

Abstract
NAFLD is a polygenic condition but the individual and cumulative contribution of identified genes remains to be established. To get additional insight into the genetic architecture of NAFLD, GWAS-identified GCKR, PPP1R3B, NCAN, LYPLAL1 and TM6SF2 genes were resequenced by next generation sequencing in a cohort of 218 NAFLD subjects and 227 controls, where PNPLA3 rs738409 and MBOAT7 rs641738 genotypes were also obtained. A total of 168 sequence variants were detected and 47 were annotated as functional. When all functional variants within each gene were considered, only those in TM6SF2 accumulate in NAFLD subjects compared to controls (P = 0.04). Among individual variants, rs1260326 in GCKR and rs641738 in MBOAT7 (recessive), rs58542926 in TM6SF2 and rs738409 in PNPLA3 (dominant) emerged as associated to NAFLD, with PNPLA3 rs738409 being the strongest predictor (OR 3.12, 95% CI, 1.8-5.5, P < 0.001). A 4-SNPs weighted genetic risk score value >0.28 was associated with a 3-fold increased risk of NAFLD. Interestingly, rs61756425 in PPP1R3B and rs641738 in MBOAT7 genes were predictors of NAFLD severity. Overall, TM6SF2, GCKR, PNPLA3 and MBOAT7 were confirmed to be associated with NAFLD and a score based on these genes was highly predictive of this condition. In addition, PPP1R3B and MBOAT7 might influence NAFLD severity.
Introduction
Non-alcoholic fatty liver disease (NAFLD), is a multifactorial disease characterized by an increased hepatic triglyceride content (>5.5% of liver weight) in the absence of an excess of alcohol consumption, HVC infection, familial hypobetalipoproteinemia or endocrine disorders1,2. NAFLD, which currently represents the leading cause of liver damage in developed countries3, has well established risk factors such as insulin resistance associated with overweight, physical inactivity and type 2 diabetes mellitus (T2DM)4. However, epidemiological, familial and twin studies have clearly indicated that the risk of NAFLD has also a strong genetic component5.
In the last few years, a large number of genetic investigations, employing single candidate gene as well as genome-wide association studies (GWAS) strategies, have provided compelling evidence that several gene variants are associated with NAFLD5. In particular, the rs738409 C > G change in the Patatin-like Phospholipase domain-containing 3 (PNPLA3) gene, coding for the I148M protein variation, has been identified as a major determinant of inter-individual and ethnicity-related differences in hepatic fat content6,7. The mechanism by which this substitution induces liver fat is related to an impaired hepatocellular triglycerides hydrolysis and increased lipogenesis associated to the 148 M allele8,9. More recently, the Transmembrane 6 Superfamily Member 2 (TM6SF2) E167K variant has also been shown to increase NAFLD susceptibility10. This effect appears to be due to an impaired mobilization of neutral lipids for very low-density lipoprotein (VLDL) assembly and secretion by the liver in E167K carriers11–13.
Furthermore, GWAS studies have indicated additional loci whose involvement in the pathogenesis of liver steatosis is less established. In particular, Speliotes E.K. et al.14 reported that variants in Protein Phosphatase 1 Regulatory Subunit 3B (PPP1R3B), Glucokinase Regulatory Protein (GCKR), Neurocan (NCAN) and Lysophospholipase Like 1 (LYPLAL1) genes were associated to the presence of NAFLD. Finally, based on results obtained in patients with alcoholic liver disease (ALD)15, Mancina et al.16 demonstrated an association between the rs641738 in the membrane Bound O-Acyltransferase Domain Containing 7 gene (MBOAT7) and the occurrence and progression of NAFLD. The minor allele (T) was significantly associated with high liver fat content only in European Americans, but not in African Americans and Hispanics. Moreover, this variant showed an additive effect with PNPLA3 and TM6SF2 single nucleotide polymorphisms (SNPs) in determining the risk of liver fibrosis17. Overall, these findings clearly indicate that the genetic predisposition to NAFLD results from a combination of several variants, which may influence different steps of hepatic lipid and carbohydrate metabolism18.
Despite this wealth of knowledge, the proportion of genetic risk of NAFLD explained by the identified loci remains modest (<5%). This might be because the majority of GWAS tag SNPs are common and/or lie in intergenic or intronic regions19. Moreover, GWAS did not capture rare or low frequency risk variants with moderate/strong effects, which could explain a part of this missing heritability. An effective way to overcome these limitations is to re-sequence the entire coding portion of candidate genes to capture all non-genotyped risk alleles. This strategy has already been successfully employed for various conditions where subjects with well distinct phenotypes were genotyped20. In addition, the candidate genes resequencing strategy, by providing a comprehensive evaluation of the polygenic architecture of NAFLD, would allow to weighing the overall as well as the individual contribution of different variants to the risk of this complex trait.
Here we provide the evaluation of genetic determinants of NAFLD using the sequencing analysis of candidate genes emerged from GWAS. To this aim, we have re-sequenced the coding regions of GCKR, PPP1R3B, LYPLAL1, NCAN and TM6SF2 genes in NAFLD and control subjects, where PNPLA3 rs738409 and MBOAT7 rs641738 genotypes were also obtained.
The association of individual variants with NAFLD has been evaluated by using logistic regression analysis. Furthermore, following the logic of recent studies that have tested in complex disorders the combined impact of multiple genetic variants21, we determined a polygenic score for NAFLD based on identified risk alleles.
Results
Subjects characteristics
Baseline characteristics of study participants are reported in Table 1. Compared with controls, NAFLD subjects were older, showed higher indices of adiposity and increased plasma triglycerides (TG) and reduced HDL-C concentrations (all P < 0.001). Also fasting plasma levels of glucose, insulin and HOMAIR values were significantly higher in NAFLD compared with control subjects (all P < 0.001). As expected, the prevalence of T2DM and metabolic syndrome (MetS) was higher in NAFLD than in controls. Moreover, subjects with NAFLD were more frequently smokers and hypertensives (P < 0.001). A statistically significant elevation of ALT (P < 0.001), AST (P = 0.008) and γGT (P < 0.001) were seen in NAFLD compared with control subjects. Among NAFLD subjects, 164 (76.3%) were classified as having moderate to severe liver steatosis according to Hamaguchi’s criteria.
Table 1.
Clinical and metabolic characteristics of study subjects.
| NAFLD | Controls | P | |
|---|---|---|---|
| N | 218 | 227 | |
| Age (years) | 54 (46–60) | 49.7 (41–58) | <0.001 |
| Gender(M/F)(%) | 69.7/30.3 | 63.0/37.0 | ns |
| BMI (kg/m2) | 29.2 (26.5–32.5) | 24.9(23.2–27.1) | <0.001 |
| Waist circumference, (cm) | 106.1 ± 13.5 | 91.2 ± 9.91 | <0.001 |
| Smokers, n (%) | 66 (30.6) | 44 (19.7) | 0.043 |
| T2DM, n (%) * | 54 (24.8) | 3 (1.3) | <0.001 |
| MetS, n (%) | 126 (57.8) | 18 (7.9) | <0.001 |
| Statins, n (%) | 50 (22.9) | 28 (12.3) | 0.003 |
| Systolic BP (mmHg) | 130 (120–140) | 120 (110–135) | <0.001 |
| Diastolic BP (mmHg) | 80 (80–85) | 80 (70–80) | <0.001 |
| TC (mg/dl) | 199.8 ± 41.1 | 201.0 ± 38.2 | ns |
| TG (mg/dl) | 134 (99–183) | 87 (68–116) | <0.001 |
| HDL-C (mg/dl) | 44 (37.5–54) | 58 (49–68) | <0.001 |
| LDL-C (mg/dl) | 125.3 ± 60.2 | 121.7 ± 33.9 | ns |
| Fasting Blood Glucose (mg/dl) | 95 (85–112) | 85 (77–90) | <0.001 |
| Fasting Insulin (UI/L) | 12.7 (8.9–19.9) | 5.7 (4.0–7.8) | <0.001 |
| HOMAIR | 3.0 (2.1–5.0) | 1.14(0.77–1.65) | <0.001 |
| ALT (UI/I) | 27 (19–42) | 15 (12–20) | <0.001 |
| AST (UI/I) | 23 (19–30) | 21 (18–26) | 0.008 |
| γ-GT (UI/I) | 27 (17–41) | 19 (15–26) | <0.001 |
Data are expressed as percentage, mean (±SD) and median (25th–75th percentile range) as appropriate.
Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase; BMI, body mass index; BP, blood pressure; y-GT, gamma glutamyl transferase; HDL, high density lipoprotein; HOMA-IR, homeostasis model of insulin resistance (fasting plasma glucose in mg/dL x fasting insulin in U/L)/405; LDL, low density lipoprotein; MetS, metabolic syndrome (defined by the NCEP-ATP III Expert Panel criteria43); NAFLD, non-alcoholic fatty liver disease; TC, total cholesterol; T2DM, type 2 diabetes mellitus.
DNA re-sequencing
Overall, 168 variants were identified, of which 100 were intronic and 68 exonic. Among exonic variants, 43 were nonsynonymous (NS), 2 nonsense, 2 frameshift and 21 synonymous (Supplementary Table S1). Thirty one (65.9%) were classifiable as rare (MAF < 0.01), 5 (10.6%) as low frequency/less common (0.01 ≤ MAF < 0.05) and 3 (6.4%) common variants (MAF ≥ 0.05). Six exonic variants (12.7%) were not been previously reported in dbSNP and thus submitted to EXAC database (http://exac.broadinstitute.org). Forty-seven variants were annotated as functional (nonsense, frameshift and nonsynonymous), 23% in GCKR, 14.8% in LYPLAL1, 34% in NCAN, 8.5% in PPP1R3B and 19.4% in TM6SF2 genes. These variants were considered for further analyses.
The list of identified variants with their in silico prediction of deleteriousness is reported in the Supplementary Table S2.
Enrichment of gene variants in NAFLD and controls
Figure 1 shows the percentage of subjects carrying at least one functional variant within each gene in study groups. Overall, 80% of subjects with NAFLD were positive for at least one variant in GCKR, 31% for LYPLAL1, 15% for NCAN, 8% for PPP1R3B and 14% for TM6SF2 genes. Among controls, 79% were positive for at least one variant in GCKR, 34% in LYPLAL1, 10% in NCAN, 4% in PPP1R3B and 8% in TM6SF2 genes. Although variants in NCAN and PPP1R3B appeared to be more frequent in cases compared with controls, only those in TM6SF2 reached the statistical significance (OR = 2.0, 95% CI, 1.0-4.0, P = 0.04). However, after correction for multiple comparisons, the association of TM6SF2 gene was no longer significant.
Figure 1.
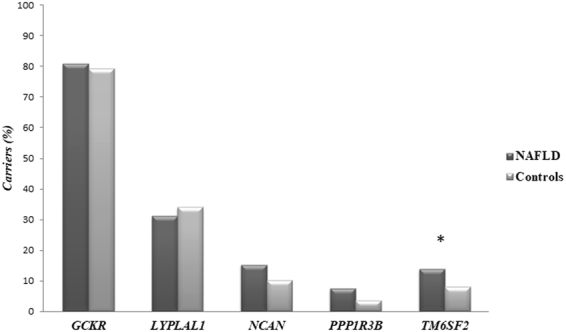
Enrichment of gene variants in NAFLD and controls. Percentage of subjects carrying at least one functional variant. In each group (NAFLD cases and controls) we count the number of subjects positive for at least one functional variant within each gene. *χ2 = 4.14, P = 0.04. *Odd Ratio unadjusted: OR = 2.0, 95% CI, 1.0-4.0, P = 0.04. In the model were included all subjects observed as carrying at least one functional variant per gene in NAFLD patients vs. controls.
Association of individual variants with NAFLD
In order to investigate the effect of genetic variants on NAFLD susceptibility, each identified sequence variation was included in a stepwise regression analysis. As all study subjects were also genotyped for the PNPLA3 rs738409 and MBOAT7 rs641738, these variants were also considered. As reported in Table 2, rs1260326 C/T in GCKR, rs58542926 C/T in TM6SF2, rs738409 C/G in PNPLA3 and MBOAT7 rs641738 T allele emerged as significantly associated with the presence of NAFLD (all P ≤ 0.05).
Table 2.
Genotype frequencies and Odds Ratios (ORs) of variants associated with NAFLD.
| Gene | SNP ID | NAFLD (N/%) | Controls (N/%) | Genotype model | |||||
|---|---|---|---|---|---|---|---|---|---|
| N = 218 | N = 227 | χ2 | P-value | OR (95% CI) | Unadjusted P-value | OR (95% CI) | Adjusted* P-value | ||
| PNPLA3 | rs738409 | ||||||||
| CC | 92 (42.2) | 123 (54.2) | |||||||
| CG | 91 (41.7) | 56 (24.7) | 2.2 (1.41–3.33) | <0.001 | 2.5 (1.30–4.85) | 0.006 | |||
| GG | 35 (16.1) | 48 (21.1) | 14.6 | 0.001 | 0.9 (0.58–1.62) | 0.9 | 4.2 (2.07–8.85) | <0.001 | |
| Dominant Model | CG + GG | 126 (57.8) | 104 (42.8) | 6.39 | 0.014 | 1.6 (1.11–2.35) | 0.012 | 3.2 (1.79–5.59) | <0.001 |
| Recessive Model | GG | 35 (16.1) | 48 (21.1) | 1.89 | 0.168 | 0.7 (0.44–1.15) | 0.16 | 3.0 (1.55–5.93) | 0.001 |
| GCKR | rs1260326 | ||||||||
| CC | 43 (19.7) | 49 (21.6) | |||||||
| CT | 90 (41.3) | 123 (54.2) | 0.8 (0.51–1.36) | 0.46 | 0.8 (0.44–1.74) | 0.69 | |||
| TT | 85 (39.0) | 55 (24.2) | 11.7 | 0.003 | 1.8 (1.03–2.99) | 0.04 | 1.8 (0.86–3.73) | 0.11 | |
| Dominant Model | CT + TT | 175 (80.3) | 178 (78.8) | 0.23 | 0.62 | 1.1 (0.70–1.77) | 0.62 | 1.2 (0.62–2.24) | 0.61 |
| Recessive Model | TT | 85 (39.0) | 55 (24.2) | 11.2 | 0.001 | 2.0 (1.32–3.00) | 0.001 | 1.9 (1.12–3.46) | 0.018 |
| TM6SF2 | rs58542926 | ||||||||
| CC | 161 (88.0) | 214 (94.3) | |||||||
| CT | 25 (11.5) | 11 (4.8) | 2.5 (1.20–5.25) | 0.014 | 2.4 (0.91–6.65) | 0.07 | |||
| TT | — | 2 (0.9) | 8.349 | 0.015 | — | ||||
| Dominant Model | CT + TT | 25 (11.5) | 13 (5.7) | 4.693 | 0.041 | 2.1 (1.06–4.28) | 0.033 | 2.2 (0.84–5.77) | 0.10 |
| MBOAT7 | rs641738 | ||||||||
| CC | 61 (28.0) | 78 (34.4) | |||||||
| CT | 105 (48.2) | 111 (48.9) | 1.21 (0.78–1.85) | 0.38 | 0.90 (0.49–1.65) | 0.74 | |||
| TT | 52 (23.9) | 38 (16.7) | 4.243 | 0.120 | 1.75 (1.02–2.99) | 0.04 | 1.75 (0.82–3.71) | 0.14 | |
| Dominant Model | CT + TT | 157 (72.0) | 149 (65.6) | 2.107 | 0.147 | 1.34 (0.90–2.02) | 0.14 | 1.09 (0.62–1.92) | 0.74 |
| Recessive Model | TT | 52 (23.9) | 38 (16.7) | 3.487 | 0.062 | 1.55 (0.97–2.48) | 0.06 | 1.86 (0.96–3.59) | 0.06 |
Data express the absolute numbers and percentages of cases and controls. Only variants showing a P value ≤ 0.06 were reported in the table.
*Models were adjusted by Age (years), Gender (M/F), BMI (kg/m2), HOMAIR, TG (mg/dl) and genotypes were considered as dominant or recessive (Logistic Regression analysis, Enter Method).
Abbreviations: BMI, Body Mass Index, GCKR, glucokinase (hexokinase 4) regulator gene, HOMAIR, homeostasis model of insulin resistance, MBOAT7, membrane-bound O-acyltransferase domain-containing 7 gene, NAFLD, non-alcoholic fatty liver disease. PNPLA3, Patatin-like phospholipase domain-containing protein 3 gene, TM6SF2, Transmembrane 6 superfamily Member 2 gene, TG, Triglycerides, OR, odd ratio, 95% CI, 95% CI confidence interval.
After adjustment for covariates such as age, gender, body mass index (BMI), HOMAIR and TG, a dominant model of inheritance best explained the association with NAFLD of rs738409 C/G PNPLA3 (OR = 3.2, 95% CI, 1.79-5.59, Padj < 0.001). Conversely, the association of rs1260326 C/T GCKR (OR = 1.9, 95% CI, 1.12-3.46, Padj = 0.018) fitted better with a recessive model of inheritance. A similar trend was observed for TM6SF2 rs58542926 T and MBOAT7 rs641738 T alleles, although with a borderline level of significance. However, it must be noted that a real estimation of the effect of TM6SF2 variant in the dominant or recessive model could not be provided because of the low frequency of the T-allele (167 K) (only two homozygous subjects). Thus, all calculations were based on the dominant model of inheritance.
When these 4 NAFLD-associated variants were tested together, they explained, overall, about 7% of the genetic risk of NAFLD and the rs738409 in PNPLA3 ranked as the strongest predictor (OR = 3.12, 95% CI, 1.8-5.5, P < 0.001) when adjusted for conventional risk factors (Table 3). Hosmer-Lomeshow goodness of fit test showed that the model combining both genetic and non-genetic variables explained the observed data (X26 = 16.45; P = 0.036) with a predictive ability of 58.2%. The results did not change even after including or excluding from the model plasma TG and glucose levels. It is worth to mention that when in this model HOMAIR was removed from covariates, TM6SF2 T allele reemerged as a significant predictor of NAFLD (OR = 3.6, 95% CI, 1.40-9.27, Padj = 0.008). This might be explained by the observation that carriers of this variant showed higher levels of HOMAIR compared to non-carriers (3.2 (2.1-6.0) vs. 1.43 (0.88-1.93), P = 0.002, respectively).
Table 3.
Independent associations of genetic variants with NAFLD.
| Gene, SNP ID | β | OR (95% CI) | P -value | *Padj-value |
|---|---|---|---|---|
|
PNPLA3, rs738409
Dominant Model |
1.14 | 3.12 (1.8–5.5) | <0.001 | 0.001 |
|
GCKR, rs1260326 Recessive Model |
0.64 | 1.90 (1.1–3.4) | 0.028 | 0.039 |
| Age (years) BMI (kg/m2) HOMAIR TG (mg/dl) |
0.02 0.19 0.71 0.01 |
1.02 (1.00–1.05) 1.21 (1.11–1.31) 2.03 (1.56–2.65) 1.01 (1.00–1.01) |
0.040 <0.001 0.001 0.001 |
0.039 0.001 0.010 0.005 |
Stepwise regression analysis (Forward-Wald Statistic) were used to test the association of clinical and genetic factors with NAFLD. In the model were included: Age (years), Gender (M/F), BMI (kg/m2), HOMAIR, TG (mg/dl), rs738409 PNPLA3 (dominant model), rs1230326 GCKR (recessive model), rs58542926 TM6SF2 (dominant model) and rs641738 MBOAT7 (recessive model). Only significant variables were reported.
*P was adjusted for multiple comparisons by using the bootstrap method.
Abbrevations: BMI, Body Mass Index, GCKR, glucokinase (hexokinase 4) regulator gene, HOMAIR, homeostasis model of insulin resistance, PNPLA3, Patatin-like phospholipase domain-containing protein 3 gene, TG, Triglycerides, OR, odd ratio, 95% CI, 95% CI confidence interval.
Next, we examined the proportion of risk of NAFLD conferred by gene-gene and gene-environment interaction. Although we did not identify any gene-gene synergy, PNPLA3 rs738409 showed a significant inverse interaction with TG (OR = 0.9, 95% CI, 0.97-0.99, P = 0.0021) and HOMAIR (OR = 0.33, 95% CI, 0.17-0.62, P = 0.001) but not with age, gender or BMI. Moreover, we observed a barely significant interaction between TM6SF2 polymorphism and BMI (OR = 0.8, 95% CI, 0.63-1.01, P = 0.07). Conversely, no interactions between GCKR rs1260326 or MBOAT7 rs641738 and conventional NAFLD risk factors were observed.
Association of genetic variants with metabolic traits
After stratifying the study population by TM6SF2 rs58542926 (dominant model), GCKR rs1260326 (recessive model), PNPLA3 rs738409 (dominant model) and MBOAT7 rs641738 (recessive model) genotypes, difference in clinical, anthropometric and biochemical indices were found across groups. NAFLD individuals carrying the minor T allele of rs58542926 (167 K) (N = 25) showed lower plasma total cholesterol (TC) (Padj = 0.021) and TG (Padj = 0.002) and higher AST (Padj = 0.006) and ALT (Padj = 0.012) levels when compared with NAFLD patients with wild-type C allele (N = 193). More importantly, the association with TC and TG levels was unchanged after adjustment for BMI, T2DM or statin therapy (all Padj < 0.05). Similarly, NAFLD patients carrying CG or GG PNPLA3 genotypes (N = 126) compared with non-carriers (N = 92) showed, lower BMI (Padj = 0.001), lower TG levels (Padj = 0.001) and HOMAIR (Padj = 0.004) and higher AST levels (Padj < 0.001). Notably, the association of [CG + GG] genotypes with TG levels persisted even after adjustment for BMI and diabetes (Padj = 0.01). On the contrary, no differences in clinical, anthropometric and biochemical indices were found across the GCKR rs1260326 or the MBOAT7 rs641738 genotypes in patients with NAFLD.
Genetic risk score (GRS) and the risk of NAFLD
The median values of weighted and unweighted 4-SNP GRS were significantly higher in NAFLD than in controls (median unweighted GRS: 3 (2–4) vs. 2 (2–3), P = 0.001; median weighted GRS: 0.38 (0.17–0.50) vs. 0.18 (0.12–0.44), P = 0.03, respectively). When weighted 4-SNP GRS values was distributed according to tertiles (Fig. 2, Panel a), the prevalence of NAFLD significantly increased along with increasing tertiles (χ2 = 14.9, P = 1 × 10-4) and the risk was significantly higher for GRS values above >0.28 (corresponding to the 2th tertile) (Fig. 2, Panel b). This trend persisted even after adjustment for age, gender, BMI, HOMAIR and TG levels.
Figure 2.

Association of weighted GRS with the risk of NAFLD. (a) Distribution of tertiles of weighted 4-SNP GRS in NAFLD patients; (b) NAFLD Odds Ratio (OR) adjusted for age, gender, BMI, HOMAIR and TG across tertiles of weighted 4-SNP GRS. The weighted 4-SNP GRS was calculated by multiplying the sum of the number of risk alleles (0–2) with the corresponding effect sizes per allele as obtained from the Dallas Heart Study22. Tertiles boundaries were defined as follow: T1 GRS ≤0.1775; T2 GRS >0.1775 and ≤0.3877; T3 GRS >0.3887. (a) Padj for trend. In the model were included age (years), gender (M/F), BMI (kg/m2), HOMAIR and TG (mg/dl) and tertiles of weighted 4-SNSP GRS (χ2 Pearson followed by Stepwise Regression analysis). (b) Adjusted NAFLD OR. In the model were included age (years), gender (M/F), BMI (kg/m2), HOMAIR and TG (mg/dl) and tertiles of weighted GRS (Stepwise Regression analysis, Forward-Wald Statistic).
Association of genetic variants with severity of NAFLD
The association of genetic variants with the ultrasound-defined severity of NAFLD is reported in Table 4. A NGS-identified variant in PPP1R3B gene (rs61756425 G/T p.S41R) emerged as more frequent in NAFLD patients than in controls (χ2 = 16.11, P < 0.001) and as the strongest independent genetic predictor of severe hepatic steatosis (OR = 32.6, 95% CI, 4.22-251.4, Padj = 0.001). This association was maintained even after bootstrap correction (two-tailed Padj = 0.001). Similarly, the rs641738 in MBOAT7 gene showed a significant effect on NAFLD severity (OR = 2.6, 95% CI, 1.10-6.28, Padj = 0.022). As expected, age, BMI and HOMAIR were detected as the non-genetic significant predictors of severity of NAFLD.
Table 4.
Predictors of NAFLD severity in the whole cohort.
| NAFLD degree | Variables | β | OR (95% CI) | P-value | *Padj-value |
|---|---|---|---|---|---|
| Severe |
PPP1R3B, rs61756425
MBOAT7, rs641738 Gender (F) BMI (kg/m2) HOMAIR |
3.48 0.96 1.26 0.23 0.60 |
32.6 (4.22–251.4) 2.6 (1.10–6.28) 0.3 (0.11–0.70) 1.2 (1.15–1.38) 1.8 (1.51–2.22) |
0.001 0.029 0.006 <0.001 <0.001 |
0.001 0.022 0.027 0.001 0.001 |
Stepwise regression analysis (Forward-Wald Statistic) were used to test the association of clinical and genetic factors with NAFLD. In the model were included: Age (years), Gender (M/F), BMI (kg/m2), HOMAIR, TG (mg/dl), rs738409 PNPLA3 (dominant model), rs1230326 GCKR (recessive model), rs58542926 TM6SF2 (dominant model) and rs641738 MBOAT7 (recessive model). Only significant variables were reported.
*P was adjusted for multiple comparisons by using the bootstrap method.
Abbrevations: BMI, Body Mass Index, HOMAIR, homeostasis model of insulin resistance, MBOAT7, membrane-bound O-acyltransferase domain-containing 7 gene, NAFLD, Non-alcoholic fatty liver disease, TG, Triglycerides, OR, odd ratio, PPP1R3B, Protein Phosphatase 1 Regulatory Subunit 3B, 95% CI, 95% CI confidence interval.
Discussion
NAFLD is a complex trait whose genetic component has been explored by many studies using different approaches18. Although several genes and genetic variants have been identified as involved in the occurrence of the disease, not all were consistently confirmed. Moreover, their combined effect on NAFLD susceptibility has rarely been explored.
In agreement with previous studies6,7,10,14,16,18,22,23, we found that PNPLA3 rs738409, GCKR rs1260326, TM6SF2 rs58542926 and MBOAT7 rs641738, but not the other GWAS identified variants14, were genetic contributors to NAFLD in our cohort.
Hepatic fat accumulation results from an imbalance between TGs acquisition, synthesis, utilization and secretion24 and, as yet described, the PNPLA3 I148M, TM6SF2 E167K and GCKR P446L polymorphisms promote steatosis through interaction with distinct metabolic mechanisms. Both genotypes in PNPLA3 and TM6SF2 influence the ability to export very low-density lipoproteins (VLDLs) from the liver8–13,17. In addition, p.P446L is a loss-of-function variant that results in increased phosphorylation of glucose25, glycolysis and fatty acid synthesis26. Similarly, recent findings indicated that the rs641738 in MBOAT7 gene, by decreasing the expression of MBOAT7 enzyme, unfavorably affects the acyl remodeling of phospholipid acyl-chain in the liver16. Taken together, these data indicate that a biologically plausible mechanism by which these gene variants directly influence the development of NAFLD exists. On the other hand, they further support the notion that the impaired lipid handling by hepatocytes has a major causal role in the pathogenesis of NAFLD.
When considered on individual basis, PNPLA3 variant ranked as the strongest genetic predictor of NAFLD followed by GCKR. In contrast, a weaker association was detected for TM6SF2 and MBOAT7. These observations could be partially explained by the low frequency of TM6SF2 risk allele as compared to PNPLA3 risk allele27. In addition, the finding that NAFLD carriers of the TM6SF2 167K allele have increased levels of HOMAIR might have masked the effect of this variant on NAFLD risk due to the predominant role of insulin resistance in the pathogenesis of NAFLD28,29. The finding on MBOAT7 is more difficult to be interpreted. However, it must be pointed out that this gene has not been consistently associated with NAFLD in all ethnic groups16 and, when a role was demonstrated, the MBOAT7 rs641738 variant showed the smallest effect in predisposing to fatty liver22.
Another interesting aspect of our findings is that, in agreement with previous studies, they do not fully support the notion that common variants are the major contributors to NAFLD susceptibility20,30. In fact, in our cohort the rs58542926 T allele (MAF ~ 7%) displayed a 2.5-fold risk of hepatic steatosis higher than the 1.9 fold risk associated to GCKR T allele (MAF ~ 42%). Thus, according to the hypothesis of Manolio T.A. et al.20, the lower frequency of rs58542926 in the general population could be the reason why we observed a larger effect of TM6SF2 T allele differently to GCKR TT genotype in the occurrence of NAFLD. Moreover, it is important to consider that in our cohort 55% of rs1260326 TT carriers were also heterozygous or homozygous carriers for the PNPLA3 rs738409, thus suggesting that the association with GCKR might be due to genetic bias.
Nevertheless, we confirmed that these genetic variants might act in an additive fashion. In fact, by using a 4-SNP GRS, we observed that the full combination of risk alleles increased the probability of hepatic steatosis up to 5 fold and this effect was present even after adjustment for traditional risk factors. These results emphasize the importance to consider a multiplicity of potentially involved gene variants when studying the genetic epidemiology of a common complex trait as NAFLD31,32. Of note, only four previous studies, carried out in different ethnic groups, have considered the combined effect of different genetic variants in determining fatty liver17,22,33,34. However, these Authors only performed genotyping tests for some of all previously GWAS-identified SNPs without exploring the entire coding region of NAFLD-associated loci. In addition, Krawczyk M. et al.17 concentrated their attention in evaluating the effect of the number of risk alleles (unweighted GRS) only on the grade of steatosis and fibrosis. Although our results need to be further evaluated in larger populations, they highlight the possibility to identify individuals at high risk of NAFLD by genotyping these genetic risk factors. Notably, EASL–EASD–EASO Clinical Practice Guidelines35 already suggest genotyping for TM6SF2 and PNPLA3 to select patients with higher risk of hepatic steatosis.
Our results indicated that the model including genetic and non-genetic variables accounts for the 58.2% of NAFLD heritability. These findings further confirm that the hepatic steatosis is a dynamic process that results from a constant interplay between genetic and environmental determinants and its heritability is not only due to the primary effect of PNPLA3, TM6SF2, GCKR and MBOAT7 genotypes but also by the secondary effects of non-genetic factors. In contrast with Stender S. et al.36, we did not find any synergy between adiposity and genotypes. The lack of interaction with BMI could be related to the fact that we did not quantify the intrahepatic triglyceride content (IHTG) but we considered hepatic steatosis as binary outcome variable in a case-control study37. Thus, while we described an inverse interaction effect between PNPLA3 genotypes HOMAIR and TG in predicting the higher risk of hepatic steatosis, the modest sample size could be the reason why we did not identified synergy with BMI37.
Finally, we found a significant association between the PPP1R3B rs61756425 and MBOAT7 rs641738 variants in predicting the severity of hepatic steatosis. Although MBOAT7 has been previously associated with progression of NAFLD16,17,38, the observation on PPP1R3B is novel. This variant has never been identified in GWAS nor associated to severity of NAFLD. This is probably due to its very low frequency in the population, thus supporting the notion that the re-sequencing of entire coding region of candidate genes may capture non-genotyped low-frequency risk alleles20,39. It must be, however, pointed out that the high value with a wide 95% CI of OR associating PPP1R3B rs61756425 with NAFLD severity may suggest a low level of accuracy of this estimate, mainly due to the very low number of carriers of this variant. Therefore, additional evaluations in much larger cohorts are needed. On the other hand, very recent observations have challenged the role of PPP1R3B as genetic factor predisposing to liver fat accumulation. In fact, Stender S. et al.40 have shown in mice that the lack of PPP1R3B was associated with reduced glycogen and unchanged fat liver content; conversely, the hepatic overexpression of PPP1R3B caused accumulation of hepatic glycogen and elevated ALT levels without affecting triglycerides accumulation. Based on this evidence, the role of PPP1R3B rs61756425 variant in liver disease surely requires additional evaluation with a direct confirmation of liver histological changes associated with this variant.
Strengths and limitations of our study must be acknowledged. To the best of ours knowledge, this is the first study reporting a comprehensive evaluation of sequence variants detectable in the entire coding regions of NAFLD-associated loci. This allowed evaluating the individual as well as the cumulative contribution of identified variants to the risk of NAFLD. However, it must be recognized that the definition of NAFLD was based on ultrasound and not on direct measurement of hepatic fat content with a more accurate methods such as the liver magnetic resonance. In addition, the size of the sequencing sample was modest so that some relevant rare variant might have been missed. To this regard, it is noteworthy that the power analysis indicated that our experimental design was able to identify low frequency variants and to detect moderate effects size. More, we did not re-sequenced the PNPLA3 and the MBOAT7 genes. For the former, we have considered the 148 M variant as it represents the only variant in this gene associated to hepatic fat content41. In fact, Donati B et al. by re-sequencing a cohort of children with early-onset histological NAFLD did not find any additional predictive rare variant in the PNPLA3 gene41. In addition, we genotyped only the MBOAT7 rs641738 variant as it was reported to display the major role in NAFLD16,42. Finally, an additional limitation of our study was the lack of replication of identified rare variants in an independent sample. However, it must be considered that in the present study we have re-sequenced well-known genetic determinants of NAFLD not requiring replication. Nevertheless, the analysis of rare variants was adjusted for multiple testing.
In conclusion, we confirmed the role of PNPLA3, TM6SF2, GCKR and MBOAT7 gene variants as genetic determinants of NAFLD and we suggested a weighted GRS based on their additive and combined effect. We believe these results point the way towards a future feasibility of creating comprehensive risk factor panels, in which applying genetic testing for the individual-level NAFLD-risk prediction. If definitely confirmed, our GRS score could offer the opportunity to exclude low-risk patients from screening tests.
Material and Methods
Study subjects
We studied 445 Caucasian, unrelated subjects, 218 with echographically defined NAFLD and 227 healthy controls. The enrollment criteria as well as the protocol for clinical and biochemical characterization of study subjects have been reported elsewhere43. In brief, NAFLD subjects, were considered eligible after exclusion of secondary causes of liver steatosis such as previous viral infection, past or present history of alcohol abuse (defined as an average daily consumption >20 g/day), use of drugs known to influence the development of hepatic steatosis as well as clinical and biochemical evidence of chronic liver diseases. Healthy controls, recruited from blood donors, were selected based on the absence of advanced liver disease at ultrasound43. Liver ultrasonography was performed with a GE Vivid S6 apparatus equipped with a 3.5-MHz convex-array probe. All examinations were done by the same hepatologist and steatosis was assessed semi-quantitatively on a scale of 0–6: 0, absent; 1, 2 mild; 3, 4 moderate; and 5, 6 severe according to the Hamaguchi criteria44.
The study protocol was reviewed and approved by the Ethics Committee of Sapienza University of Rome, Policlinico Umberto I (Rome, Italy). Written informed consent was obtained from all participants in accordance with the principles of the Helsinki Declaration. All methods were carried out in accordance with the relevant guidelines and regulations.
DNA analysis
Selection of candidate genes
The genes considered for next generation sequencing (NGS) were the following: GCKR, NCAN, PPP1R3B, LYPLAL1 and TM6SF2. They were selected because reported by GWAS to be associated with NAFLD above a significance threshold of P < 10−4 for any tagging SNPs10,14. In our screening, we have also considered the PNPLA3 rs738409 and the MBOAT7 rs641738, as previously demonstrated to be genetic determinants of NAFLD6,16. The genotyping of these latter variants was performed in duplicate by TaqMan 5′-Nucleotidase assay having a concordance rate of 100%.
Next-generation sequencing (NGS)
A custom panel was designed with the help of the AmpliSeq designer online tool (https://www.ampliseq.com), which was employed to generate optimized primer designs for the five genes present in the human reference genome (hg19). The overall coverage of the design region was 99,9%. (Pipeline version 4.2). Amplicon library preparation was performed with the Ion Ampliseq Library kit v2.0 using 10 ng of DNA (Thermo Fisher Scientific). PCR products were partially digested using FuPa reagent, followed by the ligation of barcoded sequencing adapters (Ion Xpress Barcode Adapters kit; Life Technologies, Carlsbad, CA, USA). Final libraries were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA) and quantified using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Wilmington, DE). The individual libraries were diluted to a final concentration of 100 pM and were pooled and processed to library amplification using Ion PGM Template OT2 400 kit. Unenriched libraries were quality-controlled using Ion Sphere quality control measurement on a Qubit instrument. Following library enrichment (Ion OneTouch ES), libraries were processed for sequencing by using the Ion PGM Hi-Q Sequencing Kit v2.
Data filtering and analysis
Sequencing runs were analyzed using the Torrent Suite v4.4.3 analysis. SNPs and insertion/deletions were identified across the targeted subset of the reference genome (hg19) using the analysis plug-in Torrent Variant Caller with the parameter settings optimized for germline low stringency and minimal false positive calls. The output variant call format (VCF) file was then annotated through Ion Reporter (Ion Reporter™ Software 4.6) and wANNOVAR softwares (http://wannovar.wglab.org). All sequencing variants were filtered using our custom NGS pipeline. All variants with Depth Coverage (DP) ≥30, Genotype Quality (GQ) ≥30, Allele Frequency (AF) ≥33 and ≤50 or ≥70 and ≤100, with balanced Alternate Allele Observations on the forward strand (SAF) and Alternate Allele Observations on the reverse strand (SAR) were considered as high confident variants and used for further analysis. Twenty four variants in 77 subjects with moderate quality were retested by Sanger sequencing on an ABI PRISM 3130 XL Genetic Analyzer following standard protocols. Overall, 13 variants with AF <33 and DP <20 were not confirmed.
The damaging effect of identified missense variants were evaluated by in silico prediction softwares. SIFT, PolyPhen-2, Provean, SNP&GO and Mutation T@ster Prediction softwares were used. A collective predictive score, ranging from 0–10, was calculated as the sum of individual scores of the 5 tools utilized, each being 0 (Neutral/benign/Polymorphism) or 1 (possibly damaging by Polyphen) or 2 (Disease Causing/Probably Damaging). Variants were defined as damaging if reported as deleterious in at least three of five prediction tools.
All common (MAF >5%), low frequency and rare variants (MAF ≥1 or ≤5% and <1%, respectively) annotated as functional (nonsense, frameshift, splice-region and missense) were considered for the analysis.
Power Calculation
Power analysis was performed by the Genetic Association Study (GAS) Power Calculator (© 2017 Jennifer Li Johnson | University of Michigan), commonly used to compute statistical power for one-stage genetic association studies within the setting of additive or multiplicative genetic models. The prevalence of NAFLD in our population was estimated as 0.30 and the odd ratio (OR) for each risk allele of the tested genetic variants was set at approximately 2.0, as estimated in previous studies45. Assuming an allele frequency of 0.3 (for common variants) and 0.03 (for low-frequency variants) in the general population and an additive model for disease risk, with a sample size of 218 cases and 227 controls the expected power under a significance level of 0.05 was of 100% to identify common genetic associations and 87% to identify low-frequency genetic associations. Notably our analysis had a power of 0.80 to detect genetic effects with OR of at least 1.9 for low-frequency and 1.35 for common variants.
Genetic Risk Score Computation
The Genetic Risk Score (GRS) was calculated based on the four SNPs reaching the highest levels of significance for NAFLD: rs1260326 C/T in GCKR, rs58542926 C/T in TM6SF2, rs738409 C/G in PNPLA3 and rs641738 C/T in MBOAT7 genes. Two methods were used to create the GRS: a simple count method (unweighted GRS) and a weighted method (weighted GRS)31,32. The count method assumed that each SNP contributed equally to NAFLD risk and was calculated applying a linear weighting of 0, 1 and 2 to genotypes containing 0, 1, or 2 risk alleles, respectively. While in our population we found only two homozygous subjects for rs58542926 in TM6SF2 gene this produced a score between 0 and 7, representing the maximum total number of risk alleles. The weighted 4-SNP GRS was calculated by multiplying each β -coefficient for the NAFLD phenotype obtained from the Dallas Heart Study6,22 by the number of corresponding risk alleles (0, 1, or 2) and then summing the products. The β–coefficient considered for each SNP were: 0.2653 (rs738409 PNPLA3), 0.2711 (rs58542926 TM6SF2), 0.0649 (rs1260326 GCKR) and 0.0575 (rs641738 MBOAT7). The 4-SNP GRS was modelled as a continuous variable and then categorized into tertiles.
Statistical analysis
The two-sample t-test (for parametric variables) or the Mann–Whitney test (for non-parametric variables) was used to compare the difference between case and control groups for quantitative traits, while Pearson’s χ2 test was used to compare discrete traits. Deviations of genotype frequency from the Hardy–Weinberg assumption were assessed using a χ2 test. Differences in allele and genotype frequencies between cases and controls were assessed by χ2 test under either dominant or recessive model of penetrance.
The enrichment of gene variants was evaluated by counting NAFLD cases and controls positive for at least on functional variant in each candidate gene.
Logistic regression analysis (Forward-Wald Statistic or Enter method) were adopted to assess the most significant model of inheritance for each SNP, the joint effects of genes and clinical variables and to evaluate gene-gene and gene-environmental interactions. The adequacy of the final model was assessed using Hosmer-Lameshow goodness-of-fit test. Furthermore, the Nagelkerke R2 was calculated to indicate how useful the explanatory variables in the model were in predicting NAFLD46. We further analysed the association of PNPLA3 rs738409, TM6SF2 rs58542926, GCKR rs1260326 and MBOAT7 rs641738 with biochemical indices by using General Linear Model test with bootstrap correction including age and sex as covariates. TG and TC were also adjusted for BMI, diabetes and statin therapy while HOMAIR was adjusted for BMI. For variables with skewed distributions (ALT, AST, TG, HOMAIR), a logarithm was applied before analysis to ensure that the residuals were approximately normal and had constant variance.
The effect of studied variants as well as of additional risk factors on the degree of hepatic steatosis were analyzed in logistic regression analysis (Forward-Wald Statistic) by comparing patients with severe NAFLD as having Hamaguchi score = 5–6 (N = 71) with patients without or with mild-moderate hepatic steatosis (Hamaguchi score = 0–4) (N = 362).
Associations between NAFLD risk and 4-SNP GRS were tested using Pearson correlations or logistic regression analysis.
Multiple comparisons were adjusted by bootstrap correction based on 1000 bootstrap samples with the aim to adjust raw p-value thus obtaining more robust estimates of standard errors and confidence intervals of parameters included in the models. Statistical significance was taken at nominal P-value < 0.05 for all comparisons. All analyses were performed using SPSS package (version 22.0) (SPSS, Inc., Chicago, IL, USA).
Data availability statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files). The datasets generated during and/or analyzed during the current study are not publicly available due to the lack of a specific patients’ consent but are made available by corresponding author based on reasonable request.
Electronic supplementary material
Acknowledgements
This work has been partially supported by Grant C26A14HZBX from Sapienza University of Rome.
Author Contributions
The listed authors have contributed to the work as follows: A.D.C., M.A., F.A., M.D.B. designed the study, reviewed all analyses, interpreted the data and prepared the manuscript; L.D.E., F.B., D.P., G.G., B.D.M., M.D.B., F.A. recruited subjects and performed all clinical evaluations; L.P. carried out the liver ultrasound examination; A.M., A.A. and F.C. executed the biochemical analyses; A.D.C., D.B., F.B. and G.G. performed next generation sequencing and A.D.C., D.B. and M.S. carried out genetic data filtering and analysis; A.D.C. performed genotyping tests and all statistical analyses. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-21939-0.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chalasani N, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American association for the study of liver diseases, American college of gastroenterology and the American gastroenterological association. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Nascimbeni F, et al. From NAFLD in clinical practice to answers from guidelines. J. Hepatol. 2013;59:859–871. doi: 10.1016/j.jhep.2013.05.044. [DOI] [PubMed] [Google Scholar]
- 3.Browning JD, et al. Prevalence of hepatic steatosis in an urban population in United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 4.Marchesini G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–50. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 5.Dongiovanni P, Valenti L. Genetics of nonalcoholic fatty liver disease. Metabolism. 2016;65:1026–37. doi: 10.1016/j.metabol.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 6.Romeo S, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Gene. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 8.Smagris E, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61:108–18. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pirazzi C, et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol. 2012;57:1276–82. doi: 10.1016/j.jhep.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 10.Kozlitina J, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352–6. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem. 2016;13:10659–76. doi: 10.1074/jbc.M116.719955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dongiovanni P, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61:506–14. doi: 10.1002/hep.27490. [DOI] [PubMed] [Google Scholar]
- 13.Mahdessian H, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci USA. 2014;111:8913–8918. doi: 10.1073/pnas.1323785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Speliotes EK, et al. Genome-wide association analysis identifies variants associated with non-alcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buch S, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47:1443–8. doi: 10.1038/ng.3417. [DOI] [PubMed] [Google Scholar]
- 16.Mancina RM, et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology. 2016;150:1219–1230. doi: 10.1053/j.gastro.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krawczyk M, et al. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926 and MBOAT7 rs641738 variants on NAFLD severity: a multicenter biopsy-based study. J Lipid Res. 2017;58:247–255. doi: 10.1194/jlr.P067454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol. 2017;23:1–12. doi: 10.3350/cmh.2016.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–97. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooke Bailey JN, Igo RP., Jr. Genetic Risk Scores. Curr Protoc Hum Genet. 2016;91:1.29.1–1.29.9. doi: 10.1002/cphg.20. [DOI] [PubMed] [Google Scholar]
- 22.Dongiovanni, P. et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med. 10.1111/joim.12719 (2017). [DOI] [PMC free article] [PubMed]
- 23.Wang X, Liu Z, Peng Z, Liu W. The TM6SF2 rs58542926 T Allele Is Significantly Associated with Nonalcoholic Fatty Liver Disease in Chinese. J Hepatol. 2015;62:1438–1439. doi: 10.1016/j.jhep.2015.01.040. [DOI] [PubMed] [Google Scholar]
- 24.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–23. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rees MG, et al. Cellular characterisation of the GCKR P446L variant associated with type 2 diabetes risk. Diabetologia. 2012;55:114–122. doi: 10.1007/s00125-011-2348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santoro N, et al. Hepatic de novo lipogenesis in obese youth is modulated by a common variant in the GCKR gene. J. Clin. Endocrinol. Metab. 2015;100:E1125–E1132. doi: 10.1210/jc.2015-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sookoian S, et al. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology. 2015;61:515–25. doi: 10.1002/hep.27556. [DOI] [PubMed] [Google Scholar]
- 28.Sookoian S, Pirola CJ. Systematic review with meta-analysis: risk factors for non-alcoholic fatty liver disease suggest a shared altered metabolic and cardiovascular profile between lean and obese patients. Aliment Pharmacol Ther. 2017;46:85–95. doi: 10.1111/apt.14112. [DOI] [PubMed] [Google Scholar]
- 29.Benedic M, Zhang X. Non-alcoholic fatty liver disease: An expanded review. World J Hepatol. 2017;9:715–732. doi: 10.4254/wjh.v9.i16.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Browning JD. Common genetic variants and nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2013;11:1191–3. doi: 10.1016/j.cgh.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Anderson JL, et al. Joint effects of common genetic variants from multiple genes and pathways on the risk of premature coronary artery disease. Am Heart J. 2010;160:250–256. doi: 10.1016/j.ahj.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Läll K, Mägi R, Morris A, Metspalu A, Fischer K. Personalized risk prediction for type 2 diabetes: the potential of genetic risk scores. Genet Med. 2017;19:322–329. doi: 10.1038/gim.2016.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leon-Mimila P, et al. A genetic risk score is associated with hepatic triglyceride content and non-alcoholic steatohepatitis in Mexicans with morbid obesity. Exp Mol Pathol. 2015;98:178–83. doi: 10.1016/j.yexmp.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, et al. Additive Effects of the Risk Alleles of PNPLA3 and TM6SF2 on Non-alcoholic Fatty Liver Disease (NAFLD) in a Chinese Population. Front Genet. 2016;7:140. doi: 10.3389/fgene.2016.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD) and European Association for the Study of Obesity (EASO) EASL–EASD–EASO. Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64:1388–1402. doi: 10.1016/j.jhep.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Stender S, et al. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49:842–847. doi: 10.1038/ng.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernandez LM, Blazer DG. Genes, Behavior and the Social Environment: Moving Beyond theNature/Nurture Debate. Washington (DC): National Academies Press (US). 2006;3:44–62. [PubMed] [Google Scholar]
- 38.Luukkonen PK, et al. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J Hepatol. 2016;65:1263–1265. doi: 10.1016/j.jhep.2016.07.045. [DOI] [PubMed] [Google Scholar]
- 39.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 40.Stender, S. et al. Relationship between Genetic Variation at PPP1R3B and Liver Glycogen and Triglyceride Levels. Hepatology, 10.1002/hep.29751 (2017). [DOI] [PMC free article] [PubMed]
- 41.Donati B, et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology. 2016;63:787–98. doi: 10.1002/hep.28370. [DOI] [PubMed] [Google Scholar]
- 42.Viitasalo A, et al. Association of MBOAT7 gene variant with plasma ALT levels in children: the PANIC study. Pediatr Res. 2016;80:651–655. doi: 10.1038/pr.2016.139. [DOI] [PubMed] [Google Scholar]
- 43.Di Costanzo A, et al. Non-alcoholic fatty liver disease and subclinical atherosclerosis: A comparison of metabolically- versus genetically-driven excess fat hepatic storage. Atherosclerosis. 2017;257:232–239. doi: 10.1016/j.atherosclerosis.2016.12.018. [DOI] [PubMed] [Google Scholar]
- 44.Hamaguchi M, et al. The severity of ultrasonographic findings in nonalcoholic fatty liver disease reflects the metabolic syndrome and visceral fat accumulation. Am. J. Gastroenterol. 2007;102:2708–2715. doi: 10.1111/j.1572-0241.2007.01526.x. [DOI] [PubMed] [Google Scholar]
- 45.Kahali. B, Halligan B, Speliotes EK. Insights from Genome-Wide Association Analyses of Nonalcoholic Fatty Liver Disease. Semin Liver Dis. 2015;35:375–391. doi: 10.1055/s-0035-1567870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bewick V, Cheek L, Ball J. Statistics review 14: Logistic regression. Crit Care. 2005;9:112–8. doi: 10.1186/cc3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files). The datasets generated during and/or analyzed during the current study are not publicly available due to the lack of a specific patients’ consent but are made available by corresponding author based on reasonable request.