, Gorbatyuk OS. The loss of glucose-regulated protein 78 (GRP78) during normal aging or from siRNA knockdown augments human alpha-synuclein (alpha-syn) toxicity to rat nigral neurons. Neurobiol Aging. 2015;36:2213–2223. doi: 10.1016/j.neurobiolaging.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Salman H, Bergman M, Djaldetti R, Bessler H, Djaldetti M. Decreased phagocytic function in patients with Parkinson’s disease. Biomed Pharmacother. 1999;53:146–148. doi: 10.1016/S0753-3322(99)80080-8. [DOI] [PubMed] [Google Scholar]
- 170.Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- 171.Schapira AH. Glucocerebrosidase and Parkinson disease: Recent advances. Mol Cell Neurosci. 2015;66:37–42. doi: 10.1016/j.mcn.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schapira AH, Cleeter MW, Muddle JR, Workman JM, Cooper JM, King RH. Proteasomal inhibition causes loss of nigral tyrosine hydroxylase neurons. Ann Neurol. 2006;60:253–255. doi: 10.1002/ana.20934. [DOI] [PubMed] [Google Scholar]
- 173.Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 174.Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci. 2017;18:435–450. doi: 10.1038/nrn.2017.62. doi:10.1038/nrn.2017.62 nrn.2017.62 [pii] [DOI] [PubMed] [Google Scholar]
- 175.Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L, Singh BB. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest. 2012;122:1354–1367. doi: 10.1172/JCI61332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Setsuie R, Wang YL, Mochizuki H, Osaka H, Hayakawa H, Ichihara N, Li H, Furuta A, Sano Y, Sun YJ, Kwon J, Kabuta T, Yoshimi K, Aoki S, Mizuno Y, Noda M, Wada K. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem Int. 2007;50:119–129. doi: 10.1016/j.neuint.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 177.Sevlever D, Jiang P, Yen SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry. 2008;47:9678–9687. doi: 10.1021/bi800699v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shi CH, Mao CY, Zhang SY, Yang J, Song B, Wu P, Zuo CT, Liu YT, Ji Y, Yang ZH, Wu J, Zhuang ZP, Xu YM. CHCHD2 gene mutations in familial and sporadic Parkinson’s disease. Neurobiol Aging. 2016;38:217 e219–213. doi: 10.1016/j.neurobiolaging.2015.10.040. [DOI] [PubMed] [Google Scholar]
- 179.Shimshek DR, Schweizer T, Schmid P, van der Putten PH. Excess alpha-synuclein worsens disease in mice lacking ubiquitin carboxy-terminal hydrolase L1. Sci Rep. 2012;2:262. doi: 10.1038/srep00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. New Engl J Med. 2009;361:1651–1661. doi: 10.1056/Nejmoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. doi:10.1126/science.1090278 302/5646/841 [pii] [DOI] [PubMed] [Google Scholar]
- 182.Slodzinski H, Moran LB, Michael GJ, Wang B, Novoselov S, Cheetham ME, Pearce RK, Graeber MB. Homocysteine-induced endoplasmic reticulum protein (herp) is up-regulated in parkinsonian substantia nigra and present in the core of Lewy bodies. Clin Neuropathol. 2009;28:333–343. [PubMed] [Google Scholar]
- 183.Smith GA, Isacson O, Dunnett SB. The search for genetic mouse models of prodromal Parkinson’s disease. Exp Neurol. 2012;237:267–273. doi: 10.1016/j.expneurol.2012.06.035. [DOI] [PubMed] [Google Scholar]
- 184.Sokolowski JD, Mandell JW. Phagocytic clearance in neurodegeneration. Am J Pathol. 2011;178:1416–1428. doi: 10.1016/j.ajpath.2010.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Song L, Cortopassi G. Mitochondrial complex I defects increase ubiquitin in substantia nigra. Brain Res. 2015;1594:82–91. doi: 10.1016/j.brainres.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 186.Song L, Shan Y, Lloyd KC, Cortopassi GA. Mutant Twinkle increases dopaminergic neurodegeneration, mtDNA deletions and modulates Parkin expression. Hum Mol Genet. 2012;21:5147–5158. doi: 10.1093/hmg/dds365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 188.Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Muller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Kruger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 189.Sun F, Anantharam V, Zhang D, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Proteasome inhibitor MG-132 induces dopaminergic degeneration in cell culture and animal models. Neurotoxicology. 2006;27:807–815. doi: 10.1016/j.neuro.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 190.Szondy Z, Garabuczi E, Joos G, Tsay GJ, Sarang Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol. 2014;5:354. doi: 10.3389/fimmu.2014.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tagliaferro P, Kareva T, Oo TF, Yarygina O, Kholodilov N, Burke RE. An early axonopathy in a hLRRK2(R1441G) transgenic model of Parkinson disease. Neurobiol Dis. 2015;82:359–371. doi: 10.1016/j.nbd.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Taylor TN, Caudle WM, Miller GW. VMAT2-Deficient Mice Display Nigral and Extranigral Pathology and Motor and Nonmotor Symptoms of Parkinson’s Disease. Parkinsons Dis. 2011;2011:124165. doi: 10.4061/2011/124165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Thiruchelvam M, Brockel BJ, Richfield EK, Baggs RB, Cory-Slechta DA. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: environmental risk factors for Parkinson’s disease? Brain Res. 2000;873:225–234. doi: 10.1016/s0006-8993(00)02496-3. [DOI] [PubMed] [Google Scholar]
- 194.Thiruchelvam MJ, Powers JM, Cory-Slechta DA, Richfield EK. Risk factors for dopaminergic neuron loss in human alpha-synuclein transgenic mice. Eur J Neurosci. 2004;19:845–854. doi: 10.1111/j.0953-816x.2004.03139.x. [DOI] [PubMed] [Google Scholar]
- 195.Tofaris GK, Garcia Reitbock P, Humby T, Lambourne SL, O’Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van Deerlin VM, Lee EB, Arnold SE, Duda JE, Hurtig H, Lee VM, Adler CH, Beach TG, Trojanowski JQ. Pathological alpha-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol. 2016;131:393–409. doi: 10.1007/s00401-015-1526-9. 10.1007/s00401-015-1526-9 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ungerstedt U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol. 1968;5:107–110. doi: 10.1016/0014-2999(68)90164-7. [DOI] [PubMed] [Google Scholar]
- 198.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 199.Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol. 2004;56:336–341. doi: 10.1002/ana.20256. [DOI] [PubMed] [Google Scholar]
- 200.van den Munckhof P, Luk KC, Ste-Marie L, Montgomery J, Blanchet PJ, Sadikot AF, Drouin J. Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons. Development. 2003;130:2535–2542. doi: 10.1242/dev.00464. [DOI] [PubMed] [Google Scholar]
- 201.Van Rompuy AS, Lobbestael E, Van der Perren A, Van den Haute C, Baekelandt V. Long-term overexpression of human wild-type and T240R mutant Parkin in rat substantia nigra induces progressive dopaminergic neurodegeneration. J Neuropathol Exp Neurol. 2014;73:159–174. doi: 10.1097/NEN.0000000000000039. [DOI] [PubMed] [Google Scholar]
- 202.Varma D, Sen D. Role of the unfolded protein response in the pathogenesis of Parkinson’s disease. Acta Neurobiol Exp (Wars) 2015;75:1–26. [PubMed] [Google Scholar]
- 203.Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 2011;10:1015–1025. doi: 10.1016/S1474-4422(11)70213-7. [DOI] [PubMed] [Google Scholar]
- 204.Vernon AC, Johansson SM, Modo MM. Non-invasive evaluation of nigrostriatal neuropathology in a proteasome inhibitor rodent model of Parkinson’s disease. BMC Neurosci. 2010;11:1. doi: 10.1186/1471-2202-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Villeneuve LM, Purnell PR, Boska MD, Fox HS. Early Expression of Parkinson’s Disease-Related Mitochondrial Abnormalities in PINK1 Knockout Rats. Mol Neurobiol. 2016;53:171–186. doi: 10.1007/s12035-014-8927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Villeneuve LM, Purnell PR, Stauch KL, Fox HS. Neonatal mitochondrial abnormalities due to PINK1 deficiency: Proteomics reveals early changes relevant to Parkinsons disease. Data Brief. 2016;6:428–432. doi: 10.1016/j.dib.2015.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wang X, Yen J, Kaiser P, Huang L. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal. 2010;3:ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 209.Wey MC, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease. PLoS One. 2012;7:e31522. doi: 10.1371/journal.pone.0031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yasuda T, Nihira T, Ren YR, Cao XQ, Wada K, Setsuie R, Kabuta T, Hattori N, Mizuno Y, Mochizuki H. Effects of UCH-L1 on alpha-synuclein over-expression mouse model of Parkinson’s disease. J Neurochem. 2009;108:932–944. doi: 10.1111/j.1471-4159.2008.05827.x. [DOI] [PubMed] [Google Scholar]
- 211.Zhang L, Le W, Xie W, Dani JA. Age-related changes in dopamine signaling in Nurr1 deficient mice as a model of Parkinson’s disease. Neurobiol Aging. 2012;33:1001 e1007–1016. doi: 10.1016/j.neurobiolaging.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhu JH, Guo FL, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–481. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zhu XR, Maskri L, Herold C, Bader V, Stichel CC, Gunturkun O, Lubbert H. Non-motor behavioural impairments in parkin-deficient mice. Eur J Neurosci. 2007;26:1902–1911. doi: 10.1111/j.1460-9568.2007.05812.x. [DOI] [PubMed] [Google Scholar]
- 214.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. doi:S0896627304007202 [pii] 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]