

Summary
Neurotoxic disease can mimic many common neurologic disease states, including parkinsonism, myelopathy, neuropathy, and encephalopathy. Accurate diagnosis and appropriate treatment may result in a favorable outcome. This review highlights 5 areas of neurotoxicology for which there is an emerging understanding of disease processes or patterns of exposure, including 3 specific metal toxicities (manganism, zinc-induced copper deficiency, and cobalt-chromium neuropathy). Toxin-induced posterior reversible encephalopathy syndrome is more widely recognized and reported in association with an ever-growing list of drugs. Two new categories of street drugs, synthetic cathinones and cannabinoids, have been identified as public health threats due to their popularity, availability, and severity of toxicity.
Accurate diagnosis and appropriate treatment of neurotoxic disease may result in a favorable outcome. This review highlights 5 areas of neurotoxicology for which there is an emerging understanding of disease processes or patterns of exposure.
Methcathinone abuse and manganese neurotoxicity
Exposure sources and pathophysiology
Cathinone, originally derived from the khat plant (Catha edulis), is a well-known psychostimulant. The leaves and twigs of the khat plant are chewed for their amphetamine-like euphoric effects that result from release of catecholamines from presynaptic storage sites. The designer drug methcathinone is an N-methyl analog of cathinone. Methcathinone (also called “Jeff,” “Mulka,” “Murtsovka,” “Cat,” “M-Cat,” “Ephedrone”) is manufactured by the oxidation of substances like ephedrine, pseudoephedrine, and phenylpropanolamine: constituents of various over-the-counter cough remedies. In Russia and Eastern Europe, the preferred oxidizing agent is potassium permanganate. This oxidizing agent is the source of manganese exposure.
Clinical features
A distinctive extrapyramidal syndrome, likely due to manganese toxicity, has been reported in IV methcathinone users.1-5 The duration of drug abuse does not correlate with severity of the extrapyramidal manifestations. A parkinsonian syndrome does not develop when users prepare the drug using potassium or sodium dichromate as the oxidation agent instead of potassium permanganate. Reported comorbidities include hepatitis C and HIV positivity.3 Key features of this symmetric, levodopa-unresponsive, akinetic-rigid syndrome include a gait disturbance (particularly difficulty walking backwards) and hypophonic dysarthria. A “cock walk” gait (walking on the balls of the feet), similar to that seen in mine workers with manganese toxicity, has been reported.3,4 Mild executive dysfunction or emotional lability may be present.4 Rest tremors are not common. Dystonia is commonly seen.4,5
Diagnostic testing and treatment
Blood manganese levels may be elevated but do not correlate with clinical severity or MRI findings. MRI may show a symmetric T1 hyperintensity involving the globus pallidus, striatum, and substantia nigra (figure 1). Fluorodopa PET suggests relative sparing of the nigrostriatal dopaminergic system.1 SPECT imaging in some former ephedrone users has shown a normal pattern of tracer uptake.4 Diffusion tensor imaging in some of these patients has shown widespread white matter damage with greatest severity of damage in executive motor areas.6
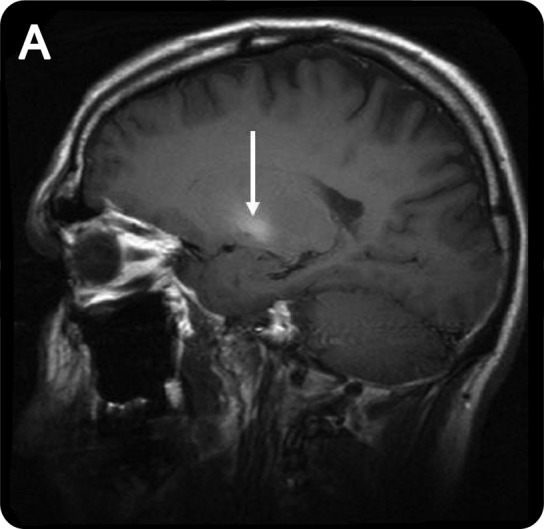
MRI in manganese neurotoxicity
Figure 1. Sagittal T1-weighted MRI shows the increased pallidal T1 signal characteristic of manganese deposition.
Cessation of methcathinone use may be accompanied by improvement or resolution of the MRI abnormalities.3 Objective clinical improvement is generally not seen, and there is usually little, if any, response to dopamine replacement. Delayed progression despite cessation of use has been reported.4
New drugs of abuse: Cathinones and cannabinoids
Within the past 2 years, the United States has seen a dramatic increase in the number of unregulated substances with psychoactive properties and abuse potential. They are frequently legal, at least for a time before the chemical substance is identified and outlawed, and are not detected on traditional urine drug screens. In addition, these “legal highs” can be obtained from gas stations and head shops and are readily available on the Internet. The packaging is frequently labeled “not intended for human consumption” in order to circumvent laws banning similar substances. Health care professionals, including neurologists, need to be aware of the acute effects of these psychoactive compounds. In many cases, the substances are too new for a clear understanding of potential chronic effects or withdrawal syndromes. The synthetic cathinones and cannabinoids are discussed here in detail, as they have been identified by the American Association of Poison Control Centers as “emerging public health threats.”7
Synthetic cathinones: The new “amphetamines”
Exposure sources and pathophysiology
Methcathinone, the first synthetic cathinone, has been discussed above in the context of manganese toxicity that can occur when methcathinone is synthesized in the presence of potassium permanganate. Newer synthetic cathinones, such as mephedrone, methylone, and 3,4-methylenedioxypyrovalerone (MDPV), are now available in the United States and are commonly called “bath salts.” These 3, the most common synthetic cathinones, have been temporarily listed by the US Drug Enforcement Agency (DEA) as schedule I substances as of October 2011. However, many other synthetic cathinones (e.g., butylone, methedrone, 3- and 4-fluoromethcathinone) remain legal. While the packaging may imply that they are to be used in a bath and the powdered contents appear similar to traditional bath salts, they are well known to contain drugs of abuse.
The synthetic cathinones are ketophenethylamines and are structurally similar to amphetamines. They increase presynaptic release of serotonin, dopamine, and norepinephrine and decrease reuptake of these same neurotransmitters.8 These substances are commonly ingested or nasally insufflated, although inhalation and injection have been described.
Clinical features
Onset of effects is typically around 30 minutes with duration of a few hours, although some effects may be prolonged. Initial effects of these substances include euphoria and agitation. User-reported neurologic symptoms include aggressiveness, bruxism, dizziness, headache, memory loss, and tremor. Medical providers additionally report dystonia, hyperreflexia, mydriasis, myoclonus, and seizures.9 The constellation of findings is typical of sympathomimetic syndrome: tachycardia, hypertension, hyperthermia, and agitation. Serotonin syndrome10 and myocarditis11 have been reported as well.
Diagnostic testing and treatment
Diagnosis is made by history and physical examination. The synthetic cathinones are not detected by usual immunoassays for amphetamines, although some may be detected as a false-positive methamphetamine screen.8 Some commercial laboratories have developed assays for quantitative testing of mephedrone, methylone, and MDPV.
There is no antidote for cathinone toxicity. Treatment is symptomatic management, with benzodiazepines for agitation, seizures, tachycardia, or hypertension. Cyproheptadine may be helpful if concern for serotonin syndrome exists. Cooling measures may be required for severe hyperthermia that does not improve with sedation.
Synthetic cannabinoids: The new “marijuana”
Exposure sources and pathophysiology
Spice and K2 are the most common brand names for the herbal marijuana alternatives that have been known drugs of abuse in Europe since the early 2000s. The neuropsychiatric effects may be due to a combination of effects of the synthetic cannabinoid itself, in addition to the herbal ingredients used as a substrate. The synthetic cannabinoids are molecules designed for research on cannabinoid receptors in an attempt to harness the potentially therapeutic benefits of tetrahydrocannabinol (Δ9-THC) and avoid the neuropsychiatric adverse effects. As a consequence of this research, hundreds of these synthetic substances have been described. In general, synthetic cannabinoids appear to bind more effectively at cannabinoid receptors (CB1 and CB2) than does Δ9-THC,12 resulting in more of a stimulant effect. In March 2011, the DEA temporarily listed 5 of the synthetic cannabinoids as schedule I drugs (JWH-018, JWH-073, JWH-200, CP 47-497, and CP 47-497 C8 homologue). Some states have banned additional compounds. Given the popularity of the herbal marijuana alternatives, it is likely that the trend will be toward use of other synthetic cannabinoids that remain legal.
Clinical features
There are scant data regarding onset and duration of symptoms, and these may vary by specific compound. Neuropsychiatric effects of use include anxiety, agitation, paranoia, and delusions, as well as psychosis. Clinical signs may include tachycardia, diaphoresis, hypokalemia, and seizures.13,14 Myocardial infarction has been reported in teenagers after stated use of K2.15
Diagnostic testing and treatment
The synthetic cannabinoids are structurally dissimilar to Δ9-THC, so will not react with the standard marijuana screen on a urine drug test. Some commercial laboratories have developed assays for testing of specific cannabinoids in blood or urine. Management is principally symptomatic, as there is no antidote for this toxicity. Benzodiazepines should be administered for agitation, anxiety, or seizure.
Zinc-induced copper deficiency and neurologic disease
Exposure sources and pathophysiology
Excess zinc ingestion is a well-recognized cause of copper deficiency (figure 2A). In addition to the common use of zinc in the prevention or treatment of common colds and sinusitis, zinc therapy has been used, often empirically, for conditions like acrodermatitis enteropathica, decubitus ulcers, sickle cell disease, celiac disease, glucagonoma, hepatic encephalopathy, psychosis, memory impairment, diarrhea, myoclonic epilepsy, and acne. A prior history of gastric surgery is an additional risk factor, as copper is absorbed in the stomach and proximal small intestine.

Zinc toxicity and copper deficiency and MRI in copper deficiency myelopathy
Figure 2. (A) Excretion of copper into the gastrointestinal tract is the major pathway that regulates copper homeostasis and prevents deficiency or toxicity. Excessive zinc ingestion is a well-recognized cause of copper deficiency. The zinc-induced inhibition of copper absorption could be the result of competition for a common transporter or a consequence of induction of metallothionein in enterocytes. Zinc causes an upregulation of metallothionein production in the enterocytes. Metallothionein has a higher binding affinity for copper than for zinc. Copper is retained within the enterocytes and lost as the intestinal cells are sloughed off. Failure to mobilize absorbed copper from intestinal cells forms the basis of Menkes disease.1 In Wilson disease there is decreased incorporation of copper into ceruloplasmin (2a) and impaired biliary excretion of copper (2b). Alb = albumin; Cp = ceruloplasmin; Cu = copper; cyt c ox = cytochrome c oxidase; M = metallothionein; SOD = superoxide dismutase; Zn = zinc. From Noseworthy JH. Fifty neurologic cases from Mayo Clinic. New York: Oxford University Press; 2004. Used with permission of Mayo Foundation for Medical Education and Research, all rights reserved. (B) Sagittal (B.1) and axial (B.2) T2-weighted MRI in a patient with copper deficiency myelopathy showing increased signal involving the dorsal column in the cervical cord. From Kumar N, Ahlskog JE, Klein CJ, Port JD. Imaging features of copper deficiency myelopathy: a study of 25 cases. Neuroradiology 2006;48:78–83.36 Used with kind permission from Springer Science+Business Media B.V.
Unusual sources of excess zinc have included patients who used excessive amounts of zinc-containing denture cream for long periods16,17 and patients swallowing zinc-containing coins.18 Excess use of zinc-containing denture creams has been implicated in patients with zinc-induced copper deficiency–related myeloneuropathy.16,17 A recent report suggests that patients with hyperzincemia of unknown cause and copper deficiency myeloneuropathy may have had denture cream use as a possible source that was not specifically evaluated.17 In this report, the use of denture fixatives in 11 patients with myeloneuropathy–hypocupremia–hyperzincemia was evaluated and the study noted a history of poorly fitting dentures and application of excessive amounts of denture adhesives in all. In some recent reports, the treatment of Wilson disease with zinc has been accompanied by neurologic manifestations related to zinc excess/copper deficiency.19,20 Individual susceptibility may play a role in hyperzincemia-induced hypocupremia and related neurologic manifestations. The presence of multiple risk factors can increase the chances of development of a clinically significant deficiency state.
Clinical features
Hematologic manifestations of copper deficiency include anemia and neutropenia. Typical bone marrow findings include a left shift in granulocytic and erythroid maturation with cytoplasmic vacuolization in erythroid and myeloid precursors and the presence of ringed sideroblasts. Copper deficiency–associated myelopathy has been well described in various animal species. Often seen in ruminants, it has been called swayback or enzootic ataxia. Neurologic manifestations of acquired copper deficiency in humans have been increasingly recognized over the past decade.21-24 Copper deficiency results in a myeloneuropathy that resembles the subacute combined degeneration seen with vitamin B12 deficiency.
Diagnostic testing and treatment
Serum and 24-hour urine specimen can be obtained for copper and zinc levels. Neuroimaging may show an increased T2 signal involving the dorsal column (figure 2B). Identification and removal of the source of zinc exposure is the primary step in management.
Neuropathy associated with cobalt-chromium metallosis after hip replacement
Exposure sources and pathophysiology
Total hip arthroplasty may be complicated by corrosion and disassembly of the components. Metallosis is a very rare complication of arthroplasty with metallic prostheses. It refers to chronic infiltration of metallic wear debris into the periprosthetic bony and soft tissues.25 Reactive chronic inflammatory changes are seen. Wear debris can rarely cause systemic intoxication by prosthetic metallic materials, mostly by cobalt-chromium. Though very rare, this topic has gained a lot of attention in recent years.
Clinical features
Neurologic complications of metallosis are restricted to a few case reports and include peripheral neuropathy, at times associated with impaired vision, hearing, or both.26-29 Hypothyroidism or cardiomyopathy may be present.28,29
Diagnostic testing and treatment
Blood cobalt and chromium levels are elevated. Improvement in neurologic manifestations may result from revision surgery and removal of prosthetic component. Chelation has been reported to reduce metal levels but not improve neurologic manifestations.27
A recent report described detailed analysis of the peripheral neuropathy seen in a patient with metallosis after hip arthroplasty with a cobalt-chromium alloy prosthesis.28 The neuropathy was painful, distally prominent, and accompanied by impaired distal position perception, absent deep tendon reflexes, and slight distal weakness. Bilateral sensorineural hearing impairment was present. Sensory nerve action potentials were absent. Motor conduction studies were normal. The patient had marked elevations of blood cobalt and chromium. A sural nerve biopsy showed moderate axonal degeneration without significant inflammation. The authors speculate that absence of inflammation may have been related to the low-dose prednisone that the patient was receiving. Cobalt and chromium levels in the sural nerve were increased compared to controls. X-ray of the hip showed a deformity in the cobalt-chromium head of the prosthesis (figure 3A). During surgery, the metallosis was found to have spread from the right hip joint to the thigh (figure 3, B and C). Revision arthroplasty and removal of soft tissue contaminated with metal debris was accompanied by gradual clinical and electrophysiologic improvement, and decrease in blood levels of cobalt and chromium.

X-ray and surgery for removal of the right hip prosthesis in a patient with cobalt-chromium metallosis-related peripheral neuropathy
Figure 3. (A) X-ray of the right hip from the patient reported in reference XX shows a deformed head dislocated from the center of the cup (arrow) and abnormal densities around the hip joint spreading to the lateral portion of the right thigh (arrowheads). (B) During surgery for removal of the right hip prosthesis, a coal tar–like fluid was obtained from the space between the right iliotibial band and lateral vastus muscle. (C) Discolored (black) right vastus lateralis. Reproduced with permission from John Wiley and Sons.29
Drug-induced posterior reversible encephalopathy syndrome
Exposure sources and pathophysiology
Posterior reversible encephalopathy syndrome (PRES) is a clinicoradiologic syndrome that may be caused by hypertension alone, but often occurs due to toxicity of immunosuppressive and chemotherapeutic drugs as well. The immunosuppressive drugs most commonly implicated in PRES are tacrolimus and cyclosporine. PRES has also been reported in association with sirolimus, methotrexate, interferon, rituximab, bevacizumab, sorafenib, sunitinib, fingolimod, and IV immunoglobulin.30 The chemotherapeutic agents reported to cause PRES are cisplatin, cyclophosphamide, cytarabine, doxorubicin, etoposide, gemcitabine, and vincristine.30 There does not seem to be a clear dose-response relationship, as PRES has been reported in patients with cyclosporine and tacrolimus levels within the therapeutic range.31 The immunosuppressive and chemotherapeutic agents predispose to PRES via endothelial dysfunction and resulting breakdown of the blood–brain barrier. Other drugs can contribute to PRES by causing acute hypertension. These drugs include the sympathomimetics (e.g., cocaine, amphetamines), serotonergics (e.g., tricyclic antidepressants and selective serotonin reuptake inhibitors), α-agonists (e.g., midodrine), and mineralocorticoids (e.g., fludrocortisone).32
Reversible vasogenic edema in the setting of relative hypertension and dysfunction of the blood–brain barrier are the hallmarks of PRES pathophysiology. Secondary effects include vasospasm, microvascular thrombosis leading to infarction, and cerebral hemorrhage. However, it is not entirely clear if the edema is primarily caused by hyperperfusion, hypoperfusion, or a combination of both. Endothelial dysfunction disrupting the integrity of the blood–brain barrier may be secondary to an immunologic process, a direct effect of hypertension, or a toxic effect of immunosuppressants and chemotherapeutic agents. Thus, if the hypertension is profound enough, it alone can result in endothelial cell damage and produce the vasogenic edema of PRES. However, if the damage to endothelial cells occurs via systemic illness or toxic effects of medications, PRES may occur at a lower blood pressure.
Clinical features
Typical symptoms of PRES include confusion, headache, vision change, and nausea. Onset occurs over hours to days. Seizures may be the first presenting symptom or may occur later in the course. About three-fourths of patients will have moderate to severe hypertension.33
Diagnostic testing and treatment
Levels of immunosuppressant drugs should be obtained to guide therapy, but the level itself does not correspond to degree of neurotoxicity.31 Brain imaging typically demonstrates symmetric occipito-parietal edema involving the white matter to a greater degree than the gray matter (figure 4) that resolves with treatment. However, the posterior regions of the frontal and temporal lobes, in addition to the thalamus, basal ganglia, brainstem, and cerebellum, may be involved as well. MRI findings are generally consistent with vasogenic edema, although focal cytotoxic edema may occur in the setting of subsequent infarction.
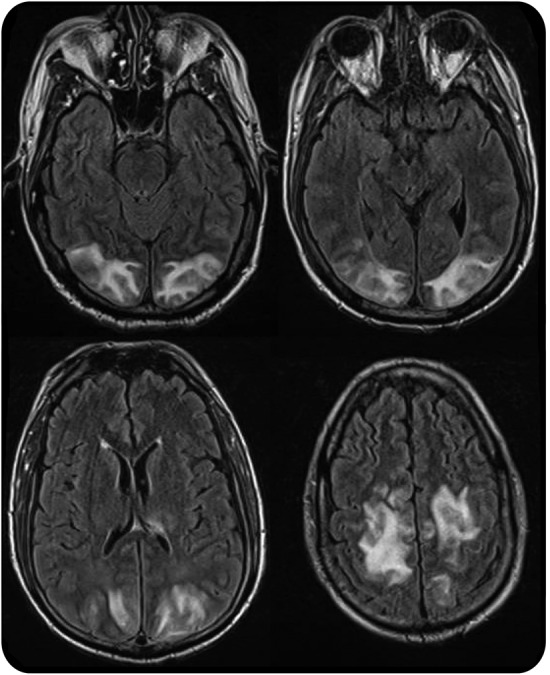
MRI in posterior reversible encephalopathy syndrome
Figure 4. Axial fluid-attenuated inversion recovery images demonstrate symmetric hyperintensities in the occipital and parietal lobes.
The initial step in treatment of PRES is identification of the etiologic factors. Decreased dose or discontinuation of immunosuppressive or chemotherapeutic agents, in addition to measured treatment of hypertension and treatment of seizures, is the key in managing toxin-induced PRES. While no data are available on optimal blood pressure management, a 20% decrease in the mean arterial pressure is a reasonable goal of initial management.34 Hypertension secondary to sympathomimetic or serotonin syndromes may respond well to the symptomatic use of benzodiazepines for agitation, motor hyperactivity, or seizures. Transplant patients requiring immunosuppression may improve with decrease in dose or transition to an alternative agent.35 Prognosis is usually good, with recovery over the course of a few days, after appropriate management.
Neurotoxicology: Five new things
Methcathinone abuse and manganese neurotoxicity
New drugs of abuse: Cathinones and cannabinoids
Zinc-induced copper deficiency myeloneuropathy
Cobalt-chromium metallosis neuropathy
Drug-induced posterior reversible encephalopathy syndrome
Correspondence to: laumjone@iupui.edu
Footnotes
Correspondence to: laumjone@iupui.edu
REFERENCES
- 1.de Bie RM, Gladstone RM, Strafella AP, Ko JH, Lang AE. Manganese-induced Parkinsonism associated with methcathinone (ephedrone) abuse. Arch Neurol 2007;64:886–889. [DOI] [PubMed]
- 2.Sanotsky Y, Lesyk R, Fedoryshyn L, Komnatska I, Matviyenko Y, Fahn S. Manganic encephalopathy due to “ephedrone” abuse. Mov Disord 2007;22:1337–1343. [DOI] [PubMed]
- 3.Stepens A, Logina I, Liguts V, et al.. A Parkinsonian syndrome in methcathinone users and the role of manganese. N Engl J Med 2008;358:1009–1017. [DOI] [PubMed]
- 4.Selikhova M, Fedoryshyn L, Matviyenko Y, et al.. Parkinsonism and dystonia caused by the illicit use of ephedrone—a longitudinal study. Mov Disord 2008;23:2224–2231. [DOI] [PubMed]
- 5.Varlibas F, Delipoyraz I, Yuksel G, Filiz G, Tireli H, Gecim NO. Neurotoxicity following chronic intravenous use of “Russian cocktail.” Clin Toxicol 2009;47:157–160. [DOI] [PubMed]
- 6.Stepens A, Stagg CJ, Platkajis A, Boudrias MH, Johansen-Berg H, Donaghy M. White matter abnormalities in methcathinone abusers with an extrapyramidal syndrome. Brain 2010;133:3676–3684. [DOI] [PMC free article] [PubMed]
- 7.Bronstein AC, Spyker DA, Cantilena LR Jr, Green JL, Rumack BH, Dart RC. 2010 Annual Report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 28th Annual Report. Clin Toxicol 2011;49:910–941. [DOI] [PubMed]
- 8.Rosenbaum CD, Carreiro SP, Babu KM. Here today, gone tomorrow: and back again? A review of herbal marijuana alternatives (K2, Spice), synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine, and piperazines. J Med Toxicol 2012;8:15–32. [DOI] [PMC free article] [PubMed]
- 9.Prosser JM, Nelson LS. The toxicology of bath salts: a review of synthetic cathinones. J Med Toxicol 2012;8:33–42. [DOI] [PMC free article] [PubMed]
- 10.Mugele J, Nanagas KA, Tormoehlen LM. Serotonin syndrome associated with MDPV use: a case report. Ann Emerg Med 2012;60:100–102. [DOI] [PubMed]
- 11.Nicholson PJ, Quinn MJ, Dodd JD. Headshop heartache: acute mephedrone “meow” myocarditis. Heart 2010;96:2051–2052. [DOI] [PubMed]
- 12.Grigoryev A, Savchuk S, Melnik A, et al.. Chromatography-mass spectrometry studies on the metabolism of synthetic cannabinoids JWH-018 and JWH-073, psychoactive components of smoking mixtures. J Chromatogr B Analyt Technol Biomed Life Sci 2006;879:1126–1136. [DOI] [PubMed]
- 13.Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol 2011;49:760–764. [DOI] [PMC free article] [PubMed]
- 14.Simmons J, Cookman L, Kang C, Skinner C. Three cases of “spice” exposure. Clin Toxicol 2011;49:431–433. [DOI] [PubMed]
- 15.Mir A, Obafemi A, Young A, Kane C. Myocardial infarction associated with use of the synthetic cannabinoid K2. Pediatrics 2011;128:e1622–e1627. [DOI] [PubMed]
- 16.Nations SP, Boyer PJ, Love LA, et al.. Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurological disease. Neurology 2008;71:639–643. [DOI] [PubMed]
- 17.Hedera P, Peltier A, Fink JK, Wilcock S, London Z, Brewer GJ. Myelopolyneuropathy and pancytopenia due to copper deficiency and high zinc levels of unknown origin II: the denture cream is a primary source of excessive zinc. Neurotoxicology 2009;30:996–999. [DOI] [PubMed]
- 18.Pawa S, Khalifa AJ, Ehrinpreis MN, Schiffer CA, Siddiqui FA. Zinc toxicity from massive and prolonged coin ingestion in an adult. Am J Med Sci 2008;336:430–433. [DOI] [PubMed]
- 19.Foubert-Samier A, Kazadi A, Rouanet M, et al.. Axonal sensory motor neuropathy in copper-deficient Wilson's disease. Muscle Nerve 2009;40:294–296. [DOI] [PubMed]
- 20.da Silva-Junior FP, Machado AA, Lucato LT, Cancado EL, Barbosa ER. Copper deficiency myeloneuropathy in a patient with Wilson disease. Neurology 2011;76:1673–1674. [DOI] [PubMed]
- 21.Kumar N, Gross JB Jr, Ahlskog JE. Myelopathy due to copper deficiency. Neurology 2003;61:273–274. [DOI] [PubMed]
- 22.Kumar N, Gross JB Jr, Ahlskog JE. Copper deficiency myelopathy produces a clinical picture like subacute combined degeneration. Neurology 2004;63:33–39. [DOI] [PubMed]
- 23.Kumar N. Copper deficiency myelopathy: human swayback. Mayo Clin Proc 2006;81:1371–1384. [DOI] [PubMed]
- 24.Jaiser SR, Winston GP. Copper deficiency myelopathy. J Neurol 2010;257:869–881. [DOI] [PMC free article] [PubMed]
- 25.Chang JD, Lee SS, Hur M, Seo EM, Chung YK, Lee CJ. Revision total hip arthroplasty in hip joints with metallosis: a single-center experience with 31 cases. J Arthroplasty 2005;20:568–573. [DOI] [PubMed]
- 26.Steens W, von Foerster G, Katzer A. Severe cobalt poisoning with loss of sight after ceramic-metal pairing in a hip: a case report. Acta Orthop 2006;77:830–832. [DOI] [PubMed]
- 27.Rizzetti MC, Liberini P, Zarattini G, et al.. Loss of sight and sound. Could it be the hip? Lancet 2009;373:1052. [DOI] [PubMed]
- 28.Oldenburg M, Wegner R, Baur X. Severe cobalt intoxication due to prosthesis wear in repeated total hip arthroplasty. J Arthroplasty 2009;24:825.e815–e820. [DOI] [PubMed]
- 29.Ikeda T, Takahashi K, Kabata T, Sakagoshi D, Tomita K, Yamada M. Polyneuropathy caused by cobalt-chromium metallosis after total hip replacement. Muscle Nerve 2010;42:140–143. [DOI] [PubMed]
- 30.Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin 2010;29:591–605. [DOI] [PubMed]
- 31.Bartynski WS, Zeigler Z, Spearman MP, Lin L, Shadduck RK, Lister J. Etiology of cortical and white matter lesions in cyclosporin-A and FK-506 neurotoxicity. AJNR Am J Neuroradiol 2001;22:1901–1914. [PMC free article] [PubMed]
- 32.Feske SK. Posterior reversible encephalopathy syndrome: a review. Semin Neurol 2011;31:202–215. [DOI] [PubMed]
- 33.Mukherjee P, McKinstry RC. Reversible posterior leukoencephalopathy syndrome: evaluation with diffusion-tensor MR imaging. Radiology 2001;219:756–765. [DOI] [PubMed]
- 34.Vaughan CJ, Delanty N. Hypertensive emergencies. Lancet 2000;356:411–417. [DOI] [PubMed]
- 35.Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol 2008;29:1036–1042. [DOI] [PMC free article] [PubMed]
- 36.Kumar N, Ahlskog JE, Klein CJ, Port JD. Imaging features of copper deficiency myelopathy: a study of 25 cases. Neuroradiology 2006;48:78–83. [DOI] [PubMed]