

Abstract
The historical diagnosis of Prader-Willi syndrome (PWS), a complex genetic disorder, in adults by clinical presentation rather than genetic testing has limited genetic subtype-specific psychometric investigations and treatment. Genetic testing and clinical psychiatric evaluation using DSM-IV-TR criteria were undertaken on 72 adult residents (34M; 38F) from the Prader-Willi Homes of Oconomowoc (PWHO), a specialty PWS group home system. Methylation specific-multiplex ligation probe amplification and high-resolution microarrays were analyzed for methylation status, 15q11-q13 deletions and maternal uniparental disomy 15 (mUPD15). Seventy (33M; 37F) of 72 residents were genetically confirmed and 36 (51%) had Type I or Type II deletions; 29 (42%) with mUPD15 and 5 (7%) with imprinting defects from three separate families. Psychiatric comorbidities were classified as anxiety disorder (38%), excoriation (skin picking) (33%), intermittent explosive disorder [(30%-predominantly among males at 45% compared with females at 16% (OR=4.3, 95%CI 1.4-13.1, p<0.008)] and psychotic features (23%). Psychiatric diagnoses did not differ between mUPD15 vs deletion, but a greater number of psychiatric diagnoses were observed for larger Type I (4.3) vs smaller Type II (3.6) deletions when age was controlled (F=5.0, p<0.04). Adults with PWS presented with uniformly higher rates of psychiatric comorbidities which differed by genetic subtype with gender-specific trends.
Keywords: Neuropsychiatric diagnoses, Prader-Willi syndrome (PWS), PWS genetic subtypes, residential care adults
Graphical abstract
Introduction
Prader-Willi syndrome (PWS) is a rare complex neurodevelopmental disorder due to errors in genomic imprinting that occurs in about 1 in 10,000 to 30,000 live births (1-5). It is characterized by multiple abnormal findings and physical changes including a characteristic facial appearance (narrow bifrontal diameter, short nose, and down- turned corners of the mouth) with sticky saliva and dental anomalies (enamel hypoplasia, caries), infantile hypotonia with a poor suck and feeding difficulties, hypogonadism/hypogenitalism, growth hormone deficiency and other endocrine problems leading to short stature, small hands/feet and infertility (6). In early childhood, food seeking and hyperphagia generally occurs leading to onset of obesity which can be life-threatening, if uncontrolled. Intellectual disability for the family background and behavioral problems including mood instability, tantrums, obsessive and compulsive behaviors, stubbornness, aggression and skin picking are common and frequently occur in childhood and continue into adulthood (4,5,7), recently supported in a cohort of 53 adults and adolescents with PWS and the overwhelming majority (89%) had at least one psychiatric diagnosis (8). The most common observation was disruptive behavior disorder (68%) followed by obsessive compulsive disorder (45%) and skin picking (35%). No correlation was found in their study between the number of psychiatric diagnoses and body mass index (BMI), IQ, hyperphagia severity, hormonal profiles or genetic subtypes but these factors strongly influenced quality of life for those with PWS.
The causation of PWS is due to loss of expression of paternally derived genes in the 15q11-q13 region generally from a de novo deletion followed by maternal uniparental disomy 15 (both copies of chromosome 15 from the mother). A small percentage of patients with PWS have an imprinting defect which may be inherited and account for a potential 50% recurrence risk for subsequent affected children (4,5,7). The PWS genetic subtypes include the paternal 15q11-q13 deletion seen in about 70% of cases and has three subtypes (larger Type I, smaller Type II and an atypical size). These deletions generally involve three common breakpoints (BPs) within the 15q11-q13 region (i.e., proximal BP1 and BP2 and distal BP3) (9, 10). Maternal uniparental disomy 15 is seen in about 30% of cases and is now recognized to occur in different subclasses such as maternal heterodisomy, segmented isodisomy or total isodisomy of chromosome 15 depending on crossing-over events and nondisjunction in meiosis I or meiosis II (5). Imprinting defects represent only a few percentage of cases and are due to either a small microdeletion of the imprinting center which controls the activity of imprinting genes in the 15q11-q13 region or from errors in methylation processing or epimutations (4, 5, 7, 11-14).
Studies in individuals with the typical Type I deletion which involves BP1 and BP3 includes TUBGCP5, CYFIP1, NIPA1 and NIPA2 genes located between BP1 and BP2, in both Prader-Willi and Angelman syndromes have been reported with more severe neurodevelopmental disturbances compared to those individuals with the smaller type II deletion involving BP2 and BP3 and the four genes intact (15). Several studies have been undertaken in the pediatric age group with limited information in adults particularly in those living in residential care. Individuals with deletions of the 15q11.2 BP1-BP2 cytogenetic region alone can present with developmental and language delay, neurobehavioral disturbances, psychiatric problems, autism, seizures, schizophrenia and mild dysmorphic features. However, not all individuals who carry the BP1-BP2 microdeletion are clinically affected, as the phenotype can show incomplete penetrance and variable expressivity (16, 17).
The type and accuracy of genetic testing for PWS has progressed over the years beginning in the 1980s with high-resolution chromosome testing to identify the 15q11-q13 deletion (18). Later, polymorphic DNA markers from chromosome 15 were used to identify the parental origin and maternal uniparental disomy 15 status (19). Fluorescence in situ hybridization (FISH) with DNA probes from the 15q11-q13 region were introduced in the early 1990s but could not distinguish between Type I or Type II deletions. DNA methylation assays of chromosome 15 were developed at about the same time and used to identify the parent of origin status and genomic imprinting errors found in 99% of subjects with PWS (20-23). However, DNA methylation studies cannot distinguish between the different genetic subtypes that cause PWS. Currently, methylation specific-multiplex ligation probe amplification (MS-MLPA) of chromosome 15 and high-resolution chromosomal single nucleotide polymorphism (SNP) microarrays are used to identify and characterize the size, class and type of genetic defect but cannot distinguish between total heterodisomy from normal biparental inheritance (5, 24-27).
Here, we utilized advances in genetic testing for the first time to characterize the specific genetic status including deletion subtypes, maternal uniparental disomy subclasses and imprinting defect status of adults with PWS residing at a syndrome-specific group home. These individuals were also assessed by an experienced psychiatrist using the Diagnostic and Statistical Manual of Mental Disorders (DSM) version DSM-IV-TR for criteria and classification of mental disorders with input from trained providers in treating adults with PWS. Detailed PWS genetic characterization and correlation with neuropsychiatric diagnostic data will be reported and discussed.
Materials and Methods
Description of Prader-Will Syndrome Specific Group Home
Prader-Willi Homes of Oconomowoc (PWHO) located in Oconomowoc, Wisconsin, is the largest USA based group home system specializing in the management of PWS in existence for 32 years. The group home is staffed 24 hours a day with residential counselors in place along with a fulltime nurse practitioner and board certified psychiatrist (Dr. Weisensel). There is complete food security at each site. Each resident has a meal plan in place and followed closely by a dietitian with expertise in PWS. The adult residents either work during the day at supervised work-sites or participate in Day RISE which is a structured day program consisting of occupational therapy, social skill activities, exercise, sensory integration and art sessions.
The majority of PWHO residents are admitted based upon self-referral or referral from the local or national chapter of the Prader-Willi Association –USA. A smaller proportion of referrals come from friends and family of prior residents, inpatient medical or psychiatric units, providers and other group homes. The reasons for admission are most often related to the physical safety of subject and/or family secondary to individual behaviors; medical complications of PWS typically related to morbid obesity or a break-down of family systems (i.e., divorce, birth of another child, family death or illness) disrupting care or aging out of school systems resulting in a loss of structure. Many individuals have failed at other residential placements prior to admission to PWHO. PWHO residents come from 11 different states with the majority from Wisconsin (N=15), Illinois (N=15) and Michigan (N=14) with their placement 100% supported by county or state funds. State/county support to reside at PWHO generally is contingent upon demonstration that the individual needs could not be met in a less restrictive environment. Thus, eighty-three percent of individuals at PWHO are followed regularly by a psychiatrist and may be psychiatrically (behaviorally, emotionally), intellectually and medically more ill then the general PWS population.
Description of Adults with Prader-Willi Syndrome
There were 84 adult residents with PWS residing at PWHO at the time of this study representing all regions of the USA. Their mean age was 37 ± 10 years and ranged from 18 to 61 years. Only 10 adults were genetically confirmed previously while 74 were diagnosed clinically based on established PWS consensus diagnostic criteria at the time of admission to PWHO (28). Seventy-three of the 84 subjects agreed to consent and participate in the study which was approved by the local human subjects research committee.
Genetic Testing Methods and Analysis
Each subject or legal guardian signed the approved consent forms, a saliva sample (2ml aliquots) was collected and DNA isolated using Norgen (Thorold, ON, Canada) following manufacturer's recommendations. The laboratory and genetic analyses were performed at the University of Kansas Medical Center utilizing the Genomics Core Facility. The PWS diagnosis was genetically confirmed using several genetic laboratory methods including methylation specific-multiplex ligation probe amplification (MS-MLPA) with DNA kits for chromosome 15 obtained from MRC-Holland (Amsterdam, Netherlands) (see Figure 1). High-resolution Affymetrix 6.0 version (Santa Clara, CA) microarrays were performed using 1.8 million copy number variant (CNV) and single nucleotide polymorphism (SNP) probes to identify chromosome 15q11-q13 deletions and maternal uniparental disomy 15 status. Genotyping of DNA microsatellite markers from chromosome 15 were also undertaken by PCR using subject and parental DNA samples to identify imprinting defects specifically in adults with an abnormal PWS methylation pattern but no recognized deletion. Chromosomal microarrays with SNP probes were used to detect maternal uniparental 15 status (heterodisomy, segmental isodisomy, total isodisomy) (see Figures 2a and 2b).
Figure 1.
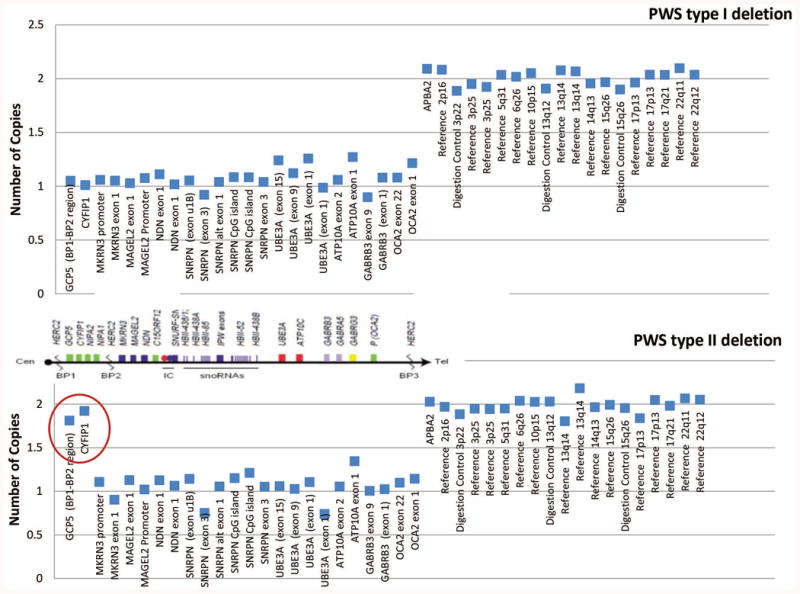
MS-MLPA results of copy number comparison with probe markers for an individual with Type I (top) and Type II (bottom) PWS deletion genetic subtypes relative to a control subject. The genomic copy number is shown on the y-axis. The circle indicates the marker probes differentiating the two deletion subtypes.
Figure 2.
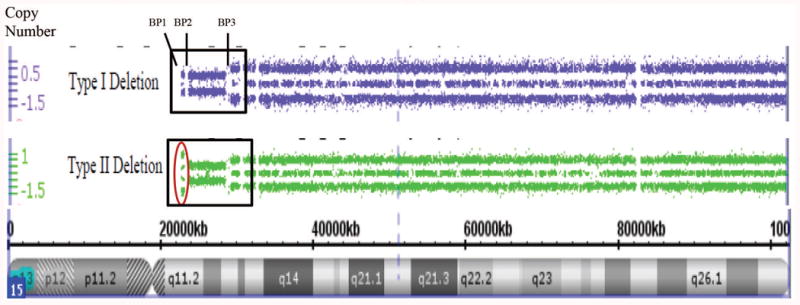
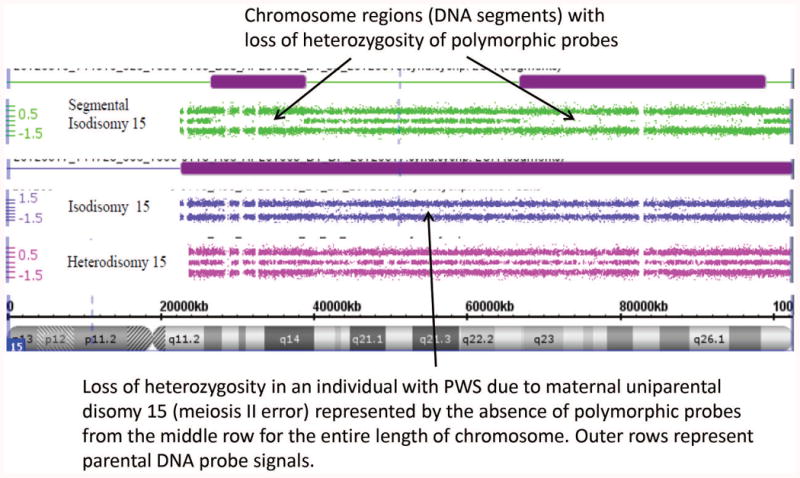
Figure 2a. High-resolution chromosome 15 ideogram and SNP microarray results for an individual with Type I (top) and Type II (bottom) PWS deletion genetic subtypes. The genomic copy number is shown on the y-axis. The box indicates the deleted 15q11-q13 region and break points (BP1, BP2 and BP3) and the circle indicates the marker probes differentiating the two deletion subtypes. Figure 2b. High-resolution chromosome microarrays using CNV and SNP probes to identify maternal uniparental disomy 15 subclasses (segmental isodisomy, isodisomy and heterodisomy) in those with an abnormal DNA methylation pattern for PWS. Heterodisomy 15 can resemble normal biparental inheritance.
Copy number variants (deletions or duplications) using the high-resolution microarrays were defined as a size ≥ 100kb with at least 50 DNA markers. Maternal disomy 15 was identified by examining chromosome regions (size and locations) and subclass status determined by the presence of loss of heterozygosity (LOH) on chromosome 15 defined as ≥8 Mb in size for segmental isodisomy 15 or total isodisomy including the entire length of chromosome 15. Maternal heterodisomy 15 is indistinguishable from normal biparental inheritance. In addition, subjects were screened for deletions or duplications of known obesity genes (N=23; e.g., FTO, BDNF, MC4R) reported by Butler et al. (29) by examining more closely the data from the high-resolution microarrays by adjusting the Affymetrix chromosome suite 1.2.2 software program cutoff for a heterozygous loss of signal from 35 DNA probes and a deletion size of 25kb at the chromosome location of the obesity-related genes [e.g., BDNF at 11p14.1, genomic coordinates of 11:27,654,292-27,722,057 (GRCh 38)] [Online Mendelian Inheritance in Man (OMIM) http://www.omim.org/]. Statistical analysis included descriptive, correlation and regression analysis of data using the SAS 9.2 version.
Neuropsychiatric Criteria and Classification Assessments Based on Diagnostic and Statistical Manual (DSM)-IV-TR
A detailed neuropsychiatric assessment was carried out by an experienced psychiatrist with particular expertise in PWS (Dr. Weisensel) familiar with the subject population using DSM-IV-TR and informed by the Diagnostic Manual-Intellectual Disability Textbook (30). This manual is referenced by clinicians and researchers engaged in the treatment, study and classification of mental illnesses or traits and for standardizing psychiatric diagnostic categories divided into five dimensions (Axis I-V) relating to different aspects of disorders or disabilities. Each category of disorder has a numeric code taken from the ICD coding system. The assessments included a history of present illness (reviewing previous psychiatric assessments, diagnoses, neuropsychiatric symptoms and behaviors, psychiatric medication and neuropsychological testing results including IQ when available); developmental history, pregnancy and birth information; medical history; allergies; current medications; review of the nutritional assessment; social and family history; and current vital signs and basic laboratory studies. This information taken together with interviews by PWHO staff intimately familiar with the subjects along with interviews with the family (if available) were combined to develop a biopsychosocial formulation leading to diagnoses based on DSM-IV-TR criteria. This included a full range of behavioral and psychiatric diagnostic codes recognized in the adult population. Statistical studies included descriptive, correlation and regression analyses of clinical and genetic data using the SAS 9.2 version.
Results
Clinical, Psychiatric and Genetic Findings
Seventy-three adult residents from PWHO participated in the study. Seventy-two subjects completed the PWS genetic subtyping process using the MS-MLPA assay initially then followed by high-resolution chromosomal microarray analysis needed if a chromosome 15q11-q13 deletion was not found. The clinical diagnosis of PWS was not confirmed by chromosomal and DNA methylation testing in two PWHO females who exhibited cardinal clinical features of PWS including hypotonia, short stature with small hands and feet along with a history of behavioral issues including impulsive aggression, obesity and food-seeking. One subject showed significant skin-picking. These two females were dropped from our analyses. Table Ia shows the summary of age at the time of the neuropsychiatric assessment for this study, gender, age at PWHO admission, body mass index (BMI, kg/m2) and number of psychiatric diagnoses for the 70 genetically confirmed PWHO residents. Table Ib shows the genetic subtype summary data of the 33 males and 37 females, age, BMI and number of psychiatric diagnoses or disorders with 36 (51%) participants having the 15q11-q13 deletion, 29 (42%) with maternal uniparental disomy and 5 (7%) with imprinting defects representing three separate families (3 siblings - 2 males and 1 female in a single family carrying an approximate 130kb microdeletion which included the PWS imprinting center). Two females had a presumed epimutation as no deletion or evidence of uniparental disomy were found on molecular testing.
Table IA. Characteristics of the PWHO Study Participants.
| A. Participants | Age (at study) Mean ±SD (range) | Age PWHO Admission Mean ±SD (range) | BMI (at study) Mean ±SD (range) | Number of Psych Diagnoses Mean ±SD (range) |
|---|---|---|---|---|
| TOTAL (N=70) | 36±10 yrs (18-60 yrs) | 24±6 yrs (17-46 yrs) | 28±5kg/m2 (20-57) | 4.2±1.2 (2-7) |
| Males (N=33) | 35±10 yrs (20-52 yrs) | 23±7 yrs (18-46 yrs) | 28±4kg/m2 (20-40) | 4.3±1.1 (3-7) |
| Females (N=37) | 37±10 yrs (18-60 yrs) | 25±6 yrs (17-41 yrs) | 28±6kg/m2 (21-57) | 4.1±1.3 (2-7) |
Seventy-three subjects were screened and 70 showed genetic confirmation of PWS with one subject excluded due to an inadequate DNA sample and two subjects excluded due to normal DNA methylation results.
Table IB. Summary of PWS Genetic Subtypes for PWHO Participants.
| PWS Subtype | Total (N=70) | Males (N=33) | Females (N=37) | Age | BMI | Number of Psychiatric Disorders |
|---|---|---|---|---|---|---|
| 15q11-q13 Deletion | 36 (51%) | 17 (51%) | 19 (51%) | 38±10yr | 28±4 | 3.9±1.1 |
| Maternal Uniparental Disomy 15 | 29 (42%) | 13 (40%) | 16 (44%) | 35±10yr | 28±7 | 4.3±1.2 |
| Imprinting Defect/Microdeletion* | 5 (7%) | 3 (9%) | 2 (5%) | 31±7yr | 27±2 | 5.0±1.6 |
Three siblings (2 males and 1 female) in a single family of five adults with PWS at the PWHO group home had the same microdeletion of the imprinting center (imprinting defects) identified with MS-MLPA and by high-resolution SNP microarray analysis. One participant was excluded due to an inability to obtain a sufficient amount of high quality DNA and two participants did not have PWS by genetic testing.
There was no evidence of consanguinity among the PWHO subjects using high-resolution SNP microarray data to identify the SNP pattern and calculate the percentage of regions of homozygosity (ROH) for all chromosomes following protocols reported previously (31, 32). Twenty-three representative obesity genes (FTO, BDNF, SIM1) (5, 29) known to contribute to obesity-related genetic disorders were examined more closely for possible microdeletions by relaxing the classification cut-off parameters of the microarray data (from 100kb to 25kb and 50 to 35 markers) utilizing the standard computer software program. No deletions were found encompassing the 23 genes studied using the less stringent copy number variant classification criteria.
Of the 36 subjects with a 15q11-q13 deletion, 14 had Type I and 22 had Type II deletions (see Figure 2a). No differences were seen between the two deletion subtypes in mean age, BMI or age at admission to PWHO. The average number of psychiatric diagnoses per subject was 4.3 +/-1.2 with a range of 2 to 7 for those with Type I deletion and 3.6 ± 1.0 with a range of 2 to 5 for the Type II deletion group. Of the 29 subjects with maternal disomy 15, 10 showed heterodisomy, 15 showed segmental isodisomy and 4 showed isodisomy for the entire chromosome 15 (see Figures 2b). Figure 3 shows the distribution of chromosome 15 bands that were found to represent segmental isodisomy in 15 individuals with PWS. The average size of the LOH region in the individuals with segmental isodisomy was 25.6 Mb ± 18.7. When divided into three arbitrary chromosome 15 segments, the average length was 12.6 Mb for the proximal segment (defined as 15q11.1-15q15.3) found in 12 subjects; 14.5 Mb in the middle segment (defined as 15q21.2-15q24.3) in nine subjects and 11.3 Mb for the distal segment (defined as 15q25.1-15q26.3) in nine subjects. Nine subjects with PWS showed only one isodisomic region, five showed two isodisomic regions and one subject showed three regions on chromosome 15. No differences were seen in the mean age, BMI, age at admission to PWHO or number of psychiatric diagnoses.
Figure 3.
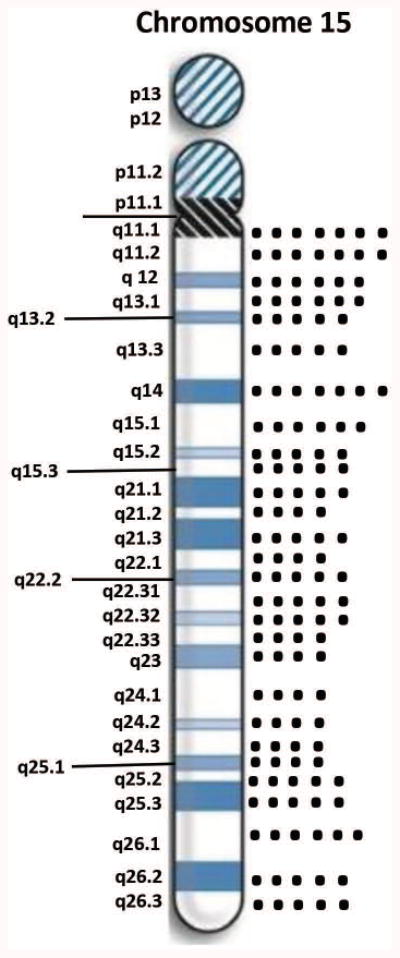
Distribution of chromosome 15 bands involved in segmental isodisomy 15 by high-resolution chromosome microarrays found in 15 adults with PWS residing at the PWHO group home.
Neuropsychiatric Diagnostic Outcomes
The frequency of primary neuropsychiatric diagnoses according to DSM-IV-TR criteria and classification is summarized in Table II. Sixteen (23% - 9F; 7M) of the total number of PWHO adults with PWS were diagnosed with any psychotic features or disorder included in the DSM-IV-TR category of “Schizophrenia and Other Psychotic Disorders” and any Bipolar or Mood Disorder with a history of psychotic features except Major Depressive Disorder with a history of psychotic features. Fifteen (21%-7F; 8M) of the total number of subjects were diagnosed with Bipolar or Mood Disorder, without a history of psychotic symptoms. Seventeen (24%- 12F; 5M) were diagnosed with any depressive disorder or an Adjustment Disorder with a preponderance of depressive symptoms. Twenty-seven (38%- 13F; 14M) were diagnosed with a disorder included in the DSM-IV-TR Anxiety Disorder category or an Adjustment Disorder with a preponderance of anxiety symptoms. Twenty-one (30%- 6F; 15M) were diagnosed with Intermittent Explosive Disorder (IED) with twice the number of males than females in this category (see Table II; OR=4.3, 95% CI 1.4,13.1, p<0.008). Twenty-three (33%- 15F; 8M) were diagnosed with Impulse-Control Disorder Not Otherwise Specified and in all cases due to recurrent skin picking. There were additional low frequency diagnoses identified including but not limited to Attention Deficit Disorder, Conduct Disorder, Tic Disorders and Learning Disorders which were excluded from analysis.
Table II. Summary of Neuropsychiatric Diagnoses for PWHO Participants with Prader-Willi Syndrome.
| Primary Psychiatric Diagnoses | Total (N=70) | Males (N=33) | Females (N=37) | Odds# Male | 95% CI | p-value |
|---|---|---|---|---|---|---|
| Any Psychotic Features | 16 (23%) | 7 (21%) | 9 (24%) | 0.8 | 0.27,2.6 | 0.76 |
| Bipolar Disorder (Nonpsychotic) | 15 (21%) | 8 (24%) | 7 (18%) | 1.4 | 0.44,4.3 | 0.59 |
| Anxiety Disorder | 27 (38%) | 14 (42%) | 13 (35%) | 1.4 | 0.52,3.6 | 0.53 |
| Major Depressive Disorder | 17 (24%) | 5 (15%) | 12 (32%) | 0.4 | 0.11,1.2 | 0.09 |
| Intermittent Explosive Disorder | 21 (30%) | 15 (45%) | 6 (16%) | 4.3 | 1.4,13.1 | 0.008* |
| Excoriation (skin picking) Disorder | 23 (33%) | 8 (24%) | 15 (41%) | 0.5 | 0.17,1.3 | 0.15 |
Odds ratio for males relative to females using Logit.
p<0.05 considered significant Additional diagnoses included: attention deficit hyperactivity disorder, conduct disorder and tic disorders. CI = confidence interval
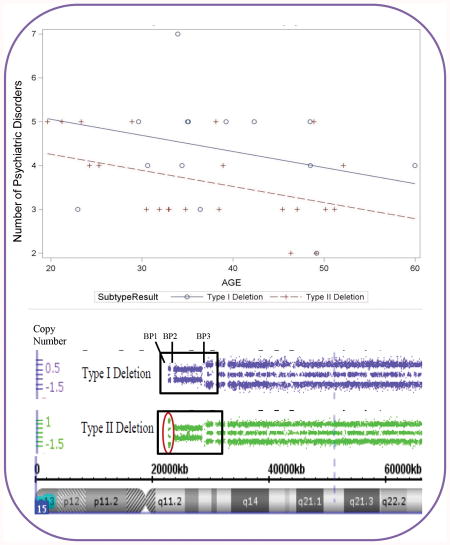
A significant positive correlation was found between age and BMI (r= 0.27, p<0.027), while a significant negative correlation was found between age and number of psychiatric disorders (r=-0.24; p<0.042). The number of psychiatric disorders was not related to BMI or age at PWHO admission for our PWS study participants. Examination of the relationship between the psychiatric diagnoses and PWS genetic subtype showed no statistically significant differences in the frequency of primary psychiatric diagnoses between those with the 15q11-q13 deletion subtypes and maternal disomy 15 subclasses (see Table III). Similarly, there were no statistically significant differences in the frequency of primary psychiatric diagnoses between those with Type I vs Type II deletion subtypes or among the three mUPD15 subtypes. The average number of psychiatric diagnoses were significantly higher for those with the Type I deletion subtype when age differences were controlled (see Figure 4a). Similarly, BMI was significantly higher for those with the Type II deletion subtype when age differences were controlled (see Figure 4b).
Table III. PWHO Neuropsychiatric Outcomes by PWS Genetic Subtype.
| Primary Psychiatric Diagnoses | UPD15 (N=29) | Deletion (N=36) | Odds# UPD15 | 95% CI | p-value* |
|---|---|---|---|---|---|
| Any Psychotic Features | 9(31%) | 7(19%) | 1.9 | 0.60,5.8 | 0.28 |
| Bipolar Disorder (Nonpsychotic) | 8(28%) | 5(14%) | 2.4 | 0.68,8.2 | 0.17 |
| Anxiety Disorder | 12(41%) | 13(36%) | 1.2 | 0.46,3.4 | 0.66 |
| Major Depressive Disorder | 5(17%) | 10(28%) | 0.5 | 0.16,1.8 | 0.32 |
| Intermittent Explosive Disorder | 8(27%) | 12(33%) | 0.8 | 0.26,2.2 | 0.62 |
| Excoriation (skin picking) Disorder | 10(34%) | 11(30%) | 1.2 | 0.42,3.4 | 0.74 |
Odds ratio for UPD15 relative to deletion subtype using Logit.
p<0.05 considered significant
Additional diagnoses included: attention deficit hyperactivity disorder, conduct disorder and tic disorders.
Figure 4.
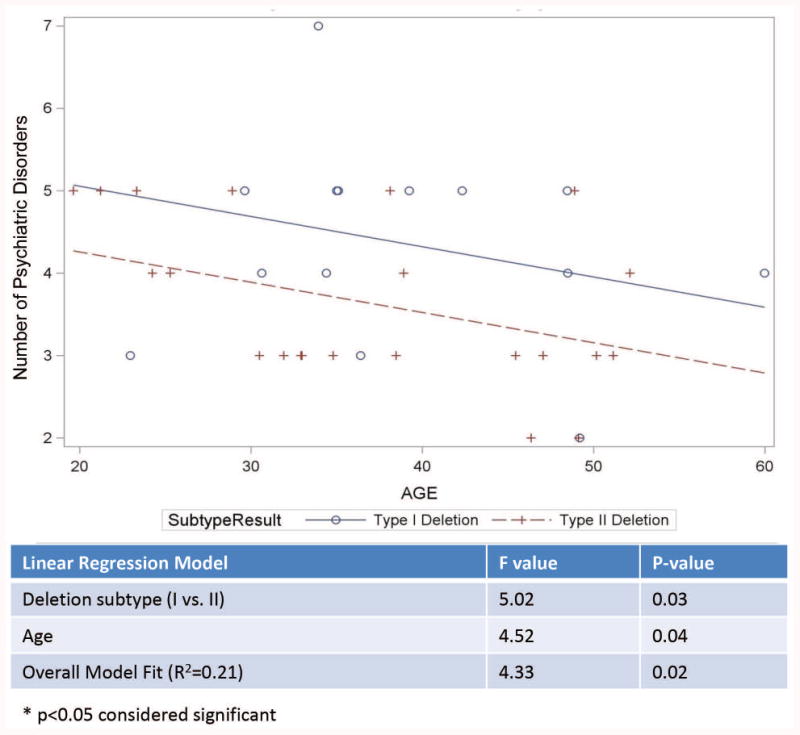
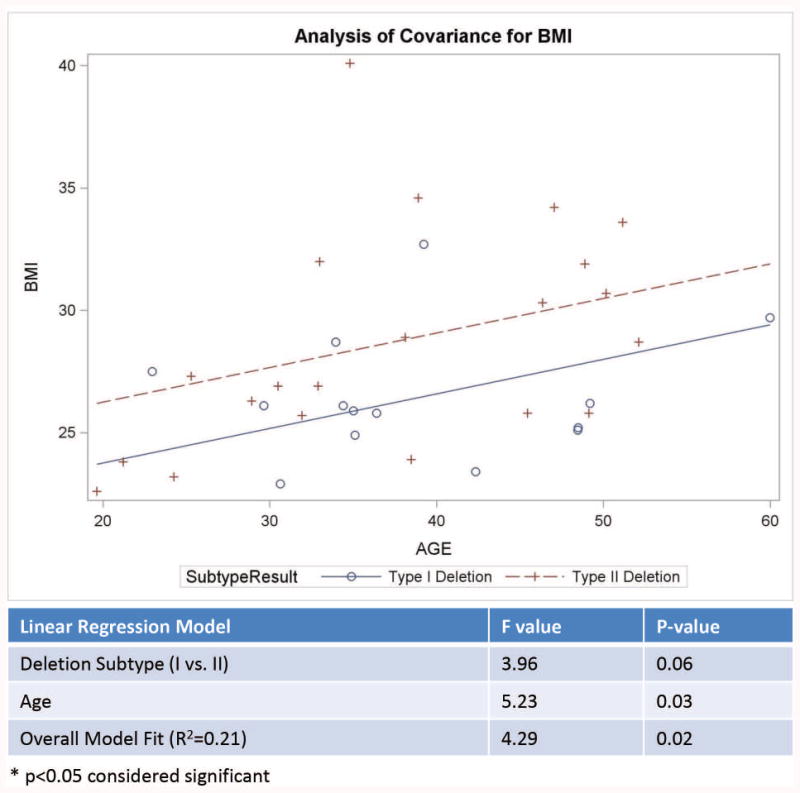
Figure 4a. Analysis of covariance model of the number of psychiatric disorders by age in years with a significant impact of age for Type I vs Type II deletion PWS genetic subtypes with more psychiatric disorders seen in the Type I deletion subgroup. Figure 4b. Analysis of covariance model of body mass index (BMI) by age in years for Type I vs Type II deletion PWS genetic subtypes.
Discussion
The frequency and pattern of our neuropsychiatric diagnoses based on DSM-IV-TR criteria and classification were described in adults with PWS in residential care. The population of individuals with PWS in our PWHO cohort is fundamentally different than previous PWS populations studied. Our residential adults showed less neuropsychiatric stability than non-residential populations at initial assessments. Psychiatric symptomatology in adults with PWS in residential care may vary over time in the presence of full food security and access to experienced clinicians. Previous studies on neuropsychiatric diagnoses or assessments of behavioral problems in adults with PWS have not included data collected during residential care by a trained psychiatrist with expertise in PWS or with updated, detailed PWS genetic subtype classification for statistical analyses. The observed difference in distribution of PWS genetic deletion subtypes and maternal disomy 15 compared with the literature is not readily explainable and may result from a variety of influences including variance associated with small sample sizes for study which adds complexity to most studies of rare diseases. Similar to general psychiatric inpatient or residential populations, the sample may be intrinsically biased towards higher severity cases which have been proposed for mUPD over deletion subtypes and thus may reflect an increased probability that PWS with maternal disomy 15 may lead to admission to residential care. Additionally, socioeconomic factors impact the decision and ability to afford placement in a specialized residential care center. There is currently no empirical evidence to support changes in the global prevalence and subtype distribution related to diagnostic, environmental or sociological factors. Our measure of the accumulated number of psychiatric diagnoses did not prove to be a strong discriminative indicator of phenotypic differences or assessment of global severity of illness in PWS. This suggests that all PWS subtypes may possess a qualitatively similar phenotype and spectrum psychiatric disorders and thus the phenotypic differences observed between PWS subtypes may arise from differences in the severity of the manifestation of psychiatric disorders rather than the type of diagnosis.
PWS individuals with the smaller Type II deletion involving breakpoints BP2 and BP3 have intact genes between breakpoints BP1 and BP2 and therefore anticipated to have fewer problems. Indeed, those individuals with PWS having the larger Type I deletion are more prone to obsessive thoughts, compulsive behaviors and self-injury (i.e., skin-picking) as well as visual processing problems with lower academic skills than those with the smaller Type II deletion or maternal disomy 15 with the four genes not deleted (14, 33, 34). Other deletions that are larger or smaller than the typical deletions vary in size and are reported in about 5% of individuals with PWS but may have unusual findings not typically seen in PWS (35, 36).
PWS and Angelman syndrome (AS), an entirely different clinical syndrome due to genomic imprinting are typically caused by a deletion of different parental origin (i.e., a paternal 15q11-q13 deletion in PWS and a maternal 15q11-q13 deletion in AS) involving the distal breakpoint BP3 and proximally placed breakpoints BP1 or BP2. While previous studies have shown that individuals with the larger typical Type I deletion present in both PWS and AS subjects have a more severe phenotype particularly neurodevelopmental problems while those with the smaller typical Type II deletion have fewer problems, our study did not see a more severe phenotype in adult individuals with Type I or Type II deletions unless corrected for age. Individuals with Type I deletions reported in predominantly pediatric literature had more compulsions and self-injurious behavior with visual perception impairment, and lower IQ, reading and math scores compared with those with Type II deletions (14, 33, 37). For example, Milner et al. (38) found significantly higher verbal IQ scores in individuals with PWS and Type II deletions than those with Type I deletions who performed more poorly on all measures of ability which may be impacted by age. The previous studies have used standardized cognitive and behavioral assessment tools while our investigation used psychiatric diagnoses based on DSM-IV-TR criteria and classification for mental disorders or illnesses.
A report by Dykens and Roof (41) examined behaviors in PWS using a mixed cohort of young and old subjects showed a relationship between genetic subtypes and ages. Negative associations were found between age and behavior in the 15q11-q13 Type I deletion subtype only which implicated non-imprinted genes between breakpoints BP1 and BP2, specifically the CYF1P1 gene as similarly reported in our PWS genetic subtype and neuropsychiatric study. Disturbed expression of CYF1P1 is reported in other developmental disabilities involving chromosome 15 disorders with significant differences in those with Type I versus Type II deletions with advancing age. Individuals with the 15q11-q13 Type I deletion consistently showed lower targeted problem behaviors and adaptive skills and externalizing symptoms with advancing age.
Psychiatric manifestations are common in PWS and represent debilitating problems that negativity impact the quality of life. The results of our novel detailed neuropsychiatric diagnostic and genetic study in adults with PWS in residential care further support the observations of psychiatric features with compulsions and skin picking as common findings (8). Several psychiatric or behavioral problems were treated with a variety of medications in our PWS adult cohort including: mood stabilizers including lithium carbonate; selective serotonin reuptake inhibitors (SSRI); serotonin/norepinephrine reuptake inhibitors (SNRI); N-acetyl cysteine (for skin picking); atypical antipsychotics; occasional benzodiazepines; rare use of stimulant and occasionally antiadrenergic agents. Differences were also seen in accumulated number of psychiatric diagnoses and BMI in those with the larger 15q11-q13 Type I deletion but only when adjusted for age. Those individuals with the larger deletion had more problems than those with the smaller Type II deletion similar to that reported in PWS cohorts in non-residential care emphasizing the importance of more research to investigate the neurodevelopmental and psychiatric impact of the four genes located in the 15q11.2 BP1 and BP2 region and their role in the PWS phenotype. Furthermore, our report is the first to describe maternal disomy 15 subclasses (heterodisomy, segmental isodisomy, total isodisomy) and examine for differences in clinical or neuropsychiatric diagnoses in individuals with PWS using the standardized DSM-IV-TR criteria and classification for mental illnesses or disorders. Precise psychiatric and behavioral phenotyping in PWS in combination with identification of genetic subtypes may aid research linking biological correlates which may lead to individualized therapeutic interventions.
Table IV. PWHO Neuropsychiatric Outcomes by PWS Genetic Subtype.
| Primary Psychiatric Diagnoses | UPD15 (N=29) | Deletion (N=36) | Odds# UPD15 | 95% CI | p-value* |
|---|---|---|---|---|---|
| Any Psychotic Features | 9(31%) | 7(19%) | 1.9 | 0.60,5.8 | 0.28 |
| Bipolar Disorder (Nonpsychotic) | 8(28%) | 5(14%) | 2.4 | 0.68,8.2 | 0.17 |
| Anxiety Disorder | 12(41%) | 13(36%) | 1.2 | 0.46,3.4 | 0.66 |
| Major Depressive Disorder | 5(17%) | 10(28%) | 0.5 | 0.16,1.8 | 0.32 |
| Intermittent Explosive Disorder | 8(27%) | 12(33%) | 0.8 | 0.26,2.2 | 0.62 |
| Excoriation (skin picking) Disorder | 10(34%) | 11(30%) | 1.2 | 0.42,3.4 | 0.74 |
Odds ratio for UPD15 relative to deletion subtype using Logit.
p<0.05 considered significant
Additional diagnoses included: attention deficit hyperactivity disorder, conduct disorder and tic disorders.
Acknowledgments
We thank the individuals with Prader-Willi syndrome and their families for participation in this study and Janice Forster, MD. We acknowledge support from the National Institute of Child Health and Human Development (NICHD) grant number HD02528 and Prayer Will Support PWS Organization (Family & Friends of Kyleigh Ellington).
Footnotes
There are no conflicts of interest to report for any of the authors.
References
- 1.Butler MG. Prader-Willi syndrome: Current understanding of cause and diagnosis. Am J Med Genet. 1990;35(3):319–332. doi: 10.1002/ajmg.1320350306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Dev Med Child Neurol. 2002;44(4):248–255. doi: 10.1017/s001216220100202x. [DOI] [PubMed] [Google Scholar]
- 3.Bittel DC, Butler MG. Prader-Willi syndrome: Clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005;7:1–20. doi: 10.1017/S1462399405009531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 5.Butler MG. Single gene and syndromic causes of obesity: Illustrative examples. Prog Mol Biol Transl Sci. 2016;140:1–45. doi: 10.1016/bs.pmbts.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler MG, Lee PDK, Whitman BY. Management of Prader-Willi Syndrome. New York: Springer-Verlag; 2006. [Google Scholar]
- 7.Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38(12):1249–1263. doi: 10.1007/s40618-015-0312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shriki-Tal L, Avrahamy H, Pollak Y, et al. Psychiatric disorders in a cohort of individuals with Prader-Willi syndrome. Eur Psychiatry. 2017;44:47–52. doi: 10.1016/j.eurpsy.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomic duplicons map to sites of instability in the Prader-Willi/Angleman syndrome chromosome region (15q11-q13) Hum Mol Genet. 1999;8(6):1025–1037. doi: 10.1093/hmg/8.6.1025. [DOI] [PubMed] [Google Scholar]
- 10.Chai JH, Locke DP, Greally JM, et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet. 2003;73(4):898–925. doi: 10.1086/378816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholls RD. Genomic imprinting and candidate genes in Prader-Willi and Angelman syndromes. Curr Opin Genet Dev. 1993;3(3):445–456. doi: 10.1016/0959-437x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- 12.Glenn CC, Driscoll DJ, Yang TP, Nicholls RD. Genomic imprinting: Potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Preprod. 1997;3(4):321–332. doi: 10.1093/molehr/3.4.321. [DOI] [PubMed] [Google Scholar]
- 13.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–75. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- 14.Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman syndromes: A molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72(3):571–577. doi: 10.1086/367926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. 2004;113:565–573. doi: 10.1542/peds.113.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox DM, Butler MG. The 15q11.2 BP1-BP2 microdeletion syndrome: a review. Int J Mol Sci. 2015;16(2):4068–4082. doi: 10.3390/ijms16024068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler MG. Clinical and genetic aspects of the 15q11.2 BP1-BP2 microdeletion disorder. J Intellect Disabil Res. 2017;61(6):568–579. doi: 10.1111/jir.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ledbetter DH, Riccardi VM, Airhart SD, et al. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304(6):325–329. doi: 10.1056/NEJM198102053040604. [DOI] [PubMed] [Google Scholar]
- 19.Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. 1989;342(6247):281–285. doi: 10.1038/342281a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuwano A, Mutirangura A, Dittrich B, et al. Molecular dissection of the Prader-Willi/Angelman syndrome region (15q11-13) by YAC cloning and FISH analysis. Hum Mol Genet. 1992;1(6):417–425. doi: 10.1093/hmg/1.6.417. [DOI] [PubMed] [Google Scholar]
- 21.Driscoll DJ, Waters MF, Williams CA, et al. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics. 1992;13(4):917–924. doi: 10.1016/0888-7543(92)90001-9. [DOI] [PubMed] [Google Scholar]
- 22.Kubota T, Sutcliffe JS, Aradhya S, et al. Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am J Med Genet. 1996;66(1):77–80. doi: 10.1002/(SICI)1096-8628(19961202)66:1<77::AID-AJMG18>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 23.Muralidhar B, Butler MG. Methylation PCR analysis of Prader-Willi syndrome, Angelman syndrome, and control subjects. Am J Med Genet. 1998;80(3):263–265. [PMC free article] [PubMed] [Google Scholar]
- 24.Procter M, Chou LS, Tang W, Jama M, Mao R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem. 2006;52(7):1276–1283. doi: 10.1373/clinchem.2006.067603. [DOI] [PubMed] [Google Scholar]
- 25.Bittel DC, Kibiryeva N, Butler MG. Methylation-specific multiplex ligation-dependent probe amplification analysis of subjects with chromosome 15 abnormalities. Genet Test. 2007;(4):467–475. doi: 10.1089/gte.2007.0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papenhausen P, Schwartz S, Risheg H, et al. UPD detection using homozygosity profiling with SNP genotyping microarray. Am J Med Genet. 2011;155(4):757–768. doi: 10.1002/ajmg.a.33939. [DOI] [PubMed] [Google Scholar]
- 27.Henkhaus RS, Kim SJ, Kimonis VE, et al. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet Test Mol Biomarkers. 2012;16(3):178–186. doi: 10.1089/gtmb.2011.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91(2):398–402. [PMC free article] [PubMed] [Google Scholar]
- 29.Butler MG, McGuire A, Manzardo AM. Clinically relevant known and candidate genes for obesity and their overlap with human infertility and reproduction. J Assist Reprod Genet. 2015;32(4):495–508. doi: 10.1007/s10815-014-0411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fletcher Robert, DSW, ACSW-Chief Editor, Loschen Earl, MD, Stavrakaki Chrissoula, MD, PhD, First Michael., MD, editors. Copyright 2007 by National Association for the Dually Diagnosed (NADD); 132 Fair Street, Kingston, NY 12401: Diagnostic Manual-Intellectual Disability: A Textbook of Diagnosis of Mental Disorders in Persons with Intellectual Disability (DM-ID) www.thenadd.org. [Google Scholar]
- 31.Butler MG, Usrey K, Roberts JL, Schroeder SR, Manzardo AM. Clinical presentation and microarray analysis of Peruvian children with atypical development and/or aberrant behavior. Genet Res Int. 2014:408516. doi: 10.1155/2014/408516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bittles AH, Black ML. The impact of consanguinity of neonatal and infant health. Early Hum Dev. 2010;86(11):737–741. doi: 10.1016/j.earlhumdev.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 33.Hartley SL, Maclean WE, Jr, Butler MG, Zarcone J, Thompson T. Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am J Med Genet A. 2005;136:140–145. doi: 10.1002/ajmg.a.30771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bittel DC, Kibiryeva N, Butler MG. Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome. Pediatrics. 2006;118:e1276–e1283. doi: 10.1542/peds.2006-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butler MG, Bittel DC, Kibiryeva N, Cooley LD, Yu S. An interstitial 15q11-q14 deletion: expanded Prader-Willi syndrome phenotype. Am J Med Genet A. 2010;152A(2):404–408. doi: 10.1002/ajmg.a.33197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim SJ, Miller JL, Kuipers PJ, et al. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. 2012;20(3):283–290. doi: 10.1038/ejhg.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zarcone J, Napolitano D, Peterson C, et al. The relationship between compulsive behaviour and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J Intellect Disabil Res. 2007;51(Pt. 6):478–487. doi: 10.1111/j.1365-2788.2006.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milner KM, Craig EE, Thompson RJ, et al. Prader-Willi syndrome: Intellectual abilities and behavioural features by genetic subtype. J Child Psychol Psychiatry. 2005;46:1089–1096. doi: 10.1111/j.1469-7610.2005.01520.x. [DOI] [PubMed] [Google Scholar]
- 39.Burnside RD, Pasion R, Mikhail FM, et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet. 2011;130(4):517–528. doi: 10.1007/s00439-011-0970-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cafferkey M, Ahn JW, Flinter F, Ogilvie C. Phenotypic features in patients with 15q11.2(BP1-BP2) deletion: further delineation of an emerging syndrome. Am J Med Genet A. 2014;164A(8):1916–1922. doi: 10.1002/ajmg.a.36554. [DOI] [PubMed] [Google Scholar]
- 41.Dykens EM, Roof E. Behavior in Prader-Willi syndrome: Relationship to genetic subtypes and age. J Child Psychol Psychiatry. 2008;49:1001–1008. doi: 10.1111/j.1469-7610.2008.01913.x. [DOI] [PubMed] [Google Scholar]