

Abstract
Significant progress has been achieved in developing precision therapies for cystic fibrosis; however, highly effective treatments that target the ion channel, CFTR, are not yet available for many patients. As numerous CFTR therapeutics are currently in the clinical pipeline, reliable screening tools capable of predicting drug efficacy to support individualized treatment plans and translational research are essential. The utilization of bronchial, nasal, and rectal tissues from individual cystic fibrosis patients for drug testing using in vitro assays such as electrophysiological measurements of CFTR activity and evaluation of fluid movement in spheroid cultures, has advanced the prediction of patient-specific responses. However, for precise prediction of drug effects, in vitro models of CFTR rescue should incorporate the inflamed cystic fibrosis airway environment and mimic the complex tissue structures of airway epithelia. Furthermore, novel assays that monitor other aspects of successful CFTR rescue such as restoration of mucus characteristics, which is important for predicting mucociliary clearance, will allow for better prognoses of successful therapies in vivo. Additional cystic fibrosis treatment strategies are being intensively explored, such as development of drugs that target other ion channels, and novel technologies including pluripotent stem cells, gene therapy, and gene editing. The multiple therapeutic approaches available to treat the basic defect in cystic fibrosis combined with relevant precision medicine models provide a framework for identifying optimal and sustained treatments that will benefit all cystic fibrosis patients.
Keywords: cystic fibrosis, precision medicine, CFTR, primary human airway epithelial cells, bronchospheroids, nasospheroids, patient-derived models
Introduction
Cystic fibrosis (CF) is characterized by abnormal epithelial ion transport resulting from mutations in the CFTR gene (1). The CFTR protein is an ion channel that mediates Cl− and HCO3− transport of secretory and absorptive epithelial cells in multiple organs including lungs and intestines. The absence of CFTR also results in enhanced Na+ uptake via the ENaC channel, leading to dehydrated airways (2–4). The majority of morbidity and mortality associated with CF is due to airway disease caused by disturbances of airway surface liquid (ASL) homeostasis that result in viscous and sticky mucus, which leads to mucus stasis, airway obstruction, persistent infection, inflammation, and a progressive decline in lung function (5, 6). Clinical advances in comprehensive treatments of CF symptoms, including strategies to improve mucociliary clearance (MCC), and antibiotics to eradicate bacterial lung infections, result in only limited capacity to delay disease progression and increase survival of CF patients (7). Discovery of the gene responsible for CF over 25 years ago (1) led to an understanding of how various CFTR mutations cause different CFTR biochemical or functional protein aberrations ranging from complete protein absence to defective protein activity. Approximately 2,000 unique CFTR variants have been identified, with F508del as the most common CF-causing mutation, and a large number of rare mutations accounting for the remainder of CF cases. Notably, the severity of CF is influenced by factors beyond CFTR mutations such as modifier genes, environment, and lifestyle, such that individuals possessing the same CFTR mutation may respond differently to the same treatment (8–10). This complexity presents an unprecedented need for relevant, predictive models for testing of CF therapies. Therapeutics are being developed that specifically target the CFTR protein, thereby alleviating CF symptoms at the molecular level. The use of small-molecular compounds that modulate CFTR (ivacaftor/VX-770 and lumacaftor/VX-809) has led to the development of two medicines for CF patients, Kalydeco (VX-770) and Orkambi (VX-770 plus VX-809). Targeted use of Kalydeco is a powerful example of how the CF community is leading the field of precision medicine. VX-770 was made available in the US in 2012 as a stand-alone therapy for individuals with CFTR gating mutations such as G551D (11, 12). Although Kalydeco has dramatically improved the lives of a small proportion of the CF population (~5%), the remaining challenge is to achieve similar success for the entire CF population. Orkambi was developed for the F508del CFTR population, but clinical gains are modest (13, 14) and adverse side effects have forced some patients to stop taking this medication. Thus, despite significant progress in developing precision therapies for CF, highly effective treatments are not yet available for most CF patients. As numerous CFTR-targeting compounds and reagents are currently in the clinical pipeline (Table 1), new screening tools capable of predicting drug efficacy in an individualized manner are needed and expected to have a profound impact on the well-being of CF patients.
Table 1.
Novel CFTR therapeutics that are currently in the drug pipeline or in clinical trials.
| Company | Compound |
|---|---|
| 4D Molecular Therapeutics | Gene therapeutic for CF using adeno-associated virus to target lung airway cells |
| AbbVie/Galapagos | C1 (GLPG2222, GLPG2851) and C2 (GLPG2737) correctors and potentiators (GLPG1837, GLPG2451, GLPG3067) |
| Arcturus Therapeutics | LUNAR-CF, mRNA therapeutic to restore CFTR expression |
| Bayer | Riociguat to improve CFTR channel expression and function |
| Calista Therapeutics | Peptide drugs that inhibit the interaction between CAL and CFTR |
| Catabasis Pharmaceuticals | CAT-5571 to activate autophagy and improve CFTR function |
| Concert Pharmaceuticals | Potentiator CTP-656, a deuterium-modified version of ivacaftor |
| CRISPR Therapeutics | CRISPR-Cas9 gene editing |
| DiscoveryBioMed | Dual acting correcting and activating CFTR ligands |
| Editas Medicine | CRISPR/Cas approaches to edit CFTR DNA |
| Flatley Discovery Lab | Correctors (FDL169) and potentiators (FDL176) |
| Genzyme/Sanofi | Non-viral gene transfer agent (gene–liposome complex pGM169/GL67A), corrector compounds |
| Homology Medicines | CFTR gene therapy and gene editing |
| Ionins Pharmaceuticals | Antisense oligonucleotides to increase CFTR and reduce ENaC expression |
| Moderna Therapeutics | mRNA therapeutic for CFTR protein expression |
| Novartis | Potentiator (QBW251) and corrector compounds |
| Parion Sciences | Corrector compounds and ENaC inhibitor (VX-371/P-1037) |
| Pfizer | Correctors (PYR-41 targeting ubiquitination) and potentiators (CP-628006) |
| ProQR Therapeutics | QR-010, 33mer antisense oligonucleotide to restore CFTR expression |
| Proteostasis Therapeutics | CFTR amplifier PTI-428; corrector (PTI-801) and potentiator (PTI-808) |
| PTC Therapeutics | Ataluren for treatment of CFTR nonsense mutations |
| Reata Pharmaceuticals | Corrector compounds |
| Shire Rare Disease | Technology to deliver normal CFTR to the lungs |
| Southern Research Institutes | Drugs for translational readthrough of CFTR nonsense mutations |
| Talee Bio | Virus-based (TL-101, TL-102) CFTR gene therapy |
| Traffick Therapeutics | Correctors NU001 and NU002 |
| Vanda Pharmaceuticals | CFTR activators discovered by Alan S. Verkman, M.D., Ph.D., (UCSF) |
| Vertex Pharmaceuticals | Corrector VX-661, next-generation correctors VX-152, VX-440, VX-445, VX-659 |
Primary cells constitute a native environment for CFTR
Since the discovery of the CFTR gene, CFTR processing has been extensively studied in cell lines and some principles of quality control and cell-surface stability were later confirmed in primary human bronchial epithelial (HBE) cells. For example, in both systems, the F508del mutant is misprocessed, and when rescued has a shortened half-life at the plasma membrane (15–17). However, CFTR displays increased cell surface stability in differentiated HBE compared to non-polarized and polarized cell lines (16). Although BHK-21, FRT, and CFBE41o- cell lines have been commonly used to screen for CFTR modulators, it has become apparent that heterologous expression systems do not always reliably predict CFTR modulator efficacy in primary cells (18–20). Establishing the therapeutic benefit of available compounds such as VX-809 and VX-770 to correct rare CFTR mutation defects is currently an active research area, and the field is moving toward development of personalized medicine, where the individual CF patient’s tissue is used (Fig. 1), and thus, each patient’s genetic makeup will be the basis for determining therapeutic options. Physiologically relevant and predictable model systems that can be utilized for screening and prediction of clinical outcomes for multiple mutations are needed. As the stringency of CFTR rescue is higher in primary airway epithelial cultures than in cell lines, to accurately assess pharmacological rescue of mutant CFTR in the airways it is critical to use patient-derived tissue to generate culture models that allow for multiple methods of examining CFTR maturation and function (Fig. 2). Translational precision medicine is therefore required for improving treatment strategies and identifying optimal pharmaceutical combinations for CF patients with F508del as well as rare CFTR mutations.
Figure 1. Precision medicine models for CF therapeutics.
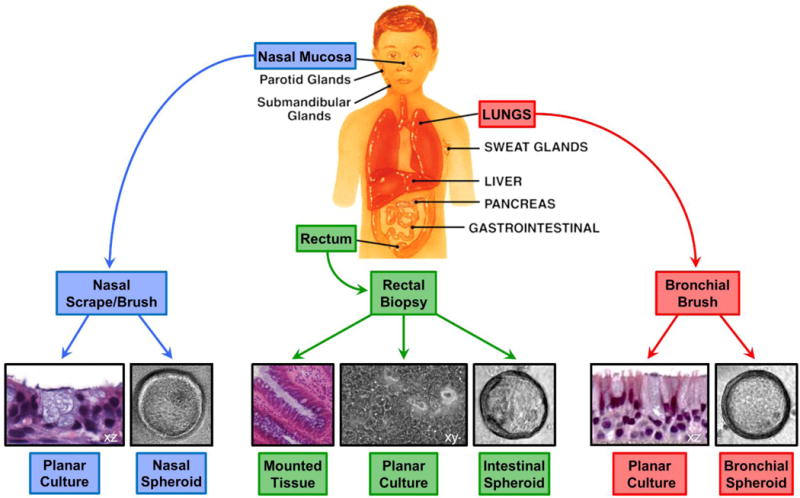
Tissue specimens collected from patients’ organs that are affected by CF can be used to generate 2D planar cultures and 3D spheroid cultures or analyzed as mounted tissue. Protocols for generating spheroid cultures from nasal, bronchial, and rectal epithelia have been developed. Planar cultures are established for human airway cells and mouse intestinal epithelial tissues and were recently developed for human rectal epithelium (61).
Figure 2. Efficient usage of donor specimens permits numerous analyses of physiological readouts for drug testing.
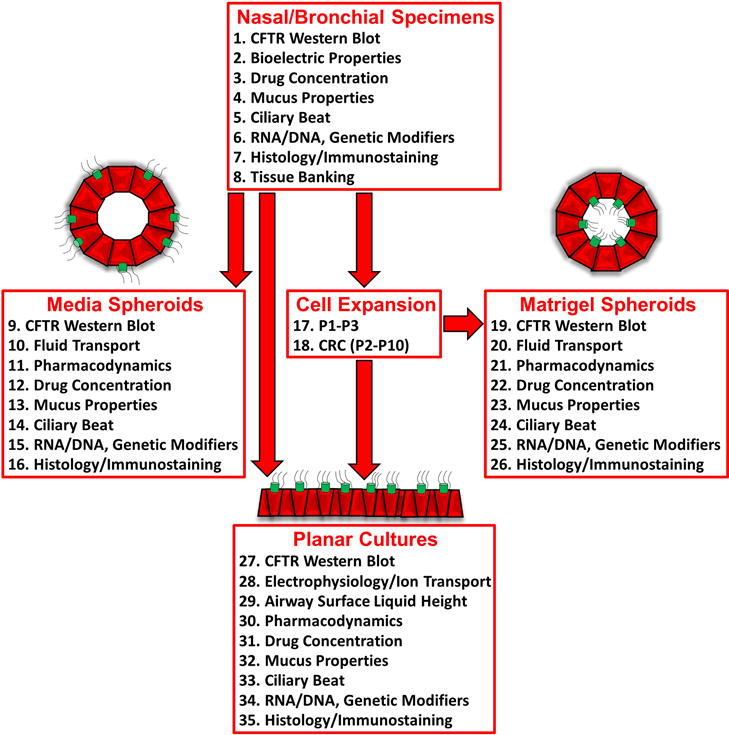
Diagram showing multiple assays that can be conducted in various culture models derived from donor specimens.
Predicting clinical outcomes by in vitro studies
Studies of CF disease have been performed using animal models or in vitro cell culture models using human or animal cells. Most animal models differ from humans in airway development and disease pathology, and furthermore, CFTR modulators may be species specific. For this reason, primary HBE cells grown on membrane supports as 2D cultures at air-liquid interface (ALI) and used in electrophysiological studies in Ussing chambers and biochemical Western blot analyses have been considered the gold standard for evaluating CFTR therapeutic rescue in vitro.
In May 2017, the US FDA approved use of Kalydeco for patients harboring one of 23 additional rare CFTR mutations. This expansion of the treatment to now 33 mutations was partially based on in vitro data, which were used together with results from earlier clinical trials. However, there are also discrepancies: in vitro, VX-809 restores F508del maturation and function up to 25% of wild-type CFTR (21), but clinical responses are less than would be predicted from this degree of correction (22, 23). In contrast, VX-770 restored function of the G551D CFTR mutation in vitro (24) and showed improvements in pulmonary function in patients harboring this mutation (11, 12). Although Orkambi treatment resulted in a slight improvement in FEV1 in F508del homozygous patients (13), F508del heterozygous patients did not show improvement with Orkambi (14). One likely cause of the limited benefit of Orkambi is destabilization of CFTR upon chronic treatment with VX-770 that we and others observed in primary HBE cells in vitro (25, 26). In contrast, acute treatment with VX-770 in these in vitro studies substantially enhanced activity of VX-809-corrected F508del CFTR (21, 25). Discrepancies between in vitro studies and clinical outcome emphasize the need for better models for drug testing. Furthermore, the cost of Kalydeco and Orkambi are extremely high (27), demonstrating the need to test more compounds.
Recent studies with primary airway (HBE and nasal epithelial, HNE) cells have exemplified the importance of considering personalized treatments for mutation-specific rescue. Interestingly in these studies, cultures from patients with R117H and P67L CFTR mutations, which are currently approved for treatment with Kalydeco (VX-770), responded even better to the VX-809/VX-770 combination treatment (Orkambi) (28, 29).
Novel models for testing of CF drugs
Relevant model systems for screening and precisely predicting clinical outcomes of CF drugs are needed to facilitate personalized treatment strategies that target specific CFTR mutations in CF individuals. We recently showed that conditionally reprogrammed variants of HBE and HNE cells exhibited electrophysiological responses similar to primary cultures (30).
Many screens were performed to identify compounds that rescue folding mutations such as F508del; however, no approach to date has been effective in advancing treatment beyond minimal or no therapeutic effects in vivo. Standard pre-clinical assays such as electrophysiological measurements may not always be suitable for rapidly predicting clinical outcomes (13, 21, 22, 25, 26). Although ASL height measurements of planar HBE cultures offer valuable insight (31), this method presents several drawbacks: fluid meniscus, culture dehydration, and variability in ASL height within each culture pose problems in reproducibility of data.
More recently, examining intestinal current measurements using CF patients’ rectal biopsy tissue has become an accepted method for studying CFTR function and responses to CFTR modulators (32, 33). In addition, 3D spheroid cultures are being employed (Fig. 1). Rectal biopsy-derived intestinal organoid models have pioneered a precision medicine approach for CF (34, 35). When CFTR is active and stimulated with forskolin, these organoids undergo forskolin-induced swelling (FIS). In the FIS assay, organoids with inside-facing functional CFTR swell when CFTR is activated by forskolin, but this response is diminished or absent in CF organoids. If patients’ genotypes are responsive to certain CFTR modulators, treatments with these drugs may restore organoid swelling. Although these assays may address gastrointestinal (GI) problems of CF patients, CF morbidity and mortality are associated with pulmonary pathogenesis. There are substantial differences in CFTR function in different tissues; in airways, CFTR plays a large role in regulating surface hydration while in intestinal tissues, CFTR plays a major role in bicarbonate (HCO3−) transport. In addition, CFTR localization, protein glycosylation, and interacting proteins, including motor proteins involved in protein trafficking, are diverse in airway and intestinal tissues. These discrepancies may affect CFTR processing and cell-surface stability and thus, cause tissue-specific differences in the effects of CFTR-targeting drugs. Furthermore, mucins expressed in airways are different from those in the colon, so modifiers acting on epithelial channels and mucins may have different effects in nasal/bronchial versus rectal tissues. Moreover, obtaining airway cells from patients’ nasal tissue is a non-invasive method compared to obtaining rectal tissue, which many patients are reluctant to provide and is typically only requested from adult CF patients.
To address the need for suitable airway precision medicine models, we developed novel spheroid assays using cultures generated from patient-derived nasal and bronchial cells (Fig. 1) to measure airway-specific fluid transport and mucus viscoelasticity (Fig. 2). Although rectal tissue may contain more CFTR than airway tissue and is therefore more sensitive to CFTR-dependent swelling assays, it is feasible to detect swelling in airway spheroids, the tissue type most severely affected in CF. These spheroid cultures mimic epithelial physiology and are cost-effectively analyzed in a high-throughput format within days of obtaining clinical tissue specimens. Such models are imperative for efficiently identifying and screening compounds before they enter clinical trials, maximizing the likelihood of achieving clinically meaningful improvements in lung and intestinal function, thus facilitating rapid progression of clinical trials toward more effective CF treatments. When cultured in matrigel, nasal and bronchial cells develop into nasospheroids and bronchospheroids, respectively, with apical membranes and cilia oriented toward the spheroid lumen. When CFTR is active, Cl− ions are secreted into the spheroid lumen, fluid follows, and the spheroids swell, similar to the rectal organoid FIS assays (34, 35). When cultured in media, airway spheroids develop in the opposite orientation, such that apical membranes and cilia face the growth medium on the outside (36–39). These explant structures spontaneously form within a few days from minimally processed airway epithelia on low-binding tissue culture (TC) plates. Because of the orientation of media spheroids, CFTR activation results in ion and fluid movement from the interior of the spheroids to the surrounding medium and therefore decreases the luminal fluid volume leading to shrinking of the spheroids.
Patient-derived airway tissue can be used to generate spheroids used in these CFTR-dependent swelling and shrinking assays to test various CFTR mutants treated with different CFTR-modifying drugs. The changes in cross-sectional area of these spheroids can be detected by microscopy. An inverted widefield fluorescence microscope, equipped with an automated XYZ stage and objectives compatible with multi-well TC plates, as well as temperature and CO2 control, is best suited for monitoring CFTR-mediated swelling and shrinking. Swelling and shrinking of spheroids are imaged over time with a sCMOS or CCD camera. Analysis of change of cross-sectional spheroid area over time can be performed with public domain or commercial software such as ImageJ/FIJI, CellProfiler (Broad Institute), and OrgSwell (Path BioAnalytics).
We designed novel assays using both nasospheroids and bronchospheroids grown in matrigel and media to monitor rescue of CFTR-mediated fluid movement. It is important to consider that CF affects not only Cl− secretion, commonly measured in CFTR therapeutic evaluation, but also movement of other ions, i.e. HCO3− and Na+, that affect fluid transport (40), which can be assessed in these spheroid cultures. Thus, it is beneficial to study fluid movement as well as ion secretion in spheroids derived from airway tissue.
Spheroid swelling assays offer the following advantages: 1) decreased cell number requirement for spheroid cultures, 2) decreased time requirement for generating differentiated spheroid cultures (10–15 days for matrigel spheroids and 2–5 days for media spheroids) vs. planar cultures (21–28 days), 3) no requirement for specialized equipment for electrophysiological analysis, besides a microscopy system, which is commonly available at many institutions, 4) high-throughput format, 5) direct evaluation of CFTR-mediated fluid movement (similar to ASL height measurements), which is affected by factors in addition to Cl− secretion (i.e. paracellular and cellular transport of other ions and water), and 6) multiple readouts can be obtained in parallel (swelling, CFTR mRNA/protein, mucus properties, ciliary activity; Fig. 2). Thus, patient-derived airway spheroids may constitute a straightforward model for measuring CFTR-mediated airway hydration utilizing a physiologically relevant ex vivo assay that detects fluid movement.
Engineering a physiological lung environment
Although numerous CF drugs are in the pharmaceutical pipeline (Table 1), current model systems for predicting clinical outcomes do not accurately represent the environment to which CF airway epithelia are chronically exposed in vivo. The development of novel in vitro models that accurately recapitulate CF airway disease may facilitate a greater understanding of disease mechanisms and aid in the development of new, more effective therapies.
Planar 2D models may not encompass all underlying mechanisms of CF, and possess several non-physiological properties including: 1) growth on cell culture insert surfaces that are stiffer than soft tissues, 2) lack of signals from the tissue microenvironment, 3) improper representation of in vivo drug pharmacodynamics, and 4) decreased ciliation of passaged cells, which substantially affects MCC. Thus, 2D models may be associated with substantial limitations in studying the mechanisms of clinically relevant CF therapies. To overcome these limitations, advanced models are in development such as biomimetic 3D airway systems comprised of three layers of diverse cell types: 1) lung microvascular endothelial cells, 2) lung fibroblasts, and 3) airway epithelial cells. These bioengineered models have an extracellular matrix biogel scaffolding, thereby promoting multicellular organization to mimic CF disease pathology to test the efficacy of CFTR modulators more accurately (41). Combination of tissue engineering and microfluidics techniques have recently yielded lung-on-a chip models that have potential for drug efficacy and toxicity testing.
The basic CFTR defect results in airways with impaired MCC, which leads to a cycle of increasing mucus obstruction, infection, and inflammation, which has a major impact on airway epithelial responses. However, because in vitro studies of CFTR rescue utilized cultures that are no longer inflamed, CFTR modulator efficacy has not been evaluated under relevant inflammatory conditions. Moreover, diminution of inflammation and its effect on CFTR rescue is of significance for CF patients receiving anti-inflammatory therapy. This issue is important because inflammation of CF HBE alters ER homeostasis and induces ER stress, which triggers the unfolded protein response that results in expansion of the ER compartment and up-regulates the expression of ER chaperone proteins, folding enzymes and lipids, thereby increasing the ER protein folding capacity (42, 43). Knowledge of the mechanisms that affect CFTR rescue in an inflamed environment may lead to better understanding of long-term treatments and new targets for improved correction. In addition, although CFTR rescue is expected to suppress CF airway inflammation, it is not known whether this effect impacts the efficacy of CFTR rescue upon long-term treatment.
Restoring mucus properties
CF airways are characterized by dehydrated mucus with an increased concentration of mucus solids and increased viscoelasticity that results in mucus plugging (44, 45). Consequently, MCC, a key element of effective host defense, is severely impaired in CF. An objective has been to determine whether rescued CFTR function directly impacts the biophysical properties of mucus (40, 46). Indeed, studies that evaluate biophysical properties of mucus (concentration, viscosity, elasticity, and adhesion), ciliary activity (beat frequency, amplitude, and coordination) or MCC transport (47) appear to be ideal measures to clarify whether restoration of mutant CFTR function by CFTR modulators can normalize aberrant CF mucus characteristics in CF in vitro models.
Efficient usage of airway specimens permits multiple physiological readouts
To make efficient usage of donor specimens, numerous analyses of physiological readouts including analysis of CFTR function and mucus properties can be performed simultaneously in vitro (Fig. 2). Outcome measures including organoid volume change, mucus viscoelasticity, and cilia activity in response to CFTR modulator compounds can be tested and compared to direct, well-accepted electrophysiological and biochemical measures of CFTR recovery. Airway specimens are obtained from CF patients by standard biopsy procedures such as brushing. Media spheroids with outside-facing cilia form directly from specimens within days. Primary cells (P0) derived from specimens are expanded up to passage 3 (P3) or conditionally reprogramed cells (CRC) can be created and expanded up to passage 10 (P10) (30). These cells can be utilized to create planar cultures on culture inserts or inside-facing spheroids in matrigel. Upon CFTR activation, change of spheroid size will indicate CFTR-mediated ion and fluid movement (48). Western blot analysis of CFTR in cell lysates allows the visualization of correction of misfolding mutations by restoring processing of CFTR to yield a mature complex glycosylated form (16, 25). Importantly, intracellular drug concentrations of CFTR modulators (i.e. VX-809, VX-661 and VX-770) can be analyzed by mass spectrometry (25, 27) and then correlated to functional readouts. Nasal and bronchial cultures can also be utilized to examine changes in mucus properties upon CFTR rescue (40, 46). Mucin concentrations in CF secretions are higher than those in non-CF secretions and can be determined utilizing native or culture mucus (45, 49–51). Changes in viscoelastic mucus properties can be analyzed by particle tracking microrheology (PMT) using fluorescent microspheres (45, 52–54). To assess alteration of mucus viscoelastic properties of planar cultures, apical mucus is collected or analyzed directly on cultures using fluorescent microbeads. For analysis of outside-facing spheroids, spheroids are dispensed in small volumes into multi-well plates and PTM is performed using the media. For analysis of inside-facing spheroids, fluorescent microbeads can be introduced into the lumen through incubation during spheroid formation or through injection. Additionally, on planar cultures, ASL height can be measured after labeling with fluorescent dextran, and MCC transport studies can be conducted by recording the transport of fluorescent beads to determine their velocity (31, 47). Cilia activity is affected by the viscoelasticity of the mucus layer. Cilia form in all culture models and thus permit analysis of ciliary beat frequency (CBF) as per established protocols (55, 56). CF mucus is more viscoelastic than non-CF mucus, and thus CBF is reduced in CF versus non-CF epithelia. RNA and DNA can be collected from cultures for analyses such as expression studies, confirmation of CFTR genotype, and determination of genetic modifier variants. Histology stains can identify cell types and ciliation (H&E stain) or indicate mucus load (Alcian blue-periodic acid-Schiff) and immunostaining allows visualization of CFTR and the major secreted mucins, MUC5AC and MUC5B (57, 58). Biobanking of tissues and expanded cells permits the utilization of specific cultures in the future, as new treatments become available. These models provide broadly applicable cost-effective diagnostic tools for evaluating multiple routes of therapeutic intervention in CF, including non-CFTR targeting therapies, genetic modifiers, and gene targeting or RNA-based approaches.
Conclusions, remaining questions, and future directions
To address patient-specific responses, the use of tissue from individual patients for CF drug testing has been established. In vitro assays of CFTR functional responses including electrophysiological measurements using CF patient-derived cells are highly valuable and have permitted the identification of CFTR-targeting drugs. In addition, novel, cost-effective 3D spheroid cultures provide a straightforward approach for predicting individual responses to CFTR-directed therapies. Airway environment and complex tissue structures should be incorporated into models of in vivo rescue of CFTR in CF lungs. Furthermore, novel assays that monitor other aspects of successful CFTR rescue such as restoration of mucus characteristics that permit MCC will allow for better prediction of successful therapies in vivo.
Although CF is considered a monogenic disorder, genetic modifiers affecting CF disease severity (8–10) have not been utilized as CF drug targets because their mechanistic roles are uncertain. Interestingly, it has been suggested that a modifier locus may affect responses of CFTR modulation in CF patients (59), raising the importance of considering CF modifiers in precision strategies to treat the disease on an individualized basis. Furthermore, recent FDA approval of Orkambi for children as young as six and Kalydeco in children as young as two raises questions about how young to start CFTR modulator therapy, since progression of severe airway obstructions and lung damage may not be reversible even with these treatments. Other questions that remain are whether these drugs should be taken during pregnancy to benefit the pregnant CF mother and potentially a CF fetus, and whether CF patients who received a donor lung should continue to take CFTR-modulating drugs to alleviate disease in other organs such as the GI tract.
A tremendously exciting development is the production of airway cultures from CF patients’ own induced pluripotent stem cells derived from skin fibroblasts or blood cells (60). Furthermore, the vast advancement of gene therapy and gene editing technologies provides new tools for CF therapy independent of the CFTR genotype. Alternative or complimentary therapies that act on other ion channels (inhibition of ENaC or activation of calcium-activated chloride channels) or that loosen mucus are being extensively explored alone and in combination with CFTR modulators. Taken together, the future for treatment of the basic defect in CF is bright and with the help of physiologically relevant precision medicine models, there is the prospect of discovering optimal and sustained treatments that are available to all CF patients.
Highlights.
Assessing pharmacological rescue of mutant CFTR requires primary CF cells.
Planar and spheroid cultures predict CFTR modulator efficacy for various mutations.
An inflamed CF lung environment can be engineered.
Mucus properties can be assayed to predict benefits of CF therapies.
Efficient usage of epithelial specimens permits multiple drug response readouts.
Acknowledgments
The authors thank Nancy L. Quinney, Susan E. Boyles, and Dr. Jennifer S. Guimbellot for implementing patient-derived models, and Drs. Charles R. Esther, Jr., Sean V. Murphy, Carla M. P. Ribeiro, and Camille Ehre for helpful discussions. We are grateful for assistance with microscopy from Drs. Michael Chua (Marsico Lung Institute Preclinical Core, supported by P30 DK065988) and Pablo Ariel (UNC Microscopy Services Laboratory, supported by P30 CA016086). This work is supported by NIH (P30 DK065988) and CFF (BOUCHE15R0, CHOLON16I0).
Author Martina Gentzsch is an inventor of the technology “airway sphere cultures as diagnostic device to monitor pharmacological responses of ion channels” which is licensed to Path BioAnalytics. Dr. Gentzsch could receive royalties related to the license in the future. These relationships have been disclosed to and are under management by the University of North Carolina at Chapel Hill.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. DOI. [DOI] [PubMed] [Google Scholar]
- 2.Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. Cftr as a camp-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. DOI. [DOI] [PubMed] [Google Scholar]
- 3.Mall M, Bleich M, Greger R, Schreiber R, Kunzelmann K. The amiloride-inhibitable na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest. 1998;102:15–21. doi: 10.1172/JCI2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mall M, Grubb BR, Harkema JR, O’Neal WK, Boucher RC. Increased airway epithelial na+ absorption produces cystic fibrosis-like lung disease in mice. Nature medicine. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 5.Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 2004;23:146–158. doi: 10.1183/09031936.03.00057003. DOI. [DOI] [PubMed] [Google Scholar]
- 6.Accurso FJ. Early pulmonary disease in cystic fibrosis. Current opinion in pulmonary medicine. 1997;3:400–403. doi: 10.1097/00063198-199711000-00002. DOI. [DOI] [PubMed] [Google Scholar]
- 7.O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 8.Corvol H, Blackman SM, Boelle PY, Gallins PJ, Pace RG, Stonebraker JR, Accurso FJ, Clement A, Collaco JM, Dang H, Dang AT, Franca A, Gong J, Guillot L, Keenan K, Li W, Lin F, Patrone MV, Raraigh KS, Sun L, Zhou YH, O’Neal WK, Sontag MK, Levy H, Durie PR, Rommens JM, Drumm ML, Wright FA, Strug LJ, Cutting GR, Knowles MR. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nature communications. 2015;6:8382. doi: 10.1038/ncomms9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun L, Rommens JM, Corvol H, Li W, Li X, Chiang TA, Lin F, Dorfman R, Busson PF, Parekh RV, Zelenika D, Blackman SM, Corey M, Doshi VK, Henderson L, Naughton KM, O’Neal WK, Pace RG, Stonebraker JR, Wood SD, Wright FA, Zielenski J, Clement A, Drumm ML, Boelle PY, Cutting GR, Knowles MR, Durie PR, Strug LJ. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nature genetics. 2012;44:562–569. doi: 10.1038/ng.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Soave D, Miller MR, Keenan K, Lin F, Gong J, Chiang T, Stephenson AL, Durie P, Rommens J, Sun L, Strug LJ. Unraveling the complex genetic model for cystic fibrosis: Pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Human genetics. 2014;133:151–161. doi: 10.1007/s00439-013-1363-7. [DOI] [PubMed] [Google Scholar]
- 11.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, Moss RB, Pilewski JM, Rubenstein RC, Uluer AZ, Aitken ML, Freedman SD, Rose LM, Mayer-Hamblett N, Dong Q, Zha J, Stone AJ, Olson ER, Ordonez CL, Campbell PW, Ashlock MA, Ramsey BW. Effect of vx-770 in persons with cystic fibrosis and the g551d-cftr mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordonez C, Elborn JS, Group VXS A cftr potentiator in patients with cystic fibrosis and the g551d mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP, Group TS. Group TS Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del cftr. N Engl J Med. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, Huang X, Waltz D, Patel NR, Rodman D, group VXs A cftr corrector (lumacaftor) and a cftr potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del cftr mutation: A phase 2 randomised controlled trial. The Lancet Respiratory medicine. 2014;2:527–538. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- 15.Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, Grinstein S. The delta f508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268:21592–21598. DOI. [PubMed] [Google Scholar]
- 16.Cholon DM, O’Neal WK, Randell SH, Riordan JR, Gentzsch M. Modulation of endocytic trafficking and apical stability of cftr in primary human airway epithelial cultures. Am J Physiol Lung Cell Mol Physiol. 2010;298:L304–314. doi: 10.1152/ajplung.00016.2009. DOI: 00016.2009 [pii]; 10.1152/ajplung.00016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gentzsch M, Chang XB, Cui L, Wu Y, Ozols VV, Choudhury A, Pagano RE, Riordan JR. Endocytic trafficking routes of wild type and deltaf508 cystic fibrosis transmembrane conductance regulator. Mol Biol Cell. 2004;15:2684–2696. doi: 10.1091/mbc.E04-03-0176. E0403-0176 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowe SM, Pyle LC, Jurkevante A, Varga K, Collawn J, Sloane PA, Woodworth B, Mazur M, Fulton J, Fan L, Li Y, Fortenberry J, Sorscher EJ, Clancy JP. Deltaf508 cftr processing correction and activity in polarized airway and non-airway cell monolayers. Pulm Pharmacol Ther. 2010;23:268–278. doi: 10.1016/j.pupt.2010.02.001. DOI: S1094-5539(10)00017-9 [pii]; 10.1016/j.pupt.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pedemonte N, Tomati V, Sondo E, Galietta LJ. Influence of cell background on pharmacological rescue of mutant cftr. Am J Physiol Cell Physiol. 2010;298:C866–874. doi: 10.1152/ajpcell.00404.2009. DOI: ajpcell.00404.2009 [pii]; 10.1152/ajpcell.00404.2009. [DOI] [PubMed] [Google Scholar]
- 20.Haggie PM, Phuan PW, Tan JA, Xu H, Avramescu RG, Perdomo D, Zlock L, Nielson DW, Finkbeiner WE, Lukacs GL, Verkman AS. Correctors and potentiators rescue function of the truncated w1282x-cystic fibrosis transmembrane regulator (cftr) translation product. J Biol Chem. 2017;292:771–785. doi: 10.1074/jbc.M116.764720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the f508del-cftr protein processing defect in vitro by the investigational drug vx-809. Proc Natl Acad Sci U S A. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, De Boeck K, Donaldson SH, Dorkin HL, Dunitz JM, Durie PR, Jain M, Leonard A, McCoy KS, Moss RB, Pilewski JM, Rosenbluth DB, Rubenstein RC, Schechter MS, Botfield M, Ordonez CL, Spencer-Green GT, Vernillet L, Wisseh S, Yen K, Konstan MW. Results of a phase iia study of vx-809, an investigational cftr corrector compound, in subjects with cystic fibrosis homozygous for the f508del-cftr mutation. Thorax. 2012;67:12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis PB. Cystic fibrosis. Pediatrics in review. 2001;22:257–264. doi: 10.1542/pir.22-8-257. DOI. [DOI] [PubMed] [Google Scholar]
- 24.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of cf airway epithelial cell function in vitro by a cftr potentiator, vx-770. Proc Natl Acad Sci U S A. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. DOI: 0904709106 [pii]; 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cholon DM, Quinney NL, Fulcher ML, Esther CR, Jr, Das J, Dokholyan NV, Randell SH, Boucher RC, Gentzsch M. Potentiator ivacaftor abrogates pharmacological correction of deltaf508 cftr in cystic fibrosis. Sci Transl Med. 2014;6:246ra296. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, Borot F, Szollosi D, Wu YS, Finkbeiner WE, Hegedus T, Verkman AS, Lukacs GL. Some gating potentiators, including vx-770, diminish deltaf508-cftr functional expression. Sci Transl Med. 2014;6:246ra297. doi: 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cholon DM, Esther CR, Jr, Gentzsch M. Efficacy of lumacaftor-ivacaftor for the treatment of cystic fibrosis patients homozygous for the f508del-cftr mutation. Expert review of precision medicine and drug development. 2016;1:235–243. doi: 10.1080/23808993.2016.1175299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gentzsch M, Ren HY, Houck SA, Quinney NL, Cholon DM, Sopha P, Chaudhry IG, Das J, Dokholyan NV, Randell SH, Cyr DM. Restoration of r117h cftr folding and function in human airway cells through combination treatment with vx-809 and vx-770. Am J Physiol Lung Cell Mol Physiol. 2016;311:L550–559. doi: 10.1152/ajplung.00186.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabusap CM, Wang W, McNicholas CM, Chung WJ, Fu L, Wen H, Mazur M, Kirk KL, Collawn JF, Hong JS, Sorscher EJ. Analysis of cystic fibrosis-associated p67l cftr illustrates barriers to personalized therapeutics for orphan diseases. JCI insight. 2016;1 doi: 10.1172/jci.insight.86581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gentzsch M, Boyles SE, Cheluvaraju C, Chaudhry IG, Quinney NL, Cho C, Dang H, Liu X, Schlegel R, Randell SH. Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. Am J Respir Cell Mol Biol. 2017;56:568–574. doi: 10.1165/rcmb.2016-0276MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tarran R, Button B, Picher M, Paradiso AM, Ribeiro CM, Lazarowski ER, Zhang L, Collins PL, Pickles RJ, Fredberg JJ, Boucher RC. Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J Biol Chem. 2005;280:35751–35759. doi: 10.1074/jbc.M505832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clancy JP, Szczesniak RD, Ashlock MA, Ernst SE, Fan L, Hornick DB, Karp PH, Khan U, Lymp J, Ostmann AJ, Rezayat A, Starner TD, Sugandha SP, Sun H, Quinney N, Donaldson SH, Rowe SM, Gabriel SE. Multicenter intestinal current measurements in rectal biopsies from cf and non-cf subjects to monitor cftr function. PLoS One. 2013;8:e73905. doi: 10.1371/journal.pone.0073905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graeber SY, Hug MJ, Sommerburg O, Hirtz S, Hentschel J, Heinzmann A, Dopfer C, Schulz A, Mainz JG, Tummler B, Mall MA. Intestinal current measurements detect activation of mutant cftr in patients with cystic fibrosis with the g551d mutation treated with ivacaftor. American journal of respiratory and critical care medicine. 2015;192:1252–1255. doi: 10.1164/rccm.201507-1271LE. [DOI] [PubMed] [Google Scholar]
- 34.Dekkers JF, Berkers G, Kruisselbrink E, Vonk A, de Jonge HR, Janssens HM, Bronsveld I, van de Graaf EA, Nieuwenhuis EE, Houwen RH, Vleggaar FP, Escher JC, de Rijke YB, Majoor CJ, Heijerman HG, de Winter-de Groot KM, Clevers H, van der Ent CK, Beekman JM. Characterizing responses to cftr-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med. 2016;8:344ra384. doi: 10.1126/scitranslmed.aad8278. [DOI] [PubMed] [Google Scholar]
- 35.Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, Brandsma AM, de Jong NW, Bijvelds MJ, Scholte BJ, Nieuwenhuis EE, van den Brink S, Clevers H, van der Ent CK, Middendorp S, Beekman JM. A functional cftr assay using primary cystic fibrosis intestinal organoids. Nature medicine. 2013;19:939–945. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- 36.Pedersen PS, Holstein-Rathlou NH, Larsen PL, Qvortrup K, Frederiksen O. Fluid absorption related to ion transport in human airway epithelial spheroids. The American journal of physiology. 1999;277:L1096–1103. doi: 10.1152/ajplung.1999.277.6.L1096. DOI. [DOI] [PubMed] [Google Scholar]
- 37.Pedersen PS, Frederiksen O, Holstein-Rathlou NH, Larsen PL, Qvortrup K. Ion transport in epithelial spheroids derived from human airway cells. The American journal of physiology. 1999;276:L75–80. doi: 10.1152/ajplung.1999.276.1.L75. DOI. [DOI] [PubMed] [Google Scholar]
- 38.Bridges MA, Walker DC, Harris RA, Wilson BR, Davidson AG. Cultured human nasal epithelial multicellular spheroids: Polar cyst-like model tissues. Biochemistry and cell biology = Biochimie et biologie cellulaire. 1991;69:102–108. doi: 10.1139/o91-016. DOI. [DOI] [PubMed] [Google Scholar]
- 39.Deslee G, Dury S, Perotin JM, Al Alam D, Vitry F, Boxio R, Gangloff SC, Guenounou M, Lebargy F, Belaaouaj A. Bronchial epithelial spheroids: An alternative culture model to investigate epithelium inflammation-mediated copd. Respir Res. 2007;8:86. doi: 10.1186/1465-9921-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birket SE, Chu KK, Houser GH, Liu L, Fernandez CM, Solomon GM, Lin V, Shastry S, Mazur M, Sloane PA, Hanes J, Grizzle WE, Sorscher EJ, Tearney GJ, Rowe SM. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. Am J Physiol Lung Cell Mol Physiol. 2016;310:L928–939. doi: 10.1152/ajplung.00395.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Konar D, Devarasetty M, Yildiz DV, Atala A, Murphy SV. Lung-on-a-chip technologies for disease modeling and drug development. Biomedical engineering and computational biology. 2016;7:17–27. doi: 10.4137/BECB.S34252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ribeiro CM, Paradiso AM, Schwab U, Perez-Vilar J, Jones L, O’Neal W, Boucher RC. Chronic airway infection/inflammation induces a ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J Biol Chem. 2005;280:17798–17806. doi: 10.1074/jbc.M410618200. DOI: M410618200 [pii]; 10.1074/jbc.M410618200. [DOI] [PubMed] [Google Scholar]
- 43.Martino ME, Olsen JC, Fulcher NB, Wolfgang MC, O’Neal WK, Ribeiro CM. Airway epithelial inflammation-induced endoplasmic reticulum ca2+ store expansion is mediated by x-box binding protein-1. J Biol Chem. 2009;284:14904–14913. doi: 10.1074/jbc.M809180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, DeMaria GC, Matsui H, Donaldson SH, Davis CW, Sheehan JK, Boucher RC, Kesimer M. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest. 2014;124:3047–3060. doi: 10.1172/JCI73469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill DB, Vasquez PA, Mellnik J, McKinley SA, Vose A, Mu F, Henderson AG, Donaldson SH, Alexis NE, Boucher RC, Forest MG. A biophysical basis for mucus solids concentration as a candidate biomarker for airways disease. PLoS One. 2014;9:e87681. doi: 10.1371/journal.pone.0087681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gianotti A, Capurro V, Scudieri P, Galietta LJ, Moran O, Zegarra-Moran O. Pharmacological rescue of mutant cftr protein improves the viscoelastic properties of cf mucus. J Cyst Fibros. 2015 doi: 10.1016/j.jcf.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. DOI. [DOI] [PubMed] [Google Scholar]
- 48.Brewington JJ, Filbrandt ET, LaRosa FJ, 3rd, Ostmann AJ, Strecker LM, Szczesniak RD, Clancy JP. Detection of cftr function and modulation in primary human nasal cell spheroids. J Cyst Fibros. 2017 doi: 10.1016/j.jcf.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill DB, Button B. Establishment of respiratory air-liquid interface cultures and their use in studying mucin production, secretion, and function. Methods Mol Biol. 2012;842:245–258. doi: 10.1007/978-1-61779-513-8_15. [DOI] [PubMed] [Google Scholar]
- 50.Esther CR, Jr, Hill DB, Button B, Shi S, Jania C, Duncan EA, Doerschuk CM, Chen G, Ranganathan S, Stick SM, Boucher RC. Sialic acid-to-urea ratio as a measure of airway surface hydration. Am J Physiol Lung Cell Mol Physiol. 2017;312:L398–L404. doi: 10.1152/ajplung.00398.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Button B, Anderson WH, Boucher RC. Mucus hyperconcentration as a unifying aspect of the chronic bronchitic phenotype. Annals of the American Thoracic Society. 2016;13(Suppl 2):S156–162. doi: 10.1513/AnnalsATS.201507-455KV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mellnik JWR, Lysy M, Vasquez PA, Pillai NS, Hill BD, Cribb J, McKinley SA, Forest MG. Maximum likelihood estimation for single particle, passive microrheology data with drift. J Rheol. 2016;60:379–392. DOI. [Google Scholar]
- 53.Lysy M, Pillai NS, Hill DB, Forest MG, Mellnik JWR, Vasquez PA, McKinley SA. Model comparison and assessment for single particle tracking in biological fluids. Journal of the American Statistical Association. 2016 doi: 10.1080/01621459.01622016.01158716. in press. DOI. [DOI] [Google Scholar]
- 54.Mellnik J, Vasquez PA, McKinley SA, Witten J, Hill DB, Forest MG. Micro-heterogeneity metrics for diffusion in soft matter. Soft matter. 2014;10:7781–7796. doi: 10.1039/c4sm00676c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knowles MR, Ostrowski LE, Leigh MW, Sears PR, Davis SD, Wolf WE, Hazucha MJ, Carson JL, Olivier KN, Sagel SD, Rosenfeld M, Ferkol TW, Dell SD, Milla CE, Randell SH, Yin W, Sannuti A, Metjian HM, Noone PG, Noone PJ, Olson CA, Patrone MV, Dang H, Lee HS, Hurd TW, Gee HY, Otto EA, Halbritter J, Kohl S, Kircher M, Krischer J, Bamshad MJ, Nickerson DA, Hildebrandt F, Shendure J, Zariwala MA. Mutations in rsph1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. American journal of respiratory and critical care medicine. 2014;189:707–717. doi: 10.1164/rccm.201311-2047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sisson JH, Stoner JA, Ammons BA, Wyatt TA. All-digital image capture and whole-field analysis of ciliary beat frequency. Journal of microscopy. 2003;211:103–111. doi: 10.1046/j.1365-2818.2003.01209.x. DOI. [DOI] [PubMed] [Google Scholar]
- 57.Kreda SM, Gentzsch M. Imaging cftr protein localization in cultured cells and tissues. Methods Mol Biol. 2011;742:15–33. doi: 10.1007/978-1-61779-120-8_2. [DOI] [PubMed] [Google Scholar]
- 58.Kreda SM, Davis CW, Rose MC. Cftr, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harbor perspectives in medicine. 2012;2:a009589. doi: 10.1101/cshperspect.a009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strug LJ, Gonska T, He G, Keenan K, Ip W, Boelle PY, Lin F, Panjwani N, Gong J, Li W, Soave D, Xiao B, Tullis E, Rabin H, Parkins MD, Price A, Zuberbuhler PC, Corvol H, Ratjen F, Sun L, Bear CE, Rommens JM. Cystic fibrosis gene modifier slc26a9 modulates airway response to cftr-directed therapeutics. Human molecular genetics. 2016;25:4590–4600. doi: 10.1093/hmg/ddw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCauley KB, Hawkins F, Serra M, Thomas DC, Jacob A, Kotton DN. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of wnt signaling. Cell stem cell. 2017;20:844–857. e846. doi: 10.1016/j.stem.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, DiSalvo M, Gunasekara DB, Dutton J, Proctor A, Lebhar MS, Williamson IA, Speer J, Howard RL, Smiddy NM, Bultman SJ, Sims CE, Magness ST, Allbritton NL. Self-renewing monolayer of primary colonic or rectal epithelial cells. Cellular and Molecular Gastroenterology and Hepatology. 2017;4:165–182.e167. doi: 10.1016/j.jcmgh.2017.02.011. DOI: https://doi.org/10.1016/j.jcmgh.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]