

Abstract
The goal of this study is to investigate the molecular pathways perturbed by in-vitro exposure of beta-methylamino-L-alanine (BMAA) to NSC-34 cells via contemporary proteomics. Our analysis of differentially regulated proteins reveals significant enrichment (p<0.01) of pathways related to ER stress, protein ubiquitination, the unfolded protein response, and mitochondrial dysfunction. Upstream regulator analysis indicates that exposure to BMAA induces activation of transcription factors (X-box binding protein 1; nuclear factor 2 erythroid like 2; promyelocytic leukemia) involved in regulation of the UPR, oxidative stress, and cellular senescence. Furthermore, we examine the hypothesis that BMAA causes protein damage via misincorporation in place of L-Serine. We are unable to detect misincorporation of BMAA into protein via analysis of cellular protein, secreted protein, targeted detection of BMAA after protein hydrolysis, or through the use of in-vitro protein translation kits.
Keywords: Amyotrophic Lateral Sclerosis, BMAA, LC-MS/MS, neurodegeneration, proteomics, exposures
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a devastatingly fatal neurodegenerative disease which is characterized by the progressive loss of upper and lower motor neurons leading to loss of voluntary muscle function and atrophy [1]. The average survival time starting from onset of symptoms in ALS patients falls between 3 and 5 years [2]. Sporadic incidences, which account for 90-95% of ALS cases, occur seemingly at random in that they are not attributable to known genetic causes. Proposed environmental factors thought to contribute to development of sporadic ALS include participation in professional sports [3], immune response to bacteria [4], military service [5], and geographical location [6].
Perhaps the strongest clue of an environmental trigger in the etiology of ALS was observed on the island of Guam in the 1950’s, at which time the prevalence of ALS was 100 times greater than the global baseline [7]. An epidemiological study linked preference for traditional Chamorro food to symptom development [8]. The traditional Chamorro diet was found to be rich in a neurotoxic noncanonical amino acid called beta-methylamino-L-alanine (BMAA) [9]
The interest in a global link between exposure to BMAA and the etiology of ALS ignited when it was discovered that BMAA was produced by a variety of cyanobacteria taxa that are found worldwide [10]. Notably, BMAA was detected in postmortem brain tissue taken from patients in the United States who had died from both sporadic ALS and Alzheimer’s disease, but not in control patients or those who had died of Huntington’s disease [11]. Additionally, individuals found to be living within close proximity to lakes containing cyanobacterial blooms in New Hampshire have been found to have a higher risk for developing ALS [12, 13]. Exposure is believed to occur through inhalation of aerosolized BMAA, ingestion of water containing cyanobacterial blooms, and consumption of fish living in and around areas containing blooms, which have been shown to be rich in BMAA [14, 15].
It has been suggested that free BMAA in the brain results in a excitotoxic effect in motor neuron cells by acting upon glutamate receptors, causing calcium influx and, ultimately, cell death [16]. A second theory is that BMAA becomes erroneously included into cellular protein in place of other natural amino acids such as L-Serine by misincorporation by tRNA synthetases [17, 18]. This hypothesis is quite intriguing since it provides a pathologic link between clinically indistinguishable familial and sporadic ALS. Misincorporation of BMAA into cellular protein could result in misfolding of protein, ER stress, and protein aggregation ultimately causing motor neuron degeneration [19, 20]; and thus, act in an analogous manner to the various missense mutations observed in familial ALS [21].
Herein, we investigate the molecular pathways perturbed by a 72-hour exposure of NSC-34 murine neuroblastoma/spinal motor neuron fusion cells to 500 μM BMAA via liquid chromatography-tandem mass spectrometry (LC-MS/MS). The following ALS related pathways were enriched as significantly affected by BMAA exposure: endoplasmic reticulum stress (enrichment p-value = 5.75E-04), protein ubiquitination (p = 1.17E-10), eIF2 signaling (4.47E-06), unfolded protein response (p = 5.89E-08), TCA cycle (p = 5.75E-09), oxidative phosphorylation (p= 1.82E-08), NRF2 oxidative stress response (2.95E-08), and mitochondrial dysfunction (p = 1.58E-12). Misincorporation of BMAA for L-serine into cellular protein was investigated by multiple methods including: 1) dynamic modification searches; 2) targeted detection of BMAA via acid hydrolysis of protein and 3) cell-free in vitro protein synthesis kits.
Methods
Materials
Fetal bovine serum (FBS), penicillin-streptomycin, Pierce® SILAC protein quantitation kit – DMEM, and 13C6-L-Arginine-HCl were purchased from ThermoFisher Scientific. DMEM high glucose cell medium, EMEM cell medium, sodium bicarbonate, D-(+)-glucose, BMAA, L-Serine (L-Ser), glycine, Dulbecco’s phosphate buffered saline (DPBS) formic Acid (FA), ammonium bicarbonate (AB), ammonium hydroxide, iodoacetamide (IAM), dithiothreitol (DTT), hydrochloric acid, and sodium deoxycholate (SDC) were obtained from Sigma Aldrich (St. Louis, MO). Sequencing grade trypsin was from Promega (Madison, WI). Vivacon500® 10,000 KDa and 30,000 KDa molecular weight cut off (MWCO) spin filters were purchased from ThermoFisher Scientific (Waltham, MA). HPLC grade acetonitrile, methanol, and water were from Burdick & Jackson (Muskegon, MI). PURExpress In-Vitro Protein Synthesis Kits Δ(aa, tRNA) (-ser, ala) were obtained from New England Biolabs. Dabsyl chloride was obtained from Supelco (Bellefonte, PA). Pico-frit columns were purchased from New Objective (Woburn, MA), and reverse phase ReproSil-Pur 120 C-18-AQ 3 μm particles were purchased from Dr. Maisch. High purity nitrogen gas was purchased from Machine & Welding Supply Co. HPLC grade water, methanol, acetone, and acetonitrile were purchased from VWR International (Morrisville, NC).
SILAC Spike-in NSC-34 Lysate Preparation
NSC-34 cells were obtained from Cedarlane (Burlington, NC). Cells were maintained, and SILAC labeled cell lysate was generated as described in Supplemental Method – SILAC Spike-in Preparation.
BMAA Exposure
NSC-34 cells were maintained in supplemented DMEM as described above. Once the cells reached 80% confluency, they were trypsinized and seeded into a 6-well plate (6 wells total) with each well receiving a 0.166 × 106 cell/well seeding density. All experimental treatments were carried out in a randomized and blocked fashion to control for plate effect. Control and 500 μM BMAA-treated samples each had three replicates to account for biological variability. A detailed description of cellular exposure and isolation can be found in Supplemental Method – BMAA Exposure and Isolation.
BMAA Dose-Response
NSC-34 cells were exposed to BMAA at different concentrations (100, 500, 1000, 10000 μM) for 72 hours. Cytotoxicity was accessed via trypan blue and MTS assays. Untreated media was used as control for comparison purposes. Three replicates per condition were carried out. For trypan blue, cells were harvested with trypsin and resuspended in PBS before addition of 0.04% trypan blue. Cell survival was assessed by direct counting. MTS reduction was measured using the Cell Titer 96 ® Aqueous One Solution Reagent. Formation of colored formazan product was measured by absorbance at 490 nm after 3 hours of incubation in the dark.
Cellular Protein Sample Preparation
Details regarding cell lysis, SILAC spike-in, sample reduction, alkylation, and finally digestion by filter aided sample preparation (FASP) can be found in Supplemental Method – Protein Digestion. Peptides were then separated into four fractions through use of a Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo, catalog number 84868) using 7.5%, 12.5%, 17.5%, and 50% ACN. Finally, the fractions were lyophilized and then reconstituted in 100 μL mobile phase A (98% water, 2% acetonitrile, 0.1% formic acid).
NanoLC MS/MS
Details regarding the data-dependent LC-MS/MS data collection are provided in Supplemental Method – LC-MS/MS Analysis. A quality control bovine serum albumin digest was run every fifth injection to ensure proper LC-MS/MS reproducibility [22], and Promega 6 × 5 LC-MS/MS Peptide Reference Mixture was run every tenth injection to further monitor general LC-MS/MS performance stability as well as stability of MS dynamic range [23]. QC data were uploaded to Panorama using Panorama AutoQC [24], and review of QC data within Panorama showed retention time, full width at half-maximum, and peak area median CV of 0.5%, 14.2%, and 18.6%, respectively throughout the experiment. Raw data files obtained in this experiment have been made available on the Chorus LC-MS data repository, and can be accessed under project ID #1357.
Database Search
Database searches were conducted using Proteome Discoverer 1.4 and the Sequest hyper-threaded algorithm. Data were searched against the Mus musculus Swiss Prot protein database (number of sequences 16,657, date accessed, 02/11/2014) [25]. Cysteine carbamidomethylation was searched as a static peptide modification, and methionine oxidation was searched as a dynamic peptide modification. Detection of BMAA misincorporation was attempted using a dynamic modification of +13.0316 Da on serine residues. SILAC label-based quantitation was carried out using the Precursor Ions Quantifier node using SILAC 2-plex (13C6-Arg and 13C6-Lys) quantification, and SILAC ratios were reported as light/heavy. Peptide spectral matches were processed using percolator [26] to enforce a peptide spectral match threshold of q-value<0.01. The law of strict parsimony was used for protein inference and grouping [27]. All quantitation was performed on the peptide level using one peptide for protein identification, and peptide identification output from Proteome Discoverer is included in supplemental information.
Data Analysis
Peptide-level experimental data were exported from Proteome Discoverer as a tab-delimited text file containing peptide sequence, protein group ID, number of protein groups assigned, peptide MS1 peak area, and peptide SILAC peak area ratio. Peptides belonging to more than one protein group were excluded. Replicates for which peak area ratio data were present in 100% of control replicates but absent in all treatment replicates, or vice versa, were removed to a separate spreadsheet. The missing values in these replicates were minimum value imputed. A two-sample t-test was then performed for calculation of p-values and log2 fold change was calculated for each peptide. P-value and fold change calculations are included in a supplemental file.
Any peptides for which peak area ratio data were missing for more than one replicate were then discarded from the original data set. Missing peak area ratios were then imputed with a random value between the minimum and maximum value for the peptide peak area. A two-sample t-test was then applied for calculation of p-values and log2 fold change was calculated for each peptide. Peptides identified as significant (p < 0.05) were subjected to gene ontology enrichment analysis by protein ID, p-value, and log2 fold change using Ingenuity Pathway Analysis (IPA), and protein interaction networks were generated using the STRING application in Cytoscape.
Cellular Lysate Acid Hydrolysis and Dabsyl Chloride Derivatization
Media and cellular lysate from BMAA-treated NSC-34 were retained for measurement of BMAA content. Detains regarding this experiment may be found in Supplemental Method – Protein Acid Hydrolysis and Analysis.
In Vitro Protein Synthesis
PURExpress In Vitro Protein Synthesis Kits Δ(aa, tRNA) –(ala, ser) were obtained from NEB (catalog number E6840Z) and were assembled according to the manufacturer’s instructions with modifications. All reactions were carried out using 250ng of either provided dihydrofolate reductase (DHFR) or hSOD1 G93A template DNA. Details about SOD1 G93A Template purchase and modification for in vitro protein synthesis may be found in Supplemental Method – SOD1 G93A Plasmid Preparation.
A control reaction was carried out, and contained only L-Ser at a concentration consistent with the manufacturer’s instructions. Further reactions were carried out with an increasing ratio (1:1, 10:1, and 100:1) of BMAA:L-Ser content by decreasing the concentration of L-Serine present in the reaction mixture. Reaction mixtures were brought to a final volume of 25 μL with DNase/RNase/Protease free water from Fisher (catalog number BP2819-1) and then incubated for two hours at 37°C and then stored overnight at −20°C prior to digestion and LC-MS/MS analysis.
Results and Discussion
A general overview of the experimental design is illustrated in Figure 1. In brief, cells were plated into a 6-well plate and treated either with L-Ser or 500 μM BMAA in triplicate (Figure 1A). To determine appropriate dosage concentration of BMAA, a dose response (Supplemental Figure 1) was conducted using a trypan blue cytotoxicity assay. The maximum concentration at which less than 20% cytotoxicity was observed was used for subsequent experiments. Cells were lysed, protein isolated, and spiked with SILAC labeled NSC-34 lysate for label-based quantitation (Figure 1B). After digestion, peptides were fractionated and analyzed by LC-MS/MS. Figure 1C shows the workflow for protein acid hydrolysis, dabsyl chloride derivatization, and SRM analysis for cellular lysate acetone precipitated protein, cellular lysate acetone precipitation supernatant, exposure media concentrate, and exposure media concentration flow-through. Finally, Figure 1D shows the experimental design for in-vitro synthesis of hSOD1-G93A protein with increasing ratio of BMAA:L-Ser.
Figure 1.
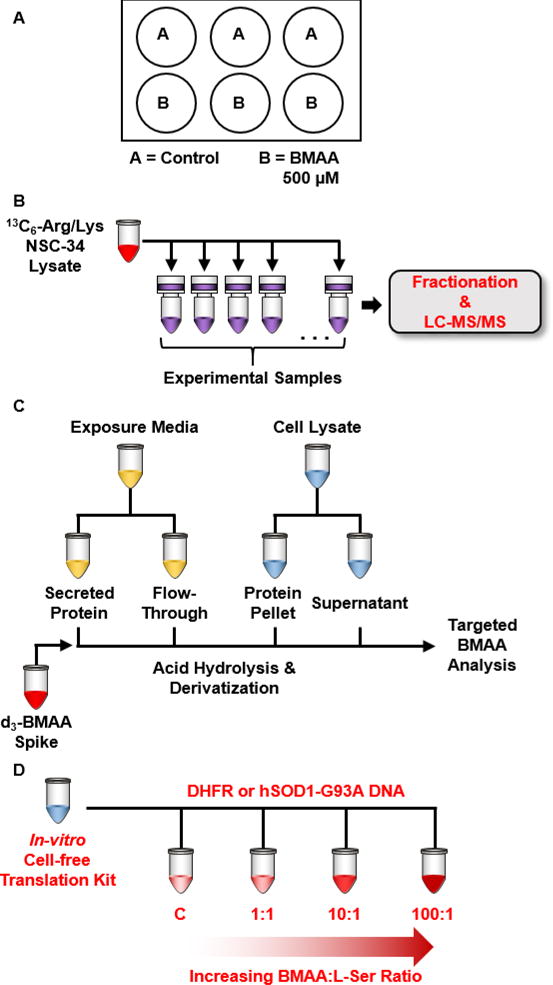
Experimental design for (A) 72-hour exposure of NSC-34 cells to 500 μM BMAA, (B) SILAC spike-in and sample preparation for untargeted proteomic analysis of BMAA-exposed NSC-34, (C) acid hydrolysis, derivatization, and SRM analysis of exposure media and cellular lysate samples, and (D) in-vitro synthesis of hSOD1 G93A protein in presence of increasing concentrations of BMAA.
Differential Protein Expression Due to BMAA Exposure
Figure 2A shows the top 24 pathways, enriched by pathway analysis of significantly differentially expressed proteins (p ≤ 0.05), which were perturbed in BMAA-exposed replicates compared to control. Also shown are the percent of proteins within each pathway that were found to be up- or down-regulated. Supplemental Table 1 lists each pathway found to be significantly perturbed by BMAA exposure along with the p-value for enrichment. Several pathways were found to be significant which are known to have overlap in cellular function, including the ER stress, protein ubiquitination, and unfolded protein response pathways as well as the TCA cycle, mitochondrial dysfunction, NRF2 oxidative stress response, and oxidative phosphorylation pathways. Interestingly, these pathways are known to play significant roles during ALS pathogenesis. Perturbation of theses listed pathways were reproduced in various other iterations of this experiment including lower exposure times (eg., 24 hour). However, exposure to other neurotoxins linked with ALS development, such as increased glutamate concentration (Supplemental Figure 2) did not reproduce perturbations in any of these described pathways. It is noteworthy that NSC-34 exposure to elevated glutamate concentration did not result in observation of protein signatures of excitotoxicity (e.g., perturbation in oxidative stress, mitochondrial dysfunction, or calcium signaling pathways). This result is corroborated by a previous study which states that the NSC-34 cell line does not serve as a good model for glutamate excitotoxicity [28]. However, lack of overlap between pathways perturbed by exposure to BMAA and exposure to elevated glutamate serves to demonstrate that the pathways perturbed by BMAA exposure are not the result of a common cellular stress response to addition of any analyte in excess to the cellular media.
Figure 2.
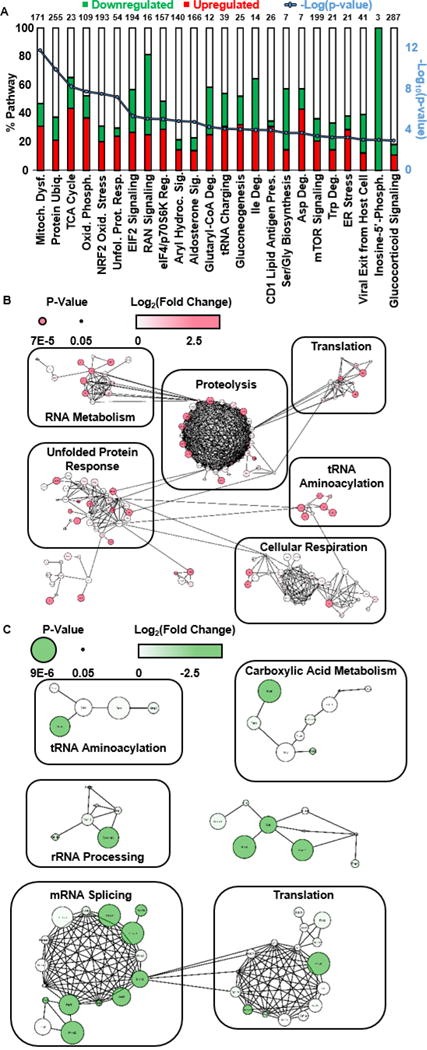
(A) Pathway enrichment results for proteins found to have significant differential regulation due to exposure to 500 μM BMAA compared to control. Upregulated (green) and downregulated (red) are plotted as a percentage of the total number of proteins within the pathway, shown on top of the bar chart. (B) Protein interaction network for proteins found to be upregulated in 500 μM BMAA exposed cells compared to control. (C) Protein interaction network for proteins found to be downregulated in 500 μM BMAA exposed cells compared to control.
To visualize the protein interactions, protein data were also analyzed within Cytoscape version 3.3.0 [29] using the stringApp application for protein interaction network analysis. Protein interaction networks were created for proteins found to be significantly upregulated (Figure 2B) and significantly downregulated (Figure 2C) in the BMAA treated samples compared to control. Groups of interacting proteins were isolated and subjected to functional annotation analysis using DAVID 6.8 [30] to determine biological function. Groups deemed to have significance (Benjamini-corrected p-value ≤ 0.05) were assigned the annotation with the lowest p-value. Multiple clusters of proteins within the interaction networks were related to ALS development: the upregulated network contains proteins related to proteolysis, unfolded protein response, and cellular respiration, whereas the downregulated network contains proteins related to translation.
Protein Ubiquitination, ER Stress, and the Unfolded Protein Response
Proteins entering the endoplasmic reticulum (ER) after synthesis undergo a series of modifications and interact with a series of molecular chaperones to facilitate proper folding [31]. Stringent quality control mechanisms prevent improperly folded proteins from exiting the ER, instead marking them for retention, refolding, and ultimately degradation. If protein refolding fails, the misfolded protein is tagged by ubiquitin ligase proteins, and is thus signaled to be delivered to the 26S proteasome for degradation [32]. Failure to remove and degrade misfolded proteins often leads to agglomeration of misfolded protein into inclusion bodies, which are known as a typical feature of various neurodegenerative disorders [33]. When accumulation of misfolded protein occurs, the cell activates a series of mechanisms known as the unfolded protein response (UPR) to alleviate ER stress. The functions encompassed by the UPR include: 1) increased expression of folding chaperones to assist in proper protein folding, 2) attenuation of translation, and 3) activation of cellular apoptosis machinery in the event of irreversible ER stress [34]. Activation of the UPR due to BMAA exposure has previously been described in exposed zebrafish by Frøyset and coworkers [35].
Significant differential regulation of the ER stress (enrichment p-value = 5.75E-04), protein ubiquitination (p = 1.17E-10) and UPR (p = 5.89E-08) pathways is indicative of a high likelihood that cellular exposure to 500 μM BMAA results in an increased incidence of misfolded protein within the cell. These results are reinforced by protein interaction network analysis (Figures 2B and 2C), which show upregulation of proteins belonging to the 26S proteasome and protein folding chaperones such as HSC70, as well as downregulation of various ribosomal proteins related to translation. Interestingly, these pathways are known to be significantly perturbed during development of various neurological diseases, including ALS [36, 37].
TCA Cycle, Oxidative Phosphorylation, NRF2, and Mitochondrial Dysfunction
The TCA cycle and oxidative phosphorylation pathway are closely linked, working together to provide energy to the cell in the form of ATP within the mitochondria [38]. The oxidative phosphorylation pathway is comprised of several protein complexes that make up the electron transfer chain, and use the energy precursors produced by the TCA cycle to create additional ATP [39]. The NRF2 oxidative stress response plays an important role in maintaining cellular redox homeostasis [40], as well as provide aid for proper mitochondrial structure and function [41]. These pathways are associated with overall mitochondrial function, and their disruption (p-value for enrichment = 5.75E-09 for TCA cycle, 1.82E-08 for oxidative phosphorylation, 2.95E-08 for NRF2, and 1.58E-12 for mitochondrial dysfunction) serves as an indication of likely mitochondrial injury as a result of BMAA exposure. Mitochondrial damage is a hallmark of ALS pathogenesis, including various facets of mitochondrial function within the cells such as altered calcium homeostasis [42] and altered electron transport chain [43]. Furthermore, these results are supported by a significant (p<0.05) reduction of mitochondrial function upon BMAA exposure using an MTS assay (Supplemental Figure 3).
Predicted Upstream Transcription Regulator Analysis
Next we aimed to identify the upstream transcription regulators which BMAA acts upon. Supplemental Table 2 lists all the upstream regulators from the Ingenuity Knowledge Base that showed significant overlap (overlap p-value ≤ 0.05) with the dataset, and an activation z-score greater than 2 or less than −2. Interestingly, XBP1 (overlap p = 3.3E-09) was predicted to be activated (z = 2.6) in this dataset. During ER stress, XBP1 mRNA becomes spliced [44] and produces the transcription factor that regulates the expression of genes related to protein folding and quality control [45], which is a potential upstream cause of ER stress and unfolded protein response disturbance as seen in the above results. While this response may be protective in certain instances of cellular stress, over activation is detrimental in ALS as depletion of XBP-1 in the nervous system activates macroautophagy and enhances survival in the SOD1 mutant mouse model of ALS [46].
NFE2L2, the gene encoding NRF2, was also predicted as an activated upstream regulator due to BMAA exposure within this dataset (overlap p-value = 2.21E-28, and activation z-score = 2.612). NRF2 is a transcription regulator which takes on a number of protective functions within the cell. Genes regulated by NRF2 have a diverse set of functions, including metabolism of electrophiles and oxidative species within the cell as well as genes involved in proteasomal degradation, such as the 26S proteasome [47]. Predicted activation of NRF2 upon exposure of NSC-34 cells to BMAA may imply the following: 1) NRF2 may be activated in response to increased oxidative stress caused by mitochondrial dysfunction, and 2) NRF2 may also activate in order to increase production of 26S proteasome units, thus increasing the cell’s capacity for degradation of misfolded proteins as part of the UPR. Interestingly, there is pronounced upregulation of 26S proteasome (proteolysis) proteins within this dataset, as noted in the protein interaction analysis (Figure 2B and 2C). Activation of NRF2 has been noted in transgenic mouse models for ALS both in astrocyte cells [48] as well as in spinal cord and muscle [49], and activation of NRF2 by activator molecules has been shown to have a neuroprotective effect in mouse ALS models [50].
BMAA Misincorporation
Based on disturbance of pathways in protein folding within the ER (e.g., unfolded protein response, ER stress, protein ubiquitination), we aimed to further investigate the hypothesis that BMAA becomes misincorporated into cellular protein in place of L-Serine. As an initial step, all peptide database searches conducted on data obtained from the control and 500 μM BMAA exposure were searched with an additional dynamic modification for BMAA replacement in place of L-Ser (Δm=+13.0316 Da). No peptides containing BMAA were deemed confidently identified after manual review of the resulting tandem mass spectra and comparison to control spectra.
Next, targeted measurement of BMAA was attempted via protein hydrolysis. Cellular lysate protein and BMAA exposure media were spiked with d3-BMAA, and subjected to acid hydrolysis followed by amino acid derivatization by dabsyl chloride and analyzed by selected reaction monitoring mass spectrometry (Figure 3). The SRM analysis found negligible levels of BMAA within acetone-precipitated cellular lysate. Supernatant from acetone precipitation of the cellular lysate was found not to contain a significantly greater amount of BMAA (p = 0.15) compared to the acetone-precipitated protein fraction. Levels of BMAA exceeding the noise signal measured within the blank were found within the protein-concentrated media sample, and a significantly greater amount (p = 0.001) of BMAA was found within the media protein concentration flow-through obtained. However, upon LC-MS/MS analysis of digested protein contained within the media, we found no evidence of BMAA misincorporation into secreted protein. It is likely that any BMAA found within the protein-concentrated fraction of exposure media is due to the presence of BMAA within residual exposure media carried over during the protein concentration procedure. These results suggest that cellular BMAA exposure has not resulted in the misincorporation of BMAA into cellular protein.
Figure 3.
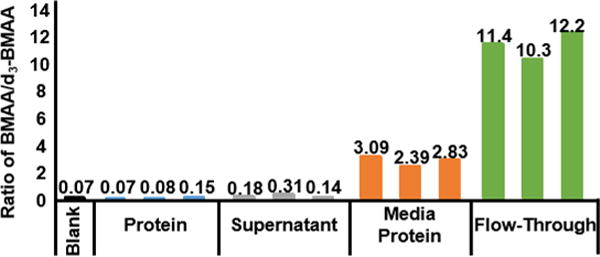
Ratio of BMAA peak area to d3-BMAA peak area in acetone precipitated experimental lysate protein, acetone precipitation supernatant, exposure media concentrate, and exposure media concentration flow through. Samples were spiked with 3.25 μM BMAA.
Finally, to test the possibility that transport of BMAA to the translational machinery was not occurring in this particular cell line, we performed a series of experiments using an in vitro protein translation kit. An initial experiment was carried out similar to one performed by Glover and coworkers [18]. A control reaction and a second reaction wherein BMAA replaced L-Ser were carried out using the provided DHFR DNA template. We found that the ability for the BMAA-containing reaction to carry out de-novo protein synthesis was drastically reduced. Although these results contradict a study reported by Glover et al., it is worth noting that these current studies were aimed at detecting BMAA in replace of L-serine in the synthesized protein where the former detected free BMAA upon acid hydrolysis of protein precipitate within the reaction mixture without targeting the newly synthesized protein. Supplemental Figure 4 shows the integrated peak area for three DHFR peptides, one of which contains no L-Ser. Each peptide was found to be present within the sample that was supplied L-Ser, but none of the three peptides was found within the sample that was supplied only BMAA. Analysis of percent protein coverage within each sample showed 66.67% protein coverage within the control reaction, and only 12.58% coverage within the BMAA containing sample. The latter synthesis was found to contain only two peptides without serine residues.
The experiment was next adapted such that no reaction would be carried out without L-Ser. We hypothesized that if the translational machinery was jumpstarted by the presence of L-serine, it may be possible that during protein translation BMAA would be misincorporated or inhibit protein translation. Furthermore this type of an experiment would more accurately depict a real life exposure to BMAA. A control reaction was carried out alongside several experimental reactions wherein the molar ratio of BMAA:L-Ser supplied during protein synthesis was increased by factors of 10 with a decreasing concentration of L-Serine. All reactions were performed with hSOD1 G93A mutant template DNA. Figure 4 shows the result of extracted MS1 peptide peak area analysis for a hSOD1 G93A peptide containing no L-Ser (Figure 4A) and a peptide containing L-Ser (Figure 4B). In the event of BMAA misincorporation into the cellular protein in place of L-Ser, peak area for the serine containing peptide would be reduced in BMAA-containing experimental samples, but peak area for the peptide containing no serine residues would remain unaffected. However, the result of the analysis shows that BMAA concentration relative to L-Ser dosage had no effect on the abundance of serine-containing peptides. In addition, no BMAA modified peptides were confidently identified in the database search. Finally, the possibility that formation of the carbamate adduct of BMAA affected its ability to misincorporate into protein during translation was explored. To this end, a control reaction containing BMAA was carried out alongside a second BMAA-containing reaction which also included 15 mM sodium bicarbonate (data not shown). No peptides containing the L-Serine to BMAA modification were identified in the newly translated BMAA protein in either reaction, suggesting that presence of bicarbonate and the resulting carbamate BMAA adduct had no effect on protein translation. Combined these data do not support BMAA misincorporation (in vitro) into cellular protein in place of L-serine.
Figure 4.
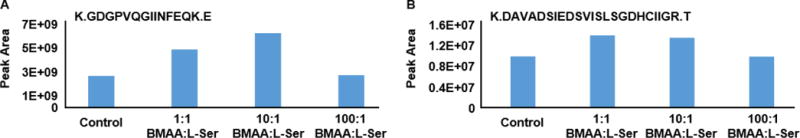
Targeted peak area analysis of (A) a peptide containing no serine, and (B) a serine-containing peptide of in-vitro synthesized hSOD-1 G93A. In-vitro synthesis kit was supplied with either no BMAA (control) or 1:1, 10:1, or 100:1 BMAA:L-Ser (decreasing amounts of L-Ser).
Conclusions
Taken together our studies indicate that exposure to BMAA induces a molecular signature that mirrors perturbed biological processes in various neurodegenerative diseases including ALS. Exposure to BMAA causes protein misfolding, culminating in ER stress and ultimately the induction of the unfolded protein response while simultaneously disrupting mitochondrial function. We hypothesize, due to disruption of the unfolded protein response as well as the eIF2 signaling (p = 4.47E-06) pathways, that upon prolonged exposure to BMAA and thus over-activation of the UPR, protein translation is attenuated via phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α). Interestingly, high levels of this translation regulator have been detected in the brains of patients with Parkinson’s and Alzheimer’s disease, supranuclear palsy, and front temporal dementia [51-53]. Inhibition of eIF2α phosphorylation has been shown to rescue TDP-43 pathology in various disease models of ALS. As a result the eif2 alpha is a promising therapeutic target and the subject of recent investigations [54].
While our results neither confirm nor exclude that BMAA is misincorporated into proteins in place of L-Serine, it is clear that this mistake in translation does not readily occur in these in vitro experiments. This observation is not completely surprising as misincorporation of endogenous amino acids does naturally occur but at an extremely low stoichiometric level (one error for 103-104 codons) [55, 56]. Assuming a similar error rate and that misincorporation of BMAA into proteins is semi-random identification of specific BMAA containing peptides would be well below the dynamic range of detection by LC MS/MS (~105). Another possibility is that individuals may have a genetic abnormality that lends them susceptible to BMAA misincorporation which would not be accurately modeled by these in vitro experiments. Reports in mice have shown motor neuron degeneration as a result of a missense mutation in the putative editing domain of alanyl-tRNA synthetase which led to heterogeneous protein production [57]. However genome-wide association studies of individuals with ALS have yet to support this hypothesis. Finally, it should be appreciated that the myriad of toxins produced by cyanobacteria [58] likely result in co-exposures with BMAA that could augment toxicity. As such, future experiments will entail investigation of molecular pathways perturbed by coexposure with BMAA and other cyanotoxins.
Supplementary Material
Statement of significance of the study.
LC-MS/MS and subsequent peptide-level SILAC spike-in quantitation of NSC-34 cells subjected to 72 hour exposure to 500 μM BMAA resulted in perturbation of various molecular pathways known to be implicated in ALS development. Pathways include protein ubiquitination, endoplasmic reticulum stress, eIF2 signaling, and unfolded protein response pathways. Perturbation of these pathways is indicative of increased incidence of misfolded protein within the cell, which is a hallmark of several neurodegenerative disease including ALS. Also perturbed were the oxidative phosphorylation, TCA cycle, NRF2 oxidative stress response, and mitochondrial dysfunction pathways. Disruption of these pathways is indicative of disrupted cellular electron transport and mitochondrial damage, which are conditions linked to ALS. The hypothesis of BMAA misincorporation into cellular protein was also tested, though no BMAA misincorporation was identified within cellular lysate or protein synthesized using an in-vitro protein synthesis kit. Our data presented here represent a novel analysis of the effects of BMAA exposure and its relation to ALS development.
Acknowledgments
MSB is thankful for startup funds provided by North Carolina State University. MSB and TEN acknowledge support from the Center for Human Health and the Environment (P30ES025128).
Footnotes
Conflicts of Interest:
The authors declare no competing financial interest.
References
- 1.Rowland LP, Shneider NA. Medical progress: Amyotrophic lateral sclerosis. New Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Chancellor AM, Slattery JM, Fraser H, Swingler RJ, et al. The Prognosis of Adult-Onset Motor-Neuron Disease - a Prospective-Study Based on the Scottish Motor-Neuron Disease Register. J Neurol. 1993;240:339–346. doi: 10.1007/BF00839964. [DOI] [PubMed] [Google Scholar]
- 3.Beghi E. Are professional soccer players at higher risk for ALS? Amyotroph Lat Scl Fr. 2013;14:501–506. doi: 10.3109/21678421.2013.809764. [DOI] [PubMed] [Google Scholar]
- 4.Halperin JJ, Kaplan GP, Brazinsky S, Tsai TF, et al. Immunologic reactivity against Borrelia burgdorferi in patients with motor neuron disease. Arch Neurol. 1990;47:586–594. doi: 10.1001/archneur.1990.00530050110021. [DOI] [PubMed] [Google Scholar]
- 5.Beard JD, Engel LS, Richardson DB, Gammon MD, et al. Military service, deployments, and exposures in relation to amyotrophic lateral sclerosis etiology. Environ Int. 2016;91:104–115. doi: 10.1016/j.envint.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyser CL, Kissel JT, Mendell JR. Three cases of amyotrophic lateral sclerosis in a common occupational environment. J Neurol. 1987;234:443–444. doi: 10.1007/BF00314096. [DOI] [PubMed] [Google Scholar]
- 7.Bradley WG, Mash DC. Beyond Guam: the cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotrophic Lateral Scler. 2009;10(Suppl 2):7–20. doi: 10.3109/17482960903286009. [DOI] [PubMed] [Google Scholar]
- 8.Reed D, Labarthe D, Chen KM, Stallones R. A Cohort Study of Amyotrophic-Lateral-Sclerosis and Parkinsonism-Dementia on Guam and Rota. Am J Epidemiol. 1987;125:92–100. doi: 10.1093/oxfordjournals.aje.a114515. [DOI] [PubMed] [Google Scholar]
- 9.Vega A, Bell EA. Alpha-Amino-Beta-Methylaminopropionic Acid a New Amino Acid from Seeds of Cycas Circinalis. Phytochemistry. 1967;6:759. [Google Scholar]
- 10.Cox PA, Banack SA, Murch SJ, Rasmussen U, et al. Diverse taxa of cyanobacteria produce beta-N-methylamino-L-alanine, a neurotoxic amino acid. Proc Natl Acad Sci U S A. 2005;102:5074–5078. doi: 10.1073/pnas.0501526102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pablo J, Banack SA, Cox PA, Johnson TE, et al. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol Scand. 2009;120:216–225. doi: 10.1111/j.1600-0404.2008.01150.x. [DOI] [PubMed] [Google Scholar]
- 12.Caller TA, Doolin JW, Haney JF, Murby AJ, et al. A cluster of amyotrophic lateral sclerosis in New Hampshire: a possible role for toxic cyanobacteria blooms. Amyotrophic Lateral Scler. 2009;10(Suppl 2):101–108. doi: 10.3109/17482960903278485. [DOI] [PubMed] [Google Scholar]
- 13.Torbick N, Hession S, Stommel E, Caller T. Mapping amyotrophic lateral sclerosis lake risk factors across northern New England. Int J Health Geogr. 2014;13:1. doi: 10.1186/1476-072X-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang LY, Kiselova N, Rosen J, Ilag LL. Quantification of neurotoxin BMAA (beta-N-methylamino-L-alanine) in seafood from Swedish markets. Sci Rep-Uk. 2014;4 doi: 10.1038/srep06931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brand LE, Pablo J, Compton A, Hammerschlag N, Mash DC. Cyanobacterial blooms and the occurrence of the neurotoxin, beta-N-methylamino-l-alanine (BMAA), in South Florida aquatic food webs. Harmful Algae. 2010;9:620–635. doi: 10.1016/j.hal.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross SM, Spencer PS. Specific Antagonism of Behavioral Action of Uncommon Amino-Acids Linked to Motor-System Diseases. Synapse. 1987;1:248–253. doi: 10.1002/syn.890010305. [DOI] [PubMed] [Google Scholar]
- 17.Dunlop RA, Cox PA, Banack SA, Rodgers KJ. The Non-Protein Amino Acid BMAA Is Misincorporated into Human Proteins in Place of L-Serine Causing Protein Misfolding and Aggregation. Plos One. 2013;8 doi: 10.1371/journal.pone.0075376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glover WB, Mash DC, Murch SJ. The natural non-protein amino acid N-β-methylamino-l-alanine (BMAA) is incorporated into protein during synthesis. Amino Acids. 2014;46:2553–2559. doi: 10.1007/s00726-014-1812-1. [DOI] [PubMed] [Google Scholar]
- 19.Bell EA. The discovery of BMAA, and examples of biomagnification and protein incorporation involving other non-protein amino acids. Amyotrophic Lateral Scler. 2009;10:21–25. doi: 10.3109/17482960903268700. [DOI] [PubMed] [Google Scholar]
- 20.Cox PA, Davis DA, Mash DC, Metcalf JS, Banack SA. Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. P Roy Soc B-Biol Sci. 2016;283 doi: 10.1098/rspb.2015.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bereman MS, Johnson R, Bollinger J, Boss Y, et al. Implementation of Statistical Process Control for Proteomic Experiments Via LC MS/MS. J Am Soc Mass Spectr. 2014;25:581–587. doi: 10.1007/s13361-013-0824-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beri J, Rosenblatt MM, Strauss E, Urh M, Bereman MS. Reagent for Evaluating Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Performance in Bottom-Up Proteomic Experiments. Anal Chem. 2015;87:11635–11640. doi: 10.1021/acs.analchem.5b04121. [DOI] [PubMed] [Google Scholar]
- 24.Bereman MS, Beri J, Sharma V, Nathe C, et al. An Automated Pipeline to Monitor System Performance in Liquid Chromatography Tandem Mass Spectrometry Proteomic Experiments. J Proteome Res. 2016 doi: 10.1021/acs.jproteome.6b00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its new supplement TREMBL. Nucleic Acids Res. 1996;24:21–25. doi: 10.1093/nar/24.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 27.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hounoum B, Vourc’h P, Felix R, Corcia P. NSC-34 Motor Neuron-Like Cells Are Unsuitable as Experimental Model for Glutamate-Mediated Excitotoxicity. 2016 doi: 10.3389/fncel.2016.00118. ncbi.nlm.nih.gov. [DOI] [PMC free article] [PubMed]
- 29.Shannon P, Markiel A, Ozier O, Baliga NS, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 31.Braakman I, Hebert DN. Protein Folding in the Endoplasmic Reticulum. Csh Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross CA, Pickart CM. The ubiquitin-proteasome pathway in Parkinson’s disease and other neurodegenerative diseases. Trends Cell Biol. 14:703–711. doi: 10.1016/j.tcb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 34.Chakrabarti A, Chen AW, Varner JD. A Review of the Mammalian Unfolded Protein Response. Biotechnology and Bioengineering. 2011;108:2777–2793. doi: 10.1002/bit.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frøyset AK, Khan EA, Fladmark KE. Quantitative proteomics analysis of zebrafish exposed to sub-lethal dosages of β-methyl-amino-L-alanine (BMAA) Sci Rep. 2016;6 doi: 10.1038/srep29631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shibata N, Asayama K, Hirano A, Kobayashi M. Immunohistochemical study on superoxide dismutases in spinal cords from autopsied patients with amyotrophic lateral sclerosis. Dev Neurosci. 1996;18:492–498. doi: 10.1159/000111445. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, et al. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- 38.Balaban RS. Regulation of Oxidative-Phosphorylation in the Mammalian-Cell. Am J Physiol. 1990;258:C377–C389. doi: 10.1152/ajpcell.1990.258.3.C377. [DOI] [PubMed] [Google Scholar]
- 39.Hatefi Y. The Mitochondrial Electron-Transport and Oxidative-Phosphorylation System. Annu Rev Biochem. 1985;54:1015–1069. doi: 10.1146/annurev.bi.54.070185.005055. [DOI] [PubMed] [Google Scholar]
- 40.Ma Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annual Review of Pharmacology and Toxicology. 2013;53:401. doi: 10.1146/annurev-pharmtox-011112-140320. 2013, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radical Bio Med. 2015;88:179–188. doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siklós L, Engelhardt J, Harati Y, Smith RG, et al. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophc lateral sclerosis. Ann Neurol. 1996;39:203–216. doi: 10.1002/ana.410390210. [DOI] [PubMed] [Google Scholar]
- 43.Bowling AC, Schulz JB, Brown RH, Beal MF. Superoxide-Dismutase Activity, Oxidative Damage, and Mitochondrial Energy-Metabolism in Familial and Sporadic Amyotrophic-Lateral-Sclerosis. J Neurochem. 1993;61:2322–2325. doi: 10.1111/j.1471-4159.1993.tb07478.x. [DOI] [PubMed] [Google Scholar]
- 44.Calfon M, Zeng H, Urano F, Till JH, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 45.Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hetz C, Thielen P, Matus S, Nassif M, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23 doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kensler TW, Wakabayash N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 48.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 Activation in Astrocytes Protects against Neurodegeneration in Mouse Models of Familial Amyotrophic Lateral Sclerosis. J Neurosci. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kraft AD, Resch JM, Johnson BDA, Johnson JA. Activation of the Nrf2-ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp Neurol. 2007;207:107–117. doi: 10.1016/j.expneurol.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neymotin A, Calingasan NY, Wille E, Naseri N, et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radical Bio Med. 2011;51:88–96. doi: 10.1016/j.freeradbiomed.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, et al. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 52.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, et al. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. The American journal of pathology. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stutzbach LD, Xie SX, Naj AC, Albin R, et al. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun. 2013;1:31. doi: 10.1186/2051-5960-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halliday M, Radford H, Zents KAM, Molloy C, et al. Repurposed drugs targeting eIF2alpha-P-mediated translational repression prevent neurodegeneration in mice. Brain. 2017 doi: 10.1093/brain/awx074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loftfield RB, Vanderjagt D. The frequency of errors in protein biosynthesis. Biochem J. 1972;128:1353–1356. doi: 10.1042/bj1281353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jakubowski H, Goldman E. Editing of errors in selection of amino acids for protein synthesis. Microbiol Rev. 1992;56:412–429. doi: 10.1128/mr.56.3.412-429.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JW, Beebe K, Nangle LA, Jang J, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 58.Codd GA, Morrison LF, Metcalf JS. Cyanobacterial toxins: risk management for health protection. Toxicol Appl Pharm. 2005;203:264–272. doi: 10.1016/j.taap.2004.02.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.