

Summary
Loss of spiral ganglion neurons (SGNs) significantly contributes to hearing loss. Otic progenitor cell transplantation is a potential strategy to replace lost SGNs. Understanding how key transcription factors promote SGN differentiation in otic progenitors accelerates efforts for replacement therapies. A pro-neural transcription factor, Neurogenin1 (Neurog1), is essential for SGN development. Using an immortalized multipotent otic progenitor (iMOP) cell line that can self-renew and differentiate into otic neurons, NEUROG1 was enriched at the promoter of cyclin-dependent kinase 2 (Cdk2) and neurogenic differentiation 1 (NeuroD1) genes. Changes in H3K9ac and H3K9me3 deposition at the Cdk2 and NeuroD1 promoters suggested epigenetic regulation during iMOP proliferation and differentiation. In self-renewing iMOP cells, overexpression of NEUROG1 increased CDK2 to drive proliferation, while knockdown of NEUROG1 decreased CDK2 and reduced proliferation. In iMOP-derived neurons, overexpression of NEUROG1 accelerated acquisition of neuronal morphology, while knockdown of NEUROG1 prevented differentiation. Our findings suggest that NEUROG1 can promote proliferation or neuronal differentiation.
Keywords: auditory, spiral ganglion neurons, inner ear, neurogenin, iMOP, progenitor, chromatin, epigenetics
Graphical Abstract
Highlights
-
•
NEUROG1 is enriched at the Cdk2 promoter
-
•
Chromatin at the Cdk2 promoter changes during proliferation and differentiation
-
•
NEUROG1 promotes expression of CDK2 to drive otic progenitor proliferation
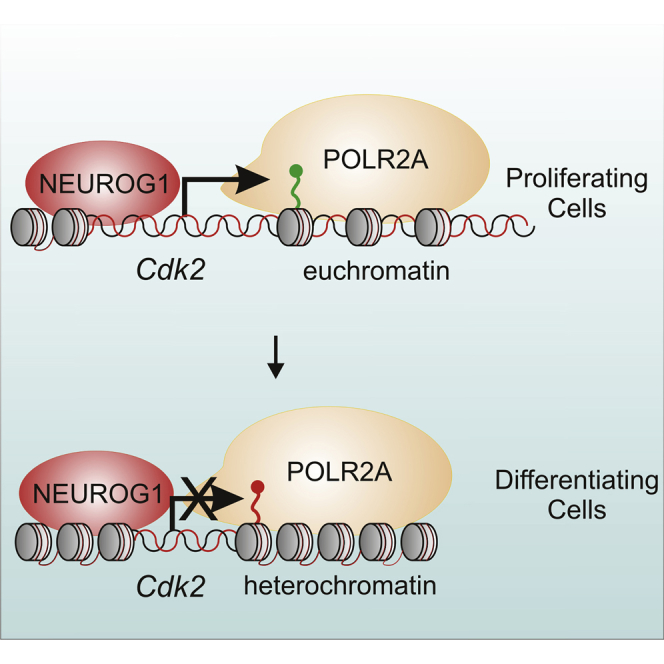
Song and colleagues showed that NEUROG1 promotes proliferation of immortalized multipotent otic progenitor (iMOP) cells by regulating CDK2 expression. The chromatin state at the Cdk2 promoter changes in proliferating and differentiating iMOP cells. NEUROG1-dependent expression of CDK2 correlates with euchromatin marks during proliferation, while heterochromatin marks associate with prevention of CDK2 expression during differentiation.
Introduction
Hair cells convert sounds into neural signals that are relayed to the brain by spiral ganglion neurons (SGNs). Sensorineural hearing loss due to noise overexposure causes hair cell loss, acute damage of synaptic terminals, and slow degeneration of the SGNs (Kujawa and Liberman, 2009). SGNs form synaptic connections with the mechanosensory hair cells (Rutherford and Moser, 2016). Once lost, SGNs do not regenerate. Different strategies have been proposed for alleviating SGN-related hearing loss. Stem cell replacement therapy for SGNs has made significant progress over the past decade (Rivolta, 2015, Shi and Edge, 2013). Using a gerbil model of SGN loss, engraftment of pluripotent stem cell-derived otic neural progenitors in the modiolus allows SGN differentiation, reforms the auditory circuit, and partially restores auditory function (Chen et al., 2012). Identifying factors that promote SGN differentiation will accelerate cell replacement therapies.
A core group of transcription factors (TFs) that include SOX2, NEUROG1, and NEUROD1 play an important role during the early development of SGNs (Evsen et al., 2013, Jahan et al., 2015a). NEUROG1 is a basic helix-loop-helix. Dimerization of NEUROG1 and E proteins allows binding of the heteromultimer to an E-box consensus DNA site (CANNTG) to promote transcription (Gamez et al., 2013, Huang et al., 2014, Ross et al., 2003). NEUROG1 affects the neural development in both the CNS and the peripheral nervous system (Ma et al., 1998, Ma et al., 2000). In the inner ear, NEUROG1 is initially expressed in the neural sensory competent domain at embryonic day 9.5 (E9.5), and fate mapping of these cells shows that NEUROG1 progenitors contribute to spiral and vestibular ganglion (Koundakjian et al., 2007). Ablation of Neurog1 leads to a complete loss of SGNs in murine inner ear (Ma et al., 2000). The presence of NEUROG1 early during neurogenesis suggests a role in progenitor expansion as well as neuronal differentiation. In addition to SGN development, NEUROG1 also affects hair cell development. Neurog1 mutant animals display smaller inner ear epithelia and fewer morphologically normal hair cells. The smaller sensory epithelia in Neurog1 mutants are due to a decrease in clonal expansion of hair cell precursors (Matei et al., 2005, Raft et al., 2007). Together, these studies suggested that NEUROG1 may have multiple cellular roles during inner ear development, including proliferation and differentiation of inner ear progenitors.
NEUROG1 is part of a TF family composed of NEUROG1–3. Expression of the NEUROG family of TFs has been used to promote neuronal differentiation of different cell types. Expression of NEUROG2 in embryonic stem cells (ESCs) results in direct lineage conversion to functional induced neuronal cells. Expression of NEUROG1 alone or with other factors has been used to induce neurogenesis in pluripotent stem cells (Lunn et al., 2012) and from fibroblasts (Blanchard et al., 2015). The neurogenin family members are potent TFs that direct differentiation of multiple cell types into neurons. However, in the inner ear NEUROG1 is unable to convert hair cells into neurons (Basch et al., 2011, Jahan et al., 2015b). Changes in the chromatin landscape during inner ear development could affect NEUROG1 transcriptional activity.
During differentiation, transcription is epigenetically modulated by deposition of post-translationally modified histones within the nucleosome (Voigt et al., 2013) Modification of different histones has significant consequences in regulating transcription. Deposition of permissive trimethylation of lysine K4 (H3K4me3) marks at the promoter region is associated with active gene expression (Heintzman et al., 2007). A broad domain of histone H3 trimethylation of lysine 27 (H3K27me3) enrichment across gene bodies corresponds to a repressed transcriptional domain, while peak enrichment of H3K27me3 at some promoters is associated with active transcription (Young et al., 2011). The simultaneous deposition of H3K27me3 and H3K4me3, known as bivalent domains, at the promoter regions are associated with genes that are transcriptionally silent, but poised for rapid expression during differentiation in vivo and in vitro (Bernstein et al., 2006, Rugg-Gunn et al., 2010, Voigt et al., 2013). As differentiation proceeds, many bivalent domains are resolved to a monovalent mark as inferred by comparing the epigenomic landscape of pluripotent and somatic cells (Mikkelsen et al., 2007). Activated genes lose the repressive H3K27me3 mark and expand the H3K4me3 mark to the gene body with enrichment at the proximal promoter and immediately downstream of transcription start sites (TSSs) (Barski et al., 2007). However, not all genes that lose the repressive H3K27me3 mark during lineage specification are activated (Rugg-Gunn et al., 2010), suggesting an additional contribution of other histone marks for transcriptional regulation.
Other histone marks are deposited in the same region as H3K4me3 and H3K27me3 during lineage specification (Barski et al., 2007) and may act sequentially or in concert to regulate transcription. H3K9ac is an epigenetic mark present at actively transcribed genes and is also present in bivalent domains (Karmodiya et al., 2012). H3K9me3, a histone mark associated with gene silencing and heterochromatin formation (Grewal and Elgin, 2002, Kouzarides, 2007, Rea et al., 2000), is also present in a subset of bivalently marked promoters in ESCs (Bilodeau et al., 2009). H3K9ac and H3K9me3 may correlate with changes in transcriptional activity as observed during lineage specification of T cells (Allan et al., 2012). To address how epigenetic changes affect the transcriptional regulatory networks during SGN differentiation (Fritzsch et al., 2010, Fritzsch et al., 2015), we used an immortalized otic progenitor cell (iMOP) (Kwan et al., 2015) and determined how epigenetic changes reflected the role of NEUROG1 in proliferation and differentiation.
Results
Differentiating iMOP Cells Are Post-mitotic and Exhibit Neuronal Markers
iMOP cells can proliferate as colony-forming otospheres or differentiate into iMOP-derived neurons (Jadali and Kwan, 2016). Incorporation of nucleotide analog (EdU [5-ethynyl-2′-deoxyuridine]) and labeling with the neuronal β-tubulin (TUBB3) marker was used to assay for proliferation and differentiation. Proliferating iMOP cultures labeled with Hoechst (Figure 1A), EdU (Figure 1B), and TUBB3 (Figure 1C) revealed a large percentage of EdU-labeled cells with no TUBB3-labeled cells (Figure 1D). In contrast, differentiating cultures labeled with Hoechst (Figure 1E), EdU (Figure 1F), and TUBB3 (Figure 1G) showed few EdU-marked cells but displayed an abundance of TUBB3-labeled cells with bipolar and pseudo-unipolar neuronal processes (Figure 1H). Quantification of EdU-labeled cells showed 27.0% in proliferating cultures and 3.7% in differentiating cultures. Quantification of TUBB3-labeled cells showed 0% in proliferating cultures and 86.5% in differentiating cultures (Figure 1I). These results suggested that the vast majority of iMOP-derived neurons are post-mitotic and express a neuronal marker.
Figure 1.

Identification of Bivalent Genes Involved in Otic Neurogenesis
(A–C) Proliferating or differentiating iMOP cells were subjected to EdU incorporation and immunostaining with TUBB3 antibodies. Fluorescence images of proliferating iMOP cells labeled with (A) Hoechst, (B) EdU, and (C) TUBB3.
(D) Merged image.
(E–G) Fluorescence image of differentiating iMOP cells labeled with (E) Hoechst, (F) EdU, and (G) TUBB3.
(H) Merged image.
(I) Percentage of EdU- and TUBB3-labeled cells in proliferating iMOP (n = 3) and iMOP-derived neurons (n = 3). Error bars denote ±SEM. ∗∗p < 0.01, ∗∗∗p < 0.001.
(J) Pairwise comparison of sequences from POLR2A, H3K4me3, and H3K27me3 ChIP-seq. Two biological replicates of each ChIP-seq sample were combined and used for analysis. Jaccard indices were listed for pairwise comparisons to show the extent of overlap.
To identify regulated genes as proliferating iMOP cells differentiate into iMOP-derived neurons, we selected genes containing bivalent promoters. Chromatin immunoprecipitation sequencing (ChIP-seq) was performed on proliferating iMOP cells and enrichment of sequence reads from POL2RA, H3K4me3, and H3K27me3 ChIP-seq ±1 kb from the TSS was used as a correlate for binding at the promoter region. Pairwise comparison was done between individual ChIP-seq reads, and the Jaccard index (JI) was used to describe the extent of overlap. Comparison of H3K4me3 with POLR2A (JI = 0.15), H3K27me3 with POLR2A (JI = 0.30), and H3K4me3 with H3K27me3 (JI = 0.17) showed a significant overlap (Figure 1J). Genes that showed binding for POLR2A, H3K4me3, and H3K27me3 in the promoter region were identified as candidate genes that may be epigenetically regulated during differentiation (Table S5).
Identification of Candidate Genes Involved in Cell-Cycle and Neuronal Differentiation
Candidate genes with bivalently marked promoters were subjected to gene ontology analysis and shown to be involved in cell-cycle checkpoint, mitotic cell cycle, chromatin organization, regulation of neuron differentiation, and regulation of neuron projection development (Figure 2A). A comprehensive list for the gene ontology analysis is provided in Table S6. Genes associated with the biological processes of mitotic cell cycle and regulation of neuron differentiation could affect the cellular decision of iMOP cells to either proliferate or differentiate into neurons (Table S7). To determine whether this subset of genes could be targets of NEUROG1, we identified a predicted NEUROG1 binding site (Mathelier et al., 2016) (Figure 2B). The predicted NEUROG1 binding site contained an E-box domain. To determine whether the promoter of these genes contained E-box sites, 1 kb of genomic sequence upstream of the TSS was obtained and curated for the presence of E-boxes for genes involved in mitotic cell cycle (Table S8) and regulation of neuron differentiation (Table S9). Genes that contained E-boxes in the promoter region were potential direct targets of NEUROG1. For the mitotic cell cycle, Cdk2, a cell-cycle-dependent kinase activated upon cyclin binding was identified. CDK2/cyclin complexes promote entry or progression into S phase. In neural stem cells, CDK2 helps the expansion of the progenitor pool prior to neuronal differentiation (Lim and Kaldis, 2012). E-box consensus sequence has been described in the cis-regulatory domains for Cdk2 (Du et al., 2004). For regulation of neuron differentiation, NeuroD1 was identified. NEUROD1 functions downstream of NEUROG1 and has been implicated in the transition from progenitor to a neuronal lineage (Evsen et al., 2013, Jahan et al., 2010, Ma et al., 1998, Rubel and Fritzsch, 2002).
Figure 2.

Changes in H3K9ac and H3K9me3 at Bivalently Marked Promoters
(A) Biological processes of genes with bivalent promoters were determined by gene ontology analysis. Pertinent biological process and associated binomial p values are listed.
(B) A consensus logo plot for NEUROG1 binding site containing an E-box (CANNTG).
(C) ChIP-seq peaks for POLR2A, H3K4me3, and H3K27me27 for Cdk2. Arrowheads indicate the approximate location of primers used for ChIP-qPCR. The number indicates the relative base-pair distance from the TSS.
(D) Enrichment of H3K9ac (active) or H3K9me3 (repressive) marks at the Cdk2 promoter region by ChIP-qPCR in proliferating iMOP (n = 3) and iMOP-derived neurons (n = 3). Background levels were determined by performing ChIP-qPCR with non-specific rabbit IgG control antibody.
(E) ChIP-seq peaks for POLR2A, H3K4me3, and H3K27me27 at NeuroD1. Arrowheads indicate the approximate location of primers relative to the TSS used for ChIP-qPCR.
(F) ChIP-qPCR of H3K9ac and H3K9me3 at the NeuroD1 promoter in proliferating progenitor (n = 3) and iMOP-derived neurons (n = 3).
(G) ChIP-seq peaks for POLR2A, H3K4me3, and H3K27me27 at Pou5f1.
(H) ChIP-qPCR of H3K9ac and H3K9me3 at the Pou5f1 promoter in proliferating progenitors (n = 3) and iMOP-derived neurons (n = 3).
Error bars denote ±SEM. ∗∗p < 0.01, ∗∗∗p < 0.001.
To visualize bivalently marked promoters, we plotted ChIP-seq reads at the genes of interest. We predicted that at bivalent promoters, the transcriptional activity of these target genes will be oppositely altered in proliferating and differentiating iMOP cells. As a correlate for transcriptional activity, H3K9ac (active) and H3K9me3 (repressive) marks were used for ChIP, and primers at H3K4me3 and H3K27me3 peaks in proximity to the TSS were used for qPCR. Enrichment of DNA after ChIP-qPCR was expressed as a percentage of input DNA. Background levels were determined by performing ChIP-qPCR using non-specific immunoglobulin G (IgG) antibodies. The largest percent enrichment of IgG antibodies was set as the baseline for ChIP-qPCR results. For Cdk2, both H3K4me3 and H3K27me3 were localized in the promoter region marked by POLR2A (Figure 2C). ChIP-qPCR of the Cdk2 promoter showed a reduction in H3K9ac when comparing proliferating iMOPs (64.2%) with iMOP-derived neurons (21.0%, p < 0.01). In contrast, an increase in H3K9me3 cells was observed when comparing proliferating iMOP (6.4%) with iMOP-derived neurons (19.3%, p < 0.01) (Figure 2D). At the NeuroD1 promoter, H3K4me3 and H3K27me3 were localized with POLR2A in two regions that could correspond to the TSS of two distinct Neurod1 variants. Two primer pairs were employed for ChIP-qPCR (Table S3) (Figure 2E). Using a primer pair (NeuroD1_1) that anneals to the 5′ TSS, ChIP-qPCR revealed an increase in H3K9ac in proliferating iMOPs (12.4%) compared with iMOP-derived neurons (42.1%, p < 0.001). In contrast, a decrease in H3K9me3 was observed in proliferating iMOPs (12.7%) compared with iMOP-derived neurons (5.6%, p < 0.01) (Figure 2F). Similarly, using a primer pair (NeuroD1_2) that annealed to the second TSS for ChIP-qPCR also showed an increase in H3K9ac mark in proliferating iMOPs (10.6%) compared with iMOP-derived neurons (36.8%, p < 0.001). A decrease in H3K9me3 was observed in proliferating iMOPs (10.6%) compared with iMOP-derived neurons (4.8%, p < 0.01) (Figure S1). Since both primer pairs showed similar results, the primer pair at the 5′ TSS (NeuroD1_1) was used for subsequent experiments.
Pou5f1 (Oct4), a pluripotent marker that is not present in lineage restricted cells such as iMOP cells (Kwan et al., 2015, Rosner et al., 1990, Scholer et al., 1990, Takahashi et al., 2007), was used as a negative control. Pou5f1 showed a broad H3K27me3 distribution, consistent with repressed expression of a pluripotency marker in iMOP cells (Figure 2G). ChIP-qPCR revealed that the Pou5f1 promoter in both proliferating and differentiating iMOP cells was essentially devoid of either H3K9ac or H3K9me3 (Figure 2H). The data suggest that the chromatin status is specific for the Cdk2 and NeuroD1 promoters and changes as iMOP cells transition from proliferation to differentiation.
Determining NEUROG1 Binding at Cdk2 and NeuroD1 Promoters
To determine whether NEUROG1 binds to the promoter of Cdk2 and NeuroD1, we performed ChIP-qPCR on endogenous and overexpressed NEUROG1. To control expression levels of NEUROG1, we used an inducible piggyBAC transposon system to generate stable cell lines (Li et al., 2013). Stable cell lines containing an inducible Neurog1 with an EGFP reporter (PB-T-Neurog1) and an inducible EGFP reporter only (PB-T-EGFP) were created (Figure 3A). To ensure that endogenous NEUROG1 could be detected, lysates from proliferating and differentiating PB-T-EGFP and PB-T-Neurog1 cultured in the absence of doxycycline (Dox) were probed for NEUROG1 (Figure S2A). All cell lines showed detectable NEUROG1 expression levels (Figure S2B). To determine the amount of Dox used to induce expression, we cultured PB-T- Neurog1 in varying amounts of Dox ranging from 0 to 2 μg/mL. The percentage of EGFP-positive cells in PB-T-Neurog1 line was also determined and plotted against different Dox concentrations (Figure 3B). Fluorescence from PB-T-Neurog1 cells with labeled with Hoechst (Figure 3C) and EGFP expression (Figure 3D) showed that the vast majority of cells (99.1%) was induced with 1 μg/mL Dox (Figure 3E). To quantify and determine the increase in Neurog1 transcript levels, we cultured PB-T-EGFP and PB-T-Neurog1 cells in the absence or presence of 1 μg/mL Dox and performed qPCR. All samples were normalized to PB-T-EGFP − Dox. PB-T-EGFP + Dox showed similar Neurog1 transcript levels. PB-T-Neurog1 − Dox showed slight elevation of Neurog1 transcript. However, NEUROG1 protein levels in PB-T-Neurog1 − Dox (Figure S2A) were not significantly different to PB-T-EGFP − Dox (Figure S2B). PB-T-Neurog1 + Dox showed a significant increase in Neurog1 transcript levels compared with all other cultures (p < 0.001) (Figure 3F). These results showed that addition of Dox increases Neurog1 transcript levels in the PB-T-Neurog1 stable cell line. To quantify NEUROG1 protein levels, we used PB-T-Neurog1 cells cultured in 0, 0.5, and 1 μg/mL Dox to detect NEUROG1 and ACTB by western blot (Figure 3G). NEUROG1 levels were normalized to ACTB and plotted relative to Dox concentrations. Increasing expression of NEUROG1 was observed with the addition of 0.5 μg/mL (5.3-fold) or 1 μg/mL (11.7-fold) Dox (Figure S2C). Together, these results showed that addition of Dox to the culture medium upregulates NEUROG1 in PB-T-Neurog1 stable cell lines.
Figure 3.

Enrichment of NEUROG1 at Cdk2 and NeuroD1 Promoter
(A) PiggyBAC constructs for generating stable cell lines allow for inducible expression of Neurog1 IRES EGFP or EGFP. Constructs with TetO driving Neurog1 IRES EGFP or EGFP were co-transfected with a rtTA construct (PB-RB). Transient expression of the transposase using the PBase allowed stable integration of plasmid sequence and generation of inducible stable iMOP cell lines PB-T-EGFP and PB-T-Neurog1.
(B) Percentage of EGFP-expressing cells were quantified in PB-T-Neurog1 cultures at different doxycycline (Dox) concentrations (n = 5 for each concentration).
(C and D) Hoechst (C) and EGFP (D) fluorescence of PB-T-Neurog1 cells after induction with 1 μg/mL Dox.
(E) Merged image showed that the vast majority of cells express EGFP (n = 3).
(F) Relative Neurog1 transcript levels in PB-T-EGFP and PB-T-Neurog1 cells cultured in the presence or absence of 1 μg/mL Dox (n = 3 for each condition).
(G) Western blot of NEUROG1 protein levels with increasing Dox concentrations (n = 3 for each condition).
(H and I) ChIP-qPCR using NEUROG1 antibody in PB-T-EGFP and PB-T-Neurog1 cells after Dox induction at the (H) Cdk2 and (I) NeuroD1 promoter regions for proliferating progenitors and iMOP-derived neurons (n = 3 for each condition).
Error bars denote ±SEM. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To determine whether endogenous and overexpressed NEUROG1 binds to the Cdk2 and NeuroD1 promoters, we performed ChIP-qPCR PB-T-EGFP and PB-T-Neurog1 cells cultured in 1 μg/mL Dox. An antibody recognizing NEUROG1 was used for ChIP, and primer pairs that annealed in the promoter regions of either Cdk2 or NeuroD1 were used for qPCR. Baseline levels for ChIP-qPCR were defined by using a non-specific IgG control. The amount of DNA enriched in each region was expressed as a percentage of input DNA. Enrichment of DNA by ChIP-qPCR using chromatin from either PB-T-EGFP or PB-T-Neurog1 cells was used to represent binding of endogenous or overexpressed NEUROG1, respectively. At the Cdk2 promoter, proliferating PB-T-EGFP (6.7%) or PB-T-Neurog1 (14.4%) cells showed enrichment above IgG control (1.5%, p < 0.001). Similarly, in iMOP-derived neurons, PB-T-EGFP (5.3%) or PB-T-Neurog1 (9.9%) cells showed enrichment above IgG control (0.22%, p < 0.001). Increased enrichment of NEUROG1 at the Cdk2 promoter was observed in PB-T-Neurog1 compared with PB-T-EGFP in proliferating iMOPs (p < 0.001) and iMOP-derived neurons (p < 0.05) (Figure 3H). At the NeuroD1 promoter, proliferating PB-T-EGFP (3.3%) or PB-T-Neurog1 (4.6%) cells showed enrichment above IgG control (0.3%, p < 0.001). Similarly, in iMOP-derived neurons, PB-T-EGFP (1.3%) or PB-T-Neurog1 (1.7%) cells showed enrichment above IgG control (0.1%, p < 0.001). Increased enrichment of NEUROG1 at the NeuroD1 promoter was observed in PB-T-Neurog1 compared with PB-T-EGFP in proliferating iMOPs (p < 0.01) (Figure 3I). At the Pou5f1 promoter, no enrichment was observed (Figure S2D). These results suggest that endogenous and overexpressed NEUROG1are enriched at both the Cdk2 and NeuroD1 promoter regions.
Overexpression of NEUROG1 Promotes Proliferation
Since NEUROG1 showed binding to the Cdk2 promoter, we wanted to determine whether overexpression of NEUROG1 could increase CDK2 levels to promote proliferation. Proliferating PB-T-EGFP and PB-T-Neurog1 cells were cultured in the presence or absence of Dox. Otospheres from cultures were collected every other day for 7 days, dissociated, labeled with Hoechst, and counted (Figure 4A). The increasing numbers of nuclei at each time point was used to calculate the doubling time for each culture. A significant decrease in doubling time (p < 0.01) was observed in PB-T-Neurog1 + Dox compared with all the other cultures (Figure 4B). Labeling with the NucRed dead cell marker did not show significant differences in dead cells between the cultures (Figures S3A–S3D). These results show that overexpression of NEUROG1 increases proliferation.
Figure 4.

NEUROG1 Overexpression in Proliferating Progenitors
(A) Automated nuclear counts with Hoechst-labeled nuclei are marked within green circles.
(B) Doubling times for PB-T-EGFP and PB-T-Neurog1 cells cultured in the absence or presence of 1 μg/mL Dox (n = 5 for each condition).
(C–F) Cells were fixed and labeled with Hoechst and subjected to EdU detection in (C) PB-T-EGFP – Dox (n = 3), (D) PB-T-EGFP + Dox (n = 3), (E) PB-T-Neurog1 – Dox (n = 3), (F) PB-T-Neurog1 + Dox (n = 3) cultures. Scale bars, 20 μm.
(G) Relative percentage of cells for PB-T-EGFP and PB-T-Neurog1 cells cultured in the absence or presence of 1 μg/mL Dox (n = 3 for each condition), and percentage of EdU-labeled cells (n = 3 for each condition).
(H) Transcript levels of NeuroD1 and Cdk2 from PB-T-EGFP and PB-T-Neurog1 cells cultured in the absence or presence of 1 μg/mL Dox (n = 3 for each condition).
(I) Western blot of CDK2 in proliferating PB-T-EGFP (n = 3) and PB-T-Neurog1 (n = 3) cultured in 1 μg/mL Dox.
(J) Quantification of CDK2 protein levels from western blots (n = 3).
Error bars denote ±SEM. ns, not significant; ∗∗p < 0.01, ∗∗∗p < 0.001.
To determine whether increased proliferation was due to increased number of cells undergoing DNA replication, we performed EdU incorporation. PB-T-EGFP and PB-T-Neurog1 cells were cultured in the presence or absence of Dox before EdU incorporation and Hoechst labeling. Hoechst- and EdU-labeled nuclei from PB-T-EGFP – Dox (Figure 4C), PB-T-EGFP + Dox (Figure 4D) and PB-T-Neurog1 – Dox (Figure 4E) cultures showed similar EdU-labeled cells while PB-T-Neurog1 + Dox cultures showed increased numbers of EdU-labeled cells (Figure 4F). Using Hoechst-labeled nuclei as an indicator of the total number of cells, the relative percentage of cells compared with PB-T-EGFP – Dox was determined. A significant increase in percentage of cells from PB-T-Neurog1 + Dox (167%, p < 0.01) cultures compared with all the other cultures was observed (Figure 4G). Hoechst- and EdU-labeled images were used to determine the percentage of EdU cells relative to PB-T-EGFP – Dox. PB-T-Neurog1 + Dox cultures showed a significant increase in the percentage of EdU-labeled cells (69%, p < 0.001) compared with other cultures (∼31%) (Figure 4G). To determine whether changes in either Cdk2 or NeuroD1 transcripts could be attributed to increased proliferation, we performed qPCR. In all the cultures, NeuroD1 levels were not significantly different. However, a significant increase in Cdk2 transcript levels in PB-T-Neurog1 + Dox cultures was observed compared with other cultures (2.12, p < 0.001) (Figure 4H). To determine whether CDK2 protein levels were upregulated after NEUROG1 overexpression, we harvested PB-T-EGFP + Dox and PB-T-Neurog1 + Dox cultures and compared CDK2 levels by western blot (Figure 4I). CDK2 levels were normalized to ACTB and compared to PB-T-EGFP + Dox. A significant increase in CDK2 levels was observed in PB-T-Neurog1 + Dox (10.4-fold, p < 0.01) (Figure 4J). Similarly, comparing PB-T-Neurog1 − Dox with PB-T-Neurog1 + Dox cells also showed an increase in CDK2 protein levels after addition of Dox (Figure S3E). A significant increase in CDK2 levels was observed in PB-T-Neurog1 + Dox (9.1-fold, p < 0.01) compared with PB-T-Neurog1 − Dox (Figure S3F). Together these results suggest that overexpression of NEUROG1 upregulates CDK2 and could increase proliferation of iMOP cells.
Knockdown of Neurog1 in Self-Renewing iMOP Cells Decreases Proliferation
To determine whether Neurog1 knockdown in proliferating iMOP cells could decrease proliferation, we transduced cells with Neurog1 short hairpin RNA (shRNA) or scrambled shRNA. Since lentiviral vectors contained a selectable blasticidin resistance cassette, cells were transduced and selected in blasticidin before performing EdU incorporation. In the scrambled shRNA control, 28.1% EdU-labeled cells were observed (Figure 5A). Neurog1 shRNA knockdown displayed a significant decrease to 5.1% of EdU-labeled cells (p < 0.001) (Figure 5B). These results suggest that NEUROG1 is required for normal S-phase entry in proliferating iMOP cells. To ensure that Neurog1 shRNA knockdown did not cause cell death, we labeled scrambled shRNA and Neurog1 shRNA cultures with a dead cell marker (NucRed). NucRed labeling showed similar percentages of dead cells in scrambled shRNA (1.2%) (Figure 5C) and Neurog1 shRNA transduced cells (1.1%) (Figure 5D). To determine the effect on cell numbers after Neurog1 knockdown, the average cell numbers per field of view (FOV) were determined. Relative to scrambled shRNA (553.5 cells/FOV), Neurog1 shRNA knockdown (178.3 cells/FOV, p < 0.01) resulted in decreased cell numbers (Figure 5E). In proliferating iMOP cultures, the size of the otospheres correlates to proliferation. Confocal images were taken from Hoechst-labeled otospheres, and the largest cross-sectional area of the otosphere was used as an indicator of sphere size. The average cross-sectional area of scrambled shRNA (5,409 μm2) was compared with Neurog1 shRNA transduced cells (1,194 μm2). Otospheres after Neurog1 shRNA knockdown showed a significant decrease in cross-sectional areas (p < 0.01) (Figure 5F). To determine relative changes in transcript levels after Neurog1 knockdown, we performed qPCR on scrambled shRNA and Neurog1 shRNA transduced cells. A significant decrease was observed in Neurog1 transcript after Neurog1 shRNA knockdown (0.09, p < 0.001) compared with scrambled shRNA. Cdk2 transcript levels were also decreased after Neurog1 shRNA (0.45, p < 0.05) while NeuroD1 was not significantly altered (Figure 5G). To determine NEUROG1 and CDK2 protein levels after transduction with Neurog1 shRNA, we performed western blotting. A decrease in both NEUROG1 and CDK2 levels was observed after Neurog1 shRNA knockdown compared with scrambled shRNA transduced cells (Figure 5H). To quantify the decrease in expression, we normalized NEUROG1 and CDK2 protein levels to ACTB. Neurog1 shRNA transduced cells showed a significant decrease in relative NEUROG1 (0.34, p < 0.05) and CDK2 (0.27, p < 0.01) protein levels relative to scrambled shRNA (Figure 5I). These results suggest that NEUROG1 regulates CDK2 expression in proliferating iMOP cells.
Figure 5.

NEUROG1 Knockdown in Proliferating Progenitors
(A–D) Hoechst- and EdU-labeled iMOP cells after transduction with (A) scrambled (scrb) shRNA (n = 3) or (B) Neurog1 shRNA (n = 3). Hoechst- and NucRed-labeled iMOP cells after transduction with (C) scrb shRNA (n = 3) or (D) Neurog1 shRNA (n = 3).
(E) Cell counts from scrb shRNA (n = 3) and Neurog1 shRNA (n = 3) transduced cultures in each field of view (FOV).
(F) Largest cross-sectional area of otospheres from scrb shRNA (n = 20 otospheres from 3 experiments) and Neurog1 shRNA transduced cultures (n = 20 otospheres from 3 experiments).
(G) Relative Neurog1, Cdk2, and NeuroD1 transcript levels in scrb shRNA (n = 3) or Neurog1 shRNA (n = 3) transduced cells.
(H) Western blot of NEUROG1, CDK2, and ACTB after transduction with scrb shRNA (n = 3) or Neurog1 shRNA (n = 3).
(I) Relative NEUROG1 (n = 3) and CDK2 (n = 3) levels from western blots.
Error bars denote ±SEM. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
CDK2 Functions Downstream of NEUROG1
To determine whether CDK2 functions downstream of NEUROG1 to promote proliferation, we overexpressed NEUROG1 while inhibiting CDK2 function. For inhibition of CDK2, the small molecule K03861 or roscovitine was employed. K03861 competes with cyclin binding to inhibit CDK2 kinase activity (Alexander et al., 2015). Roscovitine is a selective inhibitor of CDC2, CDK2, and CDK5 (Meijer et al., 1997). To determine the concentration of K03861 used, we treated unmodified iMOP cells with 0–5 μM of K03861 and determined the relative percentage of cells compared with untreated samples (Figure S4A). K03861 decreased cell numbers starting at 0.1 μM and plateaued around 1 μM (Figure S4B). Using these two concentrations, PB-T-Neurog1 + Dox cultures were treated with either 0.1 or 1 μM of K03861 before EdU incorporation was performed. Compared with PB-T-Neurog1 + Dox cultures (Figure 6A), a decrease in EdU-labeled nuclei was observed after treatment with 1 μM K03861 (Figure 6B). Relative to control cultures, a decrease in the percentage of cells was observed after treatment with 0.1 μM (43%, p < 0.001) or 1 μM (17%, p < 0.001) K03861. Similarly, the percentage of EdU-labeled nuclei decreased significantly from control (63%) after treatment with 0.1 μM (44%, p < 0.5) or 1 μM (9%, p < 0.001) K03861. To determine the concentration of roscovitine to inhibit proliferation, we treated iMOP cells with 0–1 μM roscovitine and determined the relative percentage of cells compared with untreated cultures (Figure S4C). A decrease in cell numbers was observed from 0.1 to 1 μM roscovitine. Compared with control, quantification showed a decreased percentage of cells after treatment with 0.1 μM (49%, p < 0.01), 0.5 μM (23%, p < 0.001), and 1 μM (1%, p < 0.001) roscovitine (Figure S4D). In control PB-T-Neurog1 + Dox cells, a large percentage of EdU-labeled cells was observed (58.2%) (Figure S4E). Treatment with 1 μM roscovitine decreased EdU-labeled cells (0.3%, p < 0.001) (Figure S4F). These results suggest that CDK inhibitors prevent increased proliferation after NEUROG1 overexpression.
Figure 6.

Inhibition of CDK2 Activity after NEUROG1 Overexpression
(A and B) Hoechst- and EdU-labeled PB-T-Neurog1 + Dox cells cultured (A) without K03861 (n = 3) or (B) with 1 μM K03861 (n = 3).
(C and D) Relative percentage of cells for PB-T-Neurog1 cells (C) and relative percentage of EdU-labeled cells (D) cultured with 0, 0.1, and 1 μM K03861 (n = 3 for each condition).
(E and F) Hoechst- and EdU-labeled PB-T-Neurog1 + Dox cells after transfection with (E) scrambled (scrb) siRNA (n = 3) or (F) Cdk2 siRNA (n = 3).
(G) Relative Cdk2 transcript levels from scrb siRNA (n = 3) and Cdk2 siRNA transfected cultures (n = 3).
(H) Percentage of PB-T-Neurog1 cell numbers transfected with scrb siRNA (n = 3) or Cdk2 siRNA (n = 3).
(I) Percentage of EdU-labeled cells for scrb siRNA (n = 3) or Cdk2 siRNA (n = 3) transfected cultures.
Error bars denote ±SEM. ∗p < 0.05, ∗∗∗p < 0.001.
To determine whether CDK2 specifically contributes to increased proliferation after NEUROG1 overexpression, we employed Cdk2 small interfering RNA (siRNA). PB-T-Neurog1 + Dox cells were transfected with Cdk2 siRNA or scrambled siRNA before EdU incorporation. Hoechst and EdU labeling was performed on PB-T-Neurog1 + Dox cells transduced with scrambled siRNA (Figure 6E) or Cdk2 siRNA (Figure 6F). Relative to scrambled siRNA, Cdk2 transcript levels decreased after transfection with Cdk2 siRNA (0.39, p < 0.001) (Figure 6G). Cdk2 siRNA transfected cultures showed a decrease in the percentage of cells (39%, p < 0.001) relative to scrambled siRNA (Figure 6H). Similarly, compared with scrambled siRNA control (57%), the percentage of EdU-labeled cells was lower after Cdk2 siRNA treatment (32%, p < 0.05) (Figure 6I). Together, these data suggest that CDK2 functions downstream of NEUROG1 to promote proliferation.
Overexpression of Neurog1 in Differentiating iMOPs Accelerates Differentiation
In pluripotent stem cells, overexpressing NEUROG1 robustly promotes neuronal differentiation (Blanchard et al., 2015, Kim et al., 2004, Velkey and O'Shea, 2013). To find out whether NEUROG1 overexpression promotes the acquisition of neuronal fate in differentiating iMOP cells, we compared PB-T-EGFP + Dox, PB-T-Neurog1 – Dox, and PB-T-Neurog1 + Dox cultures that were undergoing neuronal differentiation. Differentiating PB-T-EGFP + Dox cultures showed that the vast majority of cells expressed EGFP after 3 days of Dox induction. In PB-T-EGFP + Dox cultures, 65.6% of cells were marked with TUBB3 but most did not have neurites (Figure 7A). PB-T-Neurog1 – Dox showed no EGFP expression. Hoechst and TUBB3 labeling revealed that 63.3% expressed the neuronal marker but did not have neurites (Figure 7B). PB-T-Neurog1 + Dox showed EGFP expression, and 92.0% of cells labeled with TUBB3 had bipolar and pseudo-unipolar processes (Figure 7C). These results suggested that NEUROG1 may accelerate the rate at which cells acquire a neuronal cell fate. To determine the time course that cells acquire neuronal morphology, we quantified the percentage of TUBB3 cells after Dox induction for both PB-T-EGFP and PB-T-Neurog1 at different time points (days 0, 1, 3, and 7) after initiating neuronal differentiation. The percentage of TUBB3-labeled cells were determined for PB-T-EGFP + Dox and PB-T-Neurog1 – Dox and PB-T-Neurog1 + Dox cells at individual time points. PB-T-EGFP + Dox and PB-T-Neurog1 – Dox showed similar increases in TUBB3-labeled cells. Overexpression of NEUROG1 in PB-T-Neurog1 showed a faster acquisition of TUBB3-labeled cells. Comparing the cultures, overexpression of NEUROG1 in PB-T-Neurog1 + Dox cultures increased the percentage of TUBB3 labeled at day 1 (p < 0.05) and day 3 (p < 0.01) compared with PB-T-EGFP + Dox and PB-T-Neurog1 – Dox (Figure 7D). To quantify the effects on NEUROG1 downstream targets, we determined relative transcript levels for NeuroD1 and Cdk2 for PB-T-EGFP – Dox, PB-T-Neurog1 – Dox, and PB-T-Neurog1 + Dox cultures. Overexpression of NEUROG1 increased transcript levels for NeuroD1 (3.84, p < 0.05) but not Cdk2 (Figure 7E). These results suggest that overexpression of NEUROG1 accelerates neuronal differentiation.
Figure 7.

Neurog1 Overexpression and Knockdown in Differentiating Neurons
(A) EGFP expression and TUBB3 labeling of PB-T-EGFP + Dox cells (n = 3).
(B) Hoechst, EGFP and TUBB3 labeling of PB-T-Neurog1 – Dox cells (n = 3).
(C) EGFP expression and TUBB3 labeling of PB-T-Neurog1 + Dox cells (n = 3).
(D) Percent of TUBB3-positive cells in PB-T-EGFP + Dox (n = 3), PB-T-Neurog1 – Dox (n = 3), and PB-T-Neurog1 + Dox (n = 3) cells at different time points during differentiation.
(E) Relative transcript levels of NeuroD1 and Cdk2 in differentiating PB-T-EGFP + Dox (n = 3), PB-T-Neurog1 – Dox (n = 3), and PB-T-Neurog1 + Dox (n = 3) after 3 days of Dox induction.
(F and G) Hoechst-, NEUROD1-, and TUBB3-labeled differentiating iMOP cells transduced with (F) scrambled shRNA (n = 3) or (G) Neurog1 shRNA (n = 3).
(H) Percentage of NEUROD1, TUBB3, and NEUROD1/TUBB3 in scrambled (scrb) shRNA (n = 3) and Neurog1 shRNA (n = 3) transduced cells.
(I) Relative transcripts levels of Neurog1, NeuroD1, and Cdk2 after transduction with scrb shRNA (n = 3) or Neurog1 shRNA (n = 3).
Error bars denote ±SEM. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Knockdown of Neurog1 in Post-mitotic Cells Prevents Differentiation
To verify that NEUROG1 is required for neuronal differentiation, we performed Neurog1 shRNA knockdown in differentiating iMOP cells. In scrambled shRNA cells, Hoechst-labeled nuclei showed NEUROD1 expression and TUBB3-labeled cells possessed bipolar and pseudo-unipolar neurites (Figure 7F). In contrast, Neurog1 shRNA knockdown dramatically decreased the number of NEUROD1- and TUBB3-labeled cells (Figure 7G). A decrease in TUBB3-marked cells in scrambled shRNA (89.7%) compared with Neurog1 shRNA (32.1%, p < 0.01) cells was observed. A drastic decrease of NEUROD1-expressing cells was also observed when comparing scrambled shRNA (62.4%) with Neurog1 shRNA (1.7%, p < 0.01) cells. Finally, cells labeled with both TUBB3 and NEUROD1 showed a decrease in scrambled shRNA (60.3%) compared with Neurog1 shRNA (1.2%) cells (Figure 7H). To determine the changes in transcript levels after Neurog1 shRNA knockdown, we performed qPCR. Neurog1 shRNA showed a significant decrease in Neurog1 transcript (0.03, p < 0.001) compared with scrambled shRNA cells. Neurog1 shRNA knockdown also significantly decreased NeuroD1 levels (0.03, p < 0.001) compared with scrambled shRNA control. In contrast, relative Cdk2 transcript levels were similar between scrambled and Neurog1 shRNA cells (Figure 7I). These results suggest that under differentiation conditions, NEUROG1 regulates expression of NEUROD1 during neuronal differentiation, as previously shown (Pan et al., 2012).
Discussion
NEUROG1 Promotes Proliferation or Differentiation Depending on the Cellular Context
In the inner ear, expression of NEUROG1 progenitors from the neurosensory domain as well as in early differentiated neurons (Koundakjian et al., 2007, Ma et al., 1998, Ma et al., 2000) suggests that NEUROG1 functions in both cell types. Bromodeoxyuridine incorporation marks proliferating cells at E9.5 to E11.5 in the developing cochlea but did not mark developing SGNs in Neurog1 mutant animals (Matei et al., 2005). This suggests that Neurog1 plays a role in proliferation during SGN development. However, in pluripotent stem cells, overexpression of Neurog1 has been exclusively attributed to promoting neuronal differentiation (Busskamp et al., 2014, Kim et al., 2004, Velkey and O'Shea, 2013, Zhang et al., 2013). Our findings reconciled these facts, and show that NEUROG1 function can promote proliferation or neuronal differentiation. The latter may in large part be through upregulation of NEUROD1 (Ma et al., 1998, Jahan et al., 2015b)
NEUROG1 Function Correlates with Epigenetic Changes
From the ChIP-qPCR results, NEUROG1 binds to the promoter region of Cdk2 and NeuroD1 in proliferating and differentiating iMOP cells. The differences in enrichment of NEUROG1 at Cdk2 and NeuroD1 may be attributed to differences in the DNA sequence flanking E-box sites (Gordan et al., 2013). Changes in chromatin may also affect the accessibility to these E-box sites and alter binding affinity of NEUROG1. In proliferating iMOP cells, H3K4me3 and H3K27me3 histone marks are present at the promoters of Cdk2 and NeuroD1, suggesting that these genes are in a poised transcriptional state and could be subjected to additional epigenetic or transcriptional regulation. Our results suggest that changes in H3K9ac and H3K9me3 deposition at Cdk2 and NeuroD1 promoters in otic progenitors explain how NEUROG1-dependent transcription increases Cdk2 during proliferation and NeuroD1 during differentiation.
NEUROG1 Promotes Proliferation by Upregulating CDK2
Although NEUROG1 is expressed in delaminating neural precursors of the inner ear (Matei et al., 2005), its function in regulating proliferation has not been described (Doetzlhofer et al., 2004, Nagtegaal et al., 2015). Here we show that NEUROG1 contributes to cellular proliferation in otic progenitors by regulating CDK2 levels. As a member of the cyclin-dependent kinase family, CDK2 plays an important role in promoting cell-cycle progression in neural cell types. In neural stem cells, ablation of Cdk2 impairs proliferation of neural progenitor cells located in the subventricular zone of the adult brain (Jablonska et al., 2007). The involvement of NEUROG1 in proliferation agrees with the observation that some neuroectodermal tumors express NEUROG1 (White et al., 2012). We propose that overexpression of NEUROG1 can promote proliferation by increasing CDK2 levels.
ASCL1 is a proneural TF that potently converts embryonic fibroblasts into functional induced neuronal cells (Chanda et al., 2014, Pang et al., 2011). Similar to our findings, ASCL1 can act in a non-canonical manner to promote proliferation. Characterization of genome-wide ASCL1 binding sites revealed a large number of target genes involved in neuronal differentiation but also in cell-cycle progression (Castro et al., 2011). In the developing embryonic mouse brain, acute deletion of ASCL1 in the subventricular zone of the ventral telencephalon decreased proliferation of intermediate progenitors, suggesting that ASCL1 normally regulates proliferation in these cells (Castro et al., 2011), while overexpression of ASCL1 promotes proliferation (Li et al., 2014). Overexpression of NEUROG2, a member of the neurogenin family, has been extensively used to generate functional neurons from human pluripotent stem cells (Zhang et al., 2013). NEUROG2 has also been shown to transiently promote a rapidly self-renewing state in developing cortical neural progenitor cells (Hagey and Muhr, 2014). We propose that NEUROG1, like the aforementioned proneural TFs, has a non-canonical function in promoting proliferation of otic progenitors.
Decrease in CDK2 levels by NEUROG1 knockdown resulted in fewer cells entering S phase. We propose that compensation from other CDKs allows continued proliferation of iMOP cells, albeit at a much slower rate. This is similar to the observation that mouse embryos lacking all interphase CDKs (CDK2, CDK3, CDK4, and CDK6) still develop to mid-gestation because Cdk1 alone can drive the cell cycle by complexing with all the phase-specific cyclins (Santamaria et al., 2007). Although CDKs are redundant, mutations for one CDK or cyclin may show cell-type- or tissue-specific defects or abnormalities at specific stages of development. A decrease in CDK2 levels in Neurog1 mutant animals could result in decreased expansion of the progenitor pool during inner ear development and contribute to the absence of cochlear neurons in Neurog1 mutant animals.
Regulation of NEUROG1-Dependent Transcription through Epigenetic Modulation
Under different culture conditions, NEUROG1 is essential for cell proliferation and neuronal differentiation of iMOP cells. These results have implications in using NEUROG1 for therapeutic purposes. Our results suggest that the epigenetic status of NEUROG1 target genes affect transcription. Understanding how overexpression of NEUROG1 in different cell types that harbor different epigenetic landscapes could affect the design of cell conversion-based therapies. The conversion and directed differentiation of cell types into induced neuronal cells using TFs have varying conversion efficiencies (Mertens et al., 2016) which could partly be attributed to the epigenetic status of target genes. The epigenetic status of Cdk2 or NeuroD1 promoters may affect the number of proliferating or differentiating cells after NEUROG1 overexpression. Controlling the epigenetic state of target genes and changing it to either the permissive or repressive chromatin state before overexpression of NEUROG1 will help to enhance differentiation and prevent unwanted proliferation.
Experimental Procedures
Cell Culture and Generation of Stable iMOP Cell Lines
iMOP cells were cultured as described by Jadali et al. (2016). iMOP doxycycline inducible stable cell lines were generated using the piggyBAC transposon system (Li et al., 2013).
Immunofluorescence Staining
Immunostaining of iMOP cells was previously described (Kwan et al., 2015). The antibodies used for immunostaining are listed in Table S1.
ChIP-Seq and ChIP-qPCR
ChIP-seq was accomplished as described by Kwan et al. (2015) using antibodies listed in Table S1. ChIP-qPCR was done on immunoprecipitated DNA using primer pairs that amplify the promoter region in the specified genes. Primer pairs are listed in Table S2.
mRNA Expression Analysis
Total RNA was extracted using TRIzol reagent (Life Technologies) according to the manufacturer’s instructions. RNA (1 μg) was used to make cDNA using the qScript cDNA synthesis kit (Quanta Biosciences) according to the manufacturer’s instructions. Relative levels of cDNA were measured by real-time qPCR using SYBR green Taq polymerase (Life Technologies) for 40 cycles of 95°C for 15 s, and 60°C for 1 min using the StepOnePlus real-time PCR machine. Three biological replicates, each with technical triplicates were used for each qPCR sample. Primer pairs used for qPCR are listed in Table S3.
Use of shRNA, siRNA, and Small Molecule
Three different shRNAs designed to target murine Neurog1 transcript in the pLKO.1 containing puromycin resistance cassettes were purchased from Sigma and tested for their ability to knock down Neurog1 transcript. shRNA sequences against Neurog1 are listed in Table S4. CDK2 inhibitors ricovitine (Sigma) and K03861 (Selleckchem) were resuspended according to the manufacturers’ protocols. Inhibitors were used to culture cells for 3 days at the specified concentrations.
For extended and detailed procedures, see Supplemental Experimental Procedures.
Author Contributions
Z.S., A.J., and K.Y.K. designed, performed, and interpreted the experiments. K.Y.K. performed bioinformatics analysis. Z.S. and K.Y.K. wrote the manuscript. B.F. edited and amended the manuscript.
Acknowledgments
We would like to thank Dr. James Rini for providing the PiggyBAC transposon plasmids. K.Y.K. is supported in part by the Duncan and Nancy MacMillan Faculty Development Chair Endowment Fund and R01 DC015000.
Published: October 12, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and nine tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2017.09.011.
Accession Numbers
The H3K4me3 and H3K27me3 ChIP-seq data have been deposited in the GEO under GEO: GSE102733.
Supplemental Information
References
- Alexander L.T., Mobitz H., Drueckes P., Savitsky P., Fedorov O., Elkins J.M., Deane C.M., Cowan-Jacob S.W., Knapp S. Type II inhibitors targeting CDK2. ACS Chem. Biol. 2015;10:2116–2125. doi: 10.1021/acschembio.5b00398. [DOI] [PubMed] [Google Scholar]
- Allan R.S., Zueva E., Cammas F., Schreiber H.A., Masson V., Belz G.T., Roche D., Maison C., Quivy J.P., Almouzni G. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature. 2012;487:249–253. doi: 10.1038/nature11173. [DOI] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Basch M.L., Ohyama T., Segil N., Groves A.K. Canonical Notch signaling is not necessary for prosensory induction in the mouse cochlea: insights from a conditional mutant of RBPjkappa. J. Neurosci. 2011;31:8046–8058. doi: 10.1523/JNEUROSCI.6671-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bilodeau S., Kagey M.H., Frampton G.M., Rahl P.B., Young R.A. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009;23:2484–2489. doi: 10.1101/gad.1837309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard J.W., Eade K.T., Szucs A., Lo Sardo V., Tsunemoto R.K., Williams D., Sanna P.P., Baldwin K.K. Selective conversion of fibroblasts into peripheral sensory neurons. Nat. Neurosci. 2015;18:25–35. doi: 10.1038/nn.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busskamp V., Lewis N.E., Guye P., Ng A.H., Shipman S.L., Byrne S.M., Sanjana N.E., Murn J., Li Y., Li S. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014;10:760. doi: 10.15252/msb.20145508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro D.S., Martynoga B., Parras C., Ramesh V., Pacary E., Johnston C., Drechsel D., Lebel-Potter M., Garcia L.G., Hunt C. A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev. 2011;25:930–945. doi: 10.1101/gad.627811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S., Ang C.E., Davila J., Pak C., Mall M., Lee Q.Y., Ahlenius H., Jung S.W., Sudhof T.C., Wernig M. Generation of induced neuronal cells by the single reprogramming factor ASCL1. Stem Cell Reports. 2014;3:282–296. doi: 10.1016/j.stemcr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Jongkamonwiwat N., Abbas L., Eshtan S.J., Johnson S.L., Kuhn S., Milo M., Thurlow J.K., Andrews P.W., Marcotti W. Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature. 2012;490:278–282. doi: 10.1038/nature11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetzlhofer A., White P.M., Johnson J.E., Segil N., Groves A.K. In vitro growth and differentiation of mammalian sensory hair cell progenitors: a requirement for EGF and periotic mesenchyme. Dev. Biol. 2004;272:432–447. doi: 10.1016/j.ydbio.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Du J., Widlund H.R., Horstmann M.A., Ramaswamy S., Ross K., Huber W.E., Nishimura E.K., Golub T.R., Fisher D.E. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell. 2004;6:565–576. doi: 10.1016/j.ccr.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Evsen L., Sugahara S., Uchikawa M., Kondoh H., Wu D.K. Progression of neurogenesis in the inner ear requires inhibition of Sox2 transcription by neurogenin1 and neurod1. J. Neurosci. 2013;33:3879–3890. doi: 10.1523/JNEUROSCI.4030-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B., Eberl D.F., Beisel K.W. The role of bHLH genes in ear development and evolution: revisiting a 10-year-old hypothesis. Cell. Mol. Life Sci. 2010;67:3089–3099. doi: 10.1007/s00018-010-0403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B., Jahan I., Pan N., Elliott K.L. Evolving gene regulatory networks into cellular networks guiding adaptive behavior: an outline how single cells could have evolved into a centralized neurosensory system. Cell Tissue Res. 2015;359:295–313. doi: 10.1007/s00441-014-2043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamez B., Rodriguez-Carballo E., Ventura F. BMP signaling in telencephalic neural cell specification and maturation. Front. Cell. Neurosci. 2013;7:87. doi: 10.3389/fncel.2013.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan R., Shen N., Dror I., Zhou T., Horton J., Rohs R., Bulyk M.L. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013;3:1093–1104. doi: 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal S.I., Elgin S.C. Heterochromatin: new possibilities for the inheritance of structure. Curr. Opin. Genet. Dev. 2002;12:178–187. doi: 10.1016/s0959-437x(02)00284-8. [DOI] [PubMed] [Google Scholar]
- Hagey D.W., Muhr J. Sox2 acts in a dose-dependent fashion to regulate proliferation of cortical progenitors. Cell Rep. 2014;9:1908–1920. doi: 10.1016/j.celrep.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Huang C., Chan J.A., Schuurmans C. Proneural bHLH genes in development and disease. Curr. Top. Dev. Biol. 2014;110:75–127. doi: 10.1016/B978-0-12-405943-6.00002-6. [DOI] [PubMed] [Google Scholar]
- Jablonska B., Aguirre A., Vandenbosch R., Belachew S., Berthet C., Kaldis P., Gallo V. Cdk2 is critical for proliferation and self-renewal of neural progenitor cells in the adult subventricular zone. J. Cell Biol. 2007;179:1231–1245. doi: 10.1083/jcb.200702031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadali A., Kwan K.Y. Activation of PI3K signaling prevents aminoglycoside-induced hair cell death in the murine cochlea. Biol. Open. 2016;5:698–708. doi: 10.1242/bio.016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadali A., Song Z., Laureano A.S., Toro-Ramos A., Kwan K. Initiating differentiation in immortalized multipotent otic progenitor cells. J. Vis. Exp. 2016 doi: 10.3791/53692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahan I., Pan N., Kersigo J., Fritzsch B. Neurod1 suppresses hair cell differentiation in ear ganglia and regulates hair cell subtype development in the cochlea. PLoS One. 2010;5:e11661. doi: 10.1371/journal.pone.0011661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahan I., Pan N., Elliott K.L., Fritzsch B. The quest for restoring hearing: Understanding ear development more completely. Bioessays. 2015;37:1016–1027. doi: 10.1002/bies.201500044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahan I., Pan N., Kersigo J., Fritzsch B. Neurog1 can partially substitute for Atoh1 function in hair cell differentiation and maintenance during organ of Corti development. Development. 2015;142:2810–2821. doi: 10.1242/dev.123091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmodiya K., Krebs A.R., Oulad-Abdelghani M., Kimura H., Tora L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics. 2012;13:424. doi: 10.1186/1471-2164-13-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Yoon Y.S., Kim J.W., Jung M., Kim S.U., Lee Y.D., Suh-Kim H. Neurogenin1 is sufficient to induce neuronal differentiation of embryonal carcinoma P19 cells in the absence of retinoic acid. Cell. Mol. Neurobiol. 2004;24:343–356. doi: 10.1023/B:CEMN.0000022767.74774.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koundakjian E.J., Appler J.L., Goodrich L.V. Auditory neurons make stereotyped wiring decisions before maturation of their targets. J. Neurosci. 2007;27:14078–14088. doi: 10.1523/JNEUROSCI.3765-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kujawa S.G., Liberman M.C. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan K.Y., Shen J., Corey D.P. C-MYC transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem Cell Reports. 2015;4:47–60. doi: 10.1016/j.stemcr.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Michael I.P., Zhou D., Nagy A., Rini J.M. Simple piggyBac transposon-based mammalian cell expression system for inducible protein production. Proc. Natl. Acad. Sci. USA. 2013;110:5004–5009. doi: 10.1073/pnas.1218620110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Mattar P., Dixit R., Lawn S.O., Wilkinson G., Kinch C., Eisenstat D., Kurrasch D.M., Chan J.A., Schuurmans C. RAS/ERK signaling controls proneural genetic programs in cortical development and gliomagenesis. J. Neurosci. 2014;34:2169–2190. doi: 10.1523/JNEUROSCI.4077-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S., Kaldis P. Loss of Cdk2 and Cdk4 induces a switch from proliferation to differentiation in neural stem cells. Stem Cells. 2012;30:1509–1520. doi: 10.1002/stem.1114. [DOI] [PubMed] [Google Scholar]
- Lunn J.S., Pacut C., Stern E., Sakowski S.A., Velkey J.M., O'Shea S., Feldman E.L. Intraspinal transplantation of neurogenin-expressing stem cells generates spinal cord neural progenitors. Neurobiol. Dis. 2012;46:59–68. doi: 10.1016/j.nbd.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Chen Z., del Barco Barrantes I., de la Pompa J.L., Anderson D.J. neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron. 1998;20:469–482. doi: 10.1016/s0896-6273(00)80988-5. [DOI] [PubMed] [Google Scholar]
- Ma Q., Anderson D.J., Fritzsch B. Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. J. Assoc. Res. Otolaryngol. 2000;1:129–143. doi: 10.1007/s101620010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matei V., Pauley S., Kaing S., Rowitch D., Beisel K.W., Morris K., Feng F., Jones K., Lee J., Fritzsch B. Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev. Dyn. 2005;234:633–650. doi: 10.1002/dvdy.20551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A., Fornes O., Arenillas D.J., Chen C.Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016;44:D110–D115. doi: 10.1093/nar/gkv1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L., Borgne A., Mulner O., Chong J.P., Blow J.J., Inagaki N., Inagaki M., Delcros J.G., Moulinoux J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Mertens J., Marchetto M.C., Bardy C., Gage F.H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 2016;17:424–437. doi: 10.1038/nrn.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagtegaal A.P., Rainey R.N., van der Pluijm I., Brandt R.M., van der Horst G.T., Borst J.G., Segil N. Cockayne syndrome group B (Csb) and group a (Csa) deficiencies predispose to hearing loss and cochlear hair cell degeneration in mice. J. Neurosci. 2015;35:4280–4286. doi: 10.1523/JNEUROSCI.5063-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan N., Kopecky B., Jahan I., Fritzsch B. Understanding the evolution and development of neurosensory transcription factors of the ear to enhance therapeutic translation. Cell Tissue Res. 2012;349:415–432. doi: 10.1007/s00441-012-1454-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z.P., Yang N., Vierbuchen T., Ostermeier A., Fuentes D.R., Yang T.Q., Citri A., Sebastiano V., Marro S., Sudhof T.C. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raft S., Koundakjian E.J., Quinones H., Jayasena C.S., Goodrich L.V., Johnson J.E., Segil N., Groves A.K. Cross-regulation of Ngn1 and Math1 coordinates the production of neurons and sensory hair cells during inner ear development. Development. 2007;134:4405–4415. doi: 10.1242/dev.009118. [DOI] [PubMed] [Google Scholar]
- Rea S., Eisenhaber F., O'Carroll D., Strahl B.D., Sun Z.W., Schmid M., Opravil S., Mechtler K., Ponting C.P., Allis C.D. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Rivolta M.N. Developing a stem cell-based therapy for the treatment of hearing loss. Hearing Balance Commun. 2015;13:148–152. [Google Scholar]
- Rosner M.H., Vigano M.A., Ozato K., Timmons P.M., Poirier F., Rigby P.W., Staudt L.M. A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature. 1990;345:686–692. doi: 10.1038/345686a0. [DOI] [PubMed] [Google Scholar]
- Ross S.E., Greenberg M.E., Stiles C.D. Basic helix-loop-helix factors in cortical development. Neuron. 2003;39:13–25. doi: 10.1016/s0896-6273(03)00365-9. [DOI] [PubMed] [Google Scholar]
- Rubel E.W., Fritzsch B. Auditory system development: primary auditory neurons and their targets. Annu. Rev. Neurosci. 2002;25:51–101. doi: 10.1146/annurev.neuro.25.112701.142849. [DOI] [PubMed] [Google Scholar]
- Rugg-Gunn P.J., Cox B.J., Ralston A., Rossant J. Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc. Natl. Acad. Sci. USA. 2010;107:10783–10790. doi: 10.1073/pnas.0914507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford M.A., Moser T. The ribbon synapse between type I spiral ganglion neurons and inner hair cells. In: Dabdoub A., Fritsch B., Popper A.N., Fay R.R., editors. Vol. 52. Springer Handbook of Auditory Research; 2016. pp. 117–156. (The Primary Auditory Neurons of the Mammalian Cochlea). [Google Scholar]
- Santamaria D., Barriere C., Cerqueira A., Hunt S., Tardy C., Newton K., Caceres J.F., Dubus P., Malumbres M., Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- Scholer H.R., Ruppert S., Suzuki N., Chowdhury K., Gruss P. New type of POU domain in germ line-specific protein Oct-4. Nature. 1990;344:435–439. doi: 10.1038/344435a0. [DOI] [PubMed] [Google Scholar]
- Shi F., Edge A.S. Prospects for replacement of auditory neurons by stem cells. Hear. Res. 2013;297:106–112. doi: 10.1016/j.heares.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Velkey J.M., O'Shea K.S. Expression of Neurogenin 1 in mouse embryonic stem cells directs the differentiation of neuronal precursors and identifies unique patterns of down-stream gene expression. Dev. Dyn. 2013;242:230–253. doi: 10.1002/dvdy.23920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P., Tee W.W., Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White P.M., Stone J.S., Groves A.K., Segil N. EGFR signaling is required for regenerative proliferation in the cochlea: conservation in birds and mammals. Dev. Biol. 2012;363:191–200. doi: 10.1016/j.ydbio.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.D., Willson T.A., Wakefield M.J., Trounson E., Hilton D.J., Blewitt M.E., Oshlack A., Majewski I.J. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011;39:7415–7427. doi: 10.1093/nar/gkr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.