

Abstract
By regulating the accessibility of the genome, epigenetic regulators such as histone proteins and the chromatin-modifying enzymes that act upon them control gene expression. Proper regulation of this “histone code” allows for the precise control of transcriptional networks that are essential for establishing and maintaining cell fate and identity, disruption of which may drive carcinogenesis. How these dynamic epigenetic regulators contribute to both skin homeostasis and disease is only beginning to be understood. Here we provide an update of the current understanding of histone modifiers in the skin. Indeed, as one of the most innovative and rapidly expanding areas in all of medicine, it is clear that epigenome-targeting therapies hold great promise for the treatment of dermatological diseases in the coming years.
Introduction
The cell nucleus contains roughly six feet of DNA if it was stretched from end to end. This remarkable feat is accomplished through the intricate organization of that DNA into chromatin. The organization and structure of chromatin can control gene expression. The study of chromatin, the changes it undergoes, and how these changes effect transcription fall under the umbrella of epigenetics, which refers to changes in gene expression caused by factors other than alterations in the nucleotide sequence. The fundamental unit of chromatin consists of DNA wrapped around protein octamers, termed histones, in 147 base pair segments to form nucleosome subunits. Histone octamers are made of two copies of each core histone: H2A, H2B, H3, and H4. These histones have positively charged amino (N)-terminal tails which extend from the nucleosome and can undergo several modifications, which in turn affect chromatin accessibility and gene expression. These modifications, including acetylation, methylation, and ubiquitination, among others, and are regulated by various chromatin-modifying enzymes, frequently referred to as “writers” and “erasers,” which are responsible for incorporating or removing modifications, respectively. In addition to histones, writer and eraser proteins can also interact with transcription factors and other proteins, allowing for an incredibly intricate and multilayered system for the fine-tuned regulation of gene expression.
Though research on epigenetic modifiers and histone modifications has been ongoing for decades and rapidly expanded in recent years, a vast amount remains unknown about the roles and effects of these modifiers in the skin. In this review, we discuss the current evidence for the role of these histone modifiers in the skin, with a particular focus on histone methylation and acetylation, given their status as the most abundant and well-studied histone post-translational modifications. Specifically, we delve into the current knowledge of histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), and histone demethylases (HDMs), the epigenetic writers and erasers that orchestrate acetylation and methylation, respectively, and how disruption of their normal function may promote disease in the skin.
Histone Deacetylases
Zinc dependent histone deacetylases (HDACs), responsible for the removal of acetyl marks from histones and other protein substrates, are by far the most well-studied of the chromatin-modifying enzymes. They are broadly placed into four classes: Class I, including HDACs 1–3 and 8, Class II, including HDACs 4–7, 9, and 10, Class III, made up of Sirt1–7, and Class IV, containing HDAC11. The most well studied class of HDACs in the context of gene regulation are Class I deacetylases, which have been shown to have deacetylase and other unique chromatin remodeling activities. Both HDAC1 and HDAC2 have been shown to be critical during mammalian development[1]. HDAC2/3 induction, seen in HDAC1 knockout mouse embryonic stem (ES) cells, is not enough to compensate for loss of HDAC1[2], suggesting that although there is some overlap between HDAC1/2 activities, there are other unique functions that are specific to each.
HDAC1/2 is required for proper hair follicle and epidermal proliferation and differentiation[3]. Mice lacking HDAC1/2 in keratinocytes show increased expression of CDK inhibitors p21 and p16INK4A. These cell cycle inhibitors are usually repressed via p63, a p53 family transcription factor, and HDAC1/2 is found to bind to the promoters of these CDK inhibitor genes in normal, undifferentiated keratinocytes[3]. Beyond cell cycle regulation, another Class I HDAC, HDAC3, is known to contribute to glucocorticoid receptor (GR)-mediated transcriptional repression, including reducing the expression of inflammatory genes in the skin, demonstrated through keratinocyte-specific ablation of HDAC3 in mice[4]. Thus, these Class I deacetylases play important roles in maintaining skin homeostasis (Figure 1).
Figure 1.
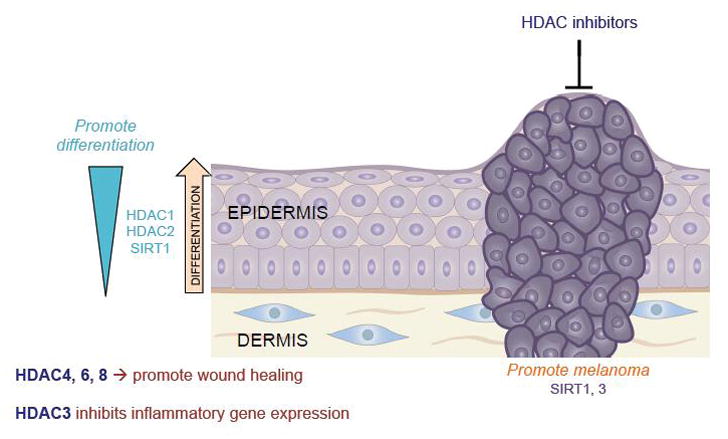
Histone deacetylases (HDACs) display a diverse and enormously complex array of roles in the skin, not only promoting differentiation and wound healing, but also cancer in other contexts. Thus HDAC inhibitors have demonstrated evidence of efficacy for a number of cutaneous neoplasms.
Regarding Class II HDACs in normal skin function, in vitro analysis of primary human skin fibroblasts has shown that HDAC4, and to a lesser extent HDAC6 and 8, are required for TGFβ1 mediated myofibroblast differentiation, an important aspect of wound healing[5]. Class III HDACs, the Sirtuin proteins, have also been associated with functions in normal skin, such as induction of keratinocyte differentiation, the inhibition of collagen degrading matrix metalloproteinases (MMPs), and decreasing DNA damage following UV exposure. Intriguingly, Sirtuin activation both by resveratrol as well as a synthetic activator decreased levels of HDAC2 and accelerated wound healing rates, an effect mimicked by HDAC inhibition by TSA[6]. These results indicate that different classes of HDACs are able to regulate the activity of others, leading to a complex interplay between deacetylases in vivo.
Considering the extensive evidence described above to demonstrate the role of HDACs in normal skin function, it is no surprise that various skin diseases are associated with the aberrant expression and function of these proteins. One of the emerging areas of study involves the roles of HDACs in skin cancer, where HDAC inhibitors are quickly becoming a well-documented therapeutic modality. In 1999, the pan-HDAC inhibitor trichostatin (TSA) was discovered to irreversibly arrest the growth of keratinocytes and squamous cell carcinoma (SCC) cells through the reduction of cyclin-dependent-kinase-1 (CDK1) transcription, indicating a potential therapeutic benefit in context of cancer[7]. Since then, HDAC inhibitors have demonstrated activity in melanoma, cutaneous T-cell lymphoma (CTCL), Merkel cell carcinoma (MCC) and other skin cancers (Figure 1). The HDAC 1–11 inhibitors suberoylanilide hydroxamic acid (SAHA), valproic acid (VPA), and romidepsin are currently used in clinical settings, although it can be argued that specific inhibition of HDAC1/2 may be sufficient to produce therapeutic advantages[8]. In addition, several other potential mechanisms of action have been proposed to explain the therapeutic advantage of HDAC inhibition. For instance, extensive studies have shown that HDAC inhibitors induce cell cycle arrest through the upregulation of p21WAF1/CIP1, the promotion of apoptosis, and inhibiting angiogenesis. In the skin, the HDAC inhibitor vorinostat was shown to inhibit tumor growth and proliferation in epidermoid carcinoma cells by reducing mTOR signaling[9], while HDAC inhibitors can also halt melanoma cell proliferation by repressing inositol polyphosphate 5-phosphatases which inhibit PI3K/Akt mediated growth[10]. In addition to the potential of HDACs 1 and 2 to promote oncogenesis, there is also some evidence that both Sirt1 and Sirt3 can promote the initiation of melanoma[11, 12]. However, the role of Sirtuins in cancer remains complicated, as Sirtuins have been categorized as both oncogenes and tumor suppressors, depending on the context[13–15]. Indeed, given the potential pleiotropic mechanisms driving HDAC-mediated tumorigenesis, further work will continue to dissect the major pathways involved and how they may be potentially targeted.
Histone Acetyltransferases
Histone acetyltransferases (HATs) are protein complexes that transfer acetyl groups onto substrates such as the amino tails of histones and transcription factors. Through this mechanism, they are able to regulate chromatin structure by neutralizing the charged interaction between histones and DNA, resulting in a loosening of the chromatin structure, and in turn, altering DNA binding affinities for transcription factors[16]. HATs can thus regulate gene expression and other processes in several tissues, including the skin.
HATs are broadly categorized into three families: p300/CREB-binding-protein (CBP), Gcn5-related N-acetyltransferases (GNAT), and MYST[17]. CBP and p300 are haplo-insufficient paralogs which contain HAT domains as well as bromodomain reader regions and transcription factor binding domains, necessary for their acetyltransferase activity and transcriptional coactivator function through formation of a transcriptional complex[18]. While histone acetylation by this family of HATs is known to have an effect on chromatin remodeling, the association between p300/CBP and other substrates has also been associated with DNA synthesis, cell-cycle progression, apoptosis, and p53 degradation. p300 has also been found to mediate the DNA damage response through the acetylation of histone 3 lysine 56 (H3K56ac), and has recently been shown to participate in epithelial differentiation through interactions with the downstream Wnt signaling molecule β-catenin[19, 20].
GNAT is an enormous superfamily consisting of several HATs found in many diverse organisms[21]. Of most relevance in mammals are GCN5 and p300/CBP-associated factor (PCAF), largely similar HATs which form complexes with other proteins in the cell[22]. The MYST family, named from their derivatives in yeast, consists of five major HATs: Tip60, MOF, MOZ, HBO1, and MORF, all of which have been associated in many cellular processes including transcriptional activation and DNA repair[23]. MOF in particular plays a role in acetylation of histone 4 lysine 16 (H4K16ac), which is required for a complete DNA damage response[24].
HATs have also been observed to have an effect on skin development and function. In human keratinocytes, the p63 isoform, ΔNp63α, undergoes p300 mediated stabilization via acetylation, the absence of which may contribute to Ectrodactyly–ectodermal dysplasia–cleft syndrome (EEC) which results in defective development of the skin and other organs[25] (Figure 2). Furthermore, p300 also plays a role in extracellular matrix (ECM) organization by upregulating collagen production via TGF-β signaling in dermal fibroblasts[26]. Beyond p300, PCAF has been shown to enhance wound repair, while ING5, a component of the MORF complex, has been implicated in a network of epidermal differentiation signaling[27]. Tip60 has also been shown to regulate p53 in response to UV damage, directly regulating the DNA damage response[28, 29]. Despite these established roles, a great deal more remains to be discovered regarding the role of the HAT proteins in skin homeostasis.
Figure 2.
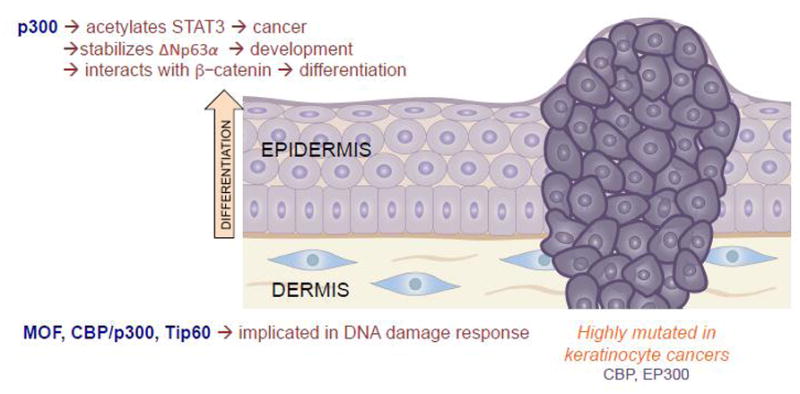
Histone acetyltransferases are dynamic activators of gene transcription, and thus play a number of diverse roles in the skin, ranging from both the promotion of development and differentiation, but also contributing to the DNA damage response and carcinogenesis.
Though the normal functions of HATs in the skin are still being elucidated, several HATs have been implicated in skin carcinogenesis. According to The Cancer Genome Atlas, both CBP and p300 (CREBBP and EP300) are frequently mutated in SCC (Figure 2). As described above, CBP/p300 has been shown to mediate the addition of H3K56ac, which is upregulated in skin cancer[30]. Further, p300 acetylates STAT3, a transcription factor known for its role in cancer, including skin cancer[31] (Figure 2). STAT3 acetylation on Lys685 can directly promote tumor growth via the decreased expression of tumor suppressors[32]. Further, p300 has been noted to be upregulated in cutaneous squamous cell carcinoma[33].
Tip60, a member of the MYST family of HATs, has also been shown to have context specific, often contradictory, roles in skin carcinogenesis. For example, increased expression of Tip60 has been observed in CTCL, which may drive increased stem cell-like properties and cancer progression[34]. This is supported by the observed increased level of Tip60 in skin tumors of mice expressing elevated ornithine decarboxylase, a marker for epithelial tumorigenesis[35]. In contrast, Tip60 is lost in metastatic melanoma, and overexpression of Tip60 in melanoma cells leads to increased sensitivity to chemotherapy[36]. Following up on this, it was reported that melanoma cells often have decreased levels of Tip60, which in turn leads to decreased p53 activation and autophagy, providing a mechanism through which melanoma cells can continue to divide[37]. Thus, it seems likely that the role of Tip60, along with most other HATs, probably relies heavily on the context and type of cancer in question.
Histone Methyltransferases
Histone methyltransferases (HMTs) and demethylases (HDMs) play a crucial role in establishing skin homeostasis by covalently and post-translationally modifying histones on amino-terminal histone tails, altering the local chromatin environment and leading to alterations in gene expression. Histone methylation can promote either the activation or the repression of transcription depending upon which histone tail residue is modified by the interacting enzymes. HMTs act on lysine residues, and some of the most well-described modifications include methylation of histone H3 on either lysine 4 (H3K4), lysine 9 (H3K9), lysine 27 (H3K27), or lysine 36 (H3K36), and of histone H4 on lysine 20 (H4K20). Common modifications that have been linked to more open regions of chromatin (euchromatin) and transcriptional activation include the trimethylation of H3K4 (H3K4me3) at gene promoters and H3K36 (H3K36me3) over gene bodies. In contrast, more compacted regions of chromatin are marked by the trimethylation of H3K27 (H3K27me3) and H3K9 (H3K9me3) and are typically associated with gene silencing, and are frequently referred to as facultative and constitutive heterochromatin, respectively. Remarkably, by regulating the accessibility of the genome to DNA repair factors, histone modifications are the most influential factor in determining both the location and density of genetic mutations in cancers[38, 39].
Of all the methylated histones, perhaps the most well-understood in the skin is H3K27me3. H3K27me3 is catalyzed by the two members of the polycomb repressive complex (PRC), PRC1 and PRC2. These complexes promote the deposition of H3K27me3 through their catalytic SET domains, made up of either EZH1 or EZH2. Seminal studies established the role of EZH2 in regulating epidermal progenitors, as it acts by simultaneously repressing differentiation through the suppression of the Ink4a/Ink4b/Arf locus and suppressing cellular growth by preventing the recruitment of the AP1 transcriptional activator[40]. Later work showed that EZH1 and EZH2 are functionally redundant, with both Ezh1- and Ezh2-deficient mouse skin upregulating similar genes in both the hair follicles and intrafollicular epidermis[41] (Figure 3). Despite this, these individual compartments displayed unique phenotypes, with the hair follicles undergoing proliferation arrest while the epidermis hyperproliferated, an effect shown to be due to differential expression of the Ink4a/Ink4b/Arf locus[41]. Similarly, recent research in mice demonstrated that Ezh1 and Ezh2 repress the transcription factor Sox2 in epidermal progenitor cells, preventing epidermal differentiation into lineage-committed Merkel cells[42]. Together, these studies establish multiple roles for EZH1 and EZH2 in epidermal development and homeostasis (Figure 3).
Figure 3.
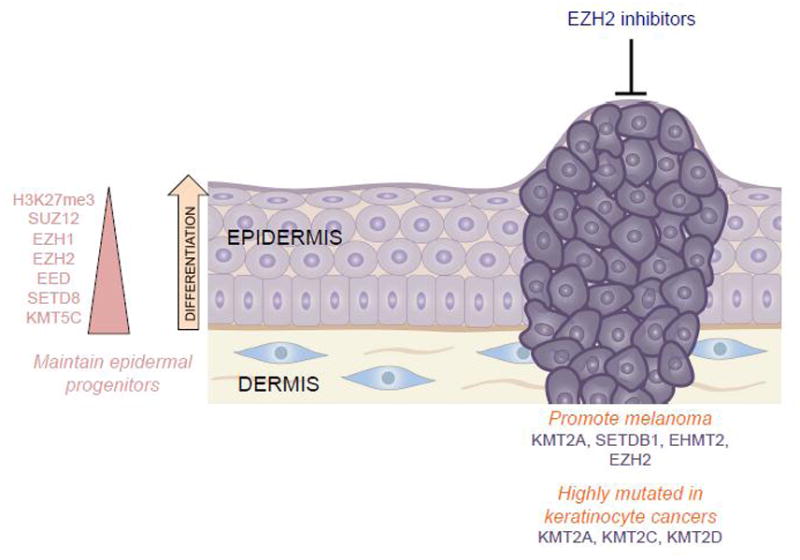
The histone methyltransferases can both activate (such as H3K4me3) and repress transcription (such as H3K27me3 and H3K9me3). In turn, they have been shown to both maintain epidermal progenitor function, as well as harbor the potential to promote cancer. Indeed, EZH2 inhibitors have demonstrated impressive results in the treatment of many types of cancer.
In addition to EZH1 and EZH2, several other histone methyltransferases have been implicated in epidermal development and differentiation. The trithorax H3K4 histone methyltransferase complexes participate through the recruitment of the differentiation-promoting transcription factor, GRHL3[43]. SETD8 (KMT5A), which catalyzes histone H4 monomethylation on lysine 20 (H4K20me1), a mark of gene activation, has similarly been demonstrated to play a role in epidermal development. Setd8 plays a role in c-Myc-induced epidermal differentiation, as deletion of Setd8 in mouse epidermis leads to apoptosis via the combined increased expression of p53 and loss of p63, a critical transcription factor required for epidermal commitment[44]. Other research established that SUV420H2 (KMT5C)-mediated Histone H4 trimethylation on lysine 20 (H4K20me3) is important for epidermal homeostasis. SUV420H2-mediated H4K20me3 levels are higher in the stem cells of the hair follicle than in other cell types of the skin and have been shown to be important in the maintenance of stem cell quiescence[45]. Collectively, these studies suggest that numerous histone methyltransferases may play crucial roles in normal human epidermal commitment and differentiation (Figure 3).
Beyond normal development, several studies have suggested a role for histone methyltransferases in skin carcinogenesis. A mouse model of multistage skin carcinogenesis revealed that the loss of H4K20me3 was associated with the hypomethylation of DNA repetitive sequences, a hallmark of cancer cells[46]. MLL family members, which methylate H3K4, include KMT2A, KMT2C, and KMT2D, are all highly mutated in squamous cell carcinoma according to The Cancer Genome Atlas (TCGA), with KMT2D being one of two genes (along with TP53) that was associated with metastatic cutaneous SCC[47] (Figure 3). KMT2A has been observed to promote the growth of melanoma via hTERT signaling[48]. Also in melanoma, gain-of-function EZH2 mutations lead to a decrease in cell contractility and an increase in cell migration[49]. Consistent with this, the EZH2 inhibitor GSK503 has been shown to stabilize melanoma and abolish metastasis formation in vivo. This same study showed that EZH2 inhibition in human melanocytes leads to the re-expression of tumor suppressors while impairing proliferation and invasiveness[50]. Several H3K9 methyltransferases have also been shown to play a role in skin carcinogenesis. SETDB1, which performs H3K9 trimethylation (H3K9me3), was established as an oncogene in melanoma after a study in a zebrafish melanoma model showed that SETDB1 significantly accelerated melanoma formation[51] (Figure 3). In a transgenic mouse model that overexpresses Suv39h1, another H3K9me3 methyltransferase, the increased global H3K9me3 levels promoted telomere homeostasis and mediated resistance to oncogenic stress such as the DMBA/TPA skin carcinogenesis model[52]. In a human study of 67 patients that employed immunohistochemistry to look at the expression of various H3K9 modifiers, it was established that overexpression of EHMT2 (G9a), which catalyzes H3K9me2, might be a marker of poor prognosis in melanoma[53]. Collectively, these studies demonstrate the importance of HMTs in skin homeostasis and their potential roles as both oncogenes and tumor suppressors in skin cancer.
Histone Demethylases
In opposition to the histone methyltransferases are the more recently discovered histone demethylases (HDMs). In 2004, LSD1 (KDM1A), which demethylates H3K4 and H3K9, was the first histone demethylase to be discovered[54]. In the skin, LSD1 has been demonstrated to interact with the transcription factor ZNF750 to repress progenitor genes during epidermal differentiation[55] (Figure 4). Beyond LSD1, over 30 other histone demethylases have since been described. In 2007, UTX (KDM6A) and JMJD3 (KDM6B) were shown to demethylate H3K27me3[56]. Since then, normal JMJD3 function has been shown to be critical for mammalian epidermal homeostasis, as JMJD3 depletion blocked differentiation, and a calcium-induced differentiation model revealed that the loss of H3K27me3 during differentiation was associated with increased binding by JMJD3[57]. Related results in a murine model of wound healing established that increased levels of Utx and Jmjd3 combined with decreased levels of the methyltransferases, Eed, Ezh2 and Suz12, led to a striking loss of H3K27me3 and upregulation of wound repair genes[58]. Further mechanistic work established that JMJD3 interacts with NF-κB at the wound edge during keratinocyte wound healing, resulting in the increased expression of inflammatory, matrix metalloproteinase, and growth factor genes via the demethylation of H3K27me3 at those gene promoters[59] (Figure 4).
Figure 4.
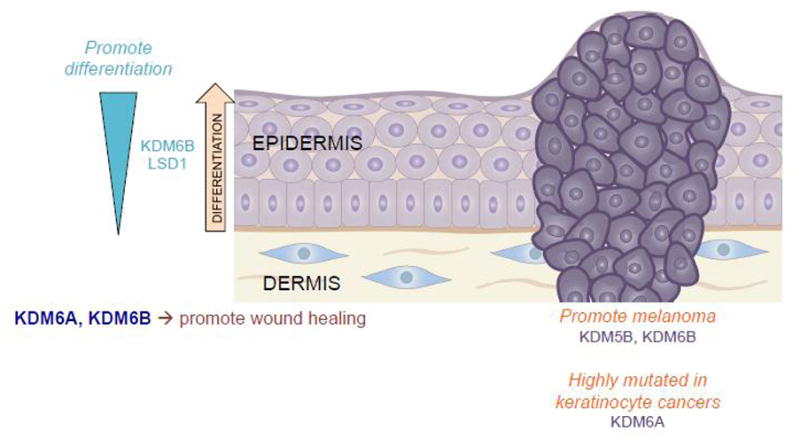
The histone demethylases are only beginning to be studied in depth in the skin. However, an accumulation of evidence thus far strongly supports their role in epidermal processes ranging from differentiation and wound healing to cancer.
Histone demethylases have similarly been implicated in skin malignancies. KDM5B, otherwise known as JARID1B, has been shown to have a potential role in promoting tumorigenesis in melanoma. One study suggested that KDM5B, which is responsible for removing tri- and di-methyl marks from H3K4, is correlated with melanoma invasiveness and that isolated KDM5B positive cells are required for continuous tumor growth[60]. A later study in human tissue supported that KDM5B is upregulated early in melanoma, however they did not find that it correlated with melanoma invasiveness[61]. Similarly, KDM6B has been shown to aid melanoma growth and metastasis in vitro and in vivo, promoting a favorable tumor microenvironment via the promotion of angiogenesis and the upregulation of PI3K signaling and NF-κB and BMP target genes[62] (Figure 4).
Conclusions
The continuous development of ever more powerful techniques for interrogating the epigenome has led epigenetics to become one of the most innovative fields within the biomedical sciences. Within dermatology, although we are only beginning to scratch the surface of our understanding, it is becoming increasingly clear that epigenetic processes exert a profound influence on a diverse array of processes ranging from development and differentiation, to cancer and aging. Histone modifications, and the chromatin-modifying enzymes that regulate them, provide an exquisite method of fine-tuning gene transcription, and in turn, directing diverse cellular processes. Given the inherent reversibility that exists in chromatin regulation, as we continue to learn more about the role of epigenetics in cutaneous disease, there is little doubt that the targeting of histone modifications and histone modifiers will provide substantial opportunities for innovation and the development of new therapies for skin disease.
Acknowledgments
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health under Award Number K08AR070289 to B.C.C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Further support was provided by Dermatology Foundation and Melanoma Research Foundation grants to B.C.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. The EMBO journal. 2002;21(11):2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes & development. 2007;21(14):1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LeBoeuf M, Terrell A, Trivedi S, Sinha S, Epstein JA, Olson EN, Morrisey EE, Millar SE. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev Cell. 2010;19(6):807–818. doi: 10.1016/j.devcel.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hua G, Ganti KP, Chambon P. Glucocorticoid-induced tethered transrepression requires SUMOylation of GR and formation of a SUMO-SMRT/NCoR1-HDAC3 repressing complex. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(5):E635–643. doi: 10.1073/pnas.1522826113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glenisson W, Castronovo V, Waltregny D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim Biophys Acta. 2007;1773(10):1572–1582. doi: 10.1016/j.bbamcr.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Abad M, Mosteiro L, Pantoja C, Canamero M, Rayon T, Ors I, Grana O, Megias D, Dominguez O, Martinez D, et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502(7471):340–345. doi: 10.1038/nature12586. [DOI] [PubMed] [Google Scholar]
- 7.Saunders N, Dicker A, Popa C, Jones S, Dahler A. Histone deacetylase inhibitors as potential anti-skin cancer agents. Cancer research. 1999;59(2):399–404. [PubMed] [Google Scholar]
- 8.Kelly RD, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41(3):741–749. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- 9.Kurundkar D, Srivastava RK, Chaudhary SC, Ballestas ME, Kopelovich L, Elmets CA, Athar M. Vorinostat, an HDAC inhibitor attenuates epidermoid squamous cell carcinoma growth by dampening mTOR signaling pathway in a human xenograft murine model. Toxicol Appl Pharmacol. 2013;266(2):233–244. doi: 10.1016/j.taap.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye Y, Jin L, Wilmott JS, Hu WL, Yosufi B, Thorne RF, Liu T, Rizos H, Yan XG, Dong L, et al. PI(4,5)P2 5-phosphatase A regulates PI3K/Akt signalling and has a tumour suppressive role in human melanoma. Nature communications. 2013;4:1508. doi: 10.1038/ncomms2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohanna M, Bonet C, Bille K, Allegra M, Davidson I, Bahadoran P, Lacour JP, Ballotti R, Bertolotto C. SIRT1 promotes proliferation and inhibits the senescence-like phenotype in human melanoma cells. Oncotarget. 2014;5(8):2085–2095. doi: 10.18632/oncotarget.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.George J, Nihal M, Singh CK, Zhong W, Liu X, Ahmad N. Pro-Proliferative Function of Mitochondrial Sirtuin Deacetylase SIRT3 in Human Melanoma. The Journal of investigative dermatology. 2016;136(4):809–818. doi: 10.1016/j.jid.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y, Huang J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell death & disease. 2014;5:e1047. doi: 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang Y, Nicholl MB. Sirtuin 1 in malignant transformation: friend or foe? Cancer Lett. 2011;306(1):10–14. doi: 10.1016/j.canlet.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 15.Ming M, Qiang L, Zhao B, He YY. Mammalian SIRT2 inhibits keratin 19 expression and is a tumor suppressor in skin. Experimental dermatology. 2014;23(3):207–209. doi: 10.1111/exd.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 17.Kaypee S, Sudarshan D, Shanmugam MK, Mukherjee D, Sethi G, Kundu TK. Aberrant lysine acetylation in tumorigenesis: Implications in the development of therapeutics. Pharmacol Ther. 2016;162:98–119. doi: 10.1016/j.pharmthera.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 18.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87(5):953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 19.Vempati RK, Jayani RS, Notani D, Sengupta A, Galande S, Haldar D. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. The Journal of biological chemistry. 2010;285(37):28553–28564. doi: 10.1074/jbc.M110.149393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rieger ME, Zhou B, Solomon N, Sunohara M, Li C, Nguyen C, Liu Y, Pan JH, Minoo P, Crandall ED, et al. p300/beta-Catenin Interactions Regulate Adult Progenitor Cell Differentiation Downstream of WNT5a/Protein Kinase C (PKC) The Journal of biological chemistry. 2016;291(12):6569–6582. doi: 10.1074/jbc.M115.706416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vetting MW, LP SdC, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433(1):212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26(37):5341–5357. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- 23.Avvakumov N, Cote J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene. 2007;26(37):5395–5407. doi: 10.1038/sj.onc.1210608. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Corsa CA, Pan PW, Wu L, Ferguson D, Yu X, Min J, Dou Y. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Molecular and cellular biology. 2010;30(22):5335–5347. doi: 10.1128/MCB.00350-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Restelli M, Molinari E, Marinari B, Conte D, Gnesutta N, Costanzo A, Merlo GR, Guerrini L. FGF8, c-Abl and p300 participate in a pathway that controls stability and function of the DeltaNp63alpha protein. Hum Mol Genet. 2015;24(15):4185–4197. doi: 10.1093/hmg/ddv151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh AK, Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J Cell Physiol. 2007;213(3):663–671. doi: 10.1002/jcp.21162. [DOI] [PubMed] [Google Scholar]
- 27.Mulder KW, Wang X, Escriu C, Ito Y, Schwarz RF, Gillis J, Sirokmany G, Donati G, Uribe-Lewis S, Pavlidis P, et al. Diverse epigenetic strategies interact to control epidermal differentiation. Nature cell biology. 2012;14(7):753–763. doi: 10.1038/ncb2520. [DOI] [PubMed] [Google Scholar]
- 28.Legube G, Linares LK, Tyteca S, Caron C, Scheffner M, Chevillard-Briet M, Trouche D. Role of the histone acetyl transferase Tip60 in the p53 pathway. The Journal of biological chemistry. 2004;279(43):44825–44833. doi: 10.1074/jbc.M407478200. [DOI] [PubMed] [Google Scholar]
- 29.Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102(4):463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 30.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459(7243):113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedranzini L, Leitch A, Bromberg J. Stat3 is required for the development of skin cancer. The Journal of clinical investigation. 2004;114(5):619–622. doi: 10.1172/JCI22800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, Hoon DS, Forman SJ, Jove R, Riggs AD, et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(20):7765–7769. doi: 10.1073/pnas.1205132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen MK, Cai MY, Luo RZ, Tian X, Liao QM, Zhang XY, Han JD. Overexpression of p300 correlates with poor prognosis in patients with cutaneous squamous cell carcinoma. Br J Dermatol. 2015;172(1):111–119. doi: 10.1111/bjd.13226. [DOI] [PubMed] [Google Scholar]
- 34.Litvinov IV, Netchiporouk E, Cordeiro B, Zargham H, Pehr K, Gilbert M, Zhou Y, Moreau L, Woetmann A, Odum N, et al. Ectopic expression of embryonic stem cell and other developmental genes in cutaneous T-cell lymphoma. Oncoimmunology. 2014;3(11):e970025. doi: 10.4161/21624011.2014.970025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hobbs CA, Wei G, DeFeo K, Paul B, Hayes CS, Gilmour SK. Tip60 protein isoforms and altered function in skin and tumors that overexpress ornithine decarboxylase. Cancer research. 2006;66(16):8116–8122. doi: 10.1158/0008-5472.CAN-06-0359. [DOI] [PubMed] [Google Scholar]
- 36.Chen G, Cheng Y, Tang Y, Martinka M, Li G. Role of Tip60 in human melanoma cell migration, metastasis, and patient survival. The Journal of investigative dermatology. 2012;132(11):2632–2641. doi: 10.1038/jid.2012.193. [DOI] [PubMed] [Google Scholar]
- 37.Lakhter AJ, Kanginakudru S, Warren S, Touloukian CE, Boissy RE, Naidu SR. Impaired PIASy-Tip60 signaling weakens activation of p53 in melanoma. Melanoma Res. 2013;23(3):213–217. doi: 10.1097/CMR.0b013e328361056d. [DOI] [PubMed] [Google Scholar]
- 38.Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488(7412):504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- 39.Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence MS, Reynolds A, Rynes E, Vlahovicek K, Stamatoyannopoulos JA, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518(7539):360–364. doi: 10.1038/nature14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 2009;136(6):1122–1135. doi: 10.1016/j.cell.2008.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, Fuchs E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes & development. 2011;25(5):485–498. doi: 10.1101/gad.2019811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bardot ES, Valdes VJ, Zhang J, Perdigoto CN, Nicolis S, Hearn SA, Silva JM, Ezhkova E. Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. The EMBO journal. 2013;32(14):1990–2000. doi: 10.1038/emboj.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopkin AS, Gordon W, Klein RH, Espitia F, Daily K, Zeller M, Baldi P, Andersen B. GRHL3/GET1 and trithorax group members collaborate to activate the epidermal progenitor differentiation program. PLoS Genet. 2012;8(7):e1002829. doi: 10.1371/journal.pgen.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Driskell I, Oda H, Blanco S, Nascimento E, Humphreys P, Frye M. The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. The EMBO journal. 2012;31(3):616–629. doi: 10.1038/emboj.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corvalan AZEA, Lowry WE, Coller HA. The Essence of Quiescence: Understanding the roles of histone modification H4K20me3 and the histone modifying enzyme Suv4–20h2 in cellular quiescence. FASEB J. 2016;30:1028.1025. [Google Scholar]
- 46.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature genetics. 2005;37(4):391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 47.Yilmaz AS, Ozer HG, Gillespie JL, Allain DC, Bernhardt MN, Furlan KC, Castro LT, Peters SB, Nagarajan P, Kang SY, et al. Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumors. Cancer. 2017;123(7):1184–1193. doi: 10.1002/cncr.30459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang C, Song C, Liu T, Tang R, Chen M, Gao F, Xiao B, Qin G, Shi F, Li W, et al. KMT2A promotes melanoma cell growth by targeting hTERT signaling pathway. Cell Death Dis. 2017;8(7):e2940. doi: 10.1038/cddis.2017.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barsotti AM, Ryskin M, Zhong W, Zhang WG, Giannakou A, Loreth C, Diesl V, Follettie M, Golas J, Lee M, et al. Epigenetic reprogramming by tumor-derived EZH2 gain-of-function mutations promotes aggressive 3D cell morphologies and enhances melanoma tumor growth. Oncotarget. 2015;6(5):2928–2938. doi: 10.18632/oncotarget.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, Arenas-Ramirez N, Haeusel J, Zhang Y, Bonalli M, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nature communications. 2015;6:6051. doi: 10.1038/ncomms7051. [DOI] [PubMed] [Google Scholar]
- 51.Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferre F, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471(7339):513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petti E, Jordi F, Buemi V, Dinami R, Benetti R, Blasco MA, Schoeftner S. Altered telomere homeostasis and resistance to skin carcinogenesis in Suv39h1 transgenic mice. Cell cycle. 2015;14(9):1438–1446. doi: 10.1080/15384101.2015.1021517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miura S, Maesawa C, Shibazaki M, Yasuhira S, Kasai S, Tsunoda K, Maeda F, Takahashi K, Akasaka T, Masuda T. Immunohistochemistry for histone h3 lysine 9 methyltransferase and demethylase proteins in human melanomas. Am J Dermatopathol. 2014;36(3):211–216. doi: 10.1097/DAD.0b013e3182964e02. [DOI] [PubMed] [Google Scholar]
- 54.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nature reviews Genetics. 2016;17(8):487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 55.Boxer LD, Barajas B, Tao S, Zhang J, Khavari PA. ZNF750 interacts with KLF4 and RCOR1, KDM1A, and CTBP1/2 chromatin regulators to repress epidermal progenitor genes and induce differentiation genes. Genes & development. 2014;28(18):2013–2026. doi: 10.1101/gad.246579.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agger K, Cloos PA, Rudkjaer L, Williams K, Andersen G, Christensen J, Helin K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009;23(10):1171–1176. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sen GL, Webster DE, Barragan DI, Chang HY, Khavari PA. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes & development. 2008;22(14):1865–1870. doi: 10.1101/gad.1673508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaw T, Martin P. Epigenetic reprogramming during wound healing: loss of polycomb-mediated silencing may enable upregulation of repair genes. EMBO Rep. 2009;10(8):881–886. doi: 10.1038/embor.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Na J, Lee K, Na W, Shin JY, Lee MJ, Yune TY, Lee HK, Jung HS, Kim WS, Ju BG. Histone H3K27 Demethylase JMJD3 in Cooperation with NF-kappaB Regulates Keratinocyte Wound Healing. The Journal of investigative dermatology. 2016;136(4):847–858. doi: 10.1016/j.jid.2015.11.029. [DOI] [PubMed] [Google Scholar]
- 60.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuzbicki L, Lange D, Straczynska-Niemiec A, Chwirot BW. JARID1B expression in human melanoma and benign melanocytic skin lesions. Melanoma Res. 2013;23(1):8–12. doi: 10.1097/CMR.0b013e32835d5d6f. [DOI] [PubMed] [Google Scholar]
- 62.Park WY, Hong BJ, Lee J, Choi C, Kim MY. H3K27 Demethylase JMJD3 Employs the NF-kappaB and BMP Signaling Pathways to Modulate the Tumor Microenvironment and Promote Melanoma Progression and Metastasis. Cancer research. 2016;76(1):161–170. doi: 10.1158/0008-5472.CAN-15-0536. [DOI] [PubMed] [Google Scholar]