

Abstract
For more than 35 years, the NOD mouse has been the primary animal model for studying autoimmune diabetes. During this time, striking similarities to the human disease have been uncovered. In both species, unusual polymorphisms in a major histocompatibility complex (MHC) class II molecule confer the most disease risk, disease is caused by perturbations by the same genes or different genes in the same biological pathways and that diabetes onset is preceded by the presence of circulating autoreactive T cells and autoantibodies that recognize many of the same islet antigens. However, the relevance of the NOD model is frequently challenged due to past failures translating therapies from NOD mice to humans and because the appearance of insulitis in mice and some patients is different. Nevertheless, the NOD mouse remains a pillar of autoimmune diabetes research for its usefulness as a preclinical model and because it provides access to invasive procedures as well as tissues that are rarely procured from patients or controls. The current article is focused on approaches to improve the NOD mouse by addressing reasons why immune therapies have failed to translate from mice to humans. We also propose new strategies for mixing and editing the NOD genome to improve the model in ways that will better advance our understanding of human diabetes. As proof of concept, we report that diabetes is completely suppressed in a knock-in NOD strain with a serine to aspartic acid substitution at position 57 in the MHC class II Aβ. This supports that similar non-aspartic acid substitutions at residue 57 of variants of the human class II HLA-DQβ homolog confer diabetes risk.
Keywords: NOD mouse, type 1 diabetes, preclinical, congenic, genetics, gene editing
Introduction
Since becoming available to the scientific community, the NOD mouse has been used extensively and has provided significant contributions to our mechanistic understanding of autoimmunity and type 1 diabetes (T1D). Indeed, the NOD mouse has been used to understand many facets of human T1D and has been the preferred model for invasive, preclinical/translational studies. While the NOD mouse has a number of critics, this model should be viewed as an important component of a comprehensive approach to understanding T1D. The NOD remains a standout model because it develops spontaneous T1D with genetic and environmental components that are relevant to the human disease. Further, as recent studies have demonstrated and as we describe below, new protocols to specifically modify single base pairs can generate loci that contain risk alleles that are orthologous to the human on the NOD background. Therefore, the NOD mouse remains a powerful and valuable implement in the investigator’s toolbox.
A major strength of the NOD model is the existence of spontaneous autoimmunity and T1D. Similar to the human condition, NOD mice develop autoantibodies (1) and exhibit increases in circulating autoreactive T cells (2, 3) prior to the onset of T1D. The β cell antigens that are targeted are also similar between these species (4). However, in the NOD mouse, the initiating antigen appears to be insulin (1), whereas in human T1D it is thought to result from several initiating antigens (5, 6). These autoimmune phenotypes are followed by the onset of hyperglycemia (7). A progressive loss of β cell function is present in both human and NOD mice suggesting similarities in β cell loss or dysfunction. While hyperglycemia in NOD females and males begins close to 12 and 15 weeks of age, respectively (8), immune infiltration into the pancreatic islets, insulitis, begins much earlier. Pathogenic T cells have been isolated from the islets of 5-week-old NOD mice (9). By 12 weeks of age, insulitis is present throughout the pancreas of NOD mice. A dissimilarity of the diabetes when comparing human and NOD mice is the appearance of insulitis. Studies from the nPOD bio-repository have been critical in defining insulitis in humans where this pathogenic lesion is less severe and less frequent than what can be observed in NOD mice (10). This may result from the fact that the autoimmunity in parental NOD mice is very aggressive and disease onset occurs over an abbreviated timeline (weeks) compared to the decidedly more attenuated onset in humans (i.e., years after the appearance of autoantibodies). Insulitis and T1D incidence in NOD mice can be reduced through genetic modification. While hundreds of variant NOD mice have been made that represent less intense forms of T1D, the idea of improving the NOD as a model for human T1D by decreasing the potency of the autoimmune response remains largely unexplored. The potential of this strategy is discussed below.
Genetics play a significant role in autoimmunity and important similarities exist when equating T1D-risk loci in human and NOD mice. The disease is polygenic in both species with over 50 loci linked to risk in human and NOD diabetes (11). However, a single locus is responsible for the majority of the risk: major histocompatibility complex (MHC) class II. Early papers were critical in establishing that NOD mice encode a T1D-risk MHC haplotype that has important resemblances to the HLA risk alleles in human. Since these publications, genetic and biochemical studies have linked risk to amino acid residue 57. The high-risk DQ2 and DQ8 alleles of human as well as the Ag7 molecule of the NOD have small polar amino acids substituted for an aspartic acid at position 57. The importance of this amino acid substitution is discussed in detail by Bettini and Bettini in this issue of Frontiers in Endocrinology (Co-published in the same edition of FiE). The genetic variations that impart risk in HLA/MHC arise from single-nucleotide polymorphisms that change the amino acid sequence. Similarly, other genes such as Ctla4 and mt-Nd2 are linked to risk in both humans and NOD mice. A single leucine to methionine substitution in mt-Nd2 as well as the human homolog, mt-ND2, provides β cells with enhanced resistance to autoimmune destruction (12, 13). While HLA/MHC and mt-ND2/mt-Nd2 represent genes with protein and biochemical differences, these non-synonymous changes in T1D are more the exception than the rule. Only seven of the >50 single-nucleotide polymorphisms associated with T1D arise in coding regions (14). The polymorphism in Ctla4 of NOD mice results in altered splicing. While the polymorphism in CTLA4 is not in an identical location, the risk variant is similarly associated with altered splicing of CTLA4 (15–17). Therefore, genes such as CTLA4 can be modeled in the NOD mouse to aid in understanding the role of non-coding genetic variation in pathogenesis of T1D. Recent advances in genetic editing have further promoted the use of NOD to understand how specific SNPs can affect protein function. Editing of the NOD genome has been used to swap T1D risk or resistance alleles allowing for the role of specific SNPs, such in MHC Class II (described below) or Ptpn22 (18), in the regulation of autoimmunity to be identified.
Another concept that holds true across species is that T1D onset results from the sum of the genetic parts. In human subjects, T1D risk increases as the haplotype of an individual contains more credible T1D susceptibility SNPs (19, 20). Similarly, by subtracting risk loci from the NOD genome through backcrossing or genetic modification, T1D risk can be altered (11). As discussed in detail below, the NOD represents a powerful tool to study epistasis.
In the current review, we highlight past contributions NOD mice have made to T1D research and outline strategies to better utilize this model in future. Included is an overview of NOD mouse’s track record as a preclinical model for developing T1D therapies and a discussion about the impact NOD congenic mice have made to understanding the genetic basis of T1D. Also discussed is a strategy to develop panels of NOD congenic mice from existing congenic stocks to better mimic the spectrum of human autoimmune diabetes subtypes. Finally, we summarize existing and emerging technologies for editing the NOD genome that should greatly enhance the NOD mouse as a research tool, especially for identifying genes that contribute to T1D development.
Preclinical Performance of the NOD Mouse
While the NOD mouse has proved useful in many preclinical research areas, significant tension has arisen over the performance of this mouse strain in bench-to-bedside efforts due to a failure to translate therapies developed in the NOD model to humans. The NOD model has been used for at least 30 years to identify agents or protocols that delay, prevent, or reverse disease. In general, investigators apply three approaches: early prevention (treatment is initiated at 3–4 weeks of age), late prevention (begin treatment at 10–12 weeks of age), or intervention after onset of T1D (reversal). Most preclinical successes in NOD have come in early prevention, where a wide array of agents or protocols can block disease. It should be noted that in most cases the impact of the drug/agent under investigation on autoimmunity (i.e., insulin autoantibodies or the presence of β cell reactive T cells) was not assessed. Further, many of these have seen little to no confirmation by independent laboratories. A recent NIH funded effort to confirm the effects of specific agents was unsuccessful at repeating the majority of the successes that were previously published (21). Late prevention represents a modality that is similar to trials established in humans where autoantibody positive individuals are identified and enrolled, such as the Diabetes Prevention Trial 1 or the recent oral insulin trial (22–24). To date, there has been a failure to translate late prevention successes in the NOD to prevention of human T1D.
At time of writing, very few therapies have resulted in T1D reversal in new-onset NOD mice and fewer still in NOD mice with established disease. Of the agents that have shown benefit, anti-CD3, and the combination of antithymocyte globulin (ATG) and granulocyte-colony stimulating factor (G-CSF) have been used in clinical trials. Preclinical studies using these modalities demonstrated an ability to reverse T1D in 39% of NOD females after onset (25). Multicenter preclinical efforts using anti-CD3 produced similar results, with less than 50% of the treated NOD mice exhibiting long-term T1D reversal (26). The rates of T1D reversal were significantly different when comparing sites, where anti-CD3 efficacy ranged from 10 to 80% among the four locations. Similarly, trials with anti-CD3 resulted in a minority of patients responding to therapy (i.e., preservation of c-peptide responses), yet none of the efforts with anti-CD3 resulted in insulin-free status for the patients (27–29). Likewise, use of ATG + G-CSF in a small multicenter clinical trial (25 total patients: 17 receiving ATG + G-CSF and 8 placebo) established that this combination did not induce T1D-remission but was effective in preventing erosion of β cell function 12 months after treatment (30). The 2-year data for ATG + G-CSF were less promising. At 24 months only 50% of the individuals who received therapy had preservation of β cell function (31). This is similar to the ATG + G-CSF reversal rates in NOD mice (32).
These data provide caution for moving agents forward for clinical trials that have been developed using NOD mice. A recent paper in Science Translational Medicine (33) has called for standards in clinical diagnosis as well as timing of therapy initiation in NOD mice. In preclinical studies, it is well established that NOD mice should be treated immediately after onset of T1D for maximal therapeutic response. Most groups have now established protocols for checking mice every other day for T1D onset allowing for initiation of therapy as soon as 1 day after diagnosis (21, 27, 28). In humans, trials enroll participants much more slowly and this delay in therapeutic administration likely postpones protection of the β cell mass allowing for further β cell loss. Additionally, it is clear that prior to agents or protocols moving to clinical trial there must be independent replication. The lack of a systematic understanding of T1D in the NOD and humans also impacts success. Comprehensive studies in comparative immunology and endocrinology are needed to mechanistically detail T1D reversal in NOD mice.
Role of NOD Congenic Mice in T1D Genetics
Since researchers first started mapping insulin-dependent diabetes (Idd) loci by outcrossing NOD mice to mouse strains that do not develop T1D [i.e., C57BL/6 (B6), C57BL/10 (B10), NOR, and C3H], considerable effort has been spent creating recombinant congenic mouse strains to delineate genetic intervals containing diabetes loci and identifying the genes within each interval that are responsible for T1D susceptibility or protection. Several regions have been refined through the generation of subcongenic stocks that encode different subregions of the original confidence interval. These strains have revealed how several of the original Idd regions are composed of multiple susceptibility and/or resistance alleles. Notable examples include Idd3 that was dissected into Idd3, Idd10, Idd17, and Idd18 (34–37), Idd5 that was dissected into Idd5.1, Idd5.2, Idd5.3, and Idd5.4 (38–41), and Idd9 that was dissected into Idd9.1, Idd9.2, Idd9.3, Idd9.4, and Idd9.5 (42–46). While many of the dominant Idd regions are now well delineated, relatively few of their underlying genes have been firmly established. This is because validation has been technically challenging, in large part because even small Idd intervals often contain large numbers of candidate genes. Slow progress in improving candidate gene identification has led to reduced support for large-scale mouse genetic studies, forcing many in the field to decommission their congenic stocks. A new generation of genetic tools discussed in Section “Strategies for Improving Candidate Gene Identification” may reverse the fortunes of some of these strains. Nevertheless, even without discovering the causative genes, congenic mice have provided valuable insight about the genetic causes of human T1D that no other resource could have delivered. Some of their most important contributions are described below.
Epistasis and Gene–Gene Interactions
Intercrossing congenic stocks has revealed that an individual’s disease risk is ultimately determined by the interactive effect of multiple Idd resistance and susceptibility loci. The challenge of disentangling these complex networks was taken up by a few courageous groups who, over decades, have detailed how different combinations of disease resistance and susceptibility loci modulate diabetes and various disease sub-phenotypes on the autoimmune-permissive NOD background. The advantage of this approach is that eliminating genetic variability between Idd loci allows for the detection of gene-masking and gene–gene interaction effects that are normally concealed in conventional genetic association studies with human subjects as well as mouse studies involving F2 and backcross one generation for segregation analysis (47).
There are several examples of how interactions between individual Idd susceptibility and resistance alleles gives rise to graded levels of diabetes on the NOD background (48–51). Among the best characterized is the interplay between the Idd3 and Idd5 congenic intervals from C57 strains when introgressed into the NOD genome. Combining Idd3 and Idd5 confers almost complete protection from T1D and insulitis on the NOD background (38). Yet, combining Idd3 with individual Idd5 subloci results in a spectrum of diabetes protective effects [reviewed elsewhere (11, 47, 52)]. At one end of the spectrum, Idd3/Idd5.1 NOD mice were found not more protected against T1D than Idd3 mice (41). Hunter et al. posited that the lack of protection in NOD-Idd3/Idd5.1 mice may result from T1D resistance alleles at Idd3 increasing the expression of CTLA-4 on the surface of CD4+ and CD8+ T cells that may render higher levels of inhibitory ligand-independent CTLA-4 induced by protective alleles at Idd5.1/Ctla-4 somewhat redundant (41, 53). On the other end of the spectrum, Idd3/Idd5.1/Idd5.3 and Idd3/Idd5.3 recombinant congenic strains were found to exhibit T1D resistance equal to NOD-Idd3/Idd5 mice. The lack of T1D initiation in the presence of severe insulitis observed in the Idd3/Idd5.1/Idd5.3 and Idd3/Idd5.3 strains indicates that the interaction between Idd5.2/Nramp1 and Idd3 is not important for T1D protection, but does contribute to the marked reduction in insulitis (41, 54). Continued studies of these strains will provide models to address the knowledge gap in additive and synergistic genetic effects.
Gene–gene interactions also exist among the various Idd5 subregions, including between Idd5.1 and Idd5.4. Idd5.4 encodes a B10-derived susceptibility allele without a known responsible gene product. Idd5.4 significantly accelerates T1D in the presence of Idd5.2 and Idd5.3, but has no impact on disease if Idd5.1 is also present (41). This suggests that Idd5.4 can neutralize the protective effects of Idd5.2 and Idd5.3 and that Idd5.4 is in turn masked by the protective effects of Idd5.1. A plausible explanation for this phenomenon is that immune events regulated by the B10-derived susceptibility allele at Idd5.4 are counteracted by Idd5.1/CTLA-4 signaling in one or more cell types. Similar masking effects have been detected among other congenic regions including between Idd19 and Idd6 on Chr.6 (49), Idd19 and Idd20 on Chr.6 (51), Idd21.2 and Idd21.1 on Chr.18 (50), and Idd14 and Idd31 on Chr.13 (55).
Evidence for epistatic interactions in humans include a study by Winkler et al. that genotyped 12 non-HLA susceptibility genes (ERBB3, PTPN2, IFIH1, PTPN22, CLEC16A, CD25, CTLA4, SH2B3, IL2, IL18RAP, IL10, and COBL) in high-risk HLA positive children of parents with T1D that were prospectively followed from birth to the development of autoantibodies and disease (19). An analysis was performed to determine the combinations of genes that most accurately predicted T1D development. The results showed that T1D progression in high-risk HLA carriers was best predicted by a collection of 8 genes (ERBB3, IFIH1, PTPN22, CLEC16A, CTLA4, SH2B3, IL18RAP, and COBL) rather than all 12 SNPs. These results suggest the presence of gene–gene interactions that mask the effect of individual diabetes susceptibility alleles. Another study searched for interactions between 38 T1D-associated non-HLA loci and different HLA class II genotypes in a large collection of T1D samples (20). It was found that SNPs within two T1D-associated genes, PTPN22 and CTLA4, alter the predicted diabetes risk of various HLA haplotypes, partly confirming earlier reports that the effect of a susceptibility allele at PTPN22 is greater in individuals expressing low-risk than high-risk HLA class II genotypes (56–58). These and other GWAS studies show how some T1D genes but not others are strongly influenced by gene-gene interactions and masking effects.
Cellular Expression of Diabetes-Associated Genes
Congenic mice offer a powerful tool to determine how different T1D genes modulate diabetogenic immune responses within specific cell types, which cannot easily be accomplished by experimentation with human samples. Previous studies have used a variety of adoptive transfer or bone marrow chimerism methods to observe that T1D genetics regulate immune dysfunction. A good illustration is the use of the B6, B10, or NOR derived Idd9/Idd11 resistance locus to inhibit diabetes. One set of studies found that complex genetic interactions within Idd9/11 regulate how B cells contribute to disease by engrafting syngeneic bone marrow and B cells purified from different Chr. 4 subcongenic donors into lethally irradiated B cell-deficient and diabetes-resistant NOD.IgHnull mice (59, 60). Diabetes development was then monitored to determine if B cells expressing separate subcongenic intervals from the NOR strain protected recipient mice from T1D compared to standard NOD B cells. The results established that at least four adjacent intervals interactively contribute to how diabetogenic B cells become tolerized or cause T1D, including processes that increase the efficiency of B cell anergy or B cell hyperresponsiveness to B cell receptor stimulation.
We used a similar strategy to show that genes within the Idd9/11 locus control pathogenic CD4 T cells responses in T1D (61). Lethally irradiated CD4-deficient NOD.CD4null mice were reconstituted with syngeneic bone marrow and CD4+ T cells isolated from NOD.NOR-(D4Mit31-D4Mit310)/DvsJ: (NOD-Idd9/11NOR) NOD mice congenic for NOR genome on Chr. 4. In this system, transfer of CD4+ T cells isolated from NOD-Idd9/11NOR mice caused less diabetes than CD4+ T cells isolated from NOD. It was also shown that CD4+ T cells from BDC2.5 TCR transgenic mice have a reduced capacity to transfer T1D to immunodeficient NOD.CB17-Prkdcscid (NOD-Scid) mice when they express protective alleles at Idd9 (62). Hamilton-Williams et al. found that CD4+ T cells that express protective B10 alleles at Idd9.2 and Idd9.3 suppress the expansion of diabetogenic CD8+ T cells (63). Their approach involved reconstituting NOD-Scid mice with purified CD4+ T cells from NOD or NOD.Idd9 congenic mice co-transferred with CD4-depleted spleen and lymph node cells from NOD donors. After reconstitution, mice were infected with a vaccinia virus encoding the H-2Kd-restricted IGRP206–214 epitope to measure the expansion of CD8 T cells specific for the islet antigen IGRP. High and low frequencies of IGRP-specific CD8 T cells were detected in mice, respectively, reconstituted with NOD and NOD.Idd9 CD4 T cells indicating that Idd9 protective alleles restore tolerance to islet IGRP through CD4 T cells.
Other cell types besides B cells and conventional CD4+ T cells have been found to regulate diabetes through Idd9. Regulatory T cells (Tregs) expressing B10-derived Idd9.1 genes have significantly higher suppressive activity than Tregs from standard NOD mice (64). The Idd9.1 sub-locus has also been reported to increase the capacity for DCs to engage and potentiate natural killer T cells, which are required for Idd9-mediated diabetes protection (65). Reciprocal transfers of NOD and NOD.Idd9 congenic mouse spleen and lymph node cells into NOD-Scid and NOD.Idd9-Scid recipients identified that non-lymphoid cells possess some component of Idd9 protection (63). Another finding was that transplanted islets from NOD-Idd9 mice are more resistant to destruction by CD8+ T cells, suggesting that an element of Idd9-mediated T1D protection maps to insulin-producing β cells (66).
Studies dissecting the effects of Idd9 and other T1D loci have demonstrated that diabetogenic immune responses develop from a complex interplay of genes in multiple cell types. Further, evidence suggests that different cell types can be affected by a single diabetes locus/gene with sometimes opposing effects on disease. Determining how individual Idd loci contribute to T1D by affecting immunoregulatory pathways in specific cells offers a useful strategy for identifying the genes underlying these regions.
Genetic Control of Insulitis
Congenic mice have revealed that non-MHC Idd loci can be separated into two classes; one that supports T1D by modulating the virulence of insulitis and/or the intrinsic resistance of β cells to cytotoxic stress, and a second class that supports T1D by regulating diabetogenic immune responses before insulitis occurs (67). In the first class, replacement of individual NOD susceptibility loci with resistance alleles from non-diabetes prone strains reduces the incidence of T1D but has no quantifiable effect on insulitis at the gross histological level compared to NOD mice of the same age. Idd loci that fall into this category include Idd9 where introgression of B10-derived resistance alleles did not alter the cellular composition of insulitis. Instead this locus changed the pathogenic properties of leukocytes that accumulated in islets and shifted cytokine production from IFNγ and TNFα to an IL-4 response (43). The overlapped B6-derived Idd11 interval also reduces the pathogenic effects of β cell-specific lymphocytes in islet infiltrates without affecting the overall amount of insulitis (44). Another example is Idd6 where C3H-derived resistance alleles confer protection against T1D but not islet infiltration. However, subtle differences exist in the invading leukocyte populations including that CD4+ T cells and B cells are slightly reduced, which is counterbalanced by an increase in non-lymphoid cells such as macrophages and dendritic cells (68).
Disease protection is highly variable among the second class of non-MHC Idd loci where resistance alleles protect against both T1D and insulitis. Some regions including Idd10/18, Idd16, and Idd21 cause a mild reduction in pancreatic infiltration but only during the early phases of insulitis (34, 50, 69, 70). Most of these loci confer relatively modest protection against T1D. In contrast, loci such as Idd3 and Idd5 that each provide substantial diabetes protection also cause a considerable delay in insulitis, although almost all NOD.Idd3 and NOD.Idd5 congenic mice eventually develop significant islet infiltration (34, 38). Other Idd loci, including Idd4 and Idd13, appear to change the distribution rather than the amount of insulitis (71, 72). NOD mice expressing either of these loci develop non-destructive peri-ductal infiltrates where invading cells remain mostly confined to the peri-islet zone until well after the age most NOD mice develop diabetes. As discussed above, none of the non-MHC Idd loci that block insulitis and T1D are sufficient on their own to substantially reduce islet inflammation. However, almost complete protection can be achieved when individual regions are combined, indicating that genetic interactions exist between specific loci that confer greater protection against islet inflammation than the collective effects of each separate region.
Together, these findings suggest that insulitis among patients is also under complex genetic control and that, in some people, combinations of T1D genes could cause high levels of non-destructive islet inflammation long before the onset of overt disease. In contrast, the degree of insulitis may correlate closely with progression to overt diabetes in patients that carry T1D genes that give rise to more virulent forms of insulitis.
Modeling the Genetic Diversity of Human T1D
A major criticism of the NOD mouse has been that this model represents the equivalent of a single human case of T1D. Consequently, immune modulation protocols developed in the NOD mouse could be limited to a few subtypes of the human disease, which may partially explain why some interventions that have shown promise in NOD mice fail to preserve β cell function in patients (73). Better predictions from mouse models might be possible if future treatment protocols were screened using multicenter efforts with heterogeneous populations of NOD-derived mice to mimic the genetic variation among patients. Such a strategy could employ a panel of NOD-related recombinant congenic strains carrying different combinations Idd loci where each strain would express a unique set of genetic variants that give rise to a specific subtype of T1D (41). This is analogous to the different subtypes of T1D that arise in patients from various segregating combinations of susceptibility and resistance alleles. The potential of this strategy is that therapies capable of inhibiting diabetes across a panel of congenic strains are more likely to be successful in genetically heterogeneous humans. There are also advantages to finding treatments that only work in congenic mice with specific combinations of Idd loci, including that this could provide valuable information about the cellular and molecular mechanisms through which an immune modulation treatment affects disease. It may also help to identify specific subsets of patients that have less or more potential for responding to a particular immune therapy.
Choosing which congenic mice to include in a future drug testing panel presents a challenge because of the large number of Idd loci it is possible to combine. It is logical that strain selection should consider the nature of the immune modulation protocol being tested. For therapies like probiotic treatment and immune suppression protocols, where the mode of action is poorly understood or where multiple cell types and molecular pathways are involved, it may be best to test mice with a diverse array of congenic intervals designed to emulate the genetic variation in human T1D. Some of the NOD-related congenic stocks described in the Section “Epistasis and Gene–Gene Interaction” may be suitable candidates, especially those that develop NOD-like levels of T1D due to introgression of susceptibility loci from non-NOD mouse strains. A more targeted panel could be employed for therapies known to act through particular cellular or molecular pathways. For instance, immune modulation protocols designed to enhance Tregs, such as low-dose IL-2 and combined ATG + G-CSF therapy, could be tested on congenic mice expressing different allelic variants of Idd3, Idd6, Idd9.1, and Idd9.3 that each separately affect the suppressive properties of Tregs (64, 68, 74, 75). Another example is antigen-specific immunotherapy where autoantigens could be screened in NOD congenic mice expressing different variants of Il2/Il21 (Idd3) (74, 76), B2m (Idd13) (72), and Ptpn22 (Idd18.2) (77) that, respectively, modulate T cell activation/effector function, peptide presentation, and TCR signaling. All of these factors contribute to the fate of self-reactive T cells that encounter autoantigen and may affect the outcome of autoantigen immunotherapy.
An obvious drawback to testing diabetes therapies using congenic mouse panels is the additional time and resources involved. Even so, the investment is worthwhile if therapies that are ineffectual in humans could be recognized before progressing to clinical trials. An example of how testing the appropriate NOD congenic strain might have produced a different result to standard NOD mice and predicted the failure of a T1D treatment is low-dose IL-2 therapy, which increases the frequency of Tregs but has not been able to produce positive effects on diabetes in patients (78). A chief reason that this treatment advanced to clinical trials is that low levels of IL-2 potently suppresses T1D development and reverses recent onset T1D in NOD mice, presumably by enhancing Treg function and/or development (79). However, it is possible that NOD mice are particularly sensitive to this type of immune modulation because this strain carries a variant of Il2 that reduces IL-2 gene expression and Treg function (74). An interesting question is whether the outcome of IL-2 treatment would be different in NOD.Idd3 mice that express the B6 variant of Il2 and results in higher levels of Il2 gene expression (74). The answer might address whether low-dose IL-2 therapy has potential for improving immune regulation and result in enhanced β cell function in patients without an IL2/IL2R signaling deficiency. This is important because it is still unclear whether defects in the IL2/IL2R pathway play a significant role in most cases of human diabetes; although a gene variant of IL2RA (CD25) has been associated with T1D risk in people, it is protective but rare (80). Furthermore, the causative gene has yet to be identified for the chromosome 4q27 region containing IL2 and IL21 that is linked with T1D susceptibility (81).
Another limitation of testing T1D therapies with NOD congenic mice is that many Idd loci strongly suppress diabetes, which will require that some experiments be performed with large numbers of animals to achieve sufficient power. Indeed, only 10–20% of female NOD.Idd3 mice develop T1D by 30 weeks of age (74, 76, 82). As mentioned above, the unique insights from congenic mice will often justify using strains with very low levels of disease. However, there is also potential to alter the genetic composition of congenic strains in ways that will increase the rate of diabetes. For instance, it may be feasible to use NOD mice heterozygous instead of homozygous for the Idd3 locus, which develop 40% diabetes (76). These mice still produce more IL-2 than standard NOD mice and would presumably be less sensitive to IL-2 therapy. Another strategy could be to breed T1D-resistant congenic strains with NOD mice carrying congenic intervals that accelerate diabetes. For example, NOD.Idd3 could be crossed to NOD mice carrying B6 alleles at Idd18.2/Ptpn22 that are more diabetogenic than the corresponding NOD alleles (77).
Strategies for Improving Candidate Gene Identification
Although genetic studies using inbred mice are costly because of the large number of mice required, they remain a powerful method of detecting rare T1D susceptibility alleles that are impractical to identify through GWAS analyses, which require tens or hundreds of thousands of human subjects (83, 84). Thus, for the reasons outlined above, the question is not whether pursuing the identity of T1D susceptibility and resistance alleles is worthwhile, but rather how to make this process more efficient by employing a comprehensive approach that utilized both human and mouse systems. Considerable encouragement comes from a new generation of genetic tools that may circumvent many of the most intractable obstacles that traditionally limited the identification of Idd candidate genes. Some of these are described in the following sections in the order of their development.
RNA Interference (RNAi)
RNA interference has proven useful for manipulating gene expression in NOD mice without introducing genetic contamination from other strains. This approach is based on a well-established transgenesis methodology that entails the direct introduction of short hairpin RNA (shRNA) containing constructs into NOD zygotes by viral transduction (85, 86). The shRNA-containing constructs are designed to silence genes that impact T1D. shRNA is a sequence of RNA that contains a tight hairpin turn. This structure is cleaved by intracellular machinery into small interfering RNA that knocks down any mRNA bearing a complementary sequence (87). Several companies are developing viral libraries that produce shRNA that integrate into the host genome and ensure stable gene silencing after integration. The silencing cassette can be incorporated into many different types of vectors, including lentiviral, adenoviral, or retroviral vectors. Using the NOD model, RNAi has already provided valuable insight into how expression of the T1D candidate genes IL17 (88), PTPN22 (89), CTLA4 (90), CLEC16A (91), RGS1 (92), and Slc11a1 (Nramp1) (93) contribute to diabetes development. It is conceivable that T1D susceptibility genes can regulate disease progression in an age-dependent manner. Establishment of inducible RNAi has also enabled temporal control of target gene knockdown to determine their functions at different stages of disease progression (94).
Zinc Finger Nuclease (ZFN)
Zinc finger nucleases are fusion proteins containing a sequence-specific DNA-binding zinc finger domain and a nuclease domain (95, 96). Engineered ZFNs specifically recognize and bind a defined target gene sequence within the nucleus of a cell and introduce a double-strand break (DSB) (97, 98). The cellular DNA repair machinery fixes these breaks, most frequently via the non-homologous end joining (NHEJ) mechanism resulting in small deletions or insertions of the gene sequence (few to hundreds of base pairs) and disruption (knockout) of the target gene (97, 98). Injected as synthetic mRNAs, ZFNs typically work at the one-cell fertilized embryo stage, resulting in single-step, whole animal gene disruption, and infrequent mosaics (99). More precise genetic engineering can be achieved as well because a DSB also stimulates DNA repair via homology-directed repair (HDR) mechanism if a homologous DNA template is co-introduced into the cell (100). Because ZFN-mediated genetic manipulation can be done directly in NOD embryos, the resulting knock-in or knockout can be generated on a pure NOD genetic background. Thus, it eliminates carryover of closely linked passenger DNA that occurs when the induced mutation is introduced in non-NOD embryonic stem cells (129 or B6) and then the targeted allele is backcrossed onto NOD. This is particularly concerning when targeting genes within known Idd regions. To study the role of the Idd9.3 candidate gene Tnfrsf9 (encoding CD137/4-1BB), we used ZFN to disrupt this gene directly in NOD embryos (101). The NOD allele of CD137 is hypofunctional when compared to the B10 protein that is expressed within the Idd9.3 congenic strain (102). Thus, it was thought that T1D development would be accelerated in the absence of CD137. Surprisingly, CD137-deficient NOD mice were less susceptible to T1D, indicating that this co-stimulatory molecule has a diabetogenic role. This conclusion could not have been made with certainty if CD137-deficient NOD mice were created by backcrossing the previously reported knockout alleles generated using 129 embryonic stem cells. We further established an important role of CD137 in promoting the accumulation of β cell autoreactive CD8+ T cells (103). In addition, CD137 had a diabetes protective function when expressed in CD4+ T cells, likely due to the immunosuppressive activity of soluble CD137 produced by Tregs (103).
As discussed earlier, the H2g7 haplotype is essential for T1D development in NOD mice. A key component of the diabetogenic H2g7 haplotype is the unique Abg7 allele. The Abg7 allele includes five nucleotide polymorphisms resulting in the conversion of two usually conserved proline and aspartic acid residues at positions 56 and 57 to histidine and serine (104). Significantly, the non-aspartic acid substitutions at residue 57 also characterize diabetogenic variants of the human class II HLA-DQβ homolog, such as DQ8 (105). While transgenic analyses have shown both histidine and serine, respectively, at positions 56 and 57 amino acid residues of Aβg7 to be important for T1D progression in NOD mice (106–108), their diabetogenic function has not been tested under a more physiological condition. To further study the role of amino acid residue at position 57 in Aβg7, we created a knock-in NOD strain by replacing the serine with an aspartic acid (Aβg7-S57D). This was achieved by co-injecting Abg7-specific ZFN coding mRNA and a plasmid construct for HDR into one-cell fertilized NOD embryos, which were subsequently transferred into pseudopregnant mothers, and live-born pups were screened for founders. We successfully established a knock-in NOD stock (formal name: NOD/ShiLtJ-H2-Ab1em2Ygch/Ygch) with the precise 3 base pair alteration resulting an aspartic acid at position 57 in the MHC class II Aβ chain. The knock-in allele was confirmed at both the genomic DNA and cDNA levels. We used two different antibody clones (AMS-32.1 and 10-3.6) to determine if MHC class II expression was altered in NOD.Abg7-S57D mice. The expression level of MHC class II on B cells and dendritic cells was found to be comparable in wild-type NOD and NOD.Abg7-S57D mice when 10-3.6 was used to stain their splenocytes (Figure 1 and not shown). Interestingly, the level of MHC class II staining was found to be lower on B cells and dendritic cells from NOD.Abg7-S57D than those from wild-type NOD mice when AMS-32.1 was used (Figure 1 and not shown). These results indicate that the aspartic acid substitution at position 57 in the Aβ chain alters the binding of AMS-32.1, presumably due to a conformational change of the antibody-binding epitope. Strikingly, diabetes development was completely suppressed in homozygous NOD.Abg7-S57D mice (Figure 2), confirming the importance of the aspartic acid residue at position 57 of the Aβ chain in T1D. The availability of this novel strain will allow studies aimed to understand how diabetogenic MHC class II molecules select and activate β-cell autoreactive CD4 T cells.
Figure 1.
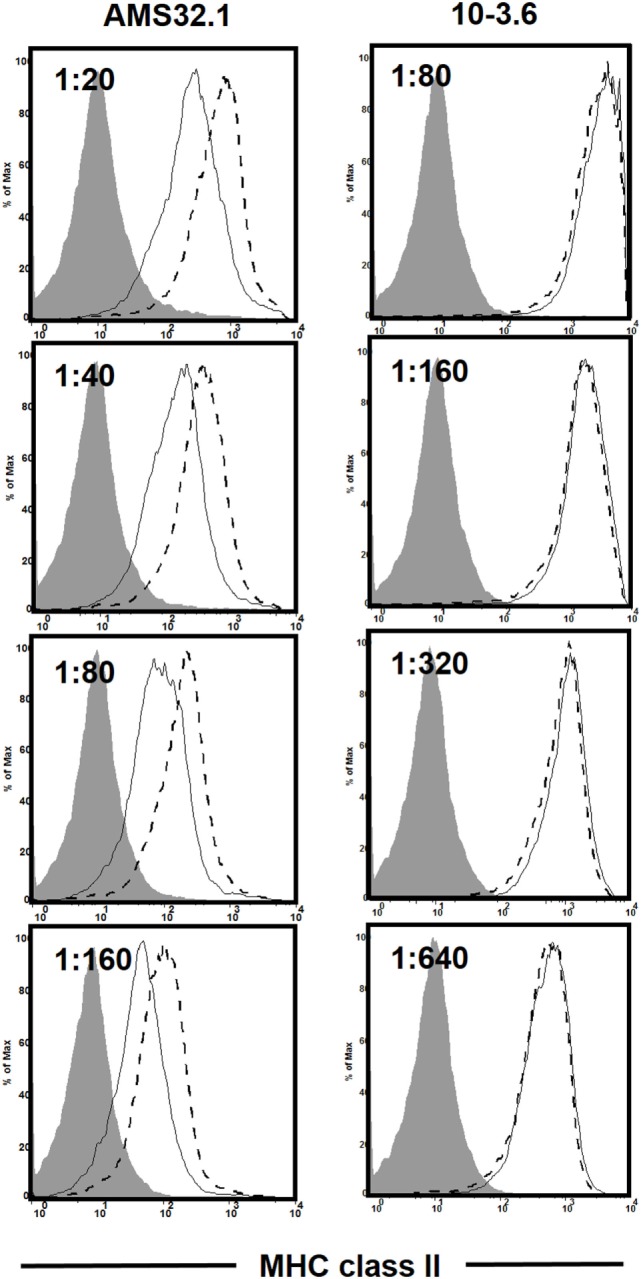
Differential staining of the major histocompatibility complex (MHC) class II molecule in wild-type NOD and NOD.Abg7-S57D mice. Total splenocytes were stained with anti-CD11c, anti-CD19, and the indicated I-Ab antibody clone at different titers. Shown is the I-Ab staining on B cells (CD19+ CD11c−) of NOD (dashed line) and NOD.Abg7-S57D (solid line) mice. The shaded area is the negative control staining using splenocytes isolated from I-Ab deficient NOD mice (NOD/ShiLtJ-H2-Ab1em1Ygch/J, JAX stock no. 027057). Similar results were obtained in two independent experiments.
Figure 2.
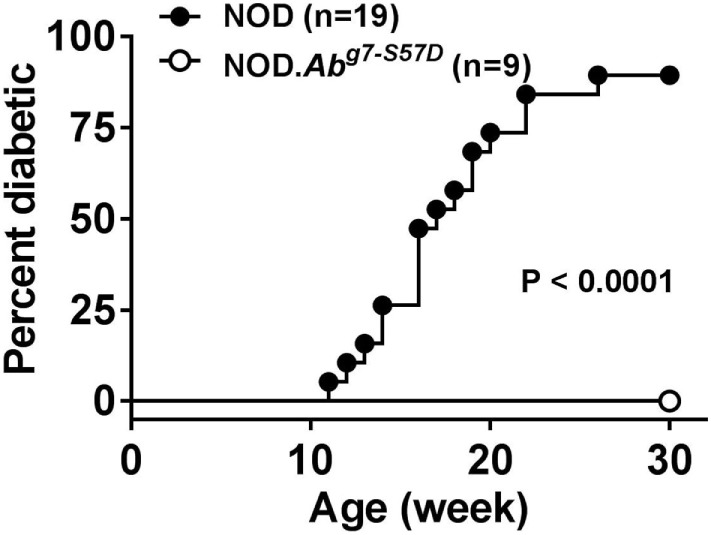
NOD.Abg7-S57D mice are completely resistant to type 1 diabetes. NOD and NOD.Abg7-S57D mice were monitored for diabetes development weekly for 30 weeks by testing urine glucose. Diabetes onset was defined by two consecutive readings of >250 mg/dl.
Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) and CRISPR-Associated Protein 9 (Cas9)
Due to its high efficiency, the CRISPR/Cas9 system has become the top choice when considering gene targeting in a variety of animal models. Similar to ZFN mediated mutagenesis, CRISPR/Cas9 also introduces a DSB, followed by repair through NHEJ and HDR dependent mechanisms (109). Cas9 nuclease is recruited to a specific DNA sequence by a single-guide RNA that can be easily designed using publically available online tools (109). Several groups, including ours, have successfully used the CRISPR/Cas9 approach to disrupt genes directly on a pure NOD genetic background (18, 110–112). The importance of affinity maturation processes of B cells (class switch recombination and somatic mutation) for T1D development in NOD mice was demonstrated by ablation of the activation-induced cytidine deaminase gene (Aicda) (111). It was recently shown that IL-2 can indirectly enhance FOXP3 expression through downregulating the level of Flicr, a long non-coding RNA (112). The function of Tregs is impaired in NOD mice partly due to reduced IL-2 production by activated T cells in this strain (74). Deletion of Flicr decreased accumulation of FOXP3low Tregs in pancreatic islets and suppressed T1D in NOD mice, likely by enhancing the stability and function of Tregs (112). Ptpn22 has also been targeted in NOD mice using the CRISPR/Cas9 system (18). A nonsynonymous single nucleotide polymorphism resulting in an amino acid substitution (R620W) in human PTPN22 has been linked to numerous autoimmune diseases, including T1D (113). Ptpn22 has been identified as a top candidate gene for the Idd18.2 region in NOD mice. To further study the role of PTPN22 in T1D, the Sherman laboratory generated a Ptpn22 knockout NOD mice as well as a knockin strain that has the R619W amino substitution to mimic the human variant (18). Ptpn22 knockout NOD females developed more rapid onset of T1D (18). Similarly, NOD females expressing the Ptpn22 KI allele (encoding 619W) also developed accelerated T1D (18), providing direct evidence to support the diabetogenic function of this variant.
Testing Human T1D Candidate Genes in NOD Mice
As noted above, the NOD mouse has been criticized for its usefulness as an animal model for human T1D, largely due to disappointing outcomes of clinical trials based on agents that showed therapeutic and/or preventive effects for mouse diabetes. The increased availability of human samples allowing direct examination of pancreata and lymphoid tissues isolated from organ donors at different stages of T1D progression has further decreased the enthusiasm of the NOD model (114). However, it remains a challenge to identify and to mechanistically study T1D susceptibility genes in human. The effect of a single gene on a phenotype is more difficult to detect due to heterogeneity in humans. Many genetic variations associated with T1D have a low phenotypic impact that overlaps when comparing carriers and non-carriers. In addition, human studies are mostly association in nature and strategies that allow investigators to directly analyze the diabetogenic function of a single SNP alone or in combination are limited. The CRISPR/Cas9 system makes it possible to engineer isogenic cell systems that can be used to specifically address the role of a SNP in gene expression and function (115). When combined with the ability to generate patient-derived iPSC and the advance of in vitro differentiation of iPSC into insulin-producing β-cells and hematopoietic stem cells, it may be possible to test the function of a SNP in cell types relevant to T1D (116, 117). However, these studies are not likely to overcome the difficulty to understand the course from altered gene expression/function to T1D development, which can only be dissected with in vivo experimental systems as disease progression is a consequence of combined effects that a variant elicits in different cell types in a time-dependent fashion. Combinational approaches using both mouse and human experimental systems are thus required to have a comprehensive understanding of the genetic control in T1D. The ability of nuclease based technology to efficiently and precisely modify the genome directly in NOD mice has opened a new door for current and future T1D genetic studies using this model.
Because T1D is a complex disease influenced by a large number of genes and ill-defined environmental factors, the NOD mouse remains an ideal animal model that provides a disease susceptible genetic background to test the diabetogenic function of a human candidate gene. For this reason, we have used both ZFNs and CRISPR/Cas9 systems to target mouse orthologs of human T1D candidate genes nominated by GWAS. As discussed above, the availability of the nuclease based technologies made it possible to do a relatively small scale but focused screening for genes that can regulate T1D progression in NOD mice. We have successfully targeted more than 40 genes directly in NOD mice. While these studies are still ongoing, the results obtained from this screening will allow us to provide additional evidence to support their roles in human T1D and prioritize them for future mechanistic studies. The eventual goal is to identify a pathway that could be pharmaceutically targeted for clinical translation.
Conclusion Remarks
Despite some shortcomings, NOD mice and NOD-derived recombinant congenic strains provide many advantages for T1D research. As discussed above, the NOD mouse continues to be an important tool for dissecting the genetic control of T1D. As will be discussed below, we also describe T1D research areas where NOD mice and related strains can provide critical information in the next decade.
Previous studies have generated NOD mice transgenically expressing HLA class I and II molecules associated with human T1D (118–121). While HLA class II molecules in NOD mice are not able to promote T1D, expression of the HLA A2.1 allele accelerates diabetes development, providing a model for identifying peptides targeted by A2.1-restricted CD8+ T cells and for testing antigen-specific immunotherapy (122, 123). When combined with various versions of the severe immunodeficient NOD mice (e.g., NSG mice), expression of HLA class I or II molecules in the absence of murine counterparts provides a superior host for primary human T cells and hematopoietic stem cell-derived immune system (124). NSG mice that also express high-risk HLA class I or II molecules have been transplanted with human peripheral blood mononuclear cells or β-cell autoreactive T-cell clones/lines isolated from T1D patients to test the diabetogenic potential of the presumably pathogenic effectors (125–127). Although much progress has been made, overt diabetes has not been observed in HLA class I or II expressing NSG mice transfused with human T cells in various settings. The eventual goal is to reconstitute a T1D prone human immune system that targets β cells derived from the same subject in a mouse for studying “human” T1D. Recent advance in differentiating human iPS cells into functional insulin-producing β-cells and hematopoietic stem cells has brought us one step closer to this goal (116, 117).
Gut microbiome has emerged as an important component that modulates the progression of T1D in both humans and NOD mice (128–134). Longitudinal studies in humans showed that alteration of the diversity and species of gut microbiota preceded T1D onset (133). Studies in NOD mice have shown that manipulation of gut microbiota by means of antibiotics, fecal transfer, or co-housing can either suppress or promote diabetes development (128, 135–138). Collectively, these studies demonstrate that the NOD mouse can provide an excellent experimental platform for understanding the roles of gut microbiota in T1D. Recent studies also suggest that T1D modulation by gut microbiota is not likely to be caused by a single species but rather due to the balance of diverse species within the bacterial community. While it remains to be tested, experiments that utilize germ-free NOD mice reconstituted with fecal samples from T1D patients, at risk individuals, and healthy subjects may provide some information regarding the “good” and “bad” gut bacterial community. This knowledge can then be used to develop methods to alter the gut microbiota for T1D prevention and set the foundation for future clinical trials.
Ethics Statement
All animal studies were approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin under protocol AUA00001863.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the manuscript was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the National Institutes of Health grants R21 AI130656 (JD), DP3 DK097605, R21 AI110963, R21 AI125879, and R01 DK107541 (Y-GC), as well as R01 DK074656, UC4 DK104194, and P01 AI042288 (CM).
References
- 1.Melanitou E, Devendra D, Liu E, Miao D, Eisenbarth GS. Early and quantal (by litter) expression of insulin autoantibodies in the nonobese diabetic mice predict early diabetes onset. J Immunol (2004) 173:6603–10. 10.4049/jimmunol.173.11.6603 [DOI] [PubMed] [Google Scholar]
- 2.You S, Belghith M, Cobbold S, Alyanakian MA, Gouarin C, Barriot S, et al. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes (2005) 54:1415–22. 10.2337/diabetes.54.5.1415 [DOI] [PubMed] [Google Scholar]
- 3.Gregori S, Giarratana N, Smiroldo S, Adorini L. Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J Immunol (2003) 171:4040–7. 10.4049/jimmunol.171.8.4040 [DOI] [PubMed] [Google Scholar]
- 4.Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127:2881–91. 10.1172/JCI94549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Regnell SE, Lernmark Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia (2017) 60:1370–81. 10.1007/s00125-017-4308-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wester A, Skärstrand H, Lind A, Ramelius A, Carlsson A, Cedervall E, et al. An increased diagnostic sensitivity of truncated GAD65 autoantibodies in type 1 diabetes may be related to HLA-DQ8. Diabetes (2017) 66:735–40. 10.2337/db16-0891 [DOI] [PubMed] [Google Scholar]
- 7.Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers (2017) 3:17016. 10.1038/nrdp.2017.16 [DOI] [PubMed] [Google Scholar]
- 8.Mathews CE, Xue S, Posgai A, Lightfoot YL, Li X, Lin A, et al. Acute versus progressive onset of diabetes in NOD mice: potential implications for therapeutic interventions in type 1 diabetes. Diabetes (2015) 64:3885–90. 10.2337/db15-0449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiLorenzo TP, Graser RT, Ono T, Christianson GJ, Chapman HD, Roopenian DC, et al. Major histocompatibility complex class I-restricted T cells are required for all but the end stages of diabetes development in nonobese diabetic mice and use a prevalent T cell receptor alpha chain gene rearrangement. Proc Natl Acad Sci U S A (1998) 95:12538–43. 10.1073/pnas.95.21.12538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and β-cell mass in the natural history of type 1 diabetes. Diabetes (2016) 65:719–31. 10.2337/db15-0779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Driver JP, Chen YG, Mathews CE. Comparative genetics: synergizing human and NOD mouse studies for identifying genetic causation of type 1 diabetes. Rev Diabet Stud (2012) 9:169–87. 10.1900/RDS.2012.9.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uchigata Y, Okada T, Gong JS, Yamada Y, Iwamoto Y, Tanaka M. A mitochondrial genotype associated with the development of autoimmune-related type 1 diabetes. Diabetes Care (2002) 25:2106. 10.2337/diacare.25.11.2106 [DOI] [PubMed] [Google Scholar]
- 13.Mathews CE, Leiter EH, Spirina O, Bykhovskaya Y, Gusdon AM, Ringquist S, et al. mt-Nd2 Allele of the ALR/Lt mouse confers resistance against both chemically induced and autoimmune diabetes. Diabetologia (2005) 48:261–7. 10.1007/s00125-004-1644-8 [DOI] [PubMed] [Google Scholar]
- 14.Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet (2015) 47:381–6. 10.1038/ng.3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vijayakrishnan L, Slavik JM, Illes Z, Greenwald RJ, Rainbow D, Greve B, et al. An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity (2004) 20:563–75. 10.1016/S1074-7613(04)00110-4 [DOI] [PubMed] [Google Scholar]
- 16.Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature (2003) 423:506–11. 10.1038/nature01621 [DOI] [PubMed] [Google Scholar]
- 17.Araki M, Chung D, Liu S, Rainbow DB, Chamberlain G, Garner V, et al. Genetic evidence that the differential expression of the ligand-independent isoform of CTLA-4 is the molecular basis of the Idd5.1 type 1 diabetes region in nonobese diabetic mice. J Immunol (2009) 183:5146–57. 10.4049/jimmunol.0802610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin X, Pelletier S, Gingras S, Rigaud S, Maine CJ, Marquardt K, et al. CRISPR-Cas9-mediated modification of the NOD mouse genome with Ptpn22R619W mutation increases autoimmune diabetes. Diabetes (2016) 65:2134–8. 10.2337/db16-0061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winkler C, Krumsiek J, Lempainen J, Achenbach P, Grallert H, Giannopoulou E, et al. A strategy for combining minor genetic susceptibility genes to improve prediction of disease in type 1 diabetes. Genes Immun (2012) 13:549–55. 10.1038/gene.2012.36 [DOI] [PubMed] [Google Scholar]
- 20.Howson JM, Cooper JD, Smyth DJ, Walker NM, Stevens H, She JX, et al. Evidence of gene-gene interaction and age-at-diagnosis effects in type 1 diabetes. Diabetes (2012) 61:3012–7. 10.2337/db11-1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant CW, Moran-Paul CM, Duclos SK, Guberski DL, Arreaza-Rubín G, Spain LM. Testing agents for prevention or reversal of type 1 diabetes in rodents. PLoS One (2013) 8:e72989. 10.1371/journal.pone.0072989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diabetes Prevention Trial-Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med (2002) 346:1685–91. 10.1056/NEJMoa012350 [DOI] [PubMed] [Google Scholar]
- 23.Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: the diabetes prevention trial – type 1. Diabetes Care (2005) 28:1068–76. 10.2337/diacare.28.5.1068 [DOI] [PubMed] [Google Scholar]
- 24.Näntö-Salonen K, Kupila A, Simell S, Siljander H, Salonsaari T, Hekkala A, et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet (2008) 372:1746–55. 10.1016/S0140-6736(08)61309-4 [DOI] [PubMed] [Google Scholar]
- 25.Sherry NA, Chen W, Kushner JA, Glandt M, Tang Q, Tsai S, et al. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology (2007) 148:5136–44. 10.1210/en.2007-0358 [DOI] [PubMed] [Google Scholar]
- 26.Gill RG, Pagni PP, Kupfer T, Wasserfall CH, Deng S, Posgai A, et al. A preclinical consortium approach for assessing the efficacy of combined anti-CD3 plus IL-1 blockade in reversing new-onset autoimmune diabetes in NOD mice. Diabetes (2016) 65:1310–6. 10.2337/db15-0492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes (2005) 54:1763–9. 10.2337/diabetes.54.6.1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med (2002) 346:1692–8. 10.1056/NEJMoa012864 [DOI] [PubMed] [Google Scholar]
- 29.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med (2005) 352:2598–608. 10.1056/NEJMoa043980 [DOI] [PubMed] [Google Scholar]
- 30.Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Rosenthal SM, Shuster JJ, et al. Anti-thymocyte globulin/G-CSF treatment preserves β cell function in patients with established type 1 diabetes. J Clin Invest (2015) 125:448–55. 10.1172/JCI78492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Perry DJ, Schultz AR, et al. Antithymocyte globulin plus G-CSF combination therapy leads to sustained immunomodulatory and metabolic effects in a subset of responders with established type 1 diabetes. Diabetes (2016) 65:3765–75. 10.2337/db16-0823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue S, Posgai A, Wasserfall C, Myhr C, Campbell-Thompson M, Mathews CE, et al. Combination therapy reverses hyperglycemia in NOD mice with established type 1 diabetes. Diabetes (2015) 64:3873–84. 10.2337/db15-0164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atkinson MA. Evaluating preclinical efficacy. Sci Transl Med (2011) 3:96cm22. 10.1126/scitranslmed.3002757 [DOI] [PubMed] [Google Scholar]
- 34.Wicker LS, Todd JA, Prins JB, Podolin PL, Renjilian RJ, Peterson LB. Resistance alleles at two non-major histocompatibility complex-linked insulin-dependent diabetes loci on chromosome 3, Idd3 and Idd10, protect nonobese diabetic mice from diabetes. J Exp Med (1994) 180:1705–13. 10.1084/jem.180.5.1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lord CJ, Bohlander SK, Hopes EA, Montague CT, Hill NJ, Prins JB, et al. Mapping the diabetes polygene Idd3 on mouse chromosome 3 by use of novel congenic strains. Mamm Genome (1995) 6:563–70. 10.1007/BF00352359 [DOI] [PubMed] [Google Scholar]
- 36.Podolin PL, Denny P, Lord CJ, Hill NJ, Todd JA, Peterson LB, et al. Congenic mapping of the insulin-dependent diabetes (Idd) gene, Idd10, localizes two genes mediating the Idd10 effect and eliminates the candidate Fcgr1. J Immunol (1997) 159:1835–43. [PubMed] [Google Scholar]
- 37.Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, Hearne CM, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature (1991) 351:542–7. 10.1038/351542a0 [DOI] [PubMed] [Google Scholar]
- 38.Hill NJ, Lyons PA, Armitage N, Todd JA, Wicker LS, Peterson LB. NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans. Diabetes (2000) 49:1744–7. 10.2337/diabetes.49.10.1744 [DOI] [PubMed] [Google Scholar]
- 39.Cornall RJ, Prins JB, Todd JA, Pressey A, DeLarato NH, Wicker LS, et al. Type 1 diabetes in mice is linked to the interleukin-1 receptor and Lsh/Ity/Bcg genes on chromosome 1. Nature (1991) 353:262–5. 10.1038/353262a0 [DOI] [PubMed] [Google Scholar]
- 40.Garchon HJ, Bedossa P, Eloy L, Bach JF. Identification and mapping to chromosome 1 of a susceptibility locus for periinsulitis in non-obese diabetic mice. Nature (1991) 353:260–2. 10.1038/353260a0 [DOI] [PubMed] [Google Scholar]
- 41.Hunter K, Rainbow D, Plagnol V, Todd JA, Peterson LB, Wicker LS. Interactions between Idd5.1/Ctla4 and other type 1 diabetes genes. J Immunol (2007) 179:8341–9. 10.4049/jimmunol.179.12.8341 [DOI] [PubMed] [Google Scholar]
- 42.Rodrigues NR, Cornall RJ, Chandler P, Simpson E, Wicker LS, Peterson LB, et al. Mapping of an insulin-dependent diabetes locus, Idd9, in NOD mice to chromosome 4. Mamm Genome (1994) 5:167–70. 10.1007/BF00352349 [DOI] [PubMed] [Google Scholar]
- 43.Lyons PA, Hancock WW, Denny P, Lord CJ, Hill NJ, Armitage N, et al. The NOD Idd9 genetic interval influences the pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and Cd137. Immunity (2000) 13:107–15. 10.1016/S1074-7613(00)00012-1 [DOI] [PubMed] [Google Scholar]
- 44.Brodnicki TC, McClive P, Couper S, Morahan G. Localization of Idd11 using NOD congenic mouse strains: elimination of Slc9a1 as a candidate gene. Immunogenetics (2000) 51:37–41. 10.1007/s002510050006 [DOI] [PubMed] [Google Scholar]
- 45.Hamilton-Williams EE, Rainbow DB, Cheung J, Christensen M, Lyons PA, Peterson LB, et al. Fine mapping of type 1 diabetes regions Idd9.1 and Idd9.2 reveals genetic complexity. Mamm Genome (2013) 24:358–75. 10.1007/s00335-013-9466-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin B, Ciecko AE, MacKinney E, Serreze DV, Chen YG. Congenic mapping identifies a novel Idd9 subregion regulating type 1 diabetes in NOD mice. Immunogenetics (2017) 69:193–8. 10.1007/s00251-016-0957-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ridgway WM, Peterson LB, Todd JA, Rainbow DB, Healy B, Burren OS, et al. Gene-gene interactions in the NOD mouse model of type 1 diabetes. Adv Immunol (2008) 100:151–75. 10.1016/S0065-2776(08)00806-7 [DOI] [PubMed] [Google Scholar]
- 48.Fraser HI, Dendrou CA, Healy B, Rainbow DB, Howlett S, Smink LJ, et al. Nonobese diabetic congenic strain analysis of autoimmune diabetes reveals genetic complexity of the Idd18 locus and identifies Vav3 as a candidate gene. J Immunol (2010) 184:5075–84. 10.4049/jimmunol.0903734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morin J, Boitard C, Vallois D, Avner P, Rogner UC. Mapping of the murine type 1 diabetes locus Idd20 by genetic interaction. Mamm Genome (2006) 17:1105–12. 10.1007/s00335-006-0076-9 [DOI] [PubMed] [Google Scholar]
- 50.Hollis-Moffatt JE, Hook SM, Merriman TR. Colocalization of mouse autoimmune diabetes loci Idd21.1 and Idd21.2 with IDDM6 (human) and Iddm3 (rat). Diabetes (2005) 54:2820–5. 10.2337/diabetes.54.9.2820 [DOI] [PubMed] [Google Scholar]
- 51.Rogner UC, Boitard C, Morin J, Melanitou E, Avner P. Three loci on mouse chromosome 6 influence onset and final incidence of type I diabetes in NOD.C3H congenic strains. Genomics (2001) 74:163–71. 10.1006/geno.2001.6508 [DOI] [PubMed] [Google Scholar]
- 52.Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol (2011) 33:67–87. 10.1007/s00281-010-0204-1 [DOI] [PubMed] [Google Scholar]
- 53.Lundholm M, Motta V, Löfgren-Burström A, Duarte N, Bergman ML, Mayans S, et al. Defective induction of CTLA-4 in the NOD mouse is controlled by the NOD allele of Idd3/IL-2 and a novel locus (Ctex) telomeric on chromosome 1. Diabetes (2006) 55:538–44. 10.2337/diabetes.55.02.06.db05-1240 [DOI] [PubMed] [Google Scholar]
- 54.Lin X, Hamilton-Williams EE, Rainbow DB, Hunter KM, Dai YD, Cheung J, et al. Genetic interactions among Idd3, Idd5.1, Idd5.2, and Idd5.3 protective loci in the nonobese diabetic mouse model of type 1 diabetes. J Immunol (2013) 190:3109–20. 10.4049/jimmunol.1203422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang N, Elso CM, Mackin L, Mannering SI, Strugnell RA, Wijburg OL, et al. Congenic mice reveal genetic epistasis and overlapping disease loci for autoimmune diabetes and listeriosis. Immunogenetics (2014) 66:501–6. 10.1007/s00251-014-0782-5 [DOI] [PubMed] [Google Scholar]
- 56.Hermann R, Lipponen K, Kiviniemi M, Kakko T, Veijola R, Simell O, et al. Lymphoid tyrosine phosphatase (LYP/PTPN22) Arg620Trp variant regulates insulin autoimmunity and progression to type 1 diabetes. Diabetologia (2006) 49:1198–208. 10.1007/s00125-006-0225-4 [DOI] [PubMed] [Google Scholar]
- 57.Smyth DJ, Cooper JD, Howson JM, Walker NM, Plagnol V, Stevens H, et al. PTPN22 Trp620 explains the association of chromosome 1p13 with type 1 diabetes and shows a statistical interaction with HLA class II genotypes. Diabetes (2008) 57:1730–7. 10.2337/db07-1131 [DOI] [PubMed] [Google Scholar]
- 58.Steck AK, Liu SY, McFann K, Barriga KJ, Babu SR, Eisenbarth GS, et al. Association of the PTPN22/LYP gene with type 1 diabetes. Pediatr Diabetes (2006) 7:274–8. 10.1111/j.1399-5448.2006.00202.x [DOI] [PubMed] [Google Scholar]
- 59.Stolp J, Chen YG, Cox SL, Henck V, Zhang W, Tsaih SW, et al. Subcongenic analyses reveal complex interactions between distal chromosome 4 genes controlling diabetogenic B cells and CD4 T cells in nonobese diabetic mice. J Immunol (2012) 189:1406–17. 10.4049/jimmunol.1200120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silveira PA, Chapman HD, Stolp J, Johnson E, Cox SL, Hunter K, et al. Genes within the Idd5 and Idd9/11 diabetes susceptibility loci affect the pathogenic activity of B cells in nonobese diabetic mice. J Immunol (2006) 177:7033–41. 10.4049/jimmunol.177.10.7033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen YG, Scheuplein F, Osborne MA, Tsaih SW, Chapman HD, Serreze DV. Idd9/11 genetic locus regulates diabetogenic activity of CD4 T-cells in nonobese diabetic (NOD) mice. Diabetes (2008) 57:3273–80. 10.2337/db08-0767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waldner H, Sobel RA, Price N, Kuchroo VK. The autoimmune diabetes locus Idd9 regulates development of type 1 diabetes by affecting the homing of islet-specific T cells. J Immunol (2006) 176:5455–62. 10.4049/jimmunol.176.9.5455 [DOI] [PubMed] [Google Scholar]
- 63.Hamilton-Williams EE, Wong SB, Martinez X, Rainbow DB, Hunter KM, Wicker LS, et al. Idd9.2 and Idd9.3 protective alleles function in CD4+ T-cells and nonlymphoid cells to prevent expansion of pathogenic islet-specific CD8+ T-cells. Diabetes (2010) 59:1478–86. 10.2337/db09-1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamanouchi J, Puertas MC, Verdaguer J, Lyons PA, Rainbow DB, Chamberlain G, et al. Idd9.1 locus controls the suppressive activity of FoxP3+CD4+CD25+ regulatory T-cells. Diabetes (2010) 59:272–81. 10.2337/db09-0648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ueno A, Wang J, Cheng L, Im JS, Shi Y, Porcelli SA, et al. Enhanced early expansion and maturation of semi-invariant NK T cells inhibited autoimmune pathogenesis in congenic nonobese diabetic mice. J Immunol (2008) 181:6789–96. 10.4049/jimmunol.181.10.6789 [DOI] [PubMed] [Google Scholar]
- 66.Hill NJ, Stotland A, Solomon M, Secrest P, Getzoff E, Sarvetnick N. Resistance of the target islet tissue to autoimmune destruction contributes to genetic susceptibility in type 1 diabetes. Biol Direct (2007) 2:5. 10.1186/1745-6150-2-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghosh S, Palmer SM, Rodrigues NR, Cordell HJ, Hearne CM, Cornall RJ, et al. Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nat Genet (1993) 4:404–9. 10.1038/ng0893-404 [DOI] [PubMed] [Google Scholar]
- 68.Rogner UC, Lepault F, Gagnerault MC, Vallois D, Morin J, Avner P, et al. The diabetes type 1 locus Idd6 modulates activity of CD4+CD25+ regulatory T-cells. Diabetes (2006) 55:186–92. 10.2337/diabetes.55.01.06.db05-0598 [DOI] [PubMed] [Google Scholar]
- 69.Robles DT, Eisenbarth GS, Dailey NJ, Peterson LB, Wicker LS. Insulin autoantibodies are associated with islet inflammation but not always related to diabetes progression in NOD congenic mice. Diabetes (2003) 52:882–6. 10.2337/diabetes.52.3.882 [DOI] [PubMed] [Google Scholar]
- 70.Pomerleau DP, Bagley RJ, Serreze DV, Mathews CE, Leiter EH. Major histocompatibility complex-linked diabetes susceptibility in NOD/Lt mice: subcongenic analysis localizes a component of Idd16 at the H2-D end of the diabetogenic H2(g7) complex. Diabetes (2005) 54:1603–6. 10.2337/diabetes.54.5.1603 [DOI] [PubMed] [Google Scholar]
- 71.Grattan M, Mi QS, Meagher C, Delovitch TL. Congenic mapping of the diabetogenic locus Idd4 to a 5.2-cM region of chromosome 11 in NOD mice: identification of two potential candidate subloci. Diabetes (2002) 51:215–23. 10.2337/diabetes.51.1.215 [DOI] [PubMed] [Google Scholar]
- 72.Serreze DV, Bridgett M, Chapman HD, Chen E, Richard SD, Leiter EH. Subcongenic analysis of the Idd13 locus in NOD/Lt mice: evidence for several susceptibility genes including a possible diabetogenic role for beta 2-microglobulin. J Immunol (1998) 160:1472–8. [PubMed] [Google Scholar]
- 73.Reed JC, Herold KC. Thinking bedside at the bench: the NOD mouse model of T1DM. Nat Rev Endocrinol (2015) 11:308–14. 10.1038/nrendo.2014.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet (2007) 39:329–37. 10.1038/ng1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kachapati K, Adams DE, Wu Y, Steward CA, Rainbow DB, Wicker LS, et al. The B10 Idd9.3 locus mediates accumulation of functionally superior CD137(+) regulatory T cells in the nonobese diabetic type 1 diabetes model. J Immunol (2012) 189:5001–15. 10.4049/jimmunol.1101013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McGuire HM, Vogelzang A, Hill N, Flodström-Tullberg M, Sprent J, King C. Loss of parity between IL-2 and IL-21 in the NOD Idd3 locus. Proc Natl Acad Sci U S A (2009) 106:19438–43. 10.1073/pnas.0903561106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fraser HI, Howlett S, Clark J, Rainbow DB, Stanford SM, Wu DJ, et al. Ptpn22 and Cd2 variations are associated with altered protein expression and susceptibility to type 1 diabetes in nonobese diabetic mice. J Immunol (2015) 195:4841–52. 10.4049/jimmunol.1402654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs β-cell function. Diabetes (2012) 61:2340–8. 10.2337/db12-0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med (2010) 207:1871–8. 10.1084/jem.20100209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet (2007) 39:1074–82. 10.1038/ng2102 [DOI] [PubMed] [Google Scholar]
- 81.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet (2007) 39:857–64. 10.1038/ng2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lyons PA, Armitage N, Argentina F, Denny P, Hill NJ, Lord CJ, et al. Congenic mapping of the type 1 diabetes locus, Idd3, to a 780-kb region of mouse chromosome 3: identification of a candidate segment of ancestral DNA by haplotype mapping. Genome Res (2000) 10:446–53. 10.1101/gr.10.4.446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41(6):703–7. 10.1038/ng.381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morahan G, Mehta M, James I, Chen WM, Akolkar B, Erlich HA, et al. Tests for genetic interactions in type 1 diabetes: linkage and stratification analyses of 4,422 affected sib-pairs. Diabetes (2011) 60:1030–40. 10.2337/db10-1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet (2003) 33:401–6. 10.1038/ng1117 [DOI] [PubMed] [Google Scholar]
- 86.Tiscornia G, Singer O, Ikawa M, Verma IM. A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci U S A (2003) 100:1844–8. 10.1073/pnas.0437912100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rao DD, Vorhies JS, Senzer N, Nemunaitis J. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev (2009) 61:746–59. 10.1016/j.addr.2009.04.004 [DOI] [PubMed] [Google Scholar]
- 88.Joseph J, Bittner S, Kaiser FM, Wiendl H, Kissler S. IL-17 silencing does not protect nonobese diabetic mice from autoimmune diabetes. J Immunol (2012) 188:216–21. 10.4049/jimmunol.1101215 [DOI] [PubMed] [Google Scholar]
- 89.Zheng P, Kissler S. PTPN22 silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes (2012) 62:896–904. 10.2337/db12-0929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gerold KD, Zheng P, Rainbow DB, Zernecke A, Wicker LS, Kissler S. The soluble CTLA-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes (2011) 60:1955–63. 10.2337/db11-0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schuster C, Gerold KD, Schober K, Probst L, Boerner K, Kim MJ, et al. The autoimmunity-associated gene CLEC16A modulates thymic epithelial cell autophagy and alters T cell selection. Immunity (2015) 42:942–52. 10.1016/j.immuni.2015.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Caballero-Franco C, Kissler S. The autoimmunity-associated gene RGS1 affects the frequency of T follicular helper cells. Genes Immun (2016) 17:228–38. 10.1038/gene.2016.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kissler S, Stern P, Takahashi K, Hunter K, Peterson LB, Wicker LS. In vivo RNA interference demonstrates a role for Nramp1 in modifying susceptibility to type 1 diabetes. Nat Genet (2006) 38:479–83. 10.1038/ng1766 [DOI] [PubMed] [Google Scholar]
- 94.Nowakowska DJ, Kissler S. Ptpn22 modifies regulatory T cell homeostasis via GITR upregulation. J Immunol (2016) 196:2145–52. 10.4049/jimmunol.1501877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jacob HJ, Lazar J, Dwinell MR, Moreno C, Geurts AM. Gene targeting in the rat: advances and opportunities. Trends Genet (2010) 26:510–8. 10.1016/j.tig.2010.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet (2010) 11:636–46. 10.1038/nrg2842 [DOI] [PubMed] [Google Scholar]
- 97.Geurts AM, Moreno C. Zinc-finger nucleases: new strategies to target the rat genome. Clin Sci (2010) 119:303–11. 10.1042/CS20100201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, et al. Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol Biol (2010) 597:211–25. 10.1007/978-1-60327-389-3_15 [DOI] [PubMed] [Google Scholar]
- 99.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science (2009) 325:433. 10.1126/science.1172447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol (2011) 29:64–7. 10.1038/nbt.1731 [DOI] [PubMed] [Google Scholar]
- 101.Chen YG, Forsberg MH, Khaja S, Ciecko AE, Hessner MJ, Geurts AM. Gene targeting in NOD mouse embryos using zinc-finger nucleases. Diabetes (2014) 63:68–74. 10.2337/db13-0192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cannons JL, Chamberlain G, Howson J, Smink LJ, Todd JA, Peterson LB, et al. Genetic and functional association of the immune signaling molecule 4-1BB (CD137/TNFRSF9) with type 1 diabetes. J Autoimmun (2005) 25:13–20. 10.1016/j.jaut.2005.04.007 [DOI] [PubMed] [Google Scholar]
- 103.Forsberg MH, Ciecko AE, Bednar KJ, Itoh A, Kachapati K, Ridgway WM, et al. CD137 plays both pathogenic and protective roles in Type 1 diabetes development in NOD mice. J Immunol (2017) 198:3857–68. 10.4049/jimmunol.1601851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class II I-A beta chain is unique. Proc Natl Acad Sci U S A (1987) 84:2435–9. 10.1073/pnas.84.8.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Todd JA, Acha-Orbea H, Bell JI, Chao N, Fronek Z, Jacob CO, et al. A molecular basis for MHC class II-associated autoimmunity. Science (1988) 240:1003–9. 10.1126/science.3368786 [DOI] [PubMed] [Google Scholar]
- 106.Lund T, O’Reilly L, Hutchings P, Kanagawa O, Simpson E, Gravely R, et al. Prevention of insulin-dependent diabetes mellitus in non-obese diabetic mice by transgenes encoding modified I-A beta-chain or normal I-E alpha-chain. Nature (1990) 345:727–9. 10.1038/345727a0 [DOI] [PubMed] [Google Scholar]
- 107.Quartey-Papafio R, Lund T, Chandler P, Picard J, Ozegbe P, Day S, et al. Aspartate at position 57 of nonobese diabetic I-Ag7 beta-chain diminishes the spontaneous incidence of insulin-dependent diabetes mellitus. J Immunol (1995) 154:5567–75. [PubMed] [Google Scholar]
- 108.Singer SM, Tisch R, Yang XD, Sytwu HK, Liblau R, McDevitt HO. Prevention of diabetes in NOD mice by a mutated I-Ab transgene. Diabetes (1998) 47:1570–7. 10.2337/diabetes.47.10.1570 [DOI] [PubMed] [Google Scholar]
- 109.Harrison MM, Jenkins BV, O’Connor-Giles KM, Wildonger J. A CRISPR view of development. Genes Dev (2014) 28:1859–72. 10.1101/gad.248252.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Qin W, Dion SL, Kutny PM, Zhang Y, Cheng AW, Jillette NL, et al. Efficient CRISPR/Cas9-mediated genome editing in mice by Zygote electroporation of nuclease. Genetics (2015) 200:423–30. 10.1534/genetics.115.176594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ratiu JJ, Racine JJ, Hasham MG, Wang Q, Branca JA, Chapman HD, et al. Genetic and small molecule disruption of the AID/RAD51 axis similarly protects nonobese diabetic mice from type 1 diabetes through expansion of regulatory B lymphocytes. J Immunol (2017) 198:4255–67. 10.4049/jimmunol.1700024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zemmour D, Pratama A, Loughhead SM, Mathis D, Benoist C. Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc Natl Acad Sci U S A (2017) 114:E3472–80. 10.1073/pnas.1700946114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bottini N, Peterson EJ. Tyrosine phosphatase PTPN22: multifunctional regulator of immune signaling, development, and disease. Annu Rev Immunol (2014) 32:83–119. 10.1146/annurev-immunol-032713-120249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pugliese A, Yang M, Kusmarteva I, Heiple T, Vendrame F, Wasserfall C, et al. The juvenile diabetes research foundation network for pancreatic organ donors with diabetes (nPOD) program: goals, operational model and emerging findings. Pediatr Diabetes (2014) 15:1–9. 10.1111/pedi.12097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wallet MA, Santostefano KE, Terada N, Brusko TM. Isogenic cellular systems model the impact of genetic risk variants in the pathogenesis of type 1 diabetes. Front Endocrinol (2017) 8:276. 10.3389/fendo.2017.00276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Millman JR, Xie C, Van Dervort A, Gurtler M, Pagliuca FW, Melton DA. Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nat Commun (2016) 7:11463. 10.1038/ncomms11463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu YF, et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature (2017) 545:432–8. 10.1038/nature22370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kudva YC, Rajagopalan G, Raju R, Abraham RS, Smart M, Hanson J, et al. Modulation of insulitis and type 1 diabetes by transgenic HLA-DR3 and DQ8 in NOD mice lacking endogenous MHC class II. Hum Immunol (2002) 63:987–99. 10.1016/S0198-8859(02)00435-4 [DOI] [PubMed] [Google Scholar]
- 119.Marron MP, Graser RT, Chapman HD, Serreze DV. Functional evidence for the mediation of diabetogenic T cell responses by HLA-A2.1 MHC class I molecules through transgenic expression in NOD mice. Proc Natl Acad Sci U S A (2002) 99:13753–8. 10.1073/pnas.212221199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Elliott JF, Liu J, Yuan ZN, Bautista-Lopez N, Wallbank SL, Suzuki K, et al. Autoimmune cardiomyopathy and heart block develop spontaneously in HLA-DQ8 transgenic IAbeta knockout NOD mice. Proc Natl Acad Sci U S A (2003) 100:13447–52. 10.1073/pnas.2235552100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Antal Z, Baker JC, Smith C, Jarchum I, Babad J, Mukherjee G, et al. Beyond HLA-A*0201: new HLA-transgenic nonobese diabetic mouse models of type 1 diabetes identify the insulin C-peptide as a rich source of CD8+ T cell epitopes. J Immunol (2012) 188:5766–75. 10.4049/jimmunol.1102930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Takaki T, Marron MP, Mathews CE, Guttmann ST, Bottino R, Trucco M, et al. HLA-A*0201-restricted T cells from humanized NOD mice recognize autoantigens of potential clinical relevance to type 1 diabetes. J Immunol (2006) 176:3257–65. 10.4049/jimmunol.176.5.3257 [DOI] [PubMed] [Google Scholar]
- 123.Niens M, Grier AE, Marron M, Kay TW, Greiner DL, Serreze DV. Prevention of “humanized” diabetogenic CD8 T-cell responses in HLA-transgenic NOD mice by a multipeptide coupled-cell approach. Diabetes (2011) 60:1229–36. 10.2337/db10-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol (2012) 12:786–98. 10.1038/nri3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Unger WW, Pearson T, Abreu JR, Laban S, van der Slik AR, der Kracht SM, et al. Islet-specific CTL cloned from a type 1 diabetes patient cause beta-cell destruction after engraftment into HLA-A2 transgenic NOD/scid/IL2RG null mice. PLoS One (2012) 7:e49213. 10.1371/journal.pone.0049213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Viehmann Milam AA, Maher SE, Gibson JA, Lebastchi J, Wen L, Ruddle NH, et al. A humanized mouse model of autoimmune insulitis. Diabetes (2014) 63:1712–24. 10.2337/db13-1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Whitfield-Larry F, Young EF, Talmage G, Fudge E, Azam A, Patel S, et al. HLA-A2-matched peripheral blood mononuclear cells from type 1 diabetic patients, but not nondiabetic donors, transfer insulitis to NOD-scid/gammac(null)/HLA-A2 transgenic mice concurrent with the expansion of islet-specific CD8+ T cells. Diabetes (2011) 60:1726–33. 10.2337/db10-1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature (2008) 455:1109–13. 10.1038/nature07336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339:1084–8. 10.1126/science.1233521 [DOI] [PubMed] [Google Scholar]
- 130.Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity (2013) 39:400–12. 10.1016/j.immuni.2013.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Endesfelder D, zu Castell W, Ardissone A, Davis-Richardson AG, Achenbach P, Hagen M, et al. Compromised gut microbiota networks in children with anti-islet cell autoimmunity. Diabetes (2014) 63:2006–14. 10.2337/db13-1676 [DOI] [PubMed] [Google Scholar]
- 132.Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD, et al. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes (2015) 64:3510–20. 10.2337/db14-1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe (2015) 17:260–73. 10.1016/j.chom.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell (2016) 165:842–53. 10.1016/j.cell.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hansen CH, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sorensen SJ, et al. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia (2012) 55:2285–94. 10.1007/s00125-012-2564-7 [DOI] [PubMed] [Google Scholar]
- 136.Peng J, Narasimhan S, Marchesi JR, Benson A, Wong FS, Wen L. Long term effect of gut microbiota transfer on diabetes development. J Autoimmun (2014) 53:85–94. 10.1016/j.jaut.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Candon S, Perez-Arroyo A, Marquet C, Valette F, Foray AP, Pelletier B, et al. Antibiotics in early life alter the gut microbiome and increase disease incidence in a spontaneous mouse model of autoimmune insulin-dependent diabetes. PLoS One (2015) 10:e0125448. 10.1371/journal.pone.0125448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hu Y, Peng J, Tai N, Hu C, Zhang X, Wong FS, et al. Maternal antibiotic treatment protects offspring from diabetes development in nonobese diabetic mice by generation of tolerogenic APCs. J Immunol (2015) 195:4176–84. 10.4049/jimmunol.1500884 [DOI] [PMC free article] [PubMed] [Google Scholar]