

Abstract
Background and Purpose
Monoacylglycerol lipase (MAGL) is an enzyme that hydrolyzes the endocannabinoid 2-arachidonoylglycerol and regulates production of arachidonic acid and prostaglandins, substances that mediate tissue inflammatory response. Here, we have studied the effects of the selective MAGL inhibitors JZL184 and MJN110 and their underlying molecular mechanisms on 3 different experimental models of focal cerebral ischemia.
Methods
Spontaneously hypertensive rats and normotensive Wistar-Kyoto rats were subject to an intracortical injection of the potent vasoconstrictor endothelin-1, permanent occlusion of a distal segment of the middle cerebral artery (MCA) via craniectomy, or transient occlusion of the MCA by the intraluminal suture method. JZL184 or MJN110 were administered 60 min after focal cerebral ischemia. Infarct volumes, hemispheric swelling, and functional outcomes were assessed between day 1 to day 28 by magnetic resonance imaging, histology, and behavioral tests.
Results
Pharmacological inhibition of MAGL significantly attenuated infarct volume and hemispheric swelling. MAGL inhibition also ameliorated sensorimotor deficits, suppressed inflammatory response, and decreased the number of degenerating neurons. These beneficial effects of MAGL inhibition were not fully abrogated by selective antagonists of cannabinoid receptors, indicating that the anti-inflammatory effects are caused by inhibition of eicosanoid production rather than by activation of cannabinoid receptors.
Conclusions
Our results suggest that MAGL may contribute to the pathophysiology of focal cerebral ischemia and is thus a promising therapeutic target for the treatment of ischemic stroke.
Keywords: endocannabinoid, MAGL, ischemic stroke, neuroprotection, JZL184, MJN110
Introduction
The endocannabinoid system, comprising the cannabinoid receptors CB1 and CB2, their lipid ligands 2-arachidonoylglycerol (2-AG) and N-arachidonoylethanolamide, and enzymes responsible for the biosynthesis and degradation of ligands, has become a subject of great interest in neuropharmacology, due to their predominant distribution in both central and peripheral nervous systems, and for its capacity to play a modulating role in diverse physiological processes1. Investigation of the endocannabinoid system has also provided new insight into the underlying pathological mechanisms of various acute and chronic neurological disorders2, 3. There is convincing evidence that the components of endocannabinoid system are altered during ischemic stroke in both animals and humans4–9, indicating that this system may contribute to the consequences of cerebral ischemia via multiple mechanisms. Although numerous studies have examined the role of the endocannabinoid system in experimental models of ischemic stroke over the past decade10, results have been rather conflicting in supporting either a beneficial or a detrimental role. Nevertheless, a recent systematic review and meta-analysis reported that cannabinoids significantly reduce infarct volume and improve functional outcome in several experimental models of cerebral ischemia11. In particular, Nomura and colleagues provided compelling evidence that the endocannabinoid and eicosanoid systems are metabolically coupled through the action of monoacylglycerol lipase (MAGL), the enzyme that hydrolyzes 2-AG to arachidonic acid for eicosanoid biosynthesis12. Pharmacological inhibition of MAGL resulted in a robust increase of the brain 2-AG levels and a concomitant reduction of arachidonic acid and downstream inflammatory eicosanoid metabolites.
While the therapeutic potential of compounds targeting the endocannabinoid system has been investigated, the effects of MAGL inhibition and the mechanisms of action in cerebral ischemia are still unclear. In the present study, we investigated the effects of two potent and selective MAGL inhibitors in three rat models of ischemic stroke.
Materials and Methods
All materials, methods, and data obtained for the current study are available from the corresponding author upon request. Detailed materials and procedures are described in the online-only Data Supplement.
Animals
Experiments were performed in adult male spontaneously hypertensive rats (SHR) and normotensive Wistar Kyoto (WKY) rats, weighing 270-300 g (Harland Laboratories, Frederick, MD). Animals were kept in a room with controlled temperature and a 12 h light/dark cycle and fed with standard food and water. SHR were selected to include chronic hypertension as a common comorbidity factor in clinical stroke, according to the recommendations of the modified Stroke Therapy Academic Industry Roundtable (STAIR) criteria13. All animal care and experimental procedures were performed in accordance with the National Institutes of Health guidelines and with an approved protocol from the National Institute of Neurological Disorders and Stroke Animal Care and Use Committee.
Induction of cerebral ischemia
Three different experimental models of ischemia were used, in which cerebral ischemia was caused by either an intracortical injection of the potent vasoconstrictor endothelin-1 (ET-1); by permanent cauterization of a distal branch of the middle cerebral artery (MCA)14; or by the standard transient MCA occlusion (tMCAO) using an intraluminal suture followed by reperfusion15.
Pharmacological treatments
All groups were performed and quantified in a randomized fashion by investigators blinded to specific treatment. Rats received an intraperitoneal administration of either JZL184 or MJN110 60 min after stroke induction. An additional set of animals received an intraperitoneal injection of either AM251 or AM630 30 min after stroke induction. Routes and dose of administration for each compound were chosen based on previous studies16–18.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) was performed 24 h after induction of stroke on a 7 T/30 cm AVIII MRI system (Bruker Biospin Inc., Billerica, MA) under inhalation anesthesia as described previously19.
Assessment of functional outcome
Functional outcome was evaluated as described previously15, 20–22, and in the online-only Data supplement.
Histology and immunohistochemistry
Free-floating coronal brain sections (30 μm) were processed 1, 3, or 7 d after ischemic stroke, immunofluorescence, and confocal microscopy were performed as described in the online-only Data Supplement.
Profiling of local cytokine expression
Local cytokine expression was performed using the rat cytokine ELISA plate array kit (Signosis, Santa Clara, CA).
RNA extraction and real-time PCR
Gene expression analysis was performed using the TaqMan Assays (Applied Biosystems).
Statistics
The statistical methods are presented in the online-only Data Supplement.
RESULTS
The MAGL inhibitor JZL184 reduces infarct volume and improves functional outcome in the ET-1 model
To investigate the MAGL inhibitor JZL184 for its therapeutic potential in animal models of stroke, we first employed the ET-1 model of focal ischemia. The ET-1 model was initially optimized in both SHR and WKY rats using an intracerebral injection of ET-1 into the right somatosensory cortex. This injection led to a significant and stable reduction of CBF (to about ~30% of baseline) over 3 h and resulted in extensive infarcts in both cortical and subcortical areas, including primary and secondary somatosensory cortex, primary motor cortex, the underlying white matter and a small region of dorsolateral striatum, as estimated by T2-weighted MRI and confirmed post-mortem using Nissl staining (Figure 1B) and immunofluorescence staining (supplementary Figure I). No lesions were noted in control rats injected with saline. Infarct volumes 24 h after ET-1-induced stroke were twice as large in SHR (99.7 ± 10.2 mm3) than in WKY (45.6 ± 5.1 mm3), indicating that hypertension significantly exacerbates the outcome after ischemic stroke (Figure 1B). The lesions were highly variable in SHR, whereas WKY exhibited small infarct size with relatively low variation. Treatment of the animals with JZL184 significantly attenuated lesion volume in both SHR (23.6 ± 5.6 mm3) and WKY (8.6 ± 2.2 mm3) compared to the animals in the respective vehicle-treated groups (P < 0.01; Figure 1B). JZL184 also significantly attenuated brain swelling in SHR versus vehicle-treated animals (P < 0.01), but the decrease in brain swelling in WKY was not significant (P > 0.05, Figure 1B). The levels of 2-AG and AEA were quantified in ipsilateral cortex of SHR 1 d after stroke induction. The MAGL inhibitor JZL184 significantly increased 2-AG levels in brain, but had no effect on AEA levels (Supplementary Figure II).
Figure 1.
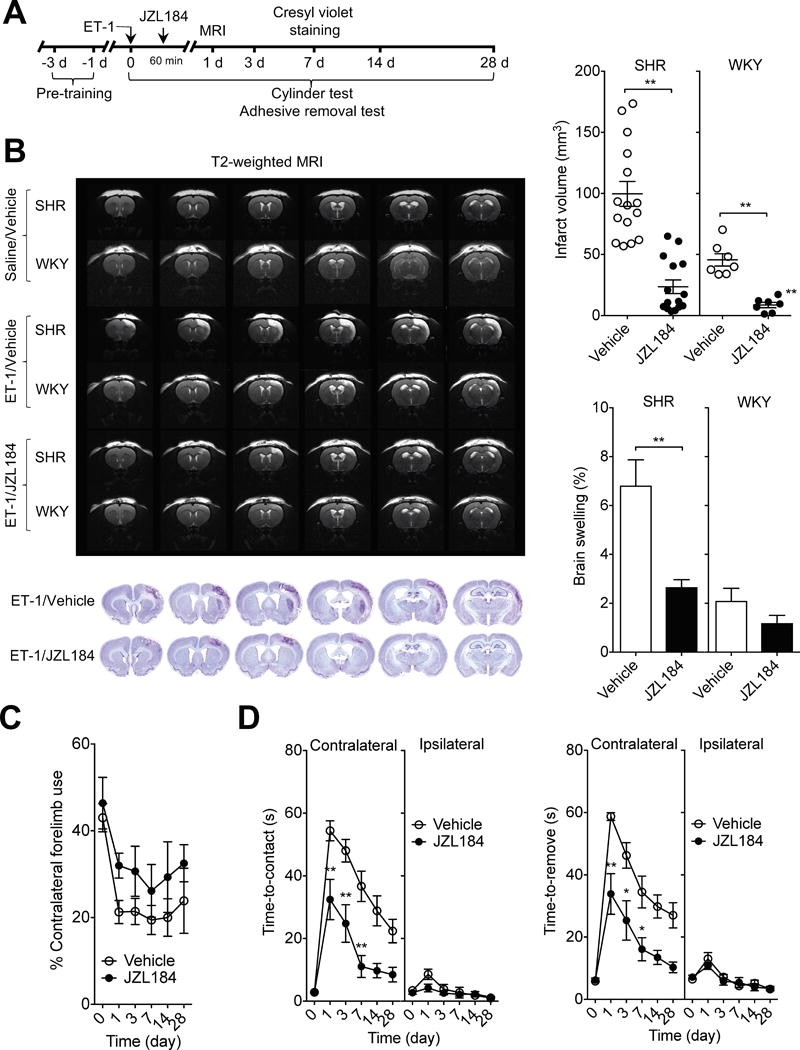
MAGL inhibitor JZL184 reduces infarct volume and improves sensorimotor impairment after ET-1-induced stroke. A, Basic experimental scheme. JZL184 (16 mg/kg) or vehicle was administered i.p. 1 h after ET-1-induced stroke and all read-out parameters were evaluated at the indicated time points. B, Representative T2-weighted MR images show smaller infarct size in JZL184-treated rats than in vehicle-treated rats at 1 d after ET-1-induced ischemic stroke (top). Representative cresyl violet-stained coronal sections in SHR at 7 d after ET-1-induced stroke (bottom). Infarct volume at 1 d after ET-1-induced stroke in SHR and WKY. Edema formation as reflected by the brain water content in the ischemic hemispheres of SHR and WKY at 1 d after ET-1-induced stroke. Data are expressed as mean ± SEM (n = 15/group for SHR and 7/group for WKY). **P < 0.01 versus vehicle, unpaired t-test. C and D, Forelimb asymmetry during spontaneous exploration assessed with cylinder test (C) and sensorimotor deficits measured by adhesive removal test (D) on day 1, 3, 7, 14 and 28 after ET-1-induced stroke in SHR. Data are expressed as mean ± SEM (n = 15/group). *P < 0.05, **P < 0.01 versus the respective vehicle group, two-way ANOVA followed by Bonferroni’s multiple comparisons test.
We then examined functional outcome in both vehicle- and JZL184-treated SHR before and on day 1, 3, 7, 14 and day 28 after stroke, using the cylinder test that assesses asymmetry in usage of the forelimbs during vertical exploration, and the adhesive removal test that assesses sensorimotor performance. In the cylinder test, animals from both groups displayed similar exploratory behavior and spontaneous forelimb usage before induction of stroke. Following stroke, vehicle-treated SHR displayed a significant increase in asymmetry of the forelimbs, and this motor deficit was maintained over the entire testing period. On the other hand, JZL184-treated SHR had a substantially improved spontaneous usage of the contralateral forelimb compared to vehicle-treated SHR (Figure 1C). In the adhesive removal test, ET-1 induced a contralateral deficit of sensorimotor performance, as evidenced by a significant increase in both time-to-contact and time-to-remove the adhesive tape, which was highest on day 1 and improved gradually with time. Treatment with JZL184 significantly alleviated the ET-1-induced contralateral deficit in sensorimotor performance observed in vehicle-treated SHR (P < 0.01; Figure 1D).
JZL184 decreases infarct size and restores neurologic deficits after permanent MCAO
To further validate the neuroprotective effects of JZL184, we tested its effect on two other models of cerebral ischemia, as recommended by the STAIR23. The permanent MCAO model, in which the distal part of the left MCA was cauterized, led to a significant and stable reduction of CBF in SHR to about ~23% of baseline, and resulted in infarct of both cortical and subcortical areas (Figure 2A). In contrast, in WKY, the lesions caused by the permanent MCAO model were limited to the cortex immediately adjacent to the craniotomy (Supplementary Figure IIIA). We found that the infarct volume after permanent MCAO was significantly smaller in JZL184-treated SHR than that in vehicle-treated SHR (P < 0.01; Figure 2A). The effect of treatment was functionally relevant, as JZL184-treated SHR had significantly better neurological scores than vehicle-treated SHR (P < 0.01; Figure 2A). However, because of the small infarct volume, there were no significant differences in infarct volumes and neurological scores between JZL184- and vehicle-treated WKY (Supplementary Figure IIIA).
Figure 2.
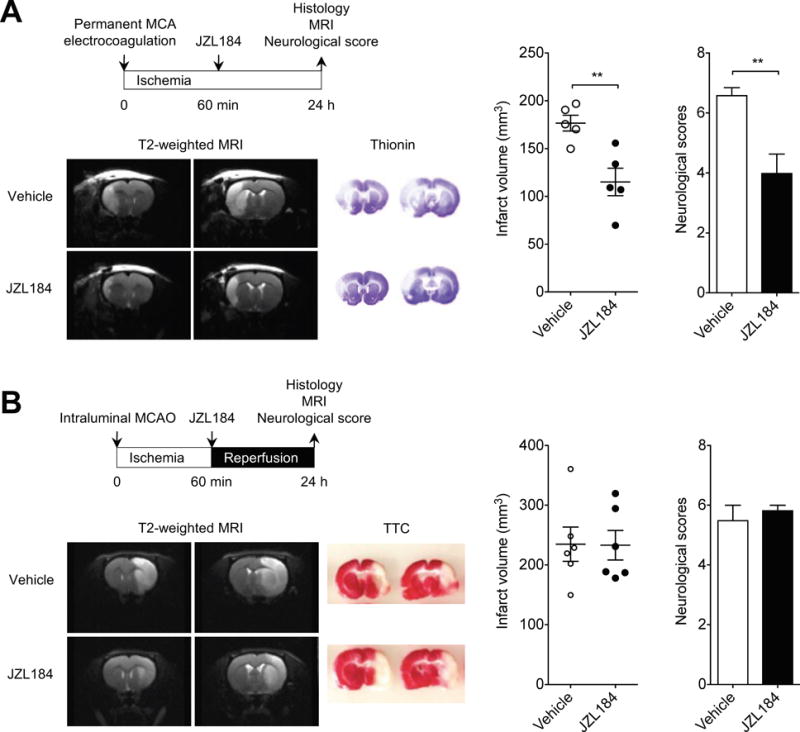
MAGL inhibitor JZL184 reduces infarct volume and improves neurologic deficits after permanent ischemic stroke. A, Basic experimental scheme of permanent MCAO model with a distal MCA electrocoagulation. JZL184 (16 mg/kg) or vehicle was administered i.p. 1 h after induction of ischemia and all read-out parameters were evaluated on day 1 after permanent occlusion of distal MCA. Representative T2-weighted MR images show smaller infarct size in JZL184-treated SHR than in vehicle-treated SHR. Representative thionin-stained coronal sections. Quantification of infarct volume. The neurological deficit was significantly decreased in JZL184-treated SHR. Data are expressed as mean ± SEM (n = 5/group). **P < 0.01 versus vehicle, unpaired t-test. B, Basic experimental scheme of transient MCAO. JZL184 (16 mg/kg, i.p.) or vehicle was administered immediately after reperfusion and all read-out parameters were evaluated on day 1 after transient MCAO. T2-weighted coronal MR images show extensive hyperintense ischemic lesions in the cortex and subcortical area at 1 d after transient MCAO in vehicle- or JZL184-treated SHR. Representative TTC-stained coronal slices. Infarct volume and neurological deficit were unchanged in JZL184-treated SHR. Data are expressed as mean ± SEM (n = 6/group).
We also studied the effect of JZL184 on the tMCAO model of ischemia/reperfusion in SHR and WKY. In SHR, the tMCAO model led to a significant reduction of CBF (to about ~10% of baseline during the ischemic phase) and resulted in extensive infarcts in both cortical and subcortical areas, as estimated by T2-weighted MRI and confirmed using standard TTC staining (Figure 2B). Unlike in the ET-1 and the permanent MCAO models of ischemia, however, JZL184 had no effect on ameliorating either infarct volume or functional outcome in SHR subjected to the tMCAO model (Figure 2B). Similarly, the differences in infarct volume and functional outcome were not statistically significant between JZL184- and vehicle-treated WKY (Supplementary Figure IIIB).
The selective MAGL inhibitor MJN110 reduces infarct volume and improves functional outcomes in hypertensive rats
To prove that the neuroprotective properties of JZL184 were specifically related to inhibition of MAGL and not due to unspecific effects, we also tested the more recently synthesized MAGL inhibitor MJN11024 in SHR after ET-1-induced stroke. We found that the infarct volume in MJN110-treated SHR was significantly smaller than that in vehicle-treated SHR (P < 0.01; Figure 3B). MJN110-treated SHR also had substantially better contralateral forelimb use than vehicle-treated controls as measured by the cylinder test, although there was no statistical differences in the forelimb asymmetry at the indicated time points (Figure 3C). On the other hand, the sensorimotor deficit of the contralateral forelimb caused by the ET-1-induced stroke was significantly less severe in MJN110-treated SHR than in vehicle-treated animals (P < 0.01; Figure 3D). These results indicate that inhibition of MAGL is an effective therapeutical approach to lessen brain damage and improve functional outcome after stroke.
Figure 3.
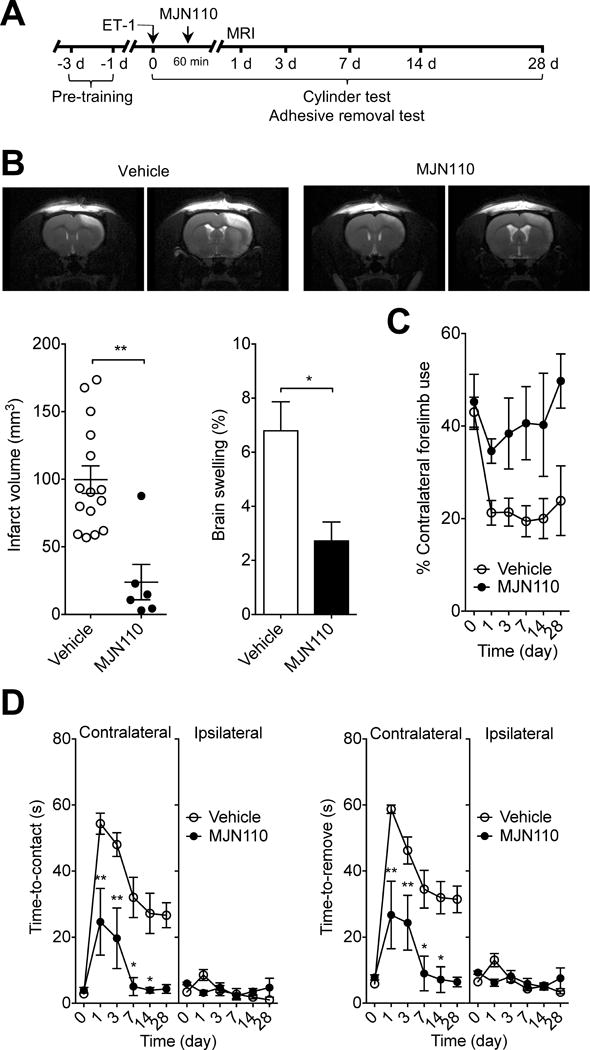
Selective MAGL inhibitor MJN110 has profound protective effects in ischemic stroke. A, Basic experimental scheme. MJN110 (20 mg/kg) or vehicle was administered i.p. 1 h after ET-1-induced stroke and all read-out parameters were evaluated at the indicated time points. B, Representative T2-weighted MR images show smaller infarct size in MJN110-treated SHR than in vehicle-treated SHR at 1 d after ET-1-induced ischemic stroke. Infarct volume at 1 d after ET-1-induced stroke in SHR. Edema formation in the ischemic hemispheres of SHR at 1 d after ET-1-induced stroke. Data are expressed as mean ± SEM (n = 15 and 6/group). **P < 0.01 versus vehicle, unpaired t-test. C and D, Forelimb asymmetry during spontaneous exploration assessed with cylinder test (C) and sensorimotor deficits measured by adhesive removal test (D) on day 1, 3, 7, 14 and 28 after ET-1-induced stroke in SHR. Data are expressed as mean ± SEM (n = 15 and 6/group). *P < 0.05, **P < 0.01 versus the respective vehicle group, two-way ANOVA followed by Bonferroni’s multiple comparisons test.
The protective effects of JZL184 are associated with reduced neuroinflammation
There is increasing evidence demonstrating the contribution of neuroinflammation to the drastic changes after ischemic stroke25. The inflammatory response not only immediately affects the infarcted tissue but also causes long-term damage in peri-infarct regions. To determine whether inhibition of MAGL prevents post-ischemic neuronal injury, we first stained fixed brain sections with active caspase-3 and NeuN in SHR that received JZL184 and observed that active caspase-3+ neurons were distributed both in the infarcted area as well as in the lesion boundary (Figure 4A). The nuclei of these neurons were densely labeled and showed morphological signs of degeneration. We found that the number of active caspase-3+ neurons in JZL184-treated SHR was significantly less than that in vehicle-treated controls 24 h after ET-1-induced stroke (P < 0.05; Figure 4A).
Figure 4.
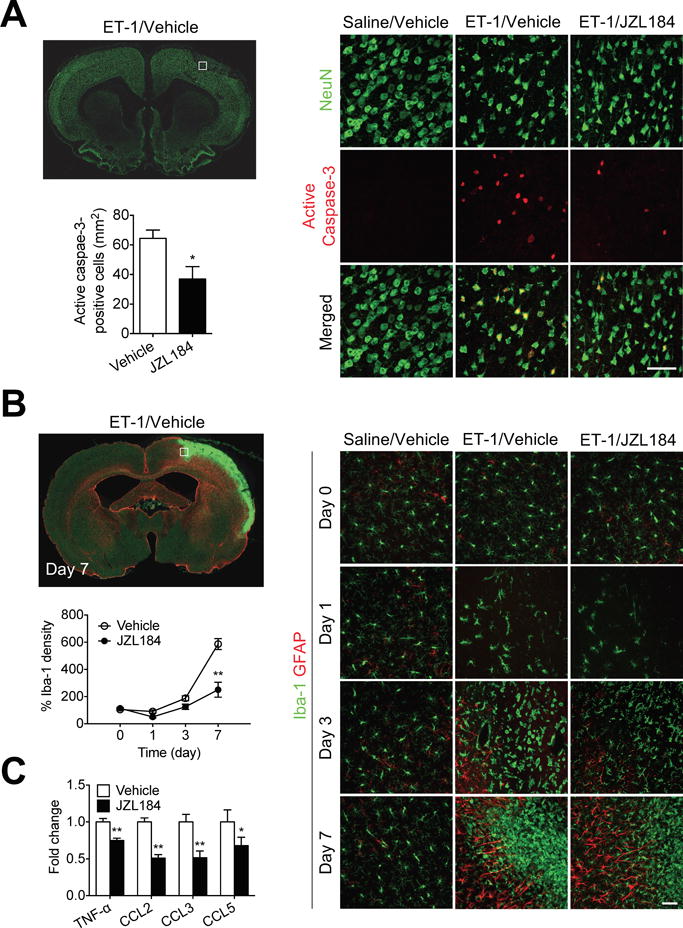
MAGL inhibitor JZL184 reduces neuronal injury and inflammation after stroke. A, Double-immunofluorescence staining of active caspase-3 (red) and neurons (NeuN, green) in the ipsilateral hemisphere of SHR at 1 d after ET-1-induced ischemic stroke. High-magnification images showing representative immunoreactivity in the cortex. Scale bar, 50 μm. Quantification of active caspase-3+ neurons in the ischemic hemisphere of vehicle- and JZL184-treated SHR. Data are expressed as mean ± SEM (n = 6/group). *P < 0.05 versus vehicle, unpaired t-test. B, Double-immunofluorescence staining of astrocytes (GFAP, red) and microglia (Iba-1, green) in the ipsilateral hemisphere of SHR at 1 d after ET-1-induced ischemic stroke. High-magnification images showing representative immunoreactivity in the cortex at 1, 3, and 7 d after ET-1-induced ischemic stroke. Scale bar, 50 μm. Quantification of the temporal density of Iba-1+ microglia in the peri-infarct regions of cortex at different time points. Data are expressed as mean ± SEM (n = 6/group). **P < 0.01 versus the respective vehicle group, two-way ANOVA followed by Bonferroni’s multiple comparison test. C, Relative protein expression of TNF-α, CCL2, CCL3, and CCL5 in the cortices of vehicle- and JZL184-treated SHR at 1 d after ET-1-induced stroke. Data are expressed as mean ± SEM (n = 6/group). *P < 0.05, **P < 0.01 versus vehicle, unpaired t-test.
To investigate whether anti-inflammatory mechanisms are involved in the neuroprotective effects of JZL184, we analyzed expression of pro-inflammatory mediators and density of Iba-1+ cells in the injured brain. Characteristic components of neuroinflammation, including marked increases in expression of Iba-1 and morphological changes of microglia, occurred in the injured hemisphere of the brain 24 h after ET-1-induced stroke (Figure 4B). Accumulation of amoeboid Iba-1+ cells persisted in the ischemic core and even more in the peri-infarct area 7 d after ET-1-induced stroke. Treatment with JZL184 significantly reduced the overall accumulation of Iba-1+ cells in both ischemic core and peri-infarct regions (P < 0.01; Figure 4B). Considering that accumulation of Iba-1+ cells occurs via multiple mechanisms, including immune cell proliferation and chemoattraction, we determined whether JZL184 affects expression of proinflammatory cytokines and chemokines. The difference in accumulation of Iba-1+ cells was biochemically relevant, as JZL184-treated SHR had significantly lower levels of CCL2, CCL3, and CCL5, major chemokines involved in chemoattractant function of immune cells26, than vehicle-treated SHR (P < 0.01; Figure 4C), indicating that the reduced density of Iba-1+ cells observed after stroke is likely due to the attenuated chemokine-mediated chemotaxis. Similarly, we also observed a significant reduction in the inflammatory cytokine TNF-α (P < 0.01; Figure 4C), suggesting that JZL184 has important immunomodulatory and anti-inflammatory roles in ischemic stroke.
Role of CB1 and CB2 receptors in the protective effect of JZL184
There are two well-known cannabinoid receptors, CB1R and CB2R, which have been previously considered as therapeutic targets for ischemic stroke10. 2-AG is a strong agonist to both CB1R and CB2R and MAGL is the main enzyme responsible for its hydrolysis12. We first analyzed expression of CB1R and CB2R in the injured cerebral cortex. While mRNA expression of CB1R slightly decreased 24 h and 7 d after ET-1-induced stroke, mRNA expression of CB2R was strongly induced on day 7 after stroke (Supplementary Figure IVA). In contrast, both CB1R and CB2R mRNA expression were not affected by JZL184 treatment on day 1 after ET-1-induced stroke (Supplementary Figure IVB). To further investigate the contribution of CB1R and CB2R to the protective effects of JZL184, AM251 (a CB1R antagonist) and AM630 (a CB2R antagonist) were administered before the JZL184 treatment as pharmacological tools. Administration of JZL184 significantly improved stroke outcome in SHR, as shown by a reduction in brain infarction and sensorimotor impairment (P < 0.01; Figure 5B). This effect was partially reversed when SHR were treated with selective CB1R antagonist AM251 (Figure 5B). Furthermore, AM251 treatment partially abrogated the JZL184-induced actions on sensorimotor impairment (Figure 5B). However, the differences in the infarct volume and functional outcome were not statistically significant (Figure 5C). Finally, we also found that the CB2R antagonist AM630 did not alter the JZL184-induced effects on infarct volume (Figure 5B) and sensorimotor performance (Figure 5D), suggesting that the beneficial effects of MAGL inhibitor might be mediated by reductions in inflammatory response, rather than enhanced action of cannabinoid receptors.
Figure 5.
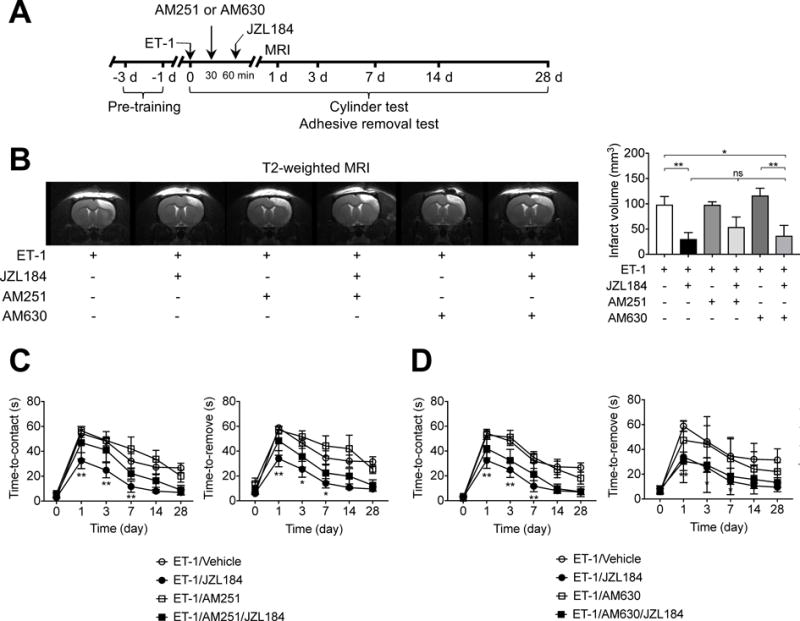
Role of CB1R and CB2R in JZL184-induced reduction of infarct volume and sensorimotor deficits. A, Basic experimental scheme. SHR were injected i.p. (1 mg/kg) with CB1R antagonist AM251 or CB2R antagonist AM630, 30 min after ET-1-induced stroke and 30 min before a single administration of JZL184 (16 mg/kg). All read-out parameters were evaluated at the indicated time points. B, Representative images of T2-weighted MRI showing the extension of hyperintense signal in vehicle-, AM251-, and AM630-treated SHR. Pharmacological blockade of CB1 and CB2 receptors with AM251 and AM630 did not altered the JZL184-induced actions on the infarct volume. Data are expressed as mean ± SEM (n = 3-5/group). *P < 0.05, **P < 0.01, one-way ANOVA followed by Bonferroni’s multiple comparisons test. C and D, Sensorimotor deficits assessed with adhesive removal test on day 1, 3, 7, 14 and 28 in SHR after ET-1-induced stroke. Both CB1 receptor antagonist (C) and CB2 receptor antagonists (D) did not significantly influenced the JZL184-induced effects on sensorimotor performance. Data are expressed as mean ± SEM (n = 3-5/group). *P < 0.05, **P < 0.01 versus the respective vehicle group, two-way ANOVA followed by Bonferroni’s multiple comparisons test.
Discussion
In the present study, we investigate the effects of pharmacological inhibition of MAGL in both normotensive and hypertensive rats exposed to three different models of ischemic stroke. We show that treatment of the animals with JZL184 or with MJN110 confers a significant amount of neuroprotection by decreasing infarct volume and improving functional outcome in both ET-1 and permanent MCAO models, but not in the transient MCAO model. We also show that the neuroprotective effects of JZL184 and MJN110 were long-lasting, as both inhibitors significantly improved sensorimotor deficits on day 28 after induction of stroke. Indeed, this improvement of functional outcome correlated with both neuroprotective and anti-inflammatory mechanisms in the sensorimotor cortex as evidenced by decreasing apoptotic pathway activation and microglial activation. These results demonstrate that MAGL blockade with JZL184 or with MJN110 significantly reduces brain damage and improves functional outcome, and pose MAGL as an important therapeutic target for neuroprotection in ischemic stroke.
JZL184 and MJN110 are potent selective inhibitors of MAGL, the primary enzyme responsible for degrading the endocannabinoid 2-AG27. They both display high selectivity for MAGL over other brain serine hydrolases, thereby making them useful for studying the effects of endogenous 2-AG. Systemic administration of JZL184 has potent neuroprotective and anti-inflammatory properties in a wide range of neurodegenerative diseases and pathological conditions, including Alzheimer’s disease28–30, Down’s syndrome31, experimental autoimmune encephalomyelitis32, Parkinson’s disease12, 33, and traumatic brain injury34.
The therapeutic potential of compounds targeting the endocannabinoid system has been extensively investigated, and neuroprotective effects have been shown in animal models of cerebral ischemia11. Furthermore, since the endocannabinoid system acts on demand with a tightly regulated spatial and temporal selectivity35, it may be possible to avoid ubiquitous cannabinoid receptor activation. Thus, it is not surprising to consider MAGL as a potential therapeutic target for improving outcomes of ischemic brain injury. 2-AG and related lipids have been shown to accumulate in a human patient following stroke4 as well as in animal models of ischemia5. Although in vitro data suggest potent neuroprotective effects of 2-AG36, 37, there has been a paucity of in vivo studies, primarily due to the lack of pharmacological agents that selectively target 2-AG metabolism. A previous study has shown a neuroprotective effect of pretreatment with MAGL inhibitor URB602 in neonatal hypoxic-ischemic brain injury in rats38. However, the low potency and lack of selectivity for MAGL over fatty acid amid hydrolase has limited the usefulness of that compound39. JZL184 robustly and selectively enhances 2-AG levels in the brain40. Importantly, Nomura and his colleagues demonstrated that the neuroprotective effects of JZL184 in the brain are not directly mediated through augmented cannabinoid receptor signaling, but rather due to decreased levels of arachidonic acid and its metabolites such as prostaglandins PGD2 and PGE212. Subsequent studies by these researchers revealed that pharmacologic inactivation of MAGL with JZL184 significantly reduced the inflammatory response and oxidative stress in a hepatic ischemia/reperfusion injury model41. More recent studies have also shown that the neuroprotective effects of JZL184 in acute and chronic animal model of traumatic brain injury and Alzheimer’s disease are primarily due to a substantial decrease in microglial activation and neuroinflammatory response30, 34. Consistently with these anti-inflammatory and neuroprotective effects, we found here that MAGL inhibition with JZL184 significantly protects against ischemic neuronal injury in our rat models of focal ischemia, concordant with suppression of the inflammatory response and microglial activation.
In this study we used both normotensive and hypertensive rats, and 3 different models of experimental ischemia. In normotensive rats, MAGL inhibition proved effective in the ET-1 model (Figure 1), but the beneficial trends observed in either permanent occlusion of a distal branch of the MCA or transient occlusion were not statistically significant (Supplementary Figure III). This could be due to the fact that normotensive rats showed small infarct sizes regardless of whether they were treated or not, making differences harder to detect without testing a much larger number of animals. In hypertensive rats, however, MAGL inhibition proved effective in both ET-1 (Figure 1) and permanent occlusion of a distal branch of the MCA models (Figure 2A), but not in the transient MCAO model (Figure 2B). Consistent with our previous report15, hypertension negatively influences the stroke outcome in different models of cerebral ischemia, and SHR showed larger infarct volumes than WKY in all 3 different ischemia models tested. A major reason for the larger infarct volume is the inadequate collateral circulation in SHR42–43. Adequate collateral circulation provides a compensatory adaptation to arterial occlusion, maintains tissue viability, and offsets infarct growth, suggesting that the collateral circulation is an important defense mechanism that could significantly extend the time window for post-ischemic treatment. Chronic hypertension impairs the development of collateral vessels in SHR and compromises the ability of the collateral circulation to safeguard rCBF values within the ischemic region. Indeed, rCBF in the ischemic region of SHR is not restored to pre-ischemic levels even after reperfusion15. Therefore, the lack of adequate collateral circulation in SHR prevents the delivery of neuroprotective treatments to the ischemic region and may explain the lack of response in SHR to MAGL inhibition in the extensive stroke caused by the tMCAO model (Figure 2B). Additional studies are needed to determine whether the lack of collateral circulation and reperfusion injury are responsible for the less beneficial effect of MAGL inhibitor observed in SHR after tMCAO.
In conclusion, the results of the present study indicate that pharmacological inhibition of MAGL confers a significant amount of neuroprotection and has long-lasting beneficial effects on functional outcome after ischemic stroke. Even though further investigation is required to elucidate the lipidomic analysis and underlying mechanisms for neuroprotective properties, the well-established effect of JZL184 on MAGL12 supports the hypothesis that the approach of preventing the degradation of 2-AG can offer a promising therapeutic approach for reducing brain damage after ischemic stroke.
Supplementary Material
Acknowledgments
The authors thank Xianfeng Zhang for excellent technical support and Dr. Dragan Maric for help with data acquisition and analysis at NINDS Flow and Imaging Cytometry Core Facility. The authors also thank the staff of the Cerebral Microcirculation Section for weekly discussions of this work.
Sources of Funding
This research was supported by the Intramural Research Program of the NIH, NINDS.
Footnotes
Disclosures
None.
References
- 1.Di Marzo V. Targeting the endocannabinoid system: To enhance or reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 2.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease successes and failures. Febs Journal. 2013;280:1918–1943. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benyo Z, Ruisanchez E, Leszl-Ishiguro M, Sandor P, Pacher P. Endocannabinoids in cerebrovascular regulation. Am J Physiol-Heart C. 2016;310:H785–H801. doi: 10.1152/ajpheart.00571.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schabitz WR, Giuffrida A, Berger C, Aschoff A, Schwaninger M, Schwab S, et al. Release of fatty acid amides in a patient with hemispheric stroke - a microdialysis study. Stroke. 2002;33:2112–2114. doi: 10.1161/01.str.0000023491.63693.18. [DOI] [PubMed] [Google Scholar]
- 5.Degn M, Lambertsen KL, Petersen G, Meldgaard M, Artmann A, Clausen BH, et al. Changes in brain levels of n-acylethanolamines and 2-arachidonoylglycerol in focal cerebral ischemia in mice. J Neurochem. 2007;103:1907–1916. doi: 10.1111/j.1471-4159.2007.04892.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhang M, Martin BR, Adler MW, Razdan RK, Ganea D, Tuma RF. Modulation of the balance between cannabinoid cb(1) and cb(2) receptor activation during cerebral ischemic/reperfusion injury. Neuroscience. 2008;152:753–760. doi: 10.1016/j.neuroscience.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brose SA, Golovko SA, Golovko MY. Brain 2-arachidonoylglycerol levels are dramatically and rapidly increased under acute ischemia-injury which is prevented by microwave irradiation. Lipids. 2016;51:487–495. doi: 10.1007/s11745-016-4144-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caruso P, Naccarato M, Faoro V, Pracella D, Borando M, Dotti I, et al. Expression of the endocannabinoid receptor 1 in human stroke: An autoptic study. J Stroke Cerebrovasc. 2016;25:2196–2202. doi: 10.1016/j.jstrokecerebrovasdis.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Naccarato M, Pizzuti D, Petrosino S, Simonetto M, Ferigo L, Grandi FC, et al. Possible anandamide and palmitoylethanolamide involvement in human stroke. Lipids Health Dis. 2010;9 doi: 10.1186/1476-511X-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pellegrini-Giampietro DE, Mannaioni G, Bagetta G. Post-ischemic brain damage: The endocannabinoid system in the mechanisms of neuronal death. Febs Journal. 2009;276:2–12. doi: 10.1111/j.1742-4658.2008.06765.x. [DOI] [PubMed] [Google Scholar]
- 11.England TJ, Hind WH, Rasid NA, O’Sullivan SE. Cannabinoids in experimental stroke: A systematic review and meta-analysis. J Cerebr Blood F Met. 2015;35:348–358. doi: 10.1038/jcbfm.2014.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishibashi S, Maric D, Mou Y, Ohtani R, Ruetzler C, Hallenbeck JM. Mucosal tolerance to e-selectin promotes the survival of newly generated neuroblasts via regulatory t-cell induction after stroke in spontaneously hypertensive rats. J Cereb Blood Flow Metab. 2009;29:606–620. doi: 10.1038/jcbfm.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang BT, Leoni RF, Silva AC. Impaired cbf regulation and high cbf threshold contribute to the increased sensitivity of spontaneously hypertensive rats to cerebral ischemia. Neuroscience. 2014;269:223–231. doi: 10.1016/j.neuroscience.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sciolino NR, Zhou W, Hohmann AG. Enhancement of endocannabinoid signaling with jzl184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol Res. 2011;64:226–234. doi: 10.1016/j.phrs.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiley JL, Walentiny DM, Wright MJ, Jr, Beardsley PM, Burston JJ, Poklis JL, et al. Endocannabinoid contribution to delta9-tetrahydrocannabinol discrimination in rodents. Eur J Pharmacol. 2014;737:97–105. doi: 10.1016/j.ejphar.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez-Rodriguez AB, Siopi E, Finn DP, Marchand-Leroux C, Garcia-Segura LM, Jafarian-Tehrani M, et al. Cb1 and cb2 cannabinoid receptor antagonists prevent minocycline-induced neuroprotection following traumatic brain injury in mice. Cereb Cortex. 2015;25:35–45. doi: 10.1093/cercor/bht202. [DOI] [PubMed] [Google Scholar]
- 19.Kang BT, Leoni RF, Kim DE, Silva AC. Phenylephrine-induced hypertension during transient middle cerebral artery occlusion alleviates ischemic brain injury in spontaneously hypertensive rats. Brain Res. 2012;1477:83–91. doi: 10.1016/j.brainres.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P, et al. The adhesive removal test: A sensitive method to assess sensorimotor deficits in mice. Nat Protoc. 2009;4:1560–1564. doi: 10.1038/nprot.2009.125. [DOI] [PubMed] [Google Scholar]
- 21.Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med. 2010;2:13. doi: 10.1186/2040-7378-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandeputte C, Taymans JM, Casteels C, Coun F, Ni Y, Van Laere K, et al. Automated quantitative gait analysis in animal models of movement disorders. BMC Neurosci. 2010;11:92. doi: 10.1186/1471-2202-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stroke Therapy Academic Industry R. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- 24.Niphakis MJ, Cognetta AB, Chang JW, Buczynski MW, Parsons LH, Byrne F, et al. Evaluation of nhs carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. Acs Chem Neurosci. 2013;4:1322–1332. doi: 10.1021/cn400116z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tobin MK, Bonds JA, Minshall RD, Pelligrino DA, Testai FD, Lazarov O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J Cereb Blood Flow Metab. 2014;34:1573–1584. doi: 10.1038/jcbfm.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolinski P, Glabinski A. Chemokines and neurodegeneration in the early stage of experimental ischemic stroke. Mediators Inflamm. 2013;2013:727189. doi: 10.1155/2013/727189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long JZ, Li WW, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP, et al. Monoacylglycerol lipase is a therapeutic target for alzheimer’s disease. Cell Rep. 2012;2:1329–1339. doi: 10.1016/j.celrep.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM, et al. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of alzheimer’s disease. Cell Rep. 2012;1:617–623. doi: 10.1016/j.celrep.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pihlaja R, Takkinen J, Eskola O, Vasara J, Lopez-Picon FR, Haaparanta-Solin M, et al. Monoacylglycerol lipase inhibitor jzl184 reduces neuroinflammatory response in apde9 mice and in adult mouse glial cells. J Neuroinflammation. 2015;12:81. doi: 10.1186/s12974-015-0305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lysenko LV, Kim J, Henry C, Tyrtyshnaia A, Kohnz RA, Madamba F, et al. Monoacylglycerol lipase inhibitor jzl184 improves behavior and neural properties in ts65dn mice, a model of down syndrome. PLoS One. 2014;9:e114521. doi: 10.1371/journal.pone.0114521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernal-Chico A, Canedo M, Manterola A, Sanchez-Gomez MV, Perez-Samartin A, Rodriguez-Puertas R, et al. Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination in vivo. Glia. 2015;63:163–176. doi: 10.1002/glia.22742. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez-Suarez D, Celorrio M, Riezu-Boj JI, Ugarte A, Pacheco R, Gonzalez H, et al. Monoacylglycerol lipase inhibitor jzl184 is neuroprotective and alters glial cell phenotype in the chronic mptp mouse model. Neurobiol Aging. 2014;35:2603–2616. doi: 10.1016/j.neurobiolaging.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 34.Katz PS, Sulzer JK, Impastato RA, Teng SX, Rogers EK, Molina PE. Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J Neurotraum. 2015;32:297–306. doi: 10.1089/neu.2014.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 36.Landucci E, Scartabelli T, Gerace E, Moroni F, Pellegrini-Giampietro DE. Cb1 receptors and post-ischemic brain damage: Studies on the toxic and neuroprotective effects of cannabinoids in rat organotypic hippocampal slices. Neuropharmacology. 2011;60:674–682. doi: 10.1016/j.neuropharm.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 37.Melis M, Pillolla G, Bisogno T, Minassi A, Petrosino S, Perra S, et al. Protective activation of the endocannabinoid system during ischemia in dopamine neurons. Neurobiol Dis. 2006;24:15–27. doi: 10.1016/j.nbd.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Carloni S, Alonso-Alconada D, Girelli S, Duranti A, Tontini A, Piomelli D, et al. Pretreatment with the monoacylglycerol lipase inhibitor urb602 protects from the long-term consequences of neonatal hypoxic-ischemic brain injury in rats. Pediatr Res. 2012;72:400–406. doi: 10.1038/pr.2012.91. [DOI] [PubMed] [Google Scholar]
- 39.Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ. Lack of selectivity of urb602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br J Pharmacol. 2007;150:186–191. doi: 10.1038/sj.bjp.0706971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oleson EB, Beckert MV, Morra JT, Lansink CS, Cachope R, Abdullah RA, et al. Endocannabinoids shape accumbal encoding of cue-motivated behavior via cb1 receptor activation in the ventral tegmentum. Neuron. 2012;73:360–373. doi: 10.1016/j.neuron.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao Z, Mulvihill MM, Mukhopadhyay P, Xu H, Erdelyi K, Hao E, et al. Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology. 2013;144:808–817 e815. doi: 10.1053/j.gastro.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coyle P, Heistad DD. Blood flow through cerebral collateral vessels in hypertensive and normotensive rats. Hypertension. 1986;8:II67–71. doi: 10.1161/01.hyp.8.6_pt_2.ii67. [DOI] [PubMed] [Google Scholar]
- 43.Tuttle JL, Sanders BM, Burkhart HM, Fath SW, Kerr KA, Watson WC, et al. Impaired collateral artery development in spontaneously hypertensive rats. Microcirculation. 2002;9:343–351. doi: 10.1038/sj.mn.7800151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.