

Abstract
Background
Human progenitor and B-cell development is a highly regulated process characterized by the ordered differential expression of numerous cell-surface and intra-cytoplasmic antigens. This study investigates the underlying coordination of these modulations by examining a series of normal bone marrow samples with the method of probability state modeling or PSM.
Results
The study is divided into two sections. The first section examines B-cell stages subsequent to CD19 up-regulation. The second section assesses an earlier differentiation stage prior to and including CD19 up-regulation.
Post CD19 antigenic Up-regulation: Statistical analyses of cytometry data derived from sixteen normal bone marrow specimens revealed that B cells have at least three distinct coordinated changes, forming four stages labeled as B1, B2, B3, and B4. At the end of B1; CD34 antigen expression down-regulates with TdT while CD45, CD81, and CD20 slightly up-regulate. At the end of B2, CD45 and CD20 up-regulate. At the end of B3 and beginning of B4; CD10, CD38, and CD81 down-regulate while CD22 and CD44 up-regulate.
Pre CD19 antigenic Up-regulation: Statistical analysis of ten normal bone marrows revealed that there are at least two measurable coordinated changes with progenitors, forming three stages labeled as P1, P2, and P3. At the end of P1, CD38 up-regulates. At the end of P2; CD19, CD10, CD81, CD22, and CD9 up-regulate while CD44 down-regulates slightly.
Conclusions
These objective results yield a clearer immunophenotypic picture of the underlying cellular mechanisms that are operating in these important developmental processes. Also, unambiguously determined stages define what is meant by “normal” B-cell development and may serve as a preliminary step for the development of highly sensitive minimum residual disease detection systems. A companion paper is simultaneously being published in Cytometry Part A that will explain in further detail the theory behind PSM.
Keywords: bone marrow ontogeny, flow cytometry, human B-cell differentiation, B-cell development, hematopoietic stem cells, bone marrow microenvironment, monoclonal antibodies, high-dimensional modeling, broadened quantile function modeling, flow cytometry, probability state modeling
Introduction
The common lymphoid progenitor (CLP) responsible for the formation of T, B and NK cells is derived from a hematopoietic stem cell (HSC) that is first identified in the embryonic aorto-gonad-mesonephros, a descendent of the mesoderm. HSCs are defined by their capacity to either self-renew or asymmetrically differentiate into committed progenitors that form all human blood cell lineages. HSCs migrate to the fetal liver and then to the bone marrow, where they reside after birth (1). During lymphoid development from the CLP, the immunophenotypic and genetic properties that distinguish mature cells are gradually acquired while those typical of less differentiated cells are lost. The signals to initiate and regulate development are due to the control imposed by a variety of marrow stromal cells, transcription factors, and coordinated regulation by the nervous system, extracellular matrix, cytokines, and adipocytes found in the bone marrow microenvironment (2).
B cells and their precursors have been extensively studied in mouse and human systems (1,3–15) and there is general agreement that antigenic markers such as CD34, CD38, CD19, CD79a/b, CD45, CD20, CD10, and others help identify the ontological steps or stages that ultimately lead to the circulation of antigen naïve B cells from bone marrow.
The general consensus of the important ontological steps leading to production of naïve B cells is summarized as follows. The earliest identifiable committed B cells derived from the CLP are called progenitor (Pro) B cells. Pro B cells arise after obligate stimulation by the transcription factor PAX-5, which engenders CD19 production. These cell surface expressing CD34+ CD19+ CD10+ CD38+ and nuclear TdT+ expressing cells lack the pre B-cell receptor or surface immunoglobulin (Ig) and characteristically initiate VDJ heavy chain rearrangements independent of any antigenic exposure. Pro B cells subsequently differentiate into CD34− CD19+ CD10+ CD38+ TdT− precursor (Pre) B cells that acquire cytoplasmic and then surface mu heavy chain complexed with a transient surrogate immunoglobulin light chain. Next, a CD19+ CD10dim/− CD38dim/− immature B cell expresses surface IgM+ and physiologic light chain. None of these aforementioned B cell stages are normally found in the circulation of healthy adults. Ultimately CD19+ CD20+ B cells co-expressing IgM and IgD heavy chains (and lacking the early differentiation markers CD34, CD10, CD38 or TdT) exit the bone marrow as transitional B cells and home to secondary lymphoid organs as fully mature naive B cells. Upon exposure to antigen, naïve B cells switch from a CD27− naïve to a CD27+ memory phenotype and undergo further Ig class switching and antibody affinity maturation (16–18).
However, many publications show conflicting definitions for the relative order and existence of these and other antigen modulations (1,4,8,11,16–21). For example, using human cord blood, Beradi et al. (19) showed that B cells and granulocytes arise from CD34+ CD38low CD19− CD10− progenitor cells in the bone marrow, suggesting the presence of a common progenitor. They also determined that the up-regulation of CD10 and CD19 committed the progenitors to the B-cell lineage. However, they did not specifically suggest the order in which CD10 and CD19 are expressed, but vaguely referred to two separate populations defined as CD34+ CD38 low CD19+ and CD34+ CD38low CD10+. Galy et al. (22) and Le Bien (8) both suggest that CD10 up-regulation is then followed by CD19 and B-cell lineage commitment. In contrast to this, Loken et al. (14) found a population of CD34+ CD10+ CD19− cells but determined that these fell outside of the lymphoid light-scattering window and were identified as granulocytes. In their hands CD34+ CD10+ CD19+ were co-expressed and constituted the very early B cells. Other reports suggested that CD19 up-regulated before CD10, and that CD38 remained negative until the differentiation of plasma cells (20). Once committed to the B-cell lineage there also appears to be a lack of agreement as to whether there are three or four stages leading to B cells exiting the bone marrow.
The contradictory nature of these references may have a number of possible causes. In many cases, the relative order of these antigenic changes has been deduced by inspection of complicated bivariate dot-plots that were produced by subjective placement of one or more visually-derived gating regions. Alternative sources of tissue, different cell-culture or study methods may yield different results and many experiments were done sorting for specific populations that also may skew results. For example, the results obtained from either cord blood or bone marrow sources could vary. Alternative antibody clones and a variety of fluorochrome conjugates were likely utilized. Some of the work in human B-cell development has been the result of studying the expression of cell-surface markers obtained from B–cell malignancies; these may or may not be an accurate reflection of normal B-cell development.
Also, it has been difficult to study a group of samples in a statistically valid manner. Recently, a new approach to modeling data called probability state modeling or PSM has been developed that allows a detailed objective analysis of the relative order of antigen intensity up and down-regulation for multi-dimensional cytometry data (23–27). This approach to modeling of complex cellular populations lends itself to reproducible and objective analyses (28–30). Intrinsic to PSM is the notion that a probability-based axis can quantify the relative order of the complex cellular changes that occur in systems like the ontogeny of immune cells.
The purpose of this study is to use probability state modeling, PSM, to quantify the locations of a set of antigen modulations during the ontological development of B cells represented in sixteen listmode files derived from human bone marrow specimens that have no known detectable hematopathological abnormalities. These critical antigenic locations are then subjected to statistical analyses to determine the discrete progenitor and B-cell stages that occur during normal differentiation and maturation (see Figure 1 for an overview of these general stage relationships and the general observations of this study). The theoretical underpinnings and statistical principles behind probability state modeling related to this study are found in our accompanying article (31).
Figure 1.
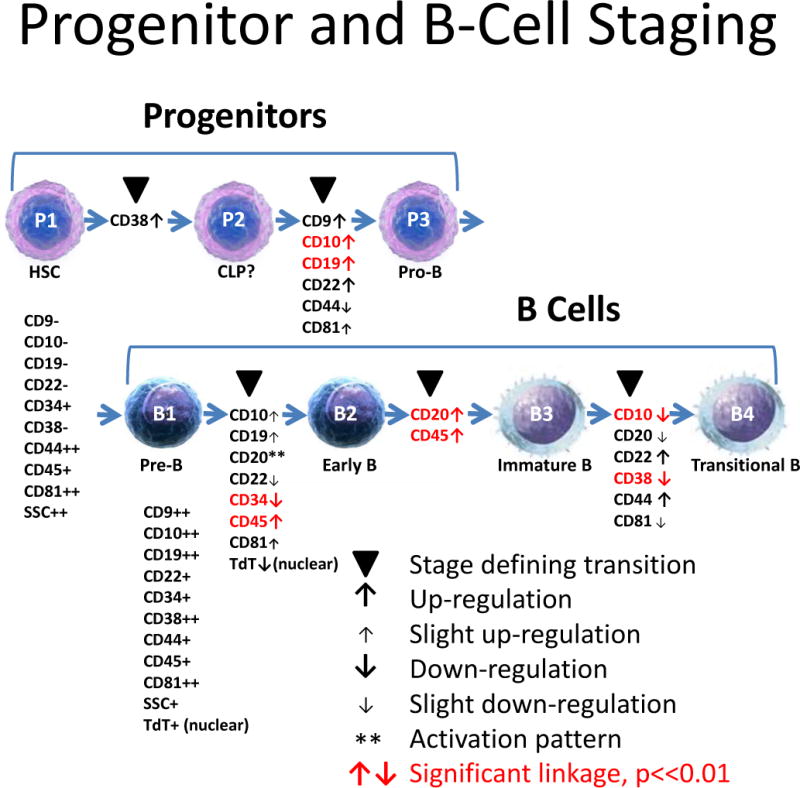
Progenitor and B-Cell Staging. This study is divided into two sections labeled as Progenitors and B cells. The first section examines B-cell stages subsequent to cell surface CD19 up-regulation. This section is presented first because the study’s antibody panels were designed primarily to elucidate this stage. The second section examines progenitor stages prior to and including CD19 up-regulation. In the context of this study, stages are defined as observable points in a cellular progression where the indicated cell-surface markers are either up-regulated (see up-arrows) or down-regulated (see down-arrows) in a coordinated manner (see solid black triangles). To be considered a stage, at least two markers must have significant correlation and any detectable separation must not be significant (see red labels). Other marker transitions not possible to statistically test were positioned to the closest stage boundaries. This study was able to distinguish three separate stages for the Progenitors, labeled as P1, P2, and P3 and four separate stages for the B cells, labeled as B1, B2, B3, and B4. Common names for these stages are shown under the cell pictures.
Our broad goal is to define an objective and statistically meaningful method for staging progressions such as B-cell ontogeny that would permit a greater understanding of the involved biological processes and mitigate the variability of earlier defined staging schemes. The other more utilitarian goal is to create highly constrained “normal” models of B-cell ontogeny in order to ultimately detect and track very low frequency aberrant cells (minimal residual disease) in patients recovering from B-cell malignancy therapies.
Materials and Methods
Immunofluorescence Staining
Samples for data files BM2-6 were prepared as previously published (32). Briefly, subsequent to obtaining IRB approval, unduplicated patient bone marrow specimens [age range 50-71; mean 61.3 years] were collected in yellow top ACD solution tubes and prepared for analysis within 24 hours [time range 16:45-21:30 hours; mean 19.37 hours]. All patient specimens were maintained at room temperature [24°C] until processing. Prior to staining, the specimen was washed three times to remove serum immunoglobulins by resuspending in 1X Dulbecco’s Phosphate Buffered Saline (DPBS) and centrifuging at 500g for five minutes at room temperature. The supernatant was aspirated and discarded. The washed specimen was resuspended to approximately its original unwashed volume with 1% Bovine Serum Albumin in DPBS. One hundred microliters (μl) of prepared specimen was mixed with 7 of the antibodies (see Table 1), vortexed and incubated for 15 minutes at room temperature. Following incubation, 2 ml 1x Becton-Dickinson (BD; San Jose, CA) FACS Lysis buffer was added, vortexed and incubated for 10 minutes at room temperature. The specimen was then centrifuged for five minutes at 500g, and the supernatant discarded. 10 μl of anti-terminal deoxynuclotidyl transferase (TdT) antibody was added and incubated for 20 minutes at room temperature. One ml of DPBS was added and then centrifuged for five minutes at 500g at room temperature, the supernatant discarded and cells resuspended in 500 μL of 1% paraformaldehyde in DPBS and vortexed.
Table 1.
Study File Summary. Sixteen bone marrow specimens from different human donors that were determined to have no observable disease were used for this study. In order to be included in the study, the listmode data had to: 1) include CD19, CD34, CD45, CD10, and CD38, 2) have good visible separation between negative and positive staining, and 3) have adequate numbers of events for staging analyses. For the B-cell staging part of the study, the files needed at least 2,500 CD19 dim/+ events and for the progenitor staging, 1,000 CD34+ events. Those files that satisfy the latter requirement have an ‘*’ suffix.
| File Name | # of Events | #CD19 dim/+ | #Prog. Events | Date Collected | Instrument | Markers Present | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD19 | CD10 | CD34 | CD38 | CD45 | CD20 | TdT | CD81 | CD22 | CD44 | CD9 | ||||||
| B_BM1.fcs* | 250000 | 16811 | 996 | 6-May-13 | Facs CantoII | x | x | x | x | x | x | x | ||||
| B_BM2.fcs | 100000 | 2589 | 23-Apr-14 | Facs CantoII | x | x | x | x | x | x | x | |||||
| B_BM3.fcs | 100000 | 4722 | 23-Apr-14 | Facs CantoII | x | x | x | x | x | x | x | |||||
| B_BM4.fcs | 100000 | 3430 | 1-May-14 | Facs CantoII | x | x | x | x | x | x | x | |||||
| B_BM5.fcs | 100000 | 8559 | 1-May-14 | Facs CantoII | x | x | x | x | x | x | x | |||||
| B_BM6.fcs | 100000 | 5583 | 1-May-14 | Facs CantoII | x | x | x | x | x | x | x | |||||
| B_BM7.fcs* | 250000 | 22012 | 15063 | 11-Jan-12 | Facs CantoII | x | x | x | x | x | x | x | ||||
| B_BM8.fcs* | 337097 | 34849 | 5450 | 25-Aug-09 | LSRII | x | x | x | x | x | x | x | ||||
| B_BM9.fcs* | 417911 | 7446 | 7448 | 3-Feb-10 | LSRII | x | x | x | x | x | x | |||||
| B_BM10.fcs* | 422432 | 11898 | 2927 | 3-Feb-10 | LSRII | x | x | x | x | x | x | |||||
| B_BM11.fcs* | 742320 | 6622 | 7179 | 4-Feb-10 | LSRII | x | x | x | x | x | x | |||||
| B_BM12.fcs* | 522838 | 23228 | 4603 | 4-Feb-10 | LSRII | x | x | x | x | x | x | |||||
| B_BM13.fcs | 171218 | 32332 | 10-Feb-10 | LSRII | x | x | x | x | x | x | x | |||||
| B_BM14.fcs* | 359633 | 30549 | 2476 | 17-Mar-10 | LSRII | x | x | x | x | x | x | x | ||||
| B_BM15.fcs* | 321185 | 6343 | 7385 | 17-Mar-10 | LSRII | x | x | x | x | x | x | x | ||||
| B_BM16.fcs* | 415403 | 10994 | 4737 | 20-Mar-10 | LSRII | x | x | x | x | x | x | x | ||||
Flow Cytometry
All the cytometry data used in this study are MIFlowCyt compliant (33). Sixteen listmode files derived from bone marrow specimens that were determined to have no observable disease were used for this study. These data were originally obtained and identified as uninvolved marrow specimens submitted for hematopoietic diagnostic evaluation. In order to be included in the study, the listmode data had to: 1) include CD19, CD34, CD45, CD10, and CD38, 2) have good visible separation between negative and positive staining, and 3) have adequate numbers of events for staging analyses. For the B-cell staging portion of the study, the files needed at least 2,500 CD19 dim/+ events and 1,000 CD34+ events for the progenitor staging. The study files are summarized in Table 1. The samples for data files BM2-6 were acquired using a standard 3-laser BD FACS Canto II. BD CS&T beads were used for daily instrument setup and standardization. Instrument setup and compensation was performed as described in Cannizzo et al., 2012 (34). Samples for data files B_BM8 −16 were prepared, described, and acquired as in Wood, 2004 (13). Samples for files B_BM1 and B_BM7 were prepared, described, and acquired as in Tario, 2014 (35) where the RBC’s were lysed by a 10 minute incubation with BD FACSLyse at room temperature. Probability State Modeling was performed using GemStone™ Software program version 1.0.115, Verity Software House, Topsham, Maine USA.
Probability State Modeling (PSM)
Probability State Modeling was performed using GemStone™ software program version 1.0.118, Verity Software House, Topsham, Maine USA. PSM is a software system that is specifically designed to model cellular progressions defined by high-dimensional data (31).
Typical modeling packages for cytometry minimize objective functions to find the best mixture of frequency-based model components to “best fit” observed data. Results from these modeling systems tend to be objective and account for measurement overlap. The most common example is the analysis of DNA content to accurately estimate percentages of G1, S, and G2M. Unfortunately, these modeling packages have been largely relegated to single-dimensional analyses because of the increasing complexity of frequency space with multi-dimensional data.
PSM avoids this modeling dimensionality barrier by not using frequency-based model components. Instead, it uses broadened cumulative percent distributions, known as expression profiles, as its model components. Because each expression profile has the same independent x-axis, cumulative percent, they can be “stacked” for any number of measurement dimensions without significantly affecting the complexity of the analysis. Similar to single-dimensional frequency-based modeling packages, PSM is also objective and accounts for measurement overlap. When applied to cellular progressions such as B-cell ontogeny in bone marrow, the analysis provides important insights into the relative timing of key antigen modulations as B cells differentiate and maturate. As shown in this manuscript, PSM also can form average models from multiple samples in order to appropriately stage the development of “normal” B cells.
Post CD19 Antigenic Up-regulation models
Two different models were constructed for this section of the analysis. Both models selected for CD19 dim to positive events that were low for side-scatter (SSC) in the lymphocyte light scatter range. The first model provided no constraints on the timing of up or down-regulation for all the stratification markers shown in Table 1. The second model constrained the up and down-regulations to be on the appropriate stage boundaries (see Tables 4A and 4B for details). All analyses were run unattended for this section.
Table 4A.
Summary of Unconstrained Model Design for B cells. The general model design for the analysis and staging of developing B cells is described above. The model comprises two cell-types, the first is the CD19− population followed by the CD19+. CD19 negative events are minimally modeled with only CD19 modeled as a constant low-pass distribution on the negative peak. Side scatter is also a constant expression profile selecting for the lymphocytes. This cell-type serves to clean-up the data in order to more cleanly select for the CD19 dim to positive events in the second cell-type. CD19 positive events are selected for with an inverse low-pass for CD19, which selects for events that are “not” CD19 negative. Again, side scatter is a constant expression profile and CD33 and CD13 are used to exclude the myeloid population. Staging, or stratification, is accomplished as shown above. The “type” defines the type of expression profile used; the “level(s)” explain how the marker modulates. Since some files do not have all markers present, the markers identified in the “comment” column may be blank for some data files.
| Post CD19 Up-Regulation Model: (Unconstrained Model) | |||||
|---|---|---|---|---|---|
| CellType | Purpose | Marker | Type | Level(s) | Comment |
| CD19− | Selection | CD19 | Low-Pass | Neg | |
| SSC-A | Constant | Lymphs | |||
| CD19+ | Selection | CD19 | Inv Low-Pass | Dim to Pos | |
| SSC-A | Constant | Lymphs | |||
| CD33 | Constant | Neg | If present | ||
| CD13 | Constant | Neg | If present | ||
| CD304 | Constant | Neg | If present | ||
| Stratification | CD10 | Step-Down | Pos to Neg | ||
| CD38 | Step-Down | Pos to Neg | |||
| CD34 | Step-Down | Pos to Neg | |||
| TdT | Step-Down | Pos to Neg | If present | ||
| CD45 | Three-Level | Dim, Int, Pos | |||
| CD20 | Three-Level | Neg, Dim, Pos | If present | ||
| CD44 | Step-Up | Neg to Pos | If present | ||
| CD22 | Step-Up | Neg to Pos | If present | ||
| CD81 | Three-Level | Dim, Pos, Dim | If present | ||
Table 4B.
Summary of Constrained Model Design for B Cells. This model uses linkages between markers to create defined stages based on the statistical analysis done for each individual data file. The general model design is the same as is summarized in Table 4A, with the addition of the linkages explained above. The second column defines the primary markers, CD10, CD34, and CD45. The control point is where the linkage is established, for example, at the point where CD10 starts to down-regulate, or “begin-down”, CD38, CD81, CD22 and CD44 are linked. The last column “Link Position” identifies the point at which CD10 (or CD34 or CD45) is linked in the expression profile of the specific Linked Marker.
| Post CD19 Up-Regulation Model: (Constrained Version) | ||||
|---|---|---|---|---|
| Cell Type | Marker | Control Point | Linked Marker | Link Position |
| CD19+ | CD10 | Begin Down | CD38 | Begin Down |
| CD81 | End Level 2 | |||
| CD22 | Begin Up | |||
| CD44 | Begin UP | |||
| CD34 | Begin Down | CD45 | End Level 1 | |
| TdT | Begin Down | |||
| CD81 | End Level 1 | |||
| CD20 | End Level 1 | |||
| CD45 | End Level 2 | CD20 | End Level 2 | |
Pre CD19 Antigenic Up-regulation models
Unconstrained and constrained models were also constructed for the progenitors as defined by CD34, CD38, CD45, and SSC. Both models selected for events that were CD34 positive, CD45 low to intermediate intensity, and SSC low to intermediate intensity. Fewer files were used in this section of the study due to inadequate numbers of CD34+ events in the omitted files (see Tables 5A and 5B for details).
Table 5A.
Summary of Unconstrained Model Design for Progenitor Cells. The general model design for the analysis and staging of progenitor cells is described above. The model comprises one cell-type which is defined or selected for by constant expression profiles for CD45, SSC-A, and CD34. Staging, or stratification, is accomplished with the markers as shown in above. The “type” defines the type of expression profile used; the “level(s)” explain how the marker modulates. Since some files do not have all markers present, the markers identified in the “comment” column may be blank for some data files.
| Progenitor Cell Model: (Unconstrained Model) | ||||
|---|---|---|---|---|
| Purpose | Marker | Type | Levels | Comment |
| Selection | CD45 | Constant | Int | |
| SSC-A | Constant | Low | ||
| CD34 | Constant | Pos | ||
| Stratification | CD19 | Step-Up | Neg to Pos | |
| CD10 | Step-Up | Neg to Pos | ||
| CD22 | Step-Up | Neg to Pos | If present | |
| CD38 | Step-Up | Neg to Pos | ||
| CD44 | Step-Down | High to Dim | If present | |
| TdT | Step-Up | Dim to Pos | If present | |
| CD9 | Step-Up | Neg to Pos | If present | |
| CD81 | Step-Up | Int to High | If present | |
Table 5B.
Summary of Constrained Model Design for Progenitor Cells. This model uses linkages between markers to create defined stages based on the statistical analysis done for each individual data file. The general model design is the same as is summarized in Table 5A, with the addition of the linkages explained above. The first column defines the primary marker, CD19. The control point is where the linkage is established, i.e., the point at which CD19 begins to up-regulate. The last column identifies the specific point at which CD19 is linked in each individual expression profile for CD10, CD44, CD22, CD81 or CD9.
| Progenitor Cell Model: (Constrained Version) | |||
|---|---|---|---|
| Marker | Control Point | Linked Marker | Link Position |
| CD19 | Begin Up | CD10 | Begin Up |
| CD44 | Begin Down | ||
| CD22 | Begin Up | ||
| CD81 | Begin Up | ||
| CD9 | Begin Up | ||
For those investigators who are interested in reproducing these findings with their own data sets, all the models are available as down-loadable files from the supplemental materials section of the journal.
Statistical Analyses
The Pearson correlation coefficients, r, t-values, and Student t-test were evaluated as described in (36)
Results
B-Cell Stage Analysis
Figure 2 is a representative summary plot of an unattended analysis for one file. The PSM overlay plot summarizes the modulation of CD34, CD45, CD20, CD10, and CD38 expression during B-cell ontogeny. The modeling process reduces the listmode data to a relatively small set of critical Control Definition Points (CDPs) for each modeled measurement. CDP’s are three-dimensional points that define measurement intensity, progression location in cumulative percent units, and line-spread for a correlated set of measurements (31). The listmode events have been selected for CD19 dim/+ expression and SSC levels consistent with lymphocytes. This example analysis shows that shortly after CD34 down-regulates, CD45 up-regulates slightly (see open triangles for control points a and b). As CD45 up-regulates for the second time, CD20 up-regulates (control points c and d). Finally, when CD38 down-regulates, CD10 also down-regulates (control points e and f). No stage information is shown in this figure because the statistically determined stages are not known at this point in the analysis.
Figure 2.
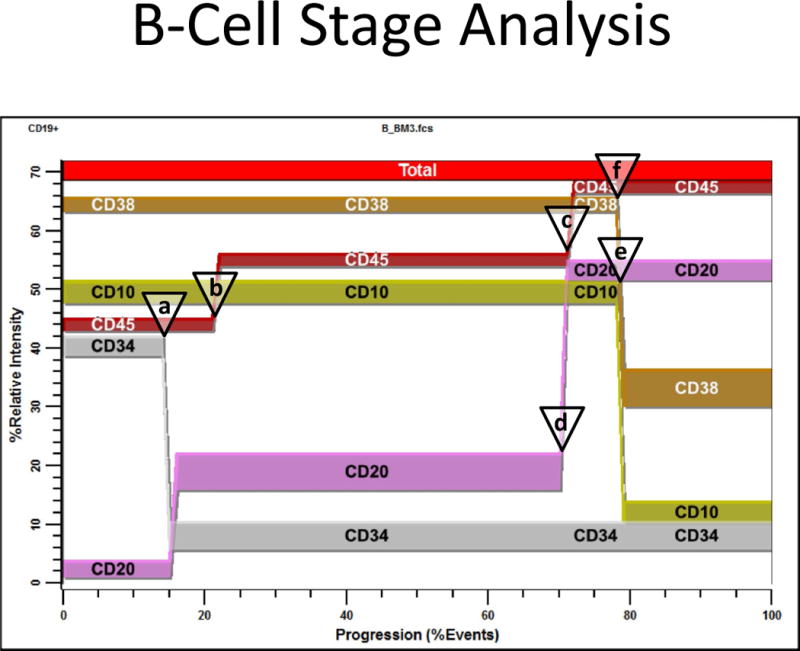
B-Cell Stage Analysis. This figure shows an unattended analysis of the study file B_BM3.fcs. The PSM overlay plot summarizes modulation of CD34, CD45, CD20, CD10, and CD38 during B-cell ontogeny. Modeling reduces the listmode data to a set of critical control points that can be used to quantify the relative order of marker changes. The listmode events have been selected for CD19 dim to positive and SSC intensity level consistent with lymphocytes (see Material & Methods for details). When CD34 down-regulates, CD45 up-regulates slightly (see open triangles for control points a and b). When CD45 up-regulates for the second time, CD20 up-regulates (control points c and d). Finally, when CD10 down-regulates, CD38 down-regulates (control points e and f). These locations are quantified by the PSM model as cumulative percentages (see (31) for details) and summarized for all files in Table 2. No stage information is shown in this figure because the statistically determined stages are not known at this point in the analysis.
All files were initially analyzed unattended with an unconstrained B-cell model to obtain critical stage locations for markers CD34, CD45, CD20, CD10, and CD38. The recorded results for all sixteen study files are shown in Table 2. Markers were deemed to be on the same stage boundary if their correlation coefficients, r, were significant and their t values were insignificant at a p value greater than 0.01 (see bottom of Table 2). Markers that had significant t values at p<0.01 were considered to be on different stage boundaries.
Table 2.
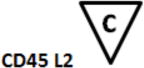
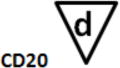
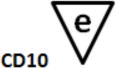
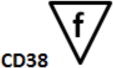
B-cell Stage Results. All files were initially analyzed unattended with a B-cell model to obtain critical stage locations for markers CD34, CD45, CD20, CD10, and CD38. The units for the locations are cumulative percentages (see (31) for details on why the cumulative percentages are useful for representing cellular progressions). The recorded results for all sixteen study files are shown above. Markers were deemed to be on the same stage boundary if their correlation coefficients, r, were significant and their t values were not (see bottom of table). Since CD34 (a) versus CD45 L2 (c) (t=9.9, p<0.00001) and CD45 L2 (c) versus CD10 (e) (t=3.5, p<0.004) were found to be significantly different, they were considered to form different stage boundaries.
| B1 | B2 | B3 and B4 | ||||
|---|---|---|---|---|---|---|
| Files |
|
|
|
|
|
|
| B_BM1 | 7.7 | 9.1 | 58.7 | 67.8 | 67.1 | |
| B_BM2 | 27.2 | 29.0 | 96.4 | 95.7 | 95.4 | 94.6 |
| B_BM3 | 14.1 | 21.3 | 71.5 | 70.8 | 78.6 | 77.8 |
| B_BM4 | 19.4 | 26.3 | 65.1 | 65.1 | 74.1 | 71.8 |
| B_BM5 | 5.1 | 7.2 | 79.4 | 76.2 | 92.3 | 92.7 |
| B_BM6 | 3.6 | 3.6 | 18.0 | 20.1 | 24.3 | 23.1 |
| B_BM7 | 7.5 | 8.2 | 73.4 | 47.5 | 79.5 | 59.3 |
| B_BM8 | 9.3 | 9.0 | 71.8 | 71.8 | 81.9 | 89.2 |
| B_BM9 | 48.6 | 50.6 | 98.8 | 100.0 | 96.7 | 95.7 |
| B_BM10 | 6.6 | 8.7 | 37.2 | 37.1 | 41.6 | 40.7 |
| B_BM11 | 11.5 | 12.3 | 83.4 | 82.0 | 83.2 | 82.2 |
| B_BM12 | 4.1 | 4.9 | 24.3 | 24.6 | 26.4 | 26.2 |
| B_BM13 | 2.4 | 6.6 | 95.4 | 97.0 | 97.5 | 95.6 |
| B_BM14 | 5.1 | 5.7 | 37.4 | 37.4 | 40.8 | 39.9 |
| B_BM15 | 41.4 | 66.6 | 81.2 | 80.6 | 78.4 | 81.7 |
| B_BM16 | 33.1 | 35.6 | 92.2 | 93.1 | 91.4 | 91.5 |
| μ | 15.4 | 19.0 | 67.8 | 66.6 | 71.9 | 70.6 |
| σ | 14.5 | 18.4 | 25.9 | 27.0 | 24.8 | 25.3 |
| r | 0.95 | 0.99 | 0.99 | |||
| H0: r=0 | <0.00001 | <0.00001 | <0.00001 | |||
| t | 2.36 | 0.21 | 1.37 | |||
| H0: t=0 | NS | NS | NS | |||
The above statistical analysis suggests the following interpretation of the B-cell stages. The location for the down-regulation of CD34 (a) and the initial up-regulation of CD45 L1 to L2 (b) represent the end of the first stage, B1 (red). The second up-regulation of CD45 L2 to L3 (c) and the up-regulation of CD20 (d) mark the end of the second stage, B2 (green). The down-regulation of CD10 and CD38 (e and f) defined the end of stage B3 (blue) and the beginning of stage B4 (purple, not shown).
The data shown in Table 2 are the model CDP locations defined with units of cumulative percent. These data can be interpreted as follows. The model specifies that CD34, CD10, and CD38 down-regulate their intensities between a positive and negative state, which can be denoted as ‘+’ and ‘−’; whereas, CD45 increases in three distinct levels of intensity, which can be denoted as L1, L2, and L3 (not shown). For the first file, the cumulative percentages were calculated to be 7.7%, 9.1%, 58.7%, 67.8%, and 67.1% for CD34, CD45 L1, CD45 L2, CD10, and CD38 respectively. The percentage, 7.7%, denotes that the model estimate for the phenotype, CD34+ CD45 L1 CD10+ CD38+, was approximately 7.7% of detected B cells. The CD45 L1 percentage is 9.1%, which is the sum of the phenotypes CD34+ CD45 L1 CD10+ CD38+ (7.7%) and CD34− CD45 L1 CD10+ CD38+ (1.4%). Note that this pattern is consistent with CD45 intensity changing from L1 to L2 slightly after CD34 down-regulates. Part of the objective for the statistics at the bottom of Table 2 is to determine whether this change is significant or not. The 58.7% in the CD45 L2 is the sum of phenotypes CD34+ CD45 L1 CD10+ CD38+ (7.7%), CD34− CD45 L1 CD10+ CD38+ (1.4%), and CD34− CD45 L2 CD10+ CD38+ (49.6%). This pattern is consistent with the second up-regulation of CD45 following the down-regulation of CD34. This type of cumulative pattern repeats for the rest of the percentages and yields a quantitative picture of the marker changes that occur during B-cell progression.
After a statistical analysis of this cumulative percentage data, the following stages were deduced from the modeling results. The down-regulation of CD34 (see a) and the initial up-regulation of CD45 L1 to L2 (b) were found to represent the end of the first stage boundary, B1 (red). The second up-regulation of CD45 L2 to L3 (c) and the up-regulation of CD20 (d) defined the end of the second stage, B2 (green). The down-regulation of CD10 and CD38 (e and f) were determined to represent the end of stage B3 (blue) and the beginning of stage B4 (purple, not shown).
Based on the B-cell staging results, a “normal” B-cell model was constructed that constrained appropriate marker modulations to the three critical control transitions shown in Figure 2 and Table 2 (a, c, and e). Markers that were not widely represented in this study (TdT, CD81, CD22, and CD44) were placed on the nearest stage boundary. The average model of the unattended analyses is presented as a PSM overlay plot in Figure 3. At the end of the B1 stage, CD34 down-regulates with TdT while CD45, CD81, and CD20 slightly up-regulate. Not shown is a slight down-regulation of CD10 and CD22 in this stage. The end of the B2 stage is defined by the up-regulation of both CD45 and CD20. The transition of B3 to B4 is marked by the down-regulation of CD10, CD38, and CD81, and the up-regulation of CD22 and CD44.
Figure 3.
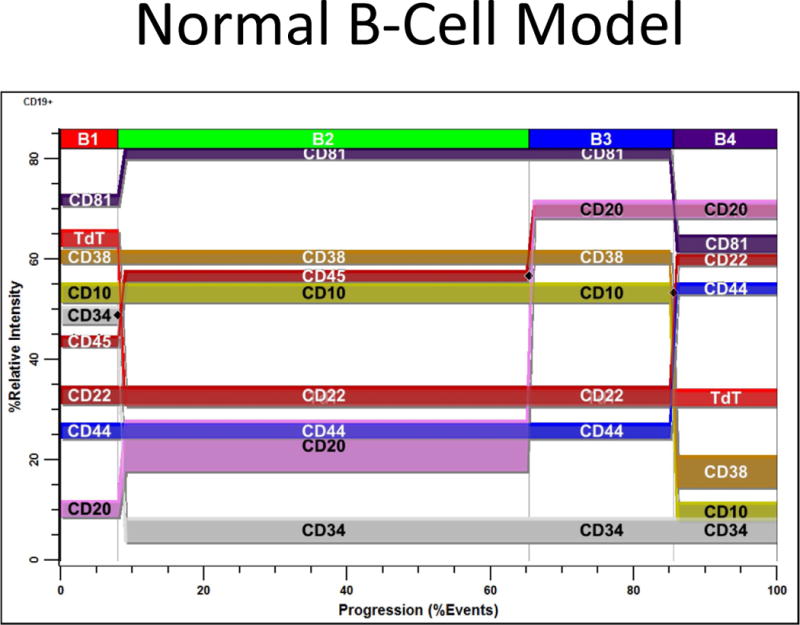
Average “Normal” B-Cell Model. Based on the B-Cell Staging results, a “normal” B-cell model was constructed that constrained appropriate marker modulations to the three critical control points shown in Figure 2 and Table 2 (a, c, and e). Those markers that were not widely represented in this study (TdT, CD81, CD22, and CD44) were placed on the nearest stage boundary. All analyses were performed unattended and the average model is presented as a PSM overlay plot in the above figure. At the end of B1; CD34 down-regulates with TdT while CD45, CD81, and CD20 slightly up-regulate. Not shown is a slight down-regulation of CD22 in this stage. At the end of B2, CD45 and CD20 up-regulate. At the end of B3; CD10, CD38, and CD81 down-regulate while CD22 and CD44 up-regulate.
A few interesting expression patterns were observed for some of the markers. Figure 4A shows the typical CD38 expression pattern. CD38 slightly up-regulates at the end of B1 and becomes very heterogeneous upon down-regulation. In Panel B, CD20 is shown to up-regulate at the end of B1 where it becomes very heterogeneous. When entering the B4 stage, it slightly down-regulates. In Panel C, CD9 staining is seen to be very heterogeneous in B1 and B4.
Figure 4.
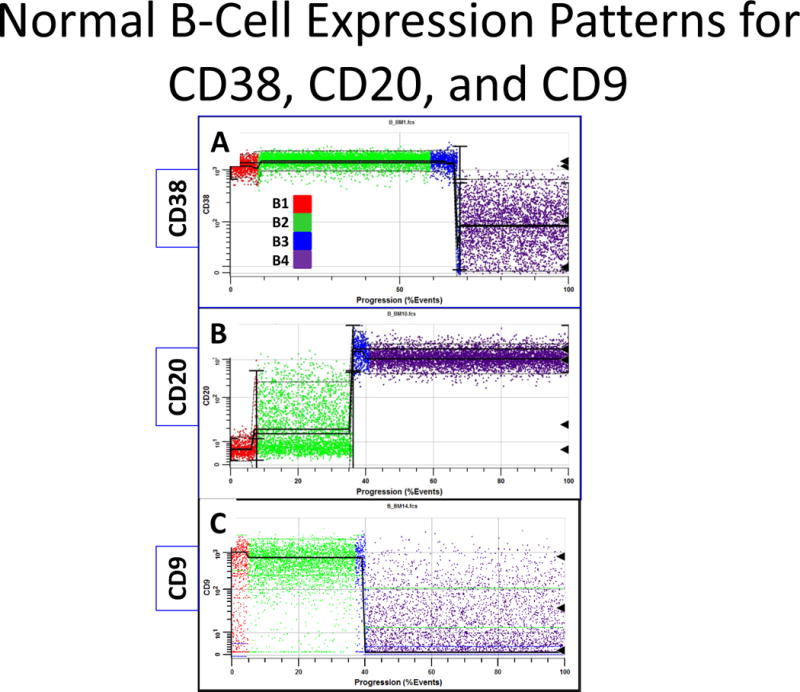
Example B-Cell Model Marker Expressions. Panel A is derived from one sample and B and C are derived from another. The black triangles to the right are detected peak locations. Red dots are events in B1, green in B2, blue in B3, and purple in B4. Panel A shows a typical expression pattern for CD38, which slightly up-regulates in B and becomes very heterogeneous after down-regulating. Panel B shows that when CD20 up-regulates at the end of B1, it becomes very heterogeneous. When entering the B4 stage, it slightly down-regulates. Panel C demonstrates that CD9 staining is normally very heterogeneous in B1 and B4.
Progenitor Stage Analysis
Figure 5 presents a summary plot for an analysis of one study file. The PSM overlay plot summarizes the modulation of CD38, CD19, and CD10 expression during very early B cell bone marrow ontogeny. The listmode events have been selected for CD45 low to intermediate intensity, SSC low to intermediate intensity, and CD34 positive expression. In this analysis CD38 up-regulates first (see a), then CD19 and CD10 up-regulate close together (b and c). These locations are quantified by the PSM model as cumulative percentages.
Figure 5.
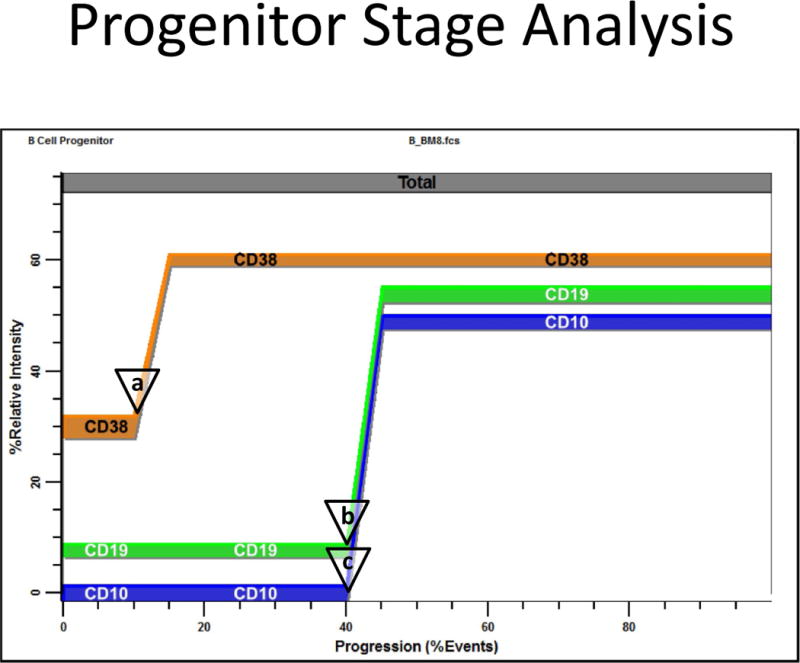
Progenitor Stage Analysis. This figure shows an analysis of the B_BM8.fcs study file. The PSM overlay plot summarizes modulation of CD38, CD19, and CD10 during very early bone marrow ontogeny. The listmode events have been selected for CD45 low to intermediate intensity, SSC low to intermediate intensity, and CD34 positive. Usually CD38 up-regulates first (see a), then CD19 and CD10 up-regulate close together (b and c). These locations are quantified by the PSM model as cumulative percentages and summarized for all suitable files in Table 3.
The progenitor stage results are summarized in Table 3. Files with enough events to analyze were modeled to obtain critical stage locations for markers CD38, CD19, and CD10. The up-regulation of CD38 (see a) was found to represent the end of the first stage boundary, P1 (red). The up-regulation of CD19 (b) and CD10 (c) were determined to occur together forming the end of stage P2 and beginning of stage P3.
Table 3.
Progenitor Stage Results. Files that had enough events to analyze were modeled to obtain critical stage locations for markers CD38, CD19, and CD10 with units of cumulative percent. The recorded results for ten study files are shown above. Markers were deemed to be on the same stage boundary if their correlation coefficients, r, were significant and their standardized differences, t, were not (see bottom of table).
| P1 | P2 and P3 | ||
|---|---|---|---|
| Files |
|
|

|
| B_BM1.fcs | 67.4 | 68.0 | 69.3 |
| B_BM7.fcs | 3.9 | 10.0 | 11.5 |
| B_BM8.fcs | 7.7 | 37.8 | 36.0 |
| B_BM9.fcs | 2.5 | 33.5 | 27.2 |
| B_BM10.fcs | 43.0 | 85.8 | 86.0 |
| B_BM11.fcs | 33.5 | 72.0 | 71.4 |
| B_BM12.fcs | 48.5 | 86.1 | 83.1 |
| B_BM14.fcs | 46.0 | 47.5 | 46.8 |
| B_BM15.fcs | 46.9 | 100.0 | 99.9 |
| B_BM16.fcs | 2.3 | 18.3 | 22.3 |
| μ | 30.2 | 55.9 | 55.4 |
| σ | 24.0 | 30.9 | 30.6 |
| r | 0.99 | ||
| H0: r=0 | <0.00001 | ||
| t | 0.62 | ||
| H0: t=0 | N.S. | ||
The up-regulation of CD38 (see a) was found to represent the end of the first stage boundary, P1 (red). The up-regulation of CD19 (see b) and CD10 (see c) were found to occur together forming the end of stage P2 and beginning of stage P3.
Based on the progenitor staging results, a normal progenitor model was constructed that constrained appropriate marker modulations to the two critical transition points shown in Figure 5 and Table 3 (see a, b). Markers that were not widely represented in this study (CD81, CD22, CD9, and CD44) were placed on the nearest stage boundary. The PSM overlay plot of the average model for all files analyzed is presented in Figure 6. The end of P1 is marked by the up-regulation of CD38. P2 ends when CD19, CD10, CD81, CD22, and CD9 up-regulate while CD44 down-regulates slightly.
Figure 6.
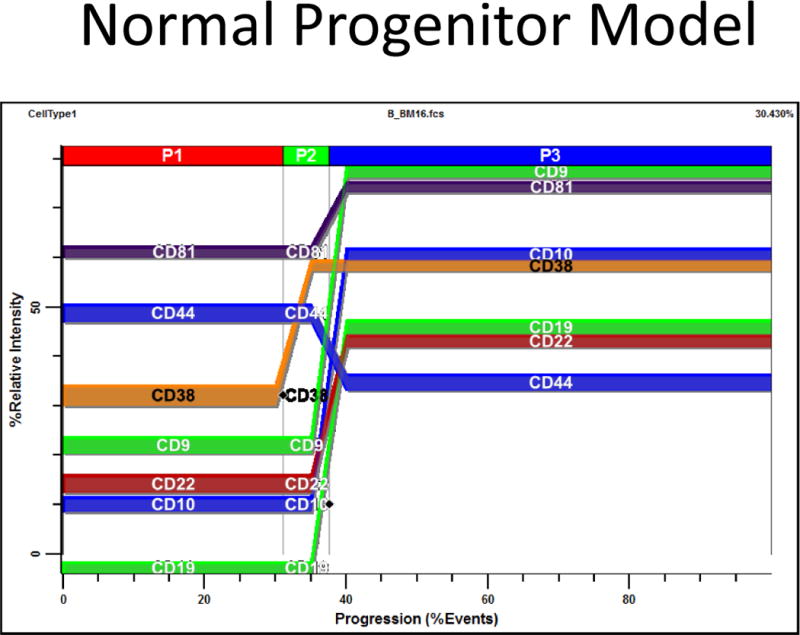
Average Progenitor Model. Based on the Progenitor Staging results, a normal progenitor model was constructed that constrained appropriate marker modulations to the two critical control points shown in Figure 5 and Table 3 (see a, b). Those markers that were not widely represented in this study (CD81, CD22, CD9, and CD44) were placed on the nearest stage boundary. Most analyses were performed unattended and the average model is presented as a PSM overlay plot in Figure 6. At the end of P1, CD38 up-regulates. At the end of P2; CD19, CD10, CD81, CD22, and CD9 up-regulate while CD44 down-regulates slightly.
A few interesting expression patterns for CD38 and CD20 were observed and are shown in Figure 7. CD38 exhibits a heterogeneous staining pattern until it up-regulates as demonstrated in Panel A. When CD20 starts to up-regulate at the end of P2, it also has a heterogeneous staining pattern with numerous positive events as shown in Panel B.
Figure 7.
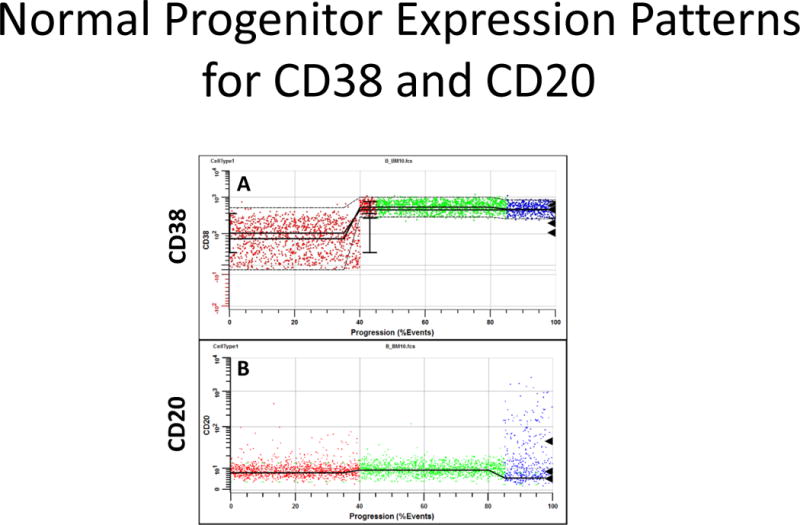
Example Progenitor Marker Patterns. Panel A shows the typical expression pattern for CD38. The black triangles to the right are detected peak locations. Usually CD38 has very heterogeneous staining until it up-regulates. Panel B shows that when CD20 up-regulates at the end of P2, it’s intensity becomes very heterogeneous.
Discussion
Post CD19 Up-Regulation
The analyses shown in Table 2 demonstrate that there are four B-cell developmental stages subsequent to CD19 up-regulation. The stage between the second up-regulation of CD45 and the down-regulation of CD10 (B3) can be either subtle or absent in some samples. In this study the average B3 stage was found to be 4.1% and reached a maximum of 12.9% in file B_BM5. In five out of twelve files, cells in B3 were not detectable. The variability of B3 and the relatively small overall percentage may explain why many investigators fuse the stages of B3 and B4 and stratify immunophenotypic B cell development in bone marrow into just three stages. In addition to the statistical evidence that B3 is a real stage, when CD20 is in the panel, its intensity is slightly higher in B3 than in B4, forming a distinctive pattern when viewed as a dot-plot. For example, the CD20 trajectory of the B cells in a CD10 versus CD20 dot-plot appears to over-shoot and then drop slightly in the later B-cell stages.
The results of modeling the five files with an antibody to TdT suggests that TdT down-regulates slightly after the reduced expression of CD34 (not shown). Unfortunately, due to an inadequate number of samples, this important observation could not be supported with appropriate statistical analyses and deserves further study.
Once the stages above were established, a “constrained” model was built to represent the normal coordinated changes that occur in B-cell ontogeny. Since a CDP can have any algebraic relationship with another CDP in PSM, it was possible to build models that constrain marker modulations to occur at the same point in the progression. There are two reasons for building a constrained model that represents normal ontogeny. The first is to summarize all stage-related expression profile modulations in the study. The second is that because of the probabilistic nature of PSM, it will classify only normal B-cell events. Therefore, it is possible to find aberrant events that for some reason do not fit into the four discrete stages defined by the model. The purposefulness and data that support this later capacity will be presented in another publication.
Many of the markers that were not well represented in this study (e.g. TdT, CD81, CD22, and CD44) were associated with the closest stage boundary. Although the quality of constrained model fitting was acceptable with most reduced chi-squares of 3.0 or less, each of these markers should eventually be subjected to the same type of statistics employed for CD34, CD45, CD10, and CD38.
The constrained model made no attempt to represent subtle marker changes with stage. For example, CD10 and CD22 down-regulate slightly at the end of stage B1 while CD38 up-regulates slightly. These subtleties are observable in the classified data after modeling, but are not represented in the model itself. The general idea of this model was to represent the major changes in B cells as they differentiate as simply as possible. Because of these factors, Figure 3 should be considered a reasonable approximation of how these markers modulate with stage. When this model is modified to find aberrant events in marrow, these subtle changes will most likely be incorporated into the model to maximize its classification power.
Figure 4 shows some interesting stage-related patterns for CD38 from file B_BM1 (Panel A) and CD20 (Panel B) and CD9 (Panel C) from file B_BM10. After CD38 down-regulates, its intensity becomes very heterogeneous as shown by the width of the expression profile in Panel A. It is unclear whether this heterogeneity is due to subsequent modulations of CD38 after down-regulation or perhaps to mature B cells not tightly regulating CD38 on their surface within this stage.
The stage-related patterns for CD20 (see Figure 4B) are even more intriguing. The heterogeneous staining pattern of CD20 in early proliferating B-cell stages such as B1 and B2 is a common finding in expanding marrows and may be helpful in understanding the efficacy of anti-CD20 therapy. In 1993, Moreau et al (37) described a population of “CD20dim” early B cells, a small fraction of which still expressed CD34, and co-expressed CD10 and CD19. These were determined to be highly proliferative as opposed to the CD20 bright cells in stages B3 and B4 that were less so. We can speculate that the CD20 up-regulation in many cells in the early B-cell stages may be associated with cell cycling. Therapies that target these cycling CD20 positive cells by inducing apoptosis may be efficacious because they are targeting actively dividing populations (32–34). This conjecture certainly deserves further study.
The expression patterns of CD9 shown in Figure 4C demonstrate a very heterogeneous staining that seems to change with stage. CD9 is generally known as a useful marker for minimum residual disease (MRD) detection (38). This heterogeneous pattern for CD9 expression may allow the visualization of malignant clones of B-cells against this distributed background, thus contributing to its diagnostic value.
Progenitor Stage Analysis
This section of the study followed the same organization as above but is presented after the more complex committed B-cell compartment, which was the primary focus of this investigation. Ten files with sufficient events to analyze the CD34+ events were modeled to obtain critical stage locations for markers CD38, CD19, and CD10 (see Table 3). All ten files were analyzed with an unconstrained version of a progenitor model (see Material & Methods and (31) for details). Unfortunately, because of the low number of CD34+ events and the heterogeneity of CD38 expression, two of the ten files needed a slight manual adjustment to the model to find the appropriate P1 to P2 stage boundaries.
The stage analysis followed the same steps as described above and is summarized at the bottom of Table 3. The up-regulation of CD38 (a) was found to represent the end of the first stage, P1 (red). The up-regulation of CD19 (b) and CD10 (c) were found to occur together, forming the end of stage P2 and beginning of stage P3.
A constrained version of this model was constructed (see Materials & Methods and Appendix for details) that forced CD10 and CD19 to have a coordinated up-regulation at the end of P2. The markers CD9, CD44, CD22, and CD81 were forced to the closest stage boundary for the same reasons as described for the B-cell model. The ten files were analyzed with this model and the averaged model is presented as an expression profile overlay in Figure 6.
The important observation in this phase of the analysis is that CD10 and CD19 seem to up-regulate at the same point in the progression (t=0.62, NS). These data suggest that the CLP stage that is normally defined as CD34+ CD10+ CD19− is either exceedingly quick and results in very small undetectable population or that this stage is not described with the correct immunophenotype. Either interpretation warrants further study.
Stage Mappings
In both sections of these analyses, simple alphanumeric labels were employed to represent all the stages found in this study (P1, P2, P3, B1, B2, B3, and B4, see Figure 1). The list below is a mapping between these labels and the more commonly used stage descriptions:
P1: HSC
P2: CLP?
P3: Pro-B
B1: Pre-B
B2: Early B
B3: Immature B
B4: Transitional B
In 60% of the samples studied, CD10 expression was up-regulated first and then shortly followed by CD19; in 40%, the reverse order was observed. When these changes for all ten samples were examined statistically, the hypothesis that there was a natural order to this progression was rejected and it was therefore concluded that they were co-expressed. Another interpretation of these data is that when stem cells commit to the B-cell lineage, the exact order of the expression of CD10 and CD19 has a stochastic element to it. In some of the study’s cases CD19 slightly precedes CD10 and in others, CD10 slightly precedes CD19. This stochastic type of hypothesis would be consistent with these analyses and might explain the variable results in the literature. Therefore, the more practical CLP phenotype might be better posed as CD38+ CD34+ CD10− CD19− (stage P2) rather than either CD38+ CD34+ CD10+ CD19− or CD38+ CD34+ CD10− CD19+. However, it may be that another marker may be found to define CLP more clearly which is why we leave the P2 stage as “CLP?” as shown above. It should also be mentioned that there is overlap between the P3 and B1 stages. Since the B-cell stages have been selected for CD19 dim to positive events, the B1 stage represents part of the P3 stage.
There are a number of potential sources of error in these analyses. Data derived from fluorescence cytometry requires compensation and inadequate compensation can make marker modulations ambiguous as to whether they are a result of signal crossover effects or real biologic coordination of the expression of antigens. All the data selected in this study were carefully screened to eliminate compensation artifacts. Since the data come from three separate laboratories running different panels of labeled antibodies, it is extremely unlikely that all three have the same compensation issues and it also strengthens the idea that the biological correlations observed are not technical in nature.
The major source of error in PSM is dictated by counting error (31). After evaluating a number of simulation studies (not shown) with similar numbers of events as found in this study, the uncertainty in estimating the location of up or down-regulations is in the order of a few tenths of a percent. This level of accuracy is about five-fold better than achieved using PSM in the past because of a relatively simple change in the design of the models. If multi-level expression profiles are designed with no transitions and the line-spreads are allowed to best fit the data, the system finds the mid-point or inflection-point of the transitions, which is considerably more accurate than either the beginning or the ending of a transition. This single change in modeling design is what gives these analyses their resolving power for quantifying the position of marker modulations along the progression axis.
Another potential source of error resides in the samples themselves. The data collected in this study were obtained from “uninvolved” bone marrow specimens submitted for diagnostic purposes. Though unlikely to show disparate results, it should be stated that these specimens might not necessarily show the same results as those obtained from normal healthy volunteer marrow specimens.
Other analysis methods have been published that examine and visualize the phenotypic changes in cells as they differentiate and mature in the marrow (39–41). Some of these methods have reported reproducibility issues and non-biological branches (39,40). One recently published method (41) generates plots that resemble expression profiles but these plots are, for the most part, not consistent with the results of this paper. The reasons for this disparity are not self-evident and deserve further study. In particular, the down-regulation of CD10 in Bendall et al. does not appear to be coordinated with CD38 down-regulation whereas in this study, the down-regulations of the two are exquisitely coordinated.
One of the main reasons for investigating progenitor and B-cell stages is their importance and prevalence in the general area of hematopathology. B cells are generally highly sensitive to apoptotic signals early in their development and when improperly triggered may lead to immunodeficiencies and other disruptions. In contrast, when mutations perturb key proliferation control checkpoints, malignancies such as B-ALL may occur. We can speculate that B-cell diversity as it has evolved is dependent upon a massive amount of somatic mutation, which despite many checkpoints is error-prone and thus potentially ‘dangerous’ – perhaps leading to one of the reasons the vast majority of hematopoietic malignancies are B-cell derived.
This study attempts to unambiguously and objectively identify the normal stages of cellular progressions involving B and progenitor cells in the marrow. Accurate staging should help investigators to better understand the cellular mechanisms that are operating in this development process. Also, once these normal stages are known, it is possible to use the same modeling techniques shown in this study to find very small numbers of aberrant B cells, which has import to sensitive minimum residual disease detection.
Appendix
Model Design Specifics
The general model design for the analysis and staging of developing B cells is described in Tables 4A and B. The model comprises two cell-types, the first is the CD19− population followed by the CD19+. CD19 negative events are minimally modeled with only CD19 modeled as a constant low-pass distribution on the negative peak. Side scatter is also a constant expression profile selecting for the lymphocytes. This cell-type serves to clean-up the data in order to more cleanly select for the CD19 dim to positive events in the second cell-type. CD19 positive events are selected for with an inverse low-pass for CD19, which selects for events that are “not” CD19 negative. Again, side scatter is a constant expression profile, CD33 and CD13 are used to exclude the myeloid population if they are present in the listmode file. Staging, or stratification, is accomplished as shown in Table 4A under the label, “stratification”. The “type” defines the type of expression profile used; the “level(s)” explain how the marker modulates. Since some files do not have all markers present, the markers identified in the “comment” column may be blank for some data files.
The “constrained” model version is summarized in Table 4B. This model uses linkages between markers to create defined stages based on the statistical analysis done for each individual data file. The general model design is the same as is summarized in Table 4A, with the addition of the linkages explained in B. The second column defines the primary markers, CD10, CD34, and CD45. The control point is where the linkage is established, for example, at the point where CD10 starts to down-regulate, or “begin-down”, CD38, CD81, CD22 and CD44 are linked. The last column “Link Position” identifies the point at which CD10 (or CD34 or CD45) is linked in the expression profile of the specific Linked Marker.
The general model design for the analysis and staging of progenitor cells is described in Tables 5A and B. The model comprises one cell-type which is defined or selected for by constant expression profiles for CD45, SSC-A, and CD34. Staging, or stratification, is accomplished with the markers as shown in Table 5A above. The “type” defines the type of expression profile used; the “level(s)” explain how the marker modulates. Since some files do not have all markers present, the markers identified in the “comment” column may be blank for some data files.
The “constrained” version of this model is summarized in Table 5B. This model uses linkages between markers to create defined stages based on the statistical analysis done for each individual data file. The general model design is the same as is summarized in Table 5A, with the addition of the linkages explained in Table 5B. The first column defines the primary marker, CD19. The control point is where the linkage is established, i.e., the point at which CD19 begins to up-regulate. The last column identifies the specific point at which CD19 is linked in each individual expression profile for CD10, CD44, CD22, CD81 or CD9.
Footnotes
Conflicting Interest Disclosure
The authors, Bagwell and Hill, are employed by Verity Software House, manufacturer of the probability state modeling software product, GemStone™.
Some of the data were recently presented in an ICCS 2014 poster session.
References
- 1.Gathings W, Lawton A, Cooper M. Immunofluorescent studies of the development of pre-B cells, B lymphocytes and immunoglobulin isotype diversity in humans. Eur J Immunol. 1977;7:804–810. doi: 10.1002/eji.1830071112. [DOI] [PubMed] [Google Scholar]
- 2.Krause D, Scadden D, Preffer F. The hematopoietic stem cell niche- home for friend and foe? Cytometry B Clin Cytom. 2013;84:78–20. doi: 10.1002/cyto.b.21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dworzak M, Fritsch G, Froschl G, Printz D, Gadner H. Four-color flow cytometric investigation of terminal deoxynucleotidyl transferase-positive lymphoid precursors in pediatric bone marrow: CD79a expression precedes CD19 in early B cell ontogeny. Blood. 1998;92:3203–3209. [PubMed] [Google Scholar]
- 4.Galy A, Travis M, Cen D, Chen B. Human T, B, natural killer, and dendritic cells arise from a common bone marrow progenitor subset. Immunity. 1995;3:459–473. doi: 10.1016/1074-7613(95)90175-2. [DOI] [PubMed] [Google Scholar]
- 5.Ghia P, ten Boekel E, Sanz E, de la Hera A, Rolink A, Melchers F. Ordering of human bone marrow B lymphocyte precursors by single-cell polymerase chain reaction analyses of the rearrangement status of the immunoglobulin H and L chain gene loci. J Exp Med. 1996;194:2217–2229. doi: 10.1084/jem.184.6.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardy R, Kayakawa K. B cell development pathways. Ann Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 7.Jackson S, Wilson P, James J, Capra J. Chapter 5 Human B Cell Subsets. 2008;98:151–224. doi: 10.1016/S0065-2776(08)00405-7. [DOI] [PubMed] [Google Scholar]
- 8.LeBien T, Womann B, Villablanca J, et al. Multiparameter flow cytometric analysis of human fetal bone marrow B cells. Leukemia. 1990;4:354–358. [PubMed] [Google Scholar]
- 9.Matthias P, Rolink A. Transcriptional networks in developing and mature B cells. Nat Rev Immunol. 2005;5:497–508. doi: 10.1038/nri1633. [DOI] [PubMed] [Google Scholar]
- 10.Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131:959–71. doi: 10.1016/j.jaci.2013.01.046. [DOI] [PubMed] [Google Scholar]
- 11.Rawlings D, Quan D, Hao Q-L, et al. Differentiation of human CD34+/CD38− cord blood stem cells into B cell progenitors in vitro. Exp Hematol. 1997;25:66–72. [PubMed] [Google Scholar]
- 12.LeBien T. Fates of human B-cell precursors. Blood. 2000;96:9–23. [PubMed] [Google Scholar]
- 13.Wood B. Multicolor immunophenotyping: human immune system hematopoiesis. Methods in Cell Biology. 2004;75:559–576. doi: 10.1016/s0091-679x(04)75023-2. [DOI] [PubMed] [Google Scholar]
- 14.Loken M, Shah V, Dattilo K, Civin C. Flow Cytometric analysis of human bone marrow II: normal lymphocyte development Blood. 1987;70:1316–1324. [PubMed] [Google Scholar]
- 15.Loken M, Wells D. Normal Antigen expression in hematopoiesis. In: Stewart Nicholson., editor. Immunophenotyping. Wiley-Liss Inc; 2000. pp. 133–160. [Google Scholar]
- 16.Campo E, Jaffe E, Harris N. Normal lymphoid organs and tissues. In: Jaffe E, Harris N, Vardiman J, Campo E, Arber D, editors. Hematopathology. Philadelphia, PA: Saunders/Elsevier; 2011. pp. 97–139. [Google Scholar]
- 17.Sanz E, Munoz A, Monserrat J, Van-Den-Rym A, Escoll P, Ranz I, Alvarez-Mon M, de-la-Hera A. Ordering human CD34+CD10−CD19+ pre/pro-B-cell and CD19− common lymphoid progenitor stages in two pro-B-cell development pathways. Proc Natl Acad Sci U S A. 2010;107:5925–30. doi: 10.1073/pnas.0907942107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warnaz K, Schlesier M. Flow cytometric phenotyping of common variable immunodeficiency Cytometry B Clin Cytom. 2008;74B:261–271. doi: 10.1002/cyto.b.20432. [DOI] [PubMed] [Google Scholar]
- 19.Beradi A, Meffre E, Pflumio F, Katz A, Vainchenker W, Schiff C, Coulombel L. Individual CD34+CD38lowCD19−CD10− progenitor cells from human cord blood generate B lymphocytes and granulocytes. Blood. 1987;89:3553–3564. [PubMed] [Google Scholar]
- 20.Macey M. Leukocyte Immunobiology. In: McCarthy D, Macey M, editors. Cytometric analysis of cell phenotype and function. Cambridge, United Kingdom: Cambridge University Press; 2001. pp. 118–123. [Google Scholar]
- 21.Sędek Ł, Bulsa J, Sonsala A, Twardoch M, Wieczorek M, Malinowska I, Derwich K, Niedźwiecki M, Sobol-Milejska G, Kowalczyk J, et al. The Immunophenotypes of Blast Cells in B-Cell Precursor Acute Lymphoblastic Leukemia: How Different Are They from Their Normal Counterparts? Cytometry B Clin Cytom. 2014;86B:41. doi: 10.1002/cyto.b.21176. [DOI] [PubMed] [Google Scholar]
- 22.Galy A, Travis M, Cen D, Chen B. Human T, B, natural killer, and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity. 1995;3:459–73. doi: 10.1016/1074-7613(95)90175-2. [DOI] [PubMed] [Google Scholar]
- 23.Bagwell C. Probability state models. USPTO. 2007;7:509. [Google Scholar]
- 24.Bagwell C. Probability state modeling: a new paradigm for cytometric analysis. In: Litwin V, Marder P, editors. Flow cytometry in Drug Discovery and Development. Hoboken NJ: John Wiley and Sons Inc; 2010. p. 281. [Google Scholar]
- 25.Bagwell C. Breaking the Dimensionality Barrier. In: Hawley TS, Hawley RG, editors. Flow Cytometry Protocols. New York City: Humana Press; 2011. p. 31. [DOI] [PubMed] [Google Scholar]
- 26.Bagwell C. A new paradigm for cytometric analysis. In: Kottke-Marchant K, Davis BH, editors. Laboratory Hematology Practice. Wiley-Blackwell Publishing Ltd; 2012. [Google Scholar]
- 27.Inokuma M, Maino V, Bagwell C. Probability state modeling of memory CD8+ T-cell differentiation. JIM. 2013;397:8–17. doi: 10.1016/j.jim.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Herbert D, Miller D, Bagwell C. Automated analysis of flow cytometric data for CD34+ stem cell enumeration using a probability state model. Cytometry B Clin Cytom. 2012;82B:313–318. doi: 10.1002/cyto.b.21032. [DOI] [PubMed] [Google Scholar]
- 29.Miller D, Hunsberger B, Bagwell C. Automated analysis of GPI-deficient leukocyte flow cytometric data using GemStone. Cytometry B Clin Cytom. 2012;82B:319–324. doi: 10.1002/cyto.b.21024. [DOI] [PubMed] [Google Scholar]
- 30.Wong L, Hill BL, Hunsberger BC, Bagwell CB, Curtis AD, Davis BH. Automated analysis of flow cytometric data for measuring neutrophil CD64 expression using a multi-instrument compatible probability state model. Cytometry B Clin Cytom. 2014 doi: 10.1002/cyto.b.21217. [DOI] [PubMed] [Google Scholar]
- 31.Bagwell C, Hunsberger B, Herbert D, Munson M, Hill B, Preffer F. Probability State Modeling Theory. Cytometry A. 2015 doi: 10.1002/cyto.a.22687. [DOI] [PubMed] [Google Scholar]
- 32.Cannizzo E, Bellio E, Sohani A, Hasserjian R, Ferry J, Dorn M, Sadowski C, Bucci J, Carulli G, Preffer F. Multiparameter immunophenotyping by flow cytometry in multiple myeloma: The diagnostic utility of defining ranges of normal antigenic expression in comparison to histology. Cytometry B Clin Cytom. 2010;78:231–8. doi: 10.1002/cyto.b.20517. [DOI] [PubMed] [Google Scholar]
- 33.Lee JA, Spidlen J, Boyce K, Cai J, Crosbie N, Dalphin M, Furlong J, Gasparetto M, Goldberg M, Goralczyk EM, et al. MIFlowCyt: the minimum information about a Flow Cytometry Experiment. Cytometry A. 2008;73:926–30. doi: 10.1002/cyto.a.20623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cannizzo E, Carulli G, Del Vecchio L, Ottaviano V, Bellio E, Zenari E, Azzara A, Petrini M, Preffer F. The role of CD19 and CD27 in the diagnosis of multiple myeloma by flow cytometry: a new statistical model. Am J Clin Pathol. 2012;137:377–86. doi: 10.1309/AJCP63TOCFNAMDMS. [DOI] [PubMed] [Google Scholar]
- 35.Tario J, Wallace P. Reagents and Cell Staining for Immunophenotyping by Flow Cytometry. In: McManus L, Mitchell R, editors. Pathobiology of Human Disease. San Diego: Elsevier; 2014. pp. 3678–3701. [Google Scholar]
- 36.Zar J. Biostatistical Analysis. New Jersey: Prentice-Hall; 1984. p. 718. [Google Scholar]
- 37.Moreau I, Duvert V, Bnachereau J, Saeland D. Culture of human fetal B-cell precursors on bone marrow stroma maintains highly proliferative CD20dim cells. Blood. 1993;81:1170–1178. [PubMed] [Google Scholar]
- 38.Borowitz M, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008;111:9. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu P. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nature Biotechnology. 2011;29:6. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linderman M. CytoSPADE: high-performance analysis and visualization of high-dimensional cytometry data. Bioinformatics. 2012;28:2. doi: 10.1093/bioinformatics/bts425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bendall S, Davis K, Amir el A, Tadmor M, Simonds E, Chen T, Shenfeld D, Nolan G, Pe’er D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–25. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]