

Abstract
Introduction
When a new drug enters the market, its full array of side effects remains to be defined. Current surveillance approaches targeting these effects remain largely reactive. There is a need for development of methods to predict specific safety events that should be sought for a given new drug during development and post-marketing activities.
Objective
We present here a safety signal identification approach applied to a new set of drug entities, inhibitors of the serine protease proprotein convertase subtilisin/kexin type 9 (PCSK9).
Methods
Using phenome-wide association study (PheWAS) methods, we analyzed available genotype and clinical data from 29,722 patients, leveraging the known effects of changes in PCSK9 to identify novel phenotypes in which this protein and its inhibitors may have impact.
Results
PheWAS revealed significantly reduced risk of hypercholesterolemia (OR 0.68, p=7.6 × 10−4) in association with a known loss-of-function variant in PCSK9, R46L. Similarly, laboratory data indicated significantly reduced beta mean LDL-C (−14.47 mg/dl, p=2.58×10−23) in individuals carrying the R46L variant. The R46L variant was also associated with increased risk of spina bifida (OR 5.90, p=2.7 × 10−4), suggesting that further investigation of potential connections between inhibition of PCSK9 and neural tube defects may be warranted.
Conclusion
This novel methodology provides an opportunity to put in place new mechanisms to assess the safety and long-term tolerability of PCSK9 inhibitors specifically and other new agents, in general, as they move into human testing and expanded clinical use.
1. Introduction
It is a virtually a given that when a new drug enters the market, the entire array of side effects remains to be defined. Important contributors to this well-recognized phenomenon include low event rates of many side effects, relatively short duration of trials for drugs intended for chronic use after approval, and heterogeneity of real world patient populations compared to trial populations. In this way, the first patient ‘users’ of a new class of drug can be likened to pre-approval clinical trial participants, in terms of the potential burden of risk they may be assuming by being among the first to use the newly-approved agent. As a result, the Food and Drug Administration (FDA) sometimes requires post-marketing investigations such as large scale post-marketing safety surveillance, Phase IV studies, or risk management and monitoring programs, though gaps persist in subsequent dissemination of postmarket analyses [1]. FDA also supports patient- and provider-driven reporting of potential adverse events via the FDA Adverse Event Reporting System (FAERS).
However, these systems either search for specific safety signals (e.g., ion channel block and arrhythmias) or remain reactive - waiting for unspecified safety events to occur, be recognized, and reported in humans - and doing so amidst an otherwise chaotic and complex healthcare environment. This challenge introduces a key question: can methods be developed to predict safety events that should be specifically sought for a given new drug during development and post-marketing activities? Early attention to such adverse drug reactions would reduce the number of patients at risk as clinical development proceeds. We present here such an approach applied to a new set of drug entities, inhibitors of the serine protease proprotein convertase subtilisin/kexin type 9 (PCSK9) [2]. Because these are new, it is likely there are side effects still yet to be identified, and if so, early detection may create the opportunity to prevent them.
1.1. Importance of pleiotropy in PCSK9
PCSK9’s involvement in lipid homeostasis is well-understood and has been the major stimulus for the development and refinement of therapeutics targeting this mechanism of action over the past decade. Since the discovery of this protein in 2003 [3], the work of understanding its function and developing agents that target PCSK9 has progressed at a rapid pace, with the first human subject treated with a PCSK9 monoclonal antibody in 2009 [4]. PCSK9 is a protein produced by the liver and other tissues and secreted into the circulation, where it binds the low-density lipoprotein receptor (LDLR), leading to intracellular degradation of the LDLR and termination of its lifecycle, and a subsequent increase in circulating low density lipoprotein cholesterol (LDL-C) levels.
Clinical trials of PCSK9 inhibitors to date have been summarized in a recent Cochrane review and have consistently shown marked reductions in LDL-C levels an average of around 50%, regardless of baseline LDL-C levels or background treatment [5]. PCSK9 has also been implicated in a wide spectrum of other biochemical and disease processes, including secretion of triglyceride-rich lipoproteins, inflammatory response in sepsis, liver regeneration following hepatectomy, susceptibility to hepatitis C virus infection, and perhaps tumor metastasis [6–8]. Thus, emerging evidence supports the possibility of pleiotropic effects of PCSK9 beyond lipid metabolism; however, there have also been a small number of patients homozygous for PCSK9 loss-of-function variants reported in the literature without obvious phenotypes beyond the expected decreases in LDL levels [9,10]. It is therefore unknown whether any pleiotropic effects, if real, would translate to effects resulting from pharmacologic PCSK9 inhibition, and whether such effects could even be considered ‘on target.’
1.2. Proposed Opportunity
This protein is particularly amenable to combined genotype and phenotype assessment, given the availability of multiple PCSK9 variants with known functional implications and the associated, deliberate objective of the monoclonal antibodies (mAbs) and other inhibitors to mimic this natural biological effect. FDA approved the first two agents in the PCSK9 inhibitor class, alirocumab and evolocumab, in mid-2015 [2]. Two additional investigational agents targeting this protein have demonstrated promising results in recently completed Phase II studies and continue in active development (LY3015014; inclisiran) [11].
In addition, PCSK9 variants include both gain-of-function and loss-of-function effects, allowing for inferences regarding directionality of biologic effects (e.g., increased or decreased risk of a given phenotype resulting from an alteration in PCSK9 function). Gain-of-function mutations in PCSK9 are rare and produce an extreme hypercholesterolemia phenotype with marked elevation of LDL-C levels and increased risk of premature coronary heart disease, whereas the more common loss-of-function variants are associated with unusually low LDL-C and decreased cardiovascular disease risk [12]. Of particular interest, the R46L loss-of-function polymorphism (rs11591147; c.137G>T; R46L) in PCSK9 has consistently been associated with 10–15% lower LDL-C across the age spectrum in the general population, and its cholesterol-lowering effect appears to be greater among normocholesterolemic individuals than among those with molecularly defined familial hypercholesterolemia (FH) [13,14]. In some studies, the magnitude of reduction in coronary heart disease or ischemic heart disease risk among R46L allele carriers was substantially higher than that predicted based on the observed reduction in LDL-C alone [15,16], consistent with the idea that genotype, as opposed to LDL-C measured during adult life, may be a more relevant measure of lifelong LDL-C levels.
We have developed a system that uses natural genetic variation to predict effects of drugs [17]. This approach leverages the known effects of changes in PCSK9 to enable the identification of other novel phenotypes in which this protein - and its inhibitors - may have impact. A recent report suggests that pursuing targets supported by human genetics could double the success rate in the clinical development of new therapeutic agents [18]. Given it is widely accepted that many existing in vitro and preclinical animal models are poor predictors of human biology [19], the method described here provides an alternative approach to accurate prediction of the physiologic effects of new therapies in humans. Thus, we can predict potential effects, both beneficial and harmful, of a pharmacological intervention in humans before any clinical trial is ever initiated, and indeed, even before a single dose is given to humans. The objective of the current study is to demonstrate the utility of this method as an approach for detecting safety signals, illustrated by exploration of the potential adverse biologic effects of PCSK9 inhibition.
2. Methods
2.1. Leveraging existing infrastructure
We use unique, large-scale resources and databases at Vanderbilt University Medical Center including BioVU, a DNA databank which houses ~250,000 DNA samples linked to a de-identified image called the Synthetic Derivative (SD) of the entire ~2.6 million subject electronic health record (EHR) [20,21]. The SD combines clinical information contained in the EHR with the data stripped of personal identifiers and modified to improve utility for research [20]. The SD contains the core elements of health-related phenotypes, e.g., diagnoses, procedures, medications, laboratory testing results, radiology reports, family history, social history, vital signs, demographics, hospitalizations, and other details extracted from clinical narratives. These resources have been used to identify diagnoses, temporality of events, sequence-based events, medication dosages, use of radiographic tests, and key concepts mined from narratives [20]. Importantly, approximately 36,000 subjects in BioVU have already been genotyped using the Illumina Infinium Exomechip, which provides the dataset for this work.
2.2. General method description
2.2.1. General method: Select the proxy single nucleotide polymorphism (SNP)
Application to PCSK9 example: Focusing on missense SNPs as more likely to have functional impact, we performed a thorough literature and database review on SNPs within our existing Exomechip data to explore concordance between our findings and known associations between PCSK9 variants and manifestations of health and disease. We employed the data gathered in this step to build a candidate hypothesis to elucidate connections between the protein, SNP, and phenotype pathway. As described further below, we selected the R46L variant in PCSK9 as our proxy SNP, given its availability on the Exomechip and literature supporting its association with PCSK9 loss of function, as well as validation of its biologic effects in our data (e.g. reduced LDL-C, reduced risk of hyperlipidemia).
2.2.2. General method: Validate known effects of the SNP in the population
Application to PCSK9 example: We conducted a phenome-wide association study (PheWAS) using our previously validated approach [22–24], including 29,722 patients of European ancestry with available Illumina Human Exome chip data and phenotype information in the SD. It is important to note that we focus on the European ancestry data because we are underpowered in other ethnicities, though efforts are underway to increase population diversity in our genotyping platforms. Odds ratios were derived from an additive logistic regression model, adjusted for age, gender, and study (whether the patient came from a specific disease cohort or genotyping set, to correct for batch effects); p values were calculated using the Wald method. Our variant of focus for the current investigation was the R46L loss-of-function variant in PCSK9.
Next, we used laboratory values collected during routine clinical care and stored within the de-identified EHR to assess function of this variant. This complementary step is intended to validate that the known effects of the SNP are seen via real-world clinical impact, i.e. association of R46L with LDL values. The analysis of distribution of laboratory testing results, also extracted from the EHR, yields data on any significant trends in increase or decrease of various laboratory test values in carriers of allele of interest as compared with non-carriers.
2.2.3. General method: Clarify orientation of efficacy and safety, and specificity of side effects
Application to PCSK9 example: As a validation step, we assessed association between R46L and its known phenotype (reduced risk of hypercholesterolemia; reduced LDL). We also explored potential novel phenotypes associated with this loss of function variant. Figure 1 provides a snapshot of this approach, including an overview of both validation data and novel phenotypes of interest.
Figure 1.
Signal detection overview: Validation and novel phenotypes
Note: Green arrows correspond with beneficial effects of reduced PCSK9; red arrows correspond with potentially harmful effects of reduced PCSK9.
Finally, we extracted the entire de-identified medical record of the patients homozygous for R46L and our novel phenotype of interest to investigate whether our candidate hypothesis has any preliminary merit in a small population.
3. Results
3.1. Validation of effect
First, we confirmed the strong association of the R46L SNP with significantly decreased LDL cholesterol and total cholesterol as noted in the laboratory analysis, corresponding with the top two hits within these results (Table 1). We believe the significant degree of concordance between these findings in a large clinical dataset with laboratory measures taken on more than 13,000–14,000 patients over time and through multiple health conditions, and the existing body of literature detailing implications of this SNP, shows validity in our datasets and approach.
Table 1.
Laboratory test results, R46L SNP in PCSK9
| SNP | Lab | Measure | Beta (mg/dl) | P-value | Cases |
|---|---|---|---|---|---|
| R46L | Cholesterol | mean | −14.23 | 1.56×10−16 | 14671 |
| R46L | LDL-C | mean | −14.47 | 2.58×10−23 | 13379 |
3.2. PheWAS
Again, PheWAS confirmed the involvement of the R46L SNP in dyslipidemias, including significantly reduced risk of hypercholesterolemia (OR 0.68, p=7.6 × 10−4, Table 2). Results also showed reduced risk of cardiovascular disease (Table 2), including coronary atherosclerosis (OR=0.79, p=0.025) and other forms of chronic heart disease (OR 0.64, p=0.042). We examined other phenotype associations with the R46L SNP in the PheWAS results to identify potential novel connections; we note a significant association between this PCSK9 loss of function SNP and significantly increased risk of spina bifida (OR 5.90, p=2.7 × 10−4, Table 2). Of note, both hypercholesterolemia and spina bifida were within the top five PheWAS results for our variant of interest. We also identified a constellation of phenotypes in our PheWAS findings related to the skeletal system: ankle and foot fractures, calcaneal spurs, vertebral fractures, as well as a compelling association with a foundational skeletal phenotype, senile osteoporosis (Table 2).
Table 2.
PheWAS results, PCSK9 variant R46L
| SNP | SNP effect | A potential safety signal OR should be: | Phenotype | Cases (n) | Controls (n) | OR (95% CI) | P-value | Case carriers (n) |
|---|---|---|---|---|---|---|---|---|
| R46L | LOF | >1 | Spina bifida | 34 | 25,268 | 5.90 (2.27–15.36) | 2.7×10−04 | 6 |
| R46L | LOF | >1 | Hypercholesterolemia | 3,840 | 14,560 | 0.68 (0.54–0.85) | 7.6×10−04 | 101 |
| R46L | LOF | >1 | Mixed hyperlipidemia* | 4,328 | 14,560 | 0.70 (0.57–0.87) | 1.3×10−03 | 114 |
| R46L | LOF | >1 | Fracture of ankle and foot* | 660 | 21864 | 1.76 (1.25–2.47) | 1.3×10−03 | 37 |
| R46L | LOF | >1 | Calcaneal spur; Exostosis NOS | 57 | 19642 | 3.43 (1.50–7.86) | 3.6×10−03 | 6 |
| R46L | LOF | >1 | Hyperlipidemia* | 9050 | 14560 | 0.79 (0.67–0.93) | 3.8×10−03 | 264 |
| R46L | LOF | >1 | Senile osteoporosis | 745 | 19123 | 1.52 (1.06–2.18) | 0.021 | 38 |
| R46L | LOF | >1 | Coronary atherosclerosis | 4960 | 18085 | 0.79 (0.65–0.97) | 0.025 | 134 |
| R46L | LOF | >1 | Fracture of foot* | 344 | 21864 | 1.69 (1.05–2.73) | 0.030 | 19 |
| R46L | LOF | >1 | Other forms of chronic heart disease | 999 | 18085 | 0.64 (0.42–0.98) | 0.042 | 23 |
| R46L | LOF | >1 | Fracture of vertebral column without mention of spinal cord injury | 617 | 21864 | 1.47 (1.01–2.15) | 0.047 | 29 |
Key: Bolded odds ratios indicate potential consequences of long-term natural PCKS9 inhibition; OR odds ratio; CI = confidence interval; LOF = loss-of-function. Note: R46L minor allele frequency is 0.01594 in the BioVU Exomechip population.
“Mixed hyperlipidemia” represents a subset of the “Hyperlipidemia” phenotype; “Fracture of foot” represents a subset of the “Fracture of ankle and foot” phenotype.
3.4. Preliminary case-level analysis
To further explore phenotypic effects of PCSK9 loss-of-function SNPs, we examined case records for the adults with the PheWAS code for spina bifida who were also homozygous for the R46L variant (n=6). Among these six individuals, two were confirmed spina bifida cases, and an additional two were affected by other neural tube defects (one patient with Chiari malformation and spinal dysraphism; one patient with encephalocele and skull base defect). One patient had no indication of spina bifida confirmed at chart review, but had another significant congenital issue involving skeletal alterations (bladder exstrophy with wide diastasis of symphysis pubis) as well as development of rare bone tumors (eosinophilic granulomas) affecting the femur and skull base in adulthood. Chart review of the remaining case revealed no indication of spina bifida, but the patient was diagnosed with another rare neurologic condition, neuromyelitis optica, in adulthood. Thus, case review revealed patient data concordant with potential involvement of reduced PCSK9 in development of spina bifida or other issues affecting the central nervous system and skeletal structure. Most of these cases unfortunately did not have LDL measurements available for assessment of the variant’s effects on LDL levels.
4. Discussion
These results demonstrate the potential utility of our approach for joining existing data sources to predict potential safety challenges, using the illustrative example of PCSK9 inhibition. In this example, the safety issue we propose is spina bifida. Because the PCSK9 inhibitors are relatively new, there is an opportunity to prevent these negative outcomes. While all of these phenotypes require considerable validation, it is encouraging to see the approved indication of the drug validated in the dataset.
This approach might provide a way of filtering to a more focused set of potential safety signals or side effects within the nearly limitless realm of human physiology. Further, evidence of concordance between the PheWAS phenotype of neural tube defects and recent literature on the involvement of PCSK9 in neurologic development and in spina bifida, specifically, suggest these associations may warrant additional investigation as data on real world use of the PCSK9 mAbs continues to accrue [25–28]. The observed association with changes affecting the skeletal system is also intriguing, given that spina bifida may often include concurrent alteration of skeletal features such as the skull and vertebrae.
These results also indicate that diagnostic and laboratory data from the EHR provide a picture that can aid in developing powering hypotheses for targeted investigations of ways in which protein changes may affect health and development of disease. Analysis of such large combined datasets can tell us how various SNPs and associated protein changes may be used to simulate the effects of a drug, and thereby how they are likely to affect the human body, with an agnostic approach that generates hypotheses regarding both therapeutic applications of relevant drugs as well as possible side effects not yet observed in humans.
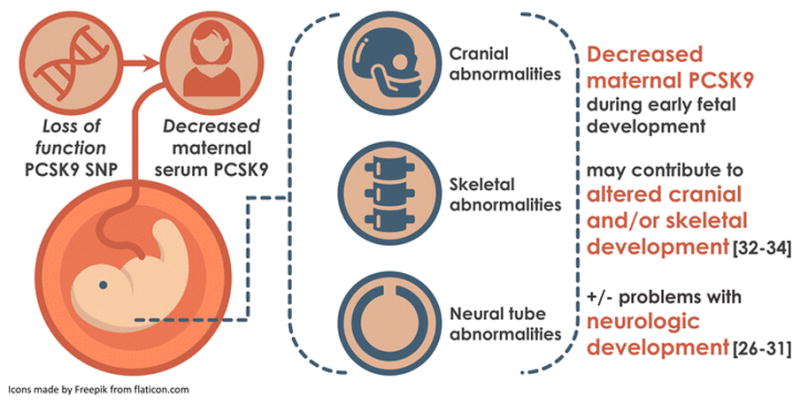
We propose it is plausible that lower levels of PCSK9, as present in individuals with the R46L variant, may lead to increased risk of development of neural tube defects. Figure 2 illustrates one set of potential pathways by which decreased PCSK9 may lead to development of a neural tube defect. PCSK9 is known to be expressed in the brain at certain critical periods during embryonic development [25–27], a key time point in the disease pathway for spina bifida. Knockdown of PCSK9 in zebrafish mRNA was associated with embryonic death at approximately 96 hours after fertilization due to central nervous system degeneration [26]. Decreased PCSK9 has also been associated with neuronal apoptosis and alterations in cortical neural differentiation.[27,29–31] Concordant with the current observations, work by An et al. also indicated a marked decrease in PCSK9 in a rat fetal model of spina bifida, as compared with normally developing fetuses, and also found a significant decrease in serum PCSK9 in women carrying a fetus affected by a neural tube defect as compared with pregnant women carrying a healthy fetus [28]. Further, this study found PCSK9 serum levels in pregnant women to be a potentially useful biomarker in humans for indicating risk of neural tube defect in the fetus, with an area under the curve of 0.763, sensitivity of 57%, and specificity of 98% [28]. Data exploring the potential contributions of decreased PCSK9 to altered development of the skeletal system itself is currently indirect remains indirect but intriguing. For example, low LDL-C, also a reasonable consequence of decreased PCSK9 levels, was associated with increased fracture risk in a case control study of patients with type 2 diabetes,[32] and inhibition of another structurally similar protein in the same family (SKI-1) has been observed to interfere with osteoblastic mineralization.[33,34] Further investigations are clearly required to explore the pathways by which alteration of PCSK9 levels may contribute to development of neural tube defects.
Figure 2.

Potential paths by which PCSK9 may contribute to development of spina bifida
A review of existing clinical trial evidence shows that trial data surrounding adverse effects of the PCSK9 inhibitors have been highly compelling in terms of the efficacy and relative safety of these agents [5]. However, these have been trials of relatively short duration as it relates to uncovering phenotypes such as those above, including risk of neural tube defects among pregnant women exposed to the PCKS9 inhibitors. Because pregnant women are often excluded from trials, this particular adverse event would have no chance of being noticed until the drug is on market. Animal models involving administration of alirocumab or evolocumab at doses ≥12 times the maximum recommended human dose from organogenesis through parturitions indicated no gross effects on pregnancy or neonatal/infant development; as with other IgG antibodies, post-natal serum samples confirmed that these two agents cross the placenta [35,36]. Clinical trials assessing short and longer term use of PCSK9 inhibitors have not noted any increase in fracture risk, though likely underpowered for such potential safety outcomes [37,38].
4.1. Limitations
Though these results are thought-provoking, we acknowledge limitations that highlight the importance of further exploration. First, while PCSK9 levels performed as a useful biomarker in one human study of spina bifida [28], additional work remains to elucidate how this protein may implicated in the developmental pathway of neural tube defects. For example, it is unclear whether alterations in lipid homeostasis are implicated in this potential phenomenon, or if another pleiotropic mechanism contributes; our study was underpowered for further covariate analysis of LDL values and genotype with occurrence of spina bifida, as only one of the case carriers had LDL data available in the EHR, which may have enabled further insights into potential pathogenesis. The current report also does not represent an exhaustive exploration of potential adverse effects of the therapeutic class; though other data suggest that alterations in glucose homeostasis may be a risk of PCSK9 loss of function,[39–41] particularly when other covariates such as serum LDL and BMI are incorporated, the R46L variant was not associated with increased risk of type 2 diabetes in our data and in another investigation.[42] Additional covariate analyses, as well as considering of postmarketing surveillance data in those using the inhibitors, will be necessary to inform a more complete evaluation of any relative contribution of PCSK9 inhibition to diabetogenesis.
Further, regarding the parallel signal involving the effects in the skeletal system, it is known that all fractures are not the same. In fact, the type of fracture has different underlying etiologies by age and other variables; for example, in one study, foot fractures in older women were commonly osteoporotic fractures, whereas ankle fractures occurred in younger women with a relatively high body mass index [43]. However, our primary focus on ICD-9 diagnostic codes for defining phenotypes in the current study precludes granular analysis of fracture etiology, as even case review may reveal general information about events leading to fracture without detailed clinician commentary on contributing factors. Additionally, the size of our observed spectrum of p-values and the limited spectrum of PCSK9 variants in our existing genotyping platform suggests our data may be underpowered for fully understanding the connections identified; further exploration in a larger dataset, including an expanded number of patients as well as a broader range of PCSK9 genotypes, would be beneficial for confirmation of this signal. Other exploratory studies will be necessary to better elucidate the involvement of PCSK9 in the proposed disease pathways.
Despite these limitations, the methodology described here is in line with the FDA’s approach to reporting potential safety signals while also explicitly acknowledging that a causal pathway has not been established [44]; further, others have also begun to explore in silico methods for predicting safety events.[45] An early safety signal uncovered in this kind of work might end up being a false positive. However, it also could provide true benefit to the design of monitoring approaches for clinical trial work as well as post-marketing surveillance. Given that these agents are approved for use in familial hypercholesterolemia, women of childbearing age are at risk of exposure. Additionally, if the constellation of skeletal symptoms observed in the current analysis did represent a real potential side effect of agents that affect PCSK9, associated risk could be prospectively preempted via a number of viable mechanisms: 1) monitoring of bone mineral density (BMD), 2) concomitant therapy using bisphosphonates should BMD changes be observed, or 3) identifying individuals who are likely less resilient to changes in bone density for closer monitoring.
5. Conclusions
The novel methodology described herein indicates that spina bifida is a potential adverse effect of PCSK9 inhibition, warranting further research into pathogenesis as well as surveillance. These methods provide an opportunity to put in place new mechanisms to assess the safety and long-term tolerability of PCSK9 inhibitors specifically and other new agents, in general, as they move into human testing and expanded clinical use. We anticipate that future research will explore the application of these methods to additional therapeutic agents to allow broader and earlier detection of safety signals for exploration and monitoring during clinical use.
Key points.
Applying phenome-wide association study (PheWAS) methods in 29,722 patients confirmed association between a proprotein convertase subtilisin/kexin type 9 (PCSK9) loss of function variant (R46L) and reduced risk of hypercholesterolemia, and revealed a potential novel association with increased risk of spina bifida.
Phenotypic effects of naturally occurring human genetic variants may provide an important opportunity for enhancing prediction of possible safety effects of novel therapeutic agents, such as the PCSK9 inhibitors.
Acknowledgments
Funding: The project described herein is supported by the Clinical and Translational Science Award award number UL1TR000445 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
We extend our sincere thanks to Nicole Zaleski, MA, MPH, for her valuable input and assistance with preparing the figures and tables for this paper, and Xinnan Niu for his assistance with preparing the data for publication.
Footnotes
COMPLIANCE WITH ETHICAL STANDARDS
Ethical approval: This project was reviewed and received a non-human subjects research determination from the Vanderbilt University Institutional Review Board (IRB #151121).
Conflicts of interest: Rebecca N. Jerome, Jill M. Pulley, Dan M. Roden, Jana K. Shirey-Rice, Lisa A. Bastarache, Gordon Bernard, Leeland Ekstrom, William J. Lancaster, and Joshua C. Denny have no conflicts of interest that are directly relevant to the content of this study
References
- 1.Cruz ML, Xu J, Kashoki M, Lurie P. Publication and Reporting of the Results of Postmarket Studies for Drugs Required by the US Food and Drug Administration, 2009 to 2013. JAMA Intern Med [Internet] 2017 doi: 10.1001/jamainternmed.2017.1313. [cited 2017 May 17]; Available from: http://jamanetwork.com/journals/jamainternalmedicine/fullarticle/2626857. [DOI] [PMC free article] [PubMed]
- 2.Noel ZR, Beavers CJ. Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors: A Brief Overview. Am J Med. 2017;130:229.e1–229.e4. doi: 10.1016/j.amjmed.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 3.Abifadel M, Varret M, Rabès J-P, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 4.Stein EA, Raal F. Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu Rev Med. 2014;65:417–31. doi: 10.1146/annurev-med-022613-090402. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt AF, Pearce LS, Wilkins JT, Overington JP, Hingorani AD, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2017;4:CD011748. doi: 10.1002/14651858.CD011748.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavori H, Rashid S, Fazio S. On the function and homeostasis of PCSK9: reciprocal interaction with LDLR and additional lipid effects. Atherosclerosis. 2015;238:264–70. doi: 10.1016/j.atherosclerosis.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walley KR. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Science Translational Medicine [Internet] 2014:6. doi: 10.1126/scitranslmed.3008782. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25320235. [DOI] [PMC free article] [PubMed]
- 8.Awan Z, Baass A, Genest J. Proprotein convertase subtilisin/kexin type 9 (PCSK9): lessons learned from patients with hypercholesterolemia. Clin Chem. 2014;60:1380–9. doi: 10.1373/clinchem.2014.225946. [DOI] [PubMed] [Google Scholar]
- 9.Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193:445–8. doi: 10.1016/j.atherosclerosis.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch L, Grishin NV, Horton JD, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79:514–23. doi: 10.1086/507488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kastelein JJP, Nissen SE, Rader DJ, Hovingh GK, Wang M-D, Shen T, et al. Safety and efficacy of LY3015014, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 (PCSK9): a randomized, placebo-controlled Phase 2 study. Eur Heart J. 2016;37:1360–9. doi: 10.1093/eurheartj/ehv707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulz R, Schlüter K-D, Laufs U. Molecular and cellular function of the proprotein convertase subtilisin/kexin type 9 (PCSK9) Basic Res Cardiol. 2015;110:4. doi: 10.1007/s00395-015-0463-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saavedra YGL, Dufour R, Davignon J, Baass A. PCSK9 R46L, lower LDL, and cardiovascular disease risk in familial hypercholesterolemia: a cross-sectional cohort study. Arterioscler Thromb Vasc Biol. 2014;34:2700–5. doi: 10.1161/ATVBAHA.114.304406. [DOI] [PubMed] [Google Scholar]
- 14.Chernogubova E, Strawbridge R, Mahdessian H, Mälarstig A, Krapivner S, Gigante B, et al. Common and low-frequency genetic variants in the PCSK9 locus influence circulating PCSK9 levels. Arterioscler Thromb Vasc Biol. 2012;32:1526–34. doi: 10.1161/ATVBAHA.111.240549. [DOI] [PubMed] [Google Scholar]
- 15.Guella I, Asselta R, Ardissino D, Merlini PA, Peyvandi F, Kathiresan S, et al. Effects of PCSK9 genetic variants on plasma LDL cholesterol levels and risk of premature myocardial infarction in the Italian population. J Lipid Res. 2010;51:3342–9. doi: 10.1194/jlr.M010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybjaerg-Hansen A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J Am Coll Cardiol. 2010;55:2833–42. doi: 10.1016/j.jacc.2010.02.044. [DOI] [PubMed] [Google Scholar]
- 17.Pulley JM, Shirey-Rice JK, Lavieri RR, Jerome RN, Zaleski NM, Aronoff DM, et al. Accelerating Precision Drug Development and Drug Repurposing by Leveraging Human Genetics. ASSAY and Drug Development Technologies. 2017;15:113–9. doi: 10.1089/adt.2016.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–60. doi: 10.1038/ng.3314. [DOI] [PubMed] [Google Scholar]
- 19.Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nature Reviews Drug Discovery. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 20.Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84:362–9. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGregor TL, Van Driest SL, Brothers KB, Bowton EA, Muglia LJ, Roden DM. Inclusion of pediatric samples in an opt-out biorepository linking DNA to de-identified medical records: pediatric BioVU. Clin Pharmacol Ther. 2013;93:204–11. doi: 10.1038/clpt.2012.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31:1102–10. doi: 10.1038/nbt.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–10. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei W-Q, Bastarache LA, Carroll RJ, Marlo JE, Osterman TJ, Gamazon ER, et al. Evaluating phecodes, clinical classification software, and ICD-9-CM codes for phenome-wide association studies in the electronic health record. In: Rzhetsky A, editor. PLOS ONE. Vol. 12. 2017. p. e0175508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rousselet E, Marcinkiewicz J, Kriz J, Zhou A, Hatten ME, Prat A, et al. PCSK9 reduces the protein levels of the LDL receptor in mouse brain during development and after ischemic stroke. J Lipid Res. 2011;52:1383–91. doi: 10.1194/jlr.M014118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poirier S, Prat A, Marcinkiewicz E, Paquin J, Chitramuthu BP, Baranowski D, et al. Implication of the proprotein convertase NARC-1/PCSK9 in the development of the nervous system. J Neurochem. 2006;98:838–50. doi: 10.1111/j.1471-4159.2006.03928.x. [DOI] [PubMed] [Google Scholar]
- 27.Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci US A. 2003;100:928–33. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.An D, Wei X, Li H, Gu H, Huang T, Zhao G, et al. Identification of PCSK9 as a novel serum biomarker for the prenatal diagnosis of neural tube defects using iTRAQ quantitative proteomics. Sci Rep. 2015;5:17559. doi: 10.1038/srep17559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerjee Y, Santos RD, Al-Rasadi K, Rizzo M. Targeting PCSK9 for therapeutic gains: Have we addressed all the concerns? Atherosclerosis. 2016;248:62–75. doi: 10.1016/j.atherosclerosis.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 30.Kysenius K, Muggalla P, Mätlik K, Arumäe U, Huttunen HJ. PCSK9 regulates neuronal apoptosis by adjusting ApoER2 levels and signaling. Cell Mol Life Sci. 2012;69:1903–16. doi: 10.1007/s00018-012-0977-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L-S, Bai X-Q, Gao Y, Wu Q, Ren Z, Li Q, et al. PCSK9 Promotes oxLDL-Induced PC12 Cell Apoptosis Through the Bcl-2/Bax-Caspase 9/3 Signaling Pathway. J Alzheimers Dis. 2017;57:723–34. doi: 10.3233/JAD-161136. [DOI] [PubMed] [Google Scholar]
- 32.Starup-Linde J, Gregersen S, Vestergaard P. Associations with fracture in patients with diabetes: a nested case-control study. BMJ Open. 2016;6:e009686. doi: 10.1136/bmjopen-2015-009686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorski JP, Huffman NT, Cui C, Henderson EP, Midura RJ, Seidah NG. Potential role of proprotein convertase SKI-1 in the mineralization of primary bone. Cells Tissues Organs (Print) 2009;189:25–32. doi: 10.1159/000151723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorski JP, Huffman NT, Chittur S, Midura RJ, Black C, Oxford J, et al. Inhibition of proprotein convertase SKI-1 blocks transcription of key extracellular matrix genes regulating osteoblastic mineralization. J Biol Chem. 2011;286:1836–49. doi: 10.1074/jbc.M110.151647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prescribing information: REPATHA(TM) (evolocumab) Amgen: 2015. [Google Scholar]
- 36.Prescribing information: PRALUENT(TM) (alirocumab) [Internet] Sanofi Regeneron; 2015. [cited 2017 May 22]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125559Orig1s000lbledt.pdf. [Google Scholar]
- 37.Sullivan D, Olsson AG, Scott R, Kim JB, Xue A, Gebski V, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA. 2012;308:2497–506. doi: 10.1001/jama.2012.25790. [DOI] [PubMed] [Google Scholar]
- 38.Blom DJ, Hala T, Bolognese M, Lillestol MJ, Toth PD, Burgess L, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–19. doi: 10.1056/NEJMoa1316222. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt AF, Swerdlow DI, Holmes MV, Patel RS, Fairhurst-Hunter Z, Lyall DM, et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97–105. doi: 10.1016/S2213-8587(16)30396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saavedra YGL, Dufour R, Baass A. Familial hypercholesterolemia: PCSK9 InsLEU genetic variant and prediabetes/diabetes risk. J Clin Lipidol. 2015;9:786–793. e1. doi: 10.1016/j.jacl.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 41.Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N Engl J Med. 2016;375:2144–53. doi: 10.1056/NEJMoa1604304. [DOI] [PubMed] [Google Scholar]
- 42.Bonnefond A, Yengo L, May CL, Fumeron F, Marre M, Balkau B, et al. The loss-of-function PCSK9 p.R46L genetic variant does not alter glucose homeostasis. Diabetologia. 2015;58:2051–5. doi: 10.1007/s00125-015-3659-8. [DOI] [PubMed] [Google Scholar]
- 43.Hasselman CT, Vogt MT, Stone KL, Cauley JA, Conti SF. Foot and ankle fractures in elderly white women. Incidence and risk factors. J Bone Joint Surg Am. 2003;85–A:820–4. doi: 10.2106/00004623-200305000-00008. [DOI] [PubMed] [Google Scholar]
- 44.Research C for DE and. FDA Adverse Events Reporting System (FAERS) Potential Signals of Serious Risks/New Safety Information Identified from the FDA Adverse Event Reporting System (FAERS) [Internet] [cited 2017 May 12]. Available from: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/UCM082196.
- 45.Walker VM, Davey Smith G, Davies NM, Martin RM. Mendelian randomization: a novel approach for the prediction of adverse drug events and drug repurposing opportunities. Int J Epidemiol. 2017 doi: 10.1093/ije/dyx207. [DOI] [PMC free article] [PubMed] [Google Scholar]