

CONSPECTUS
Protons are vastly abundant in a wide range of exciting macromolecules and thus can be a powerful probe to investigate the structure and dynamics at atomic resolution using solid-state NMR (ssNMR) spectroscopy. Unfortunately, the high signal sensitivity, afforded by the high natural-abundance and high gyromagnetic ratio of protons, is greatly compromised by severe line broadening due to the very strong 1H–1H dipolar couplings. As a result, protons are rarely used, in spite of the desperate need for enhancing the sensitivity of ssNMR to study a variety of systems that are not amenable for high resolution investigation using other techniques including X-ray crystallography, cryo-electron microscopy, and solution NMR spectroscopy. Thanks to the remarkable improvement in proton spectral resolution afforded by the significant advances in magic-angle-spinning (MAS) probe technology, 1H ssNMR spectroscopy has recently attracted considerable attention in the structural and dynamics studies of various molecular systems. However, it still remains a challenge to obtain narrow 1H spectral lines, especially from proteins, without resorting to deuteration. In this Account, we review recent proton-based ssNMR strategies that have been developed in our laboratory to further improve proton spectral resolution without resorting to chemical deuteration for the purposes of gaining atomistic-level insights into molecular structures of various crystalline solid systems, using small molecules and peptides as illustrative examples. The proton spectral resolution enhancement afforded by the ultrafast MAS frequencies up to 120 kHz is initially discussed, followed by a description of an ensemble of multidimensional NMR pulse sequences, all based on proton detection, that have been developed to obtain in-depth information from dipolar couplings and chemical shift anisotropy (CSA). Simple single channel multidimensional proton NMR experiments could be performed to probe the proximity of protons for structure determination using 1H–1H dipolar couplings and to evaluate the changes in chemical environments as well as the relative orientation to the external magnetic field using proton CSA. Due to the boost in signal sensitivity enabled by proton detection under ultrafast MAS, by virtue of high proton natural abundance and gyromagnetic ratio, proton-detected multidimensional experiments involving low-γ nuclei can now be accomplished within a reasonable time, while the higher dimension also offers additional resolution enhancement. In addition, the application of proton-based ssNMR spectroscopy under ultrafast MAS in various challenging and crystalline systems is also presented. Finally, we briefly discuss the limitations and challenges pertaining to proton-based ssNMR spectroscopy under ultrafast MAS conditions, such as the presence of high-order dipolar couplings, friction-induced sample heating, and limited sample volume. Although there are still a number of challenges that must be circumvented by further developments in radio frequency pulse sequences, MAS probe technology and approaches to prepare NMR-friendly samples, proton-based ssNMR has already gained much popularity in various research domains, especially in proteins where uniform or site-selective deuteration can be relatively easily achieved. In addition, implementation of the recently developed fast data acquisition approaches would also enable further developments in the design and applications of proton-based ultrafast MAS multidimensional ssNMR techniques.
Graphical abstract
INTRODUCTION
Solid-state NMR spectroscopy has been playing significant roles in providing piercing insights into the molecular structures and dynamics for a wide variety of systems, mainly due to its capability of selectively manipulating various anisotropic spin interactions in solids.1 In particular, it has evolved as a routine approach over the past decades for studying low-γ nuclei (13C, 15N, etc.) due to their large chemical shift spans; however, a large sample amount is often required to overcome the sensitivity issue in these NMR experiments, unless isotopic labeling or other sensitivity enhancement methods such as dynamic nuclear polarization (DNP) are adopted.2 Herein, protons become one of the most straightforward nuclei for detection in multidimensional spectroscopy to overcome the sensitivity issue, mainly due to 1H high natural abundance, high gyromagnetic ratio, and prevalence in various molecular systems. However, the application of proton-based ssNMR spectroscopy has often been hampered by the poor spectral resolution, which stems from the presence of various strong anisotropic spin interactions. Generally, three approaches are often utilized to improve the proton spectral resolution: deuteration, CRAMPS (combined rotation and multiple pulse spectroscopy), and ultrafast magic angle spinning (MAS). Deuteration of a sample chemically reduces the amount of protons in the system and therefore suppresses 1H–1H dipolar couplings, which, for example, has been demonstrated to significantly reduce the line widths of proton peaks to ∼15 Hz in a microcrystalline sample of the SH3 domain from chicken α-spectrin at a moderate spinning frequency.3 However, the level of deuteration has to be optimized for achieving best resolution,4 and it can be difficult and expensive. CRAMPS is another approach that involves a combined manipulation of the spin part of the dipolar coupling Hamiltonian with radio frequency (RF) pulses and the spatial part with MAS.5 However, the setup of CRAMPS experiments is cumbersome, and the signal-to-noise ratio is often compromised due to the large receiver bandwidth and low probe quality factor. Fortunately, tremendous advances in MAS probe technology have recently enabled spinning up to 130 kHz using small rotors of diameters less than 0.75 mm, which could effectively suppress the strong 1H–1H dipolar couplings that cause severe line broadening of 1H spectral lines for solids, and thus ultrafast MAS enhances the spectral resolution. In fact, the maximum achievable spinning frequency (νmax) is inversely proportional to the rotor diameter (d) according to a simple relationship νmax ≈ 80 kHz·mm/d, considering the influences of bearing load, strength of rotor materials, turbine torques, and other factors as well.6 In further combination with appropriate deuteration and high magnetic fields, it has been possible to achieve solution-like proton NMR spectra even in proteins, which has attracted much attention in recent years.7–11 In fact, the discovery and demonstration of new phenomena under ultrafast MAS has also greatly simplified the design of pulse sequences and experimental setup.12–14 Consequently, the development of proton-based ssNMR methodology has become the focus of recent studies.
In this Account, we present and discuss recent developments in proton-based ssNMR methods under ultrafast MAS. We begin by showing the remarkable enhancement in proton spectral resolution enabled by ultrafast MAS and then proceed to discuss the development of multidimensional proton-based ssNMR techniques for extracting valuable information, such as the proton proximity, proton chemical shift anisotropy, 1H–1H distances, and heteronuclear proximity. Although several excellent reviews on the application of proton-detected ssNMR in proteins have been published in the last several years,8,9,11 the main focus of these reports was on perdeuterated proteins and paramagnetic systems. Here, we focus on the recent developments and applications of multidimensional proton-based ssNMR spectroscopy on rigid crystalline solids without any chemical pretreatment.
ENHANCED 1H SPECTRAL RESOLUTION BY ULTRAFAST MAS
A major obstacle for the widespread application of proton NMR spectroscopy on solids is the poor chemical shift resolution, which is quite limited by the narrow chemical shift span of protons (∼15 ppm). Therefore, the only approach to improve proton spectral resolution is to reduce the line widths of proton peaks, which are dominated by chemical shift dispersion and inherent anisotropic interactions, primarily the 1H–1H dipolar couplings and 1H chemical shift anisotropy (CSA). On the basis of average Hamiltonian theory,15,16 the “inhomogeneous” CSA Hamiltonian commutes with itself at all times, and all the commutator terms will vanish. In addition, even under the currently available highest magnetic-field strength of 24 T (1020 MHz 1H Larmor frequency),17 the maximum chemical shift anisotropy for the protons would be around 20 kHz (∼20 ppm), which is still much smaller than the ultrafast spinning frequency (>60 kHz). Therefore, the CSA of protons will be completely averaged out by MAS frequency of 60 kHz and higher. However, in contrast to proton CSA, the ubiquitous presence of 1H–1H dipolar couplings cause severe “homogeneous” line broadening, rendering the peak line widths much larger than their chemical shift differences. In particular, the high-order dipolar interaction Hamiltonian terms (either multispin dipolar interactions or cross-terms between dipolar coupling and chemical shift difference tensor) do not vanish completely, but rather become smaller with increasing the MAS frequency.10 Therefore, higher spinning speeds will greatly benefit the suppression of higher-order dipolar terms, and thus improve the proton spectral resolution, as shown in Figure 1A. By increase of the spinning speed from 10 to 110 kHz, the aliphatic side-chain protons in the bone organic matrix and the structural OH groups in bone mineral are progressively resolved, demonstrating the capability of ultrafast MAS for suppressing the strong dipolar couplings.18
Figure 1.
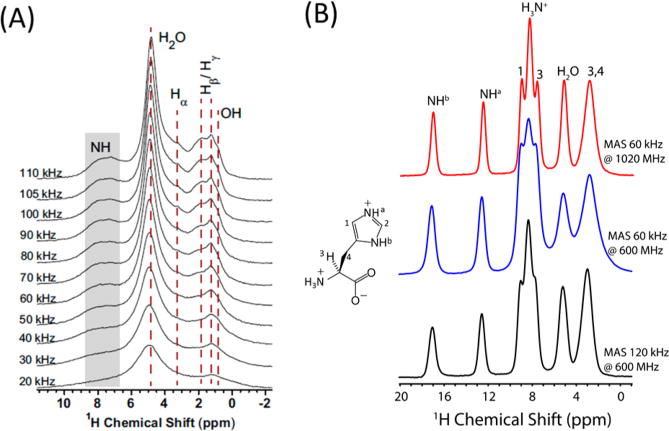
Faster sample spinning increases spectral resolution of protons. (A) 1H MAS NMR spectra recorded at 14.1 T from bovine cortical bone under multiple MAS rates. Reproduced with permission from ref 18. Copyright 2015 Nature Publishing Group. (B) 1D ultrafast MAS proton spectra of L-histidine·HCl·H2O. Adapted with permission from ref 17. Copyright 2015 Elsevier.
Chemical shift dispersion (distribution) is another important cause for the frequently observed line broadening of proton peaks. Chemical shift dispersion could result from disordered arrangements of molecular chains in the system or amorphous arrangements of microcrystals in the NMR rotors. On the other hand, the presence of anisotropic bulk magnetic susceptibility (ABMS) could also result in chemical shift dispersions and thus induce inhomogeneous broadening on the order of 1 to 2 ppm, particularly for π-electron solid systems.6,17 A typical example is shown in Figure 1B for L-histidine·HCl·H2O, where the resolution of imidazole ring protons does not improve significantly with the increasing spinning speed from 60 to 120 kHz under 600 MHz magnetic-field strength. However, the resolution is greatly improved when the magnetic field increases from 600 to 1020 MHz due to the increased chemical shift range. It should be noted here that ABMS could not be averaged out completely by MAS; however, since the ABMS Hamiltonian has similar dependence on the spin part as the isotropic chemical shift Hamiltonian,19 significant line-narrowing might still be achieved using multiple-echo pulse trains like CPMG.20 Thus, for fully protonated systems, the combination of high external magnetic fields and ultrafast MAS is the most straightforward and effective approach for enhancing proton spectral resolution. Further combination with appropriate deuteration and microcrystallization would definitely help in narrowing proton line widths similar to solution NMR.3
1H/1H HOMONUCLEAR CORRELATION EXPERIMENTS UNDER ULTRAFAST MAS
1H/1H chemical shift correlation experiments are quite challenging on solids because of the low spectral resolution of proton spectra. However, with the recent remarkable developments in ultrafast MAS technology, the proton spectral resolution has been greatly enhanced, rendering it possible to study molecular structures with proton site resolution. As the dipolar couplings are greatly suppressed by ultrafast MAS, coherent magnetization transfer based on flip–flop or flip–flip/flop–flop spin diffusion has been greatly slowed down. Therefore, 1H–1H dipolar couplings have to be reintroduced for fast proton magnetization transfer among protons to establish homonuclear correlations. Finite-pulse radiofrequency driven dipolar recoupling (fp-RFDR) is one of the simplest rotor-synchronized sequences (τ−π−τ) for recoupling zero-quantum dipolar interactions under fast MAS.21,22 In fact, our recent studies have demonstrated the merits of using RFDR on mobile solids to recover motionally averaged dipolar couplings among protons.23,24 We have also applied fp-RFDR on rigid solids for recoupling the dipolar couplings averaged by ultrafast MAS to accelerate the coherent polarization transfer among protons,25,26 as shown in Figure 2A. A new phase cycling scheme, XY414, was proposed due to its superior performance over other phase cycling schemes in overcoming the RF field inhomogeneity as well as chemical shift offsets, regardless of the strength of the applied RF field.25,27 As the XY8-based phase cycling28 removes the dipolar × dipolar cross-terms of the Hamiltonian that involve zero-quantum (ZQ) and double-quantum (DQ) homonuclear terms, which could assist the magnetization transfer,27 the dipolar recoupling and magnetization transfer efficiency of XY414 is better than that of XY814 (a supercycling scheme of XY8).30 However, XY414 and XY814 both have similar performances when used for studies on low-γ nuclei (like 13C, 15N) due to their weak homonuclear dipolar couplings (i.e., the high-order dipolar–dipolar cross terms could be ignored). As also shown in Figure 2B, a 2.84 ms fp-RFDR mixing time is sufficient to achieve a total correlation among all proton resonances in the N-acetyl-15N-L-valyl-15N-L-leucine (NAVL) sample.29 Significant sensitivity and resolution enhancements are also observed when the spinning frequency is increased from 40 to 90 kHz, particularly in the 0–5 ppm region. In particular, the two peaks observed around 4 ppm are well resolved under >60 kHz MAS, which is more clearly revealed from the 1D slices along the F2 dimension taken at 8.9 ppm of the F1 dimension (as shown in Figure 2C). By changing the fp-RFDR mixing time, the time-dependent behavior of cross-peak intensities could be potentially used to quantify the dipolar couplings recovered under ultrafast MAS, and thus the 1H–1H distances.23 Consequently, the build-up rates of the cross-peak intensities as a function of fp-RFDR mixing time could directly provide proximity information on the proton pairs in rigid solids.23,27 Thus, the 1H/1H fp-RFDR-based chemical shift correlation experiment is a robust method for probing proton–proton proximities due to its high sensitivity and enhanced spectral resolution under ultrafast MAS. In case of mobile solids, however, care must be taken in the analysis of the build-up curves of cross-peaks intensities for extracting accurate information on proton proximity due to the mobility-induced averaging of dipolar couplings. In addition, by incorporating constant-time 13C chemical shift evolution into the pulse sequence, 1H/1H correlation among protons bonded to two neighboring carbons could also be obtained for further improving proton spectral resolution as well as resonance assignments.31
Figure 2.
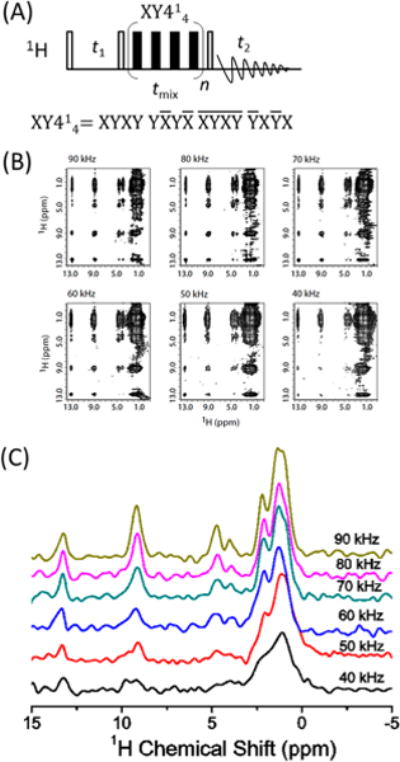
Chemical shift correlation of protons enabled by ultrafast MAS. (A) 2D 1H/1H chemical shift correlation experiment using fp-RFDR to recouple proton dipolar couplings. XY414 scheme was used for the fp-RFDR sequence. (B) 2D 1H/1H fp-RFDR spectra of N-acetyl-15N-L-valyl-15N-L-leucine (NAVL) obtained at different sample spinning speeds as indicated. As seen from the spectra, enhancement of spectral resolution is achieved by increasing the MAS speed. The fp-RFDR mixing time was 2.84 ms; 64 t1 increments were used. Reproduced with permission from ref 29. Copyright 2015 Nature Publishing Group. (C) 1D 1H F2 slices at the chemical shift of 8.9 ppm of F1 dimension at different spinning speeds.
DETERMINATION OF PROTON CHEMICAL SHIFT TENSORS
Proton chemical shift tensor is a very sensitive probe of the local inter- and intramolecular interactions, such as the ring current effect and hydrogen bonds that play important roles in controlling the three-dimensional molecular conformations and packing in solid-state structures of various organic and functional materials. Recently, R-symmetry-based two-dimensional (2D) 1H/1H anisotropic/isotropic chemical shift correlation experiments for the accurate measurement of the 1H CSA under ultrafast MAS have been well reported.32–34 However, even at the highest currently achievable spinning frequency of ∼130 kHz, 1H MAS NMR spectra of densely proton-coupled rigid solids still suffer from poor resolution and severe peak overlapping. In such cases and to a large extent, it is difficult to determine the CSA of overlapped proton sites with high accuracy in the standard 1H/1H chemical shift anisotropy/ single-quantum chemical shift (CS/SQ) correlation experiments. However, the double quantum (DQ) chemical shift spectrum has a doubled spectral width compared to the single quantum (SQ) chemical shift spectrum, indicating a higher spectral resolution along the DQ dimension. By correlating DQ chemical shift with chemical shift anisotropy, it is thus possible to extract chemical shift tensors of proton sites whose signals are not well resolved along the SQ chemical shift dimension.35 The pulse sequence is shown in Figure 3A. Broadband Back-to-Back (BABA) sequence36,37 is initially employed to excite DQ coherence for recording the DQ chemical shift evolution, and then the DQ coherence is reconverted to zero-quantum (ZQ) followed by chemical shift anisotropy evolution. As shown in Figure 3B for ibuprofen, the peak at 0.8 ppm originates from three different methyl groups. Therefore, it is not possible to specifically measure the CSA tensor of each methyl group from the standard 2D CSA/CS correlation experiment. Instead, it is noteworthy that the peak at a DQ frequency of 13.4 ppm is induced by the hydroxyl proton (proton 1) and its nearby methyl protons (proton 11). By taking the CSA slice at the DQ chemical shift of 13.4 ppm, as shown in Figure 3C, the CSA tensors of protons 11 and 1 could be extracted with the help of numerical simulations. Similarly, the DQ peak at DQ frequency of 19.7 ppm is induced by hydroxyl (proton 1) and aromatic (protons 2, 3, 4, and 5) protons, and the CSA slice taken at the DQ frequency of 19.7 ppm contains the CSA information for both hydroxyl and aromatic protons. Therefore, when the proton SQ resolution is relatively poor, valuable information can be obtained if the DQ dimension could be well utilized and correlated with other spin interactions. Recently, a proton-detected 3D 15N/1H/1H isotropic/anisotropic/isotropic chemical shift correlation experiment was also proposed to obtain site-specific CSA of amide protons by using the broader span of 15N isotropic chemical shifts.38 It is worth noting that the frequency separation between the observed singularities in the proton CSA line shape is insensitive to the asymmetry parameter (η) but increases linearly with increasing anisotropic chemical shift parameter (δaniso). However, the line shapes around the singularities vary significantly with η.35 Therefore, the premise for extracting accurate CS parameters is to record reliable and undistorted experimental powder CS line shapes.
Figure 3.
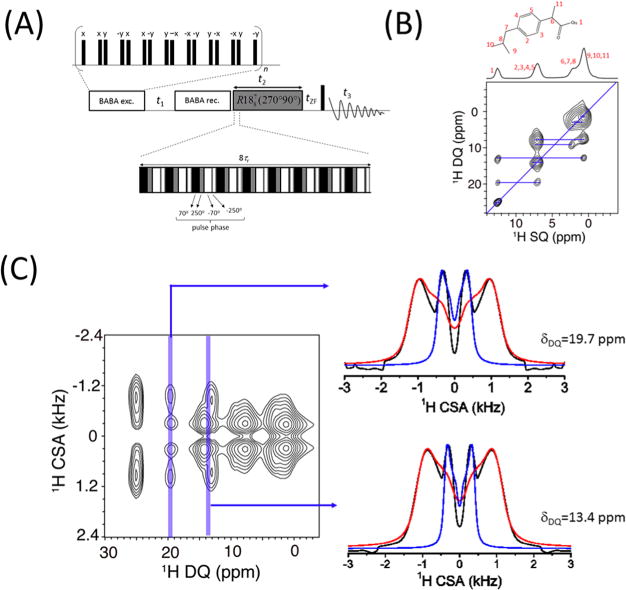
3D ultrafast MAS technique enables the measurement of proton CSAs. (A) Pulse sequence used for the 3D DQ/CSA/SQ correlation experiment. Broadband BABA sequence is used for DQ excitation and reconversion, while a composite-180° (namely, 270°0–90°180) based symmetry sequence R1887 is used for recoupling proton CSA. (B) Molecular structure of ibuprofen and 2D DQ/SQ (F1/F3) chemical shift correlation spectrum extracted from the 3D spectrum. (C) 2D CSA/DQ correlation spectrum extracted from the 3D spectrum of ibuprofen. The CSA slices at the DQ frequency of 19.7 ppm and that at 13.4 ppm are shown on the right. Experimental line shapes are indicated in black, while the simulated ones are indicated in red and blue. Reprinted from ref 35, with the permission of AIP Publishing.
1H–1H DISTANCE MEASUREMENT
It has been a long-standing challenge to measure 1H–1H distances in solids because of the presence of homogeneous and strong 1H–1H dipolar couplings. On one hand, the widespread presence of a multispin 1H–1H dipolar coupling network renders it difficult to average out the 1H–1H dipolar couplings even under ultrafast MAS above 100 kHz. On the other hand, the dipolar truncation effect generally makes the measurement of weak 1H–1H dipolar couplings difficult or impossible.39,40 To overcome these difficulties, we have proposed a method for 1H–1H distance measurements by taking advantage of ultrafast MAS and selective excitation of specific proton peaks.41 On this basis, the magnetization transfer between the selected protons and the nearby ones could be coherently studied by incorporating fp-RFDR,41 as depicted by the pulse sequence shown in Figure 4A. The first cross-polarization (CP) is used to enhance the overall 13C magnetization, while DANTE (delays alternating with nutations for tailored excitation)42 is used to selectively flip the magnetization at a specific 13C chemical shift. As a result, the second short-contact-time CP enables the selection of signals that correspond to protons bonded to the initial selectively excited carbons. Note that if the fp-RFDR mixing time is set to zero, this method could also be used for signal assignment of proton spectra.41 By varying the fp-RFDR mixing time, the signal intensities of nearby protons increase, as shown in Figure 4B, while the signals of selected protons decay. By use of numerical simulations adopting the same parameters used in the experiments, the linear relationship between the signal intensity build-up rate and the dipolar couplings could be obtained, as shown in Figure 4C. The build-up rate of signal intensity is defined as the inverse of the time needed to reach the maximum signal intensity. Once the experimental build-up rate is determined, the dipolar couplings as well as the distance between the proton pair could be extracted from the linear relationship between build-up rate and dipolar couplings. As determined, the H5−H8 and H5−H9 distances are around 2.33 and 2.43 Å, respectively, which are both in good agreement with the X-ray crystal structure.43
Figure 4.
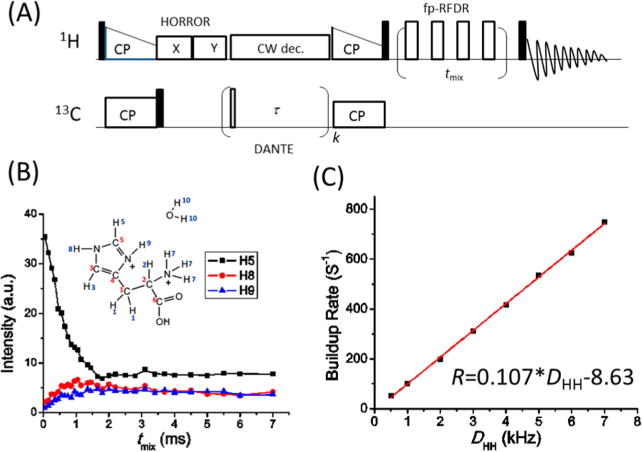
Measurement of proton–proton distances under ultrafast MAS. (A) RF pulse sequence for selective detection of proton resonances and 1H–1H distance measurements. (B) Decay of H5 peak intensity and intensity buildup of H8 and H9 peaks as a function of fp-RFDR mixing time following the selective excitation of the H5 peak in L-histidine·HCl·H2O. The contact time for the first CP is 2 ms, while it is 100 μs for the second CP. (C) Simulated build-up rate (R) as a function of the 1H–1H dipolar couplings (DHH) in a two-spin system. The 1H 90° pulse was 1.4 μs, and the MAS frequency was 60 kHz. Reprinted from ref 41, with the permission of AIP Publishing.
3D EXPERIMENTS FOR HIGHER RESOLUTION AND SPECTRAL EDITING
High-resolution spectra can be obtained either by line width reduction or by rendering the spectra less crowded by filtering undesired signals in multidimensional experiments. The first approach has been discussed above, while the second approach for obtaining high-resolution spectra and extracting atomic-level structural and dynamical information from 3D experiments is discussed in this section.
3D Proton SQ/DQ/SQ Correlation Experiment Provides Three-Spin Proximity Information
The regular 2D DQ/SQ correlation experiment provides information about two-spin proximity only. For probing multispin proximity, one has to resort to multiple-quantum (MQ) NMR experiments; however, the obtained MQ spectra, like the TQ (triple-quantum)/SQ correlation spectrum, are usually more difficult to analyze than DQ/SQ spectra, especially for systems with multiple proton sites. Recently, we have demonstrated that a SQ/DQ/SQ three-dimensional experiment29 (Figure 5A) can provide valuable information about the proximity between a spin and a certain other dipolar-coupled pair of spins, in addition to regular SQ/DQ and SQ/SQ correlations. This approach successfully combines proton detection, ultrafast MAS, and multiple frequency dimensions to yield very high-resolution proton-based 3D NMR spectra. In particular, the DQ/SQ1 (F2/F1) correlation spectrum could provide information about the proximity between a spin and another pair of spins. For example, if proton A has magnetization exchange with proton B through fp-RFDR, while protons B and C are close enough to induce a DQ coherence signal, there will be a cross peak with a chemical shift of proton A in the SQ1 dimension and with the sum of the chemical shifts of protons B and C in the DQ dimension. Furthermore, the DQ/SQ1 (F2/F1) spectra sliced at the isotropic chemical shift of SQ2 (F3) dimension could further distinguish different molecular structures that the regular 2D SQ/DQ spectrum fails to provide, as shown in Figure 5B–D. For example, assume that there are three peaks with chemical shifts of wA, wB, and wC, corresponding to protons A, B1/B2, and C, respectively. If a regular 2D DQ/SQ spectrum gives a DQ cross peaks between chemical shifts wA and wB, as well as between wB and wC, as shown in Figure 5B, it could result from (a) two isolated spin pairs that are far from each other, A−B1 and B2−C, or (b) a “tight” structure with protons B1 and B2 close to each other, A−B1−B2−C or B1−A−B2−C. However, these two structures correspond to two different DQ/SQ1 (F2/F1) spectra if they are sliced at wA from the 3D SQ/DQ/SQ correlation spectrum. The DQ/SQ1 spectrum of structure a would be identical to the regular 2D DQ/SQ spectrum as shown in Figure 5C; whereas for structure b, there is a cross peak between the chemical shifts of wC and wA + wB in the DQ/SQ1 (F2/F1) slice taken at wA, as shown in Figure 5D. More details can be found in ref 29, where the usefulness of such experiment is well demonstrated on the NAVL sample.
Figure 5.
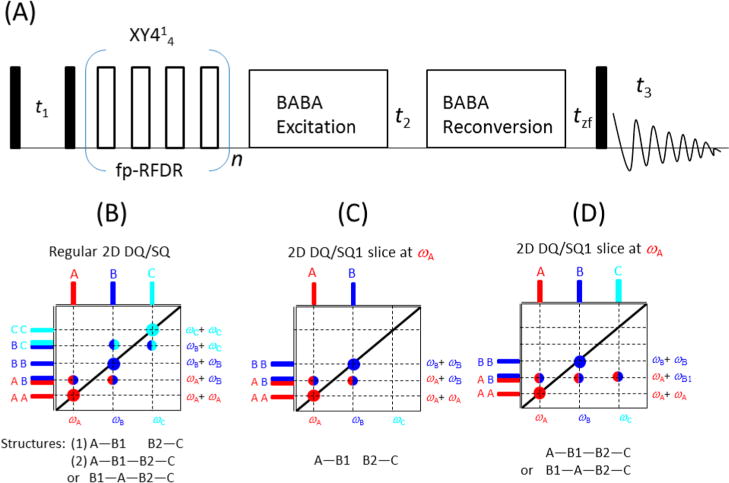
Enhancing proton spectral resolution by using double quantum coherence under ultrafast MAS. (A) Pulse sequence for 3D SQ/DQ/SQ correlation experiment. Reproduced with permission from ref 29. Copyright 2015 Nature Publishing Group. (B) Schematic spectrum for the regular 2D DQ/SQ correlation experiment and the corresponding molecular structures, while panels C and D indicate the 2D DQ/SQ1 (F2/F1) sliced at the isotropic chemical shift of ωA (in the F3 dimension) corresponding to two different structures shown below the spectra.
3D Proton-Detected Experiment for Low-γ Homonuclear/Heteronuclear Correlations
Low-γ homonuclear (e.g., 13C, 15N) correlation experiments are widely used in high-resolution structural studies of proteins. Although 13C/13C chemical shift correlation experiments using various dipolar recoupling techniques are commonly used under MAS, 15N/15N homonuclear correlation experiments have always been challenging due to the very weak (almost negligible) 15N−15N dipolar couplings in peptides and proteins.44,45 However, by utilizing protons to transfer magnetization among 15N nuclei, 15N/15N correlations could be easily achieved.46 In addition, proton-detection under ultrafast MAS generally enables a higher sensitivity and thus reduces the experimental time.47 The pulse sequence for a proton-detected 3D 15N/15N/1H correlation experiment48 is shown in Figure 6A, where four CP periods are used for polarization transfer between 1H and 15N. By taking a slice at the isotropic chemical shift of the amide proton, the 15N/15N homonuclear correlation spectrum could be easily obtained (Figure 6B). The cross peaks between two different 15N chemical shifts can be clearly observed, which is not seen in the regular 2D 15N/15N correlation experiment using 15N−15N dipolar couplings for polarization transfer. While it is possible to correlate intra- or intermolecular 15N nuclei depending on the molecular structure under investigation, the 1H–1H mixing time determines whether 15N−15N correlation is intramolecular or intermolecular in peptides or proteins, which is important for structural analysis. Similarly, 14N/14N correlation can be accomplished from a 3D 14N/14N/1H chemical shift correlation experiment through fp-RFDR using four simple J-HMQC polarization transfer periods.49
Figure 6.
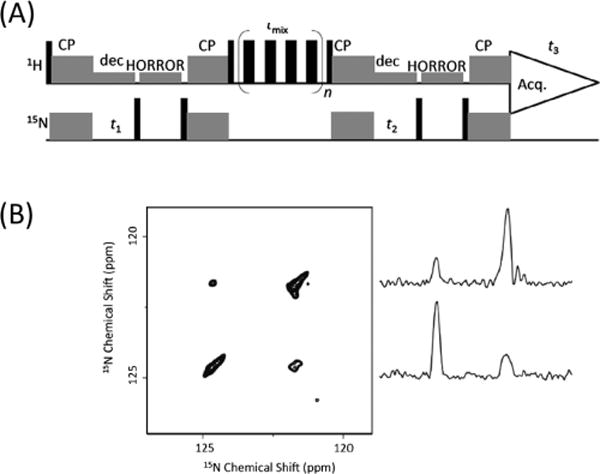
Chemical shift correlation of 15N nuclei enabled by proton mixing under ultrafast MAS. (A) Proton-detected 3D 15N/15N/1H experiment that correlates the isotropic chemical shifts of 15N, 15N, and 1H nuclei. (B) A 15N/15N correlation spectrum sliced at the amide proton chemical shift from the 3D 15N/15N/1H spectrum with a 6.4 ms RFDR mixing time at 100 kHz MAS on powdered NAVL. 1D spectral slices extracted from the 2D 15N/15N spectrum are shown (right). Adapted with permission from ref 48. Copyright 2014 Elsevier.
Although CP is widely used for heteronuclear polarization, RINEPT pulse sequence can also be employed for polarization transfer in rigid solids.14,50 However, such transfer requires the simultaneous presence of 1H–1H and 1H–13C dipolar couplings. Herein, the quaternary carbon signal is generally filtered out by 1H → 13C INEPT under ultrafast MAS due to the weak remote 13C−1H dipolar couplings and absence of 1H−1H dipolar coupling nearby.50,51 In the same manner, the non-carbon-bonded proton signals will also be filtered out by the 13C → 1H INEPT transfer, which could in principle improve the proton spectral resolution.51 Finally, it is worth noting that a multidimensional experiment is always challenging for natural-abundance samples when low-γ nuclei are involved. However, as we recently demonstrated, an incorporation of multiple-contact CP could significantly enhance the sensitivity of 2D proton-detected 13C/1H HETCOR experiment; the achieved signal enhancement is more impressive when natural-abundance 13C spectrum is directly recorded.52
PERSPECTIVES AND OUTLOOK
By virtue of enhanced resolution and high sensitivity of proton detection under ultrafast MAS, proton-based NMR spectroscopy has gained much popularity in investigating the molecular structures and dynamics in various molecular systems. One-dimensional 1H MAS NMR spectra could be used to identify and distinguish the different polymorphs of solid compounds due to the sensitivity of chemical shift environment to subtle changes in the structures, while a 2D 1H/1H fp-RFDR-based SQ/SQ chemical shift correlation experiment directly provides information on site-specific proton proximity. The 2D 1H/1H DQ/SQ correlation experiment is a good alternative to the SQ/SQ chemical shift correlation experiment due to the doubled spectral width along the DQ dimension. A 3D single-channel proton-based 1H NMR experiment, by using the high resolution advantage of the DQ dimension, could be further employed to extract the structural information that was missing in the regular 2D experiments. Furthermore, by involving other nuclei, such as 13C or 15N, site-specific proton signals could be obtained or filtered out and thus enabled the extraction of 1H–1H distances. The proton-detected HETCOR experiment has also been widely used for rapid resonance assignment and determination of heteronuclear proximity. Also, low-γ nuclei homonuclear correlation could be achieved by using protons as the polarization transfer media and proton detection for signal enhancement. Therefore, an ensemble of key proton-based experiments under ultrafast MAS conditions could offer significant insights into molecular structures and dynamics. With the rapid developments in ultrafast MAS probe technology, high resolution proton-based ssNMR spectroscopy will become more user-friendly and popular in the near future to investigate the structures and dynamics of a wide variety of molecular systems, including fully pronated proteins53 or labeled peptides in cell membrane.54 However, proton resolution obtained so far is still limited by the unaveraged higher-order dipolar coupling terms and anisotropic bulk magnetic susceptibility, particularly in densely proton-coupled solids. In this case, microcrystallization can reduce the chemical shift dispersion due to conformational heterogeneity and therefore can render resolution enhancement; further developments in the chemical and biological approaches to prepare NMR-friendly samples would greatly assist the further developments and widespread applications of ultrafast MAS ssNMR. In addition, the friction-induced sample heating could become a significantly limiting factor for the functional heat-sensitive biological samples as the spinning speed becomes >100 kHz. It should also be pointed out that the decreasing sample volume rendered by the decreasing rotor dimension to accomplish ultrafast spinning speeds is another significant factor to be dealt with particularly when proton-detected multidimensional experiments involving low abundant, low-γ nuclei on naturally abundant macromolecules or biopolymers are performed. Nevertheless, there are still considerable perspectives for further improving the proton spectral resolution by increasing MAS frequency, boosting the static magnetic field, appropriate deuteration schemes, approaches to enhance sample quality, implementation of fast data acquisition methods, or a combination of these approaches. Furthermore, the resolution and sensitivity enhancements offered by ultrafast MAS 1H-based NMR spectroscopy can provide new prospects for studying insoluble noncrystalline solids that cannot be studied by solution NMR spectroscopy or diffraction techniques.
Acknowledgments
We acknowledge financial support from the National Institutes of Health (GM084018 to A.R.). We thank Dr. Yusuke Nishiyama and JEOL RESONANCE for fruitful collaborations in the development of ultrafast-MAS techniques using a 0.75 mm MAS probe.
Biographies
Rongchun Zhang was a postdoctoral research fellow working with Prof. Ayyalusamy Ramamoorthy in the Department of Chemistry and Biophysics Program at the University of Michigan, Ann Arbor, focusing on the development of solid-state NMR methodology for the study of materials. He received his Ph. D. from Nankai University studying the microstructure and dynamics of polymers with high- and low-field NMR in the laboratories of Prof. Pingchuan Sun. He recently joined the State Key Laboratory of Medicinal Chemical Biology at Nankai University, China.
Kamal H. Mroue was a postdoctoral research and teaching fellow working with Prof. Ayyalusamy Ramamoorthy in the Department of Chemistry and Biophysics Program at the University of Michigan, Ann Arbor. His current research interests focus on applying and further developing modern multidimensional NMR spectroscopy to understand the structure and dynamics of complex biological materials, such as bone and related tissues, and how they are related to biological function. He received his Ph.D. in Chemistry from the University of Waterloo, Ontario, Canada, where he worked in the laboratory of Prof. William P. Power on the application of solid-state NMR techniques for studying half-integer quadrupolar nuclei in organometallic complexes.
Ayyalusamy Ramamoorthy is Robert W. Parry Collegiate Professor of Chemistry and Biophysics at the University of Michigan, where he has been a faculty member since 1996. His current research interests are in the development and applications of NMR spectroscopy to study the structure, dynamics, and function of membrane protein complexes, amyloid proteins, nanomaterials, and biomineralized tissues. Details about his current research can be found at http://www.umich.edu/~ramslab.
Footnotes
ORCID
Rongchun Zhang: 0000-0002-2480-2652
Ayyalusamy Ramamoorthy: 0000-0003-1964-1900
Notes
The authors declare no competing financial interest.
References
- 1.Ernst RR, Bodenhausen G, Wokaun A. Principles of nuclear magnetic resonance in one and two dimensions. Clarendon Press; Oxford: 1987. [Google Scholar]
- 2.Maly T, Debelouchina GT, Bajaj VS, Hu K-N, Joo C-G, Mak-Jurkauskas ML, Sirigiri JR, van der Wel PCA, Herzfeld J, Temkin RJ, Griffin RG. Dynamic nuclear polarization at high magnetic fields. J Chem Phys. 2008;128:052211. doi: 10.1063/1.2833582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chevelkov V, Rehbein K, Diehl A, Reif B. Ultrahigh Resolution in Proton Solid-State NMR Spectroscopy at High Levels of Deuteration. Angew Chem, Int Ed. 2006;45:3878–3881. doi: 10.1002/anie.200600328. [DOI] [PubMed] [Google Scholar]
- 4.Akbey Ü, Lange S, Trent Franks W, Linser R, Rehbein K, Diehl A, van Rossum B-J, Reif B, Oschkinat H. Optimum levels of exchangeable protons in perdeuterated proteins for proton detection in MAS solid-state NMR spectroscopy. J Biomol NMR. 2010;46:67–73. doi: 10.1007/s10858-009-9369-0. [DOI] [PubMed] [Google Scholar]
- 5.Madhu PK. High-resolution solid-state NMR spectroscopy of protons with homonuclear dipolar decoupling schemes under magic-angle spinning. Solid State Nucl Magn Reson. 2009;35:2–11. doi: 10.1016/j.ssnmr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Nishiyama Y. Fast magic-angle sample spinning solid-state NMR at 60–100 kHz for natural abundance samples. Solid State Nucl Magn Reson. 2016;78:24–36. doi: 10.1016/j.ssnmr.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Bertini I, Emsley L, Lelli M, Luchinat C, Mao J, Pintacuda G. Ultrafast MAS Solid-State NMR Permits Extensive 13C and 1H Detection in Paramagnetic Metalloproteins. J Am Chem Soc. 2010;132:5558–5559. doi: 10.1021/ja100398q. [DOI] [PubMed] [Google Scholar]
- 8.Reif B. Ultra-high resolution in MAS solid-state NMR of perdeuterated proteins: Implications for structure and dynamics. J Magn Reson. 2012;216:1–12. doi: 10.1016/j.jmr.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 9.Parthasarathy S, Nishiyama Y, Ishii Y. Sensitivity and Resolution Enhanced Solid-State NMR for Paramagnetic Systems and Biomolecules under Very Fast Magic Angle Spinning. Acc Chem Res. 2013;46:2127–2135. doi: 10.1021/ar4000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Böckmann A, Ernst M, Meier BH. Spinning proteins, the faster, the better? J Magn Reson. 2015;253:71–79. doi: 10.1016/j.jmr.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Andreas LB, Le Marchand T, Jaudzems K, Pintacuda G. High-resolution proton-detected NMR of proteins at very fast MAS. J Magn Reson. 2015;253:36–49. doi: 10.1016/j.jmr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Ernst M, Samoson A, Meier BH. Low-power decoupling in fast magic-angle spinning NMR. Chem Phys Lett. 2001;348:293–302. [Google Scholar]
- 13.Laage Sgn, Marchetti A, Sein J, Pierattelli R, Sass HJ, Grzesiek S, Lesage A, Pintacuda G, Emsley L. Band-Selective 1H− 13C Cross-Polarization in Fast Magic Angle Spinning Solid-State NMR Spectroscopy. J Am Chem Soc. 2008;130:17216–17217. doi: 10.1021/ja805926d. [DOI] [PubMed] [Google Scholar]
- 14.Holland GP, Cherry BR, Jenkins JE, Yarger JL. Proton-detected heteronuclear single quantum correlation NMR spectroscopy in rigid solids with ultra-fast MAS. J Magn Reson. 2010;202:64–71. doi: 10.1016/j.jmr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haeberlen U, Waugh JS. Coherent Averaging Effects in Magnetic Resonance. Phys Rev. 1968;175:453–467. [Google Scholar]
- 16.Maricq MM, Waugh JS. NMR in rotating solids. J Chem Phys. 1979;70:3300–3316. [Google Scholar]
- 17.Pandey MK, Zhang R, Hashi K, Ohki S, Nishijima G, Matsumoto S, Noguchi T, Deguchi K, Goto A, Shimizu T, Maeda H, Takahashi M, Yanagisawa Y, Yamazaki T, Iguchi S, Tanaka R, Nemoto T, Miyamoto T, Suematsu H, Saito K, Miki T, Ramamoorthy A, Nishiyama Y. 1020 MHz single-channel proton fast magic angle spinning solid-state NMR spectroscopy. J Magn Reson. 2015;261:1–5. doi: 10.1016/j.jmr.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mroue KH, Nishiyama Y, Kumar Pandey M, Gong B, McNerny E, Kohn DH, Morris MD, Ramamoorthy A. Proton-Detected Solid-State NMR Spectroscopy of Bone with Ultrafast Magic Angle Spinning. Sci Rep. 2015;5:11991. doi: 10.1038/srep11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samoson A, Tuherm T, Gan Z. High-Field High-Speed MAS Resolution Enhancement in 1H NMR Spectroscopy of Solids. Solid State Nucl Magn Reson. 2001;20:130–136. doi: 10.1006/snmr.2001.0037. [DOI] [PubMed] [Google Scholar]
- 20.Garroway AN. Homogeneous and inhomogeneous nuclear spin echoes in organic solids: Adamantane. J Magn Reson. 1977;28:365–371. [Google Scholar]
- 21.Bennett AE, Griffin RG, Ok JH, Vega S. Chemical shift correlation spectroscopy in rotating solids: Radio frequency-driven dipolar recoupling and longitudinal exchange. J Chem Phys. 1992;96:8624–8627. [Google Scholar]
- 22.Ishii Y. 13C−13C dipolar recoupling under very fast magic angle spinning in solid-state nuclear magnetic resonance: Applications to distance measurements, spectral assignments, and high-throughput secondary-structure determination. J Chem Phys. 2001;114:8473–8483. [Google Scholar]
- 23.Ramamoorthy A, Xu J. 2D 1H/1H RFDR and NOESY NMR Experiments on a Membrane-Bound Antimicrobial Peptide Under Magic Angle Spinning. J Phys Chem B. 2013;117:6693–6700. doi: 10.1021/jp4034003. [DOI] [PubMed] [Google Scholar]
- 24.Pandey MK, Vivekanandan S, Yamamoto K, Im S, Waskell L, Ramamoorthy A. Proton-detected 2D radio frequency driven recoupling solid-state NMR studies on micelle-associated cytochrome-b5. J Magn Reson. 2014;242:169–179. doi: 10.1016/j.jmr.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishiyama Y, Zhang R, Ramamoorthy A. Finite-pulse radio frequency driven recoupling with phase cycling for 2D 1H/1H correlation at ultrafast MAS frequencies. J Magn Reson. 2014;243:25–32. doi: 10.1016/j.jmr.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang R, Ramamoorthy A. Dynamics-based selective 2D 1H/1H chemical shift correlation spectroscopy under ultrafast MAS conditions. J Chem Phys. 2015;142:204201. doi: 10.1063/1.4921381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang R, Nishiyama Y, Sun P, Ramamoorthy A. Phase cycling schemes for finite-pulse-RFDR MAS solid state NMR experiments. J Magn Reson. 2015;252:55–66. doi: 10.1016/j.jmr.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gullion T, Baker DB, Conradi MS. New, compensated Carr-Purcell sequences. J Magn Reson. 1990;89:479–484. [Google Scholar]
- 29.Zhang R, Pandey MK, Nishiyama Y, Ramamoorthy A. A Novel High-Resolution and Sensitivity-Enhanced Three-Dimensional Solid-State NMR Experiment Under Ultrafast Magic Angle Spinning Conditions. Sci Rep. 2015;5:11810. doi: 10.1038/srep11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen M, Hu B, Lafon O, Trébosc J, Chen Q, Amoureux J-P. Broadband finite-pulse radio-frequency-driven recoupling (fp-RFDR) with (XY8)41 super-cycling for homo-nuclear correlations in very high magnetic fields at fast and ultra-fast MAS frequencies. J Magn Reson. 2012;223:107–119. doi: 10.1016/j.jmr.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 31.Zhang R, Ramamoorthy A. Constant-time 2D and 3D through-bond correlation NMR spectroscopy of solids under 60 kHz MAS. J Chem Phys. 2016;144:034202. doi: 10.1063/1.4940029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levitt MH. Symmetry-Based Pulse Sequences in Magic-Angle Spinning Solid-State NMR. In: Grant DM, Harris RK, editors. Encyclopedia of Nuclear Magnetic Resonance. John Wiley & Sons; Chichester U.K: 2012. [Google Scholar]
- 33.Miah HK, Bennett DA, Iuga D, Titman JJ. Measuring proton shift tensors with ultrafast MAS NMR. J Magn Reson. 2013;235:1–5. doi: 10.1016/j.jmr.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Pandey MK, Malon M, Ramamoorthy A, Nishiyama Y. Composite-180° Pulse-Based Symmetry Sequences to Recouple Proton Chemical Shift Anisotropy Tensors under Ultrafast MAS Solid-State NMR Spectroscopy. J Magn Reson. 2015;250:45–64. doi: 10.1016/j.jmr.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang R, Mroue KH, Ramamoorthy A. Proton chemical shift tensors determined by 3D ultrafast MAS double-quantum NMR spectroscopy. J Chem Phys. 2015;143:144201. doi: 10.1063/1.4933114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feike M, Demco DE, Graf R, Gottwald J, Hafner S, Spiess HW. Broadband multiple-quantum NMR spectroscopy. J Magn Reson Ser A. 1996;122:214–221. [Google Scholar]
- 37.Saalwächter K, Lange F, Matyjaszewski K, Huang C-F, Graf R. BaBa-xy16: Robust and broadband homonuclear DQ recoupling for applications in rigid and soft solids up to the highest MAS frequencies. J Magn Reson. 2011;212:204–215. doi: 10.1016/j.jmr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Pandey MK, Yarava JR, Zhang R, Ramamoorthy A, Nishiyama Y. Proton-detected 3D 15N/1H/1H isotropic/anisotropic/isotropic chemical shift correlation solid-state NMR at 70 kHz MAS. Solid State Nucl Magn Reson. 2016;76–77:1–6. doi: 10.1016/j.ssnmr.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramamoorthy A, Wei Y, Lee DK. PISEMA Solid-State NMR Spectroscopy. Annu Rep NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- 40.Bayro MJ, Huber M, Ramachandran R, Davenport TC, Meier BH, Ernst M, Griffin RG. Dipolar truncation in magic-angle spinning NMR recoupling experiments. J Chem Phys. 2009;130:114506. doi: 10.1063/1.3089370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang R, Ramamoorthy A. Selective excitation enables assignment of proton resonances and 1H-1H distance measurement in ultrafast magic angle spinning solid state NMR spectroscopy. J Chem Phys. 2015;143:034201. doi: 10.1063/1.4926834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bodenhausen G, Freeman R, Morris GA. A simple pulse sequence for selective excitation in Fourier transform NMR. J Magn Reson. 1976;23:171–175. [Google Scholar]
- 43.Oda K, Koyama H. A refinement of the crystal structure of histidine hydrochloride monohydrate. Acta Crystallogr, Sect B: Struct Crystallogr Cryst Chem. 1972;B28:639–642. [Google Scholar]
- 44.Lewandowski JzR, Paëpe GlD, Eddy MT, Griffin RG. 15N−15N Proton Assisted Recoupling in Magic Angle Spinning NMR. J Am Chem Soc. 2009;131:5769–5776. doi: 10.1021/ja806578y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramamoorthy A, Gierasch LM, Opella SJ. Four-Dimensional Solid-State NMR Experiment That Correlates the Chemical-Shift and Dipolar-Coupling Frequencies of Two Hetero-nuclei with the Exchange of Dilute-Spin Magnetization. J Magn Reson Ser B. 1995;109:112–116. doi: 10.1006/jmrb.1995.1157. [DOI] [PubMed] [Google Scholar]
- 46.Wei Y, Ramamoorthy A. 2D 15N−15N isotropic chemical shift correlation established by 1H−1H dipolar coherence transfer in biological solids. Chem Phys Lett. 2001;342:312–316. [Google Scholar]
- 47.Paulson EK, Morcombe CR, Gaponenko V, Dancheck B, Byrd RA, Zilm KW. Sensitive High Resolution Inverse Detection NMR Spectroscopy of Proteins in the Solid State. J Am Chem Soc. 2003;125:15831–15836. doi: 10.1021/ja037315+. [DOI] [PubMed] [Google Scholar]
- 48.Nishiyama Y, Malon M, Ishii Y, Ramamoorthy A. 3D 15N/15N/1H chemical shift correlation experiment utilizing an RFDR-based 1H/1H mixing period at 100kHz MAS. J Magn Reson. 2014;244:1–5. doi: 10.1016/j.jmr.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pandey MK, Nishiyama Y. Proton-detected 3D 14N/14N/1H isotropic shift correlation experiment mediated through 1H−1H RFDR mixing on a natural abundant sample under ultrafast MAS. J Magn Reson. 2015;258:96–101. doi: 10.1016/j.jmr.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 50.Zhang R, Ramamoorthy A. Performance of RINEPT is amplified by dipolar couplings under ultrafast MAS conditions. J Magn Reson. 2014;243:85–92. doi: 10.1016/j.jmr.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang R, Nishiyama Y, Ramamoorthy A. Proton-Detected 3D 1H/13C/1H Correlation Experiment for Structural Analysis in Rigid Solids Under Ultrafast-MAS above 60 kHz. J Chem Phys. 2015;143:164201. doi: 10.1063/1.4933373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang R, Chen Y, Rodriguez-Hornedo N, Ramamoorthy A. Enhancing NMR Sensitivity of Natural-Abundance Low-γ Nuclei by Ultrafast Magic-Angle-Spinning Solid-State NMR Spectroscopy. ChemPhysChem. 2016;17:2962–2966. doi: 10.1002/cphc.201600637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stanek J, Andreas LB, Jaudzems K, Cala D, Lalli D, Bertarello A, Schubeis T, Akopjana I, Kotelovica S, Tars K, Pica A, Leone S, Picone D, Xu Z-Q, Dixon NE, Martinez D, Berbon M, El Mammeri N, Noubhani A, Saupe S, Habenstein B, Loquet A, Pintacuda G. NMR Spectroscopic Assignment of Backbone and Side-Chain Protons in Fully Protonated Proteins: Microcrystals, Sedimented Assemblies, and Amyloid Fibrils. Angew Chem, Int Ed. 2016;55:15504–15509. doi: 10.1002/anie.201607084. [DOI] [PubMed] [Google Scholar]
- 54.Sani MA, Separovic F. Progression of NMR studies of membrane-active peptides from lipid bilayers to live cells. J Magn Reson. 2015;253:138–142. doi: 10.1016/j.jmr.2014.11.016. [DOI] [PubMed] [Google Scholar]