


Abstract
Bifidobacterium breve represents one of the most abundant bifidobacterial species in the gastro-intestinal tract of breast-fed infants, where their presence is believed to exert beneficial effects. In the present study whole genome sequencing, employing the PacBio Single Molecule, Real-Time (SMRT) sequencing platform, combined with comparative genome analysis allowed the most extensive genetic investigation of this taxon. Our findings demonstrate that genes encoding Restriction/Modification (R/M) systems constitute a substantial part of the B. breve variable gene content (or variome). Using the methylome data generated by SMRT sequencing, combined with targeted Illumina bisulfite sequencing (BS-seq) and comparative genome analysis, we were able to detect methylation recognition motifs and assign these to identified B. breve R/M systems, where in several cases such assignments were confirmed by restriction analysis. Furthermore, we show that R/M systems typically impose a very significant barrier to genetic accessibility of B. breve strains, and that cloning of a methyltransferase-encoding gene may overcome such a barrier, thus allowing future functional investigations of members of this species.
INTRODUCTION
The human gut microbiota represents an extremely complex microbial assembly of an estimated 10–100 trillion symbiotic bacteria per individual (1). Common representatives of this microbiota include members of the genus Bifidobacterium, which are particularly prevalent in the gastrointestinal tract (GIT) of healthy, breast-fed infants, where they are among the first colonizers of the newborn gut (2). Low abundance of bifidobacteria has been associated with a number of diseases, e.g. inflammatory bowel disease, metabolic syndrome, and colorectal cancer, thereby presenting possibilities for their use as biomarkers or treatments (3–5). Members of this genus have therefore been incorporated as live ingredients of functional foods, while they have also enjoyed intense scientific scrutiny, including comparative and functional genomic efforts aimed at unraveling the molecular mechanisms underlying their beneficial activity.
Bifidobacterium breve (next to B. longum and B. bifidum) is abundantly present in the gut microbiota of healthy infants (2,6–10), and has been subject of genetic studies, including insertional mutagenesis (11–13). In silico comparative and pan-genome analysis of 25 B. breve genomes revealed that ∼70% of the gene content of a given strain is common to all members of this species (designated the core genome), while the remaining genes (i.e. the variome) represent intra-species diversity (14).
Genes encoding Restriction/Modification (R/M) and CRISPR/Cas (Clustered Regularly Interspaced Short Palindromic Repeats) systems are part of the B. breve variome, and represent defense systems against invasion by exogenous DNA (14–17). R/M systems are relevant for the study of bifidobacteria as they obstruct genetic accessibility (18–20). In bifidobacteria three types of base modification are predicted: N6-methyladenine (m6A), N4-methylcytosine (m4C) and 5-methylcytosine (m5C) (21), which can be detected using a combination of PacBio SMRT and Illumina bisulfite sequencing (BS-seq) (22,23). Methyltransferases (MTases) are typically found as part of R/M systems, which use base methylation to discriminate ‘self’ from ‘non-self’ DNA, protecting methylated host DNA from cleavage while targeting unmethylated exogenous DNA (16,24). Individual R/M systems are generally composed of one or more MTases and restriction endonucleases (REases), and are classified into four Types (I, II, III, IV) based on the number of subunits, co-factor requirements, mechanism of action, target sequence and type of cleavage (24,25).
A typical Type I R/M system is multi-subunit complex, which is comprised of an REase (named HsdR or R) responsible for ATP-dependent DNA cleavage, an MTase (named HsdM or M) producing m6A or m4C methylated bases, and a specificity determinant (HsdS or S) responsible for recognition of (in most cases) asymmetric (rather than palindromic) sites. DNA cleavage by a Type I REase occurs at a variable distance from the recognition sequence (25,26). Type II R/M systems typically consist of two distinct enzymes (MTase and REase) recognizing palindromic sequences, within which the REase introduces a symmetrical double-stranded cut. The Type II family represents the most heterogeneous group of R/Ms in bacteria, further divided in 11 subtypes (IIA, IIB, IIC, IIE, IIF, IIG, IIH, IIM, IIP, IIS and IIT), based on the genetic arrangement of its constituent REase and MTase components, and the target or type of cleavage (25).
To date >200 bifidobacterial R/Ms have been predicted (http://rebase.neb.com/cgi-bin/pacbiolist; http://tools.neb.com/genomes) (21). For only a few of these systems the recognition sequence of the REase has been determined by classical methods involving restriction analysis (19,27–30), while in recent years their characterization has been assisted by Pacbio SMRT technology (18,20). Methylase activity of three R/M systems has been described for B. breve UCC2003 (BbrUI, BbrUII and BbrUIII), where M.BbrUI and M.BbrUIII possess cytosine-specific MTase activity, while M.BbrUII is responsible for N6-adenosine methylation (19). Improvement in transformation efficiencies of bifidobacterial strains have been shown to be achievable using plasmid modification which would elude cleavage by the host-encoded R-M systems (31). This approach applied to B. breve UCC2003 showed that transformation efficiency can be increased by more than 1000-fold when the endogenous R/M systems are by-passed by the use of pre-methylated plasmid DNA, obtained from an E. coli strain expressing the M.BbrUII and M.BbrUIII MTases (19).
The current work reports on the sequencing of a large B. breve collection employing PacBio SMRT sequencing, generating an updated view on strain diversity of this species. Furthermore, SMRT-mediated methylome analysis, combined with Illumina BS-seq and comparative genome analysis, allowed us to determine the methylome of this species, thereby providing an overview of the harbored R/M systems with corresponding recognition sites. Therefore, the information generated will constitute a solid reference for future improvement of genetic accessibility of members of this species.
MATERIALS AND METHODS
DNA isolation and CGH experiments
Bifidobacterial strains used in this study (Supplementary Table S1) were routinely cultivated at 37°C in an anaerobic chamber (Davidson and Hardy, Belfast, Ireland) in Reinforced Clostridial Medium (RCM Oxoid, Ltd., Basingstoke, Hampshire, United Kingdom). Where appropriate, tetracycline (Tc; 10 μg ml−1), chloramphenicol (Cm; 3 μg ml−1), erythromycin (Em; 1 μg ml−1) or spectinomycin (Sp; 100 μg ml−1) was included in the growth medium. Following growth, total bifidobacterial DNA was isolated as previously described (32) and then labelled using the Kreatech ULS labelling kit according to the manufacturer's instructions (Kreatech, Amsterdam, The Netherlands). Labelled total gDNA was hybridized as described in the Agilent manual ‘Two-Color Microarray-Based Gene Expression Analysis’ (v4.0) (publication no. G4140–90050) to the B. breve UCC2003 microarray slides (feature format: 4 × 44K) overnight at 65°C (NB. B. breve UCC2003 was used as the reference strain and as a positive control). Following hybridization, microarrays were washed in accordance with Agilent standard procedures and scanned using an Agilent DNA microarray scanner (model G2565A). Generated scans were converted to data files with Agilent Feature Extraction software (Version 9.5). DNA microarray data sets were processed as previously described (33–35). Differential signal intensity tests were performed with the Cyber-T implementation of a variant of the t-test (36). Data were analysed and visualized using the Multi-experiment Viewer Tmev v.4.9 with hierarchical clustering analysis based on covariance method and complete linkage.
SMRT sequencing and sequence annotation
Genome sequencing of B. breve strains was performed by GATC Biotech Ltd. (Germany) using Pacific Biosciences SMRT RSII technology. Raw sequencing reads were de novo assembled using the Hierarchical Genome Assembly Process (HGAP) protocol RS_Assembly.2 implemented in the SMRT Analysis software v.2.3 with default parameters (https://github.com/PacificBiosciences/SMRT-Analysis).
Open Reading Frame (ORF) prediction and automatic annotation was performed using Prodigal v2.0 (http://prodigal.ornl.gov) for gene predictions, BLASTP v2.2.26 (cut-off E-value of 0.0001) (37) for sequence alignments against a combined bifidobacterial genome-based database, and MySQL relational database to assign annotations. Predicted functional assignments were manually revised and edited using similarity searches against the non-redundant protein database curated by the National Centre for Biotechnology Information (ftp://ftp.ncbi.nih.gov/blast/db/) and PFAM database (http://pfam.sanger.ac.uk), which allowed a more detailed, in silico characterization of hypothetical proteins. GenBank editing and manual inspection was performed using Artemis v18 (http://www.sanger.ac.uk/resources/soft-ware/artemis/). Transfer RNA genes were identified employing tRNAscan-SE v1.4 and ribosomal RNA genes were detected based on the software package Rnammer v1.2 (38) supported by BLASTN v2.2.26.
Comparative genomics
Genomic synteny among individual B. breve genomes was inspected using whole-genome DNA sequence alignments using the software package MUMmer v3.0 (39). Comparisons and alignments at protein and nucleotide levels were performed on sequences deduced using the same ORF prediction pipeline Prodigal v2.0 (http://prodigal.ornl.gov). Comparative genome analyses were performed using a combination of all-against-all, bi-directional BLAST alignments (37) (cut-off: E-value < 0.0001, with at least 50% identity across at least 50% of either protein sequence), and the Markov Cluster Algorithm (MCL) implemented in the mclblastline pipeline v12-0678 (40) in order to group corresponding genes into families that share a particular function. As part of the comparative genome analysis all identified gene families were assigned to either the core genome or to the dispensable genome based on their presence in all analysed strains or in a subset, respectively.
Phylogenomic analyses
Phylogenomic relationships between strains were assessed based on nucleotide alignments of the core genome gene content, which included only single-copy orthologues. An additional filter for paralogues was applied to the core genome in order to exclude families represented by more than a single member, as they do not represent robust evolutionary markers (41). Gene alignments were conducted using MUSCLE v.3.8.31 (42), followed by construction of a phylogenetic tree for each single-copy gene using the maximum-likelihood in PhyML v3.0 (43) and tree concatenation. A final consensus tree was computed using the Consense module from Phylip package v3.69 (http://evolution.genetics.washington.edu/phylip.html) using the majority rule method.
Methylome analysis (SMRT and BS-seq sequencing)
Methylome analysis was performed using a combination of SMRT sequencing and comparative genome analysis. Base modification and methylated motif detection was performed employing the SMRT Analysis portal following de novo genome assembly using the ‘RS_Modification_and_Motif_Analysis.1’ protocol. Methylation motifs with a score of 40 (corresponding to a P-value of 0.0001) or higher were considered specific and were taken for further analysis (https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/Methylome-Analysis-Technical-Note).
The deduced protein products of all identified ORFs were searched for similarity to known R/M systems (including orphan methylases) using BLASTP (37) alignments against the REBASE database (http://rebase.neb.com/rebase/rebase.html) (21). Significant BLASTP hits were selected using a cut-off E-value of <0.0001 and exhibiting over 30% similarity across at least 80% of the sequence length. Further manual refinement of these predictions included discarding of false positive BLASTP hits and refinement using PFAM database (http://pfam.sanger.ac.uk). A comparative genome analysis was employed to associate the presence of R/M system-encoding genes with the presence of methylation motif(s).
In selected cases, Illumina sequencing of bisulfite-treated chromosomal DNA (termed BS-seq) was performed in order to reliably detect m5C-methylated bases. The strains subjected to BS-seq were B. breve NRBB01, B. breve NRBB02, B. breve NRBB11, B. breve NRBB56, B. breve NRBB50, B. breve DRBB26, B. breve DRBB28 and B. breve 215W4–47a. BS-seq was performed by GATC Biotech Ltd. (Germany) using a HiSeq 2500 platform employing a paired-end library and generating sequencing read lengths of 125 bp. The obtained BS-seq raw data sets were subjected to Bismark software (v.0.14.5) analysis in order to reveal the presence of m5C modifications and the identification of corresponding methylation (recognition) motifs. Raw reads were low-quality filtered using NGSQCToolkit_v2.3.3 (http://www.nipgr.res.in/ngsqctoolkit.html) and mapped on the finished reference genome using Bismark v0.14.5 developed by Babraham Bioinformatics (http://www.bioinformatics.babraham.ac.uk/projects/bismark/) in a non-directional mode. Methylated cytosines were extracted from a BedGraph output using 80% of m5C>T conversion as a cut-off value and sequence context was analysed by extracting +/- 10 bp around each identified methylated position. Meme suite (http://meme-suite.org/) was employed to determine a context consensus at positions where methylations were observed and Integrative Genomics Viewer v2.3 (https://www.broadinstitute.org/igv/) was used for data visualization and inspection.
Plasmid DNA isolation and transformation of B. breve strains
Mini-preparation of plasmid DNA from E. coli or B. breve (Table 1) was performed using the Qiaprep spin plasmid miniprep kit (Qiagen GmBH, Hilden, Germany). For isolation of bifidobacterial DNA, an initial lysis step was incorporated into the plasmid isolation procedure, cells were resuspended in lysis buffer supplemented with lysozyme (30 mg ml−1) and incubated at 37°C for 30 min. For transformation of B. breve strains 50 ml of modified Rogosa medium mMRS prepared from first principles (44) supplemented with 0.05% cysteine, and 1% glucose, lactose or maltose, was inoculated with 4 ml of a B. breve overnight culture and incubated anaerobically at 37°C. Once an optical density (OD600nm) value between 0.6–0.8 was reached, bacterial cells were collected by centrifugation at 4500 rpm for 10 min at 4°C. The obtained cell pellet was washed twice with chilled sucrose citrate buffer (1 mM citrate [pH 5.8], 0.5 M sucrose), and then resuspended in 200 μl of ice-cold sucrose citrate buffer. Fifty μl of the cell suspension was used for each electro-transformation; cells and plasmid DNA were mixed and held on ice prior to applying an electrical pulse at 25 μF, 200 ohms and 2 kV. Following this, 1 ml of reinforced clostridial medium (RCM) (Oxoid, Hampshire, England) was added to the cell suspension, which was then incubated anaerobically for 2.5 h at 37°C. Serial dilutions were plated on reinforced clostridial agar (RCA) containing the appropriate antibiotic and plates were incubated anaerobically at 37°C for 48 h, after which the transformation frequency was calculated as the number of transformants obtained per μg of plasmid DNA.
Table 1. Plasmids used in this study.
| Plasmid | Relevant characteristics | Reference or Source |
|---|---|---|
| pNZ8048 | Gene expression vector PnisA, Cmr | De Ruyter et al. (1996) |
| pDM1 | pAM5 derivative containing spectinomycin resistance cassette | O’Connell Motherway and Watson et al. (2014) |
| pAM5 | pBC1-puC19-Tc | Alvarez-Martín et al. (2007) |
Methylase cloning
For construction of the methylase gene-harbouring plasmid pNZ-M.NRBB52, the predicted methyltransferase-encoding gene NRBB52_0014 including its presumed promoter region was amplified by PCR employing chromosomal DNA of B. breve NRBB52 as a template, and using Q5 DNA polymerase. To facilitate cloning XbaI/HindIII restriction sites were incorporated at the 5′ ends of the forward and reverse primers, respectively. The generated NRBB52_0014-encompassing amplicon was digested with XbaI and HindIII, and ligated to pNZ8048 restricted with the same enzymes. The resulting ligation mixture was introduced into Escherichia coli EC101 (45) by electrotransformation, after which transformants were selected based on chloramphenicol resistance. The plasmid content of a number of transformants was screened by restriction analysis and the integrity of positively identified clones was verified by sequencing prior to introduction into the methylase-negative E. coli strain ER2796 (46).
The effect of methylation of plasmid DNA on the transformation efficiency of B. breve NRBB52 was subsequently assessed by introduction of the shuttle vector pAM5 into E. coli ER2796 pNZ-M.NRBB52. The corresponding plasmid preparations were then used to determine how efficiently pAM5 isolated from E. coli ER2796 pNZ-M.NRBB52 could be introduced into B. breve NRBB52 by electrotransformation.
RESULTS AND DISCUSSION
General genome features and comparative genomics of B. breve
In order to determine intra-species diversity of B. breve, a collection of 71 B. breve strains (Supplementary Table S1) was assessed by Comparative Genome Hybridization (CGH) using micro arrays that were based on the annotated genes of the B. breve UCC2003 genome as a reference. As controls we included six bifidobacterial strains that did not belong to B. breve as verified by (partial) 16S rRNA gene sequencing (Supplementary Table S1). Raw data analysis of the CGH results allowed the construction of a gene presence/absence matrix, which was used to perform a two-way hierarchical clustering, resulting in the identification of 24 distinct B. breve genome groups (Figure 1). Based on this CGH-directed grouping, 27 strains were selected for PacBio SMRT sequencing in order to obtain full genome sequences and to capture a wide range of B. breve genomic diversity. Table 2 outlines the salient features of the obtained B. breve genomes, being in line with what was previously described for (a smaller number of) members of this species and bifidobacteria in general (14,47).
Figure 1.

CGH and hierarchical clustering of bifidobacterial strains. Heatmap representing presence (black) or absence (yellow) of B. breve variable genes obtained by comparative genomics hybridization (CGH) analyses across 71 B. breve and 6 non-B. breve strains. Clusters obtained using a two-way hierarchical clustering analysis are highlighted with blue boxes and relative cluster numbers are indicated.
Table 2. General genome features of the 27 newly sequenced B. breve representatives (clonal strains are indicated in *).
| Genome | Size (bp) | CGH GROUP | ANI % (B. breve UCC2003) | Contigs | Pacbio Coverage | ORFs number | rRNA loci | Selected for Comparative analysis | TUG | Mobile regions | CRISPR | Accession number |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B. breve NRBB56 | 2425122 | 2 | 98.9 | 1 | 179 | 1985 | 2 | Yes | 10 | 3 | 13 kb | CP021394 |
| B. breve NRBB52 | 2379672 | 6 | 98.6 | 1 | 354 | 1989 | 3 | Yes | 9 | 3 | 11 kb | CP021393 |
| B. breve DRBB28 | 2462170 | 24 | 98.6 | 1 | 391 | 2096 | 2 | Yes | 51 | 5 | No | CP021553 |
| B. breve NRBB57 | 2510381 | 12 | 98.7 | 1 | 206 | 2125 | 3 | Yes | 48 | 4 | No | CP021389 |
| B. breve DRBB27* | 2435083 | 7 | 98.2 | 1 | 349 | 2079 | 2 | Yes | 167 | 4 | No | CP021552 |
| B. breve DRBB29* | 2435086 | 7 | 98.2 | 1 | 362 | 2084 | 2 | No | - | 4 | No | CP023198 |
| B. breve NRBB09 | 2265557 | 10 | 98.5 | 1 | 445 | 1881 | 3 | Yes | 38 | 3 | 11 kb | CP021387 |
| B. breve NRBB11 | 2377562 | 11 | 98.6 | 1 | 397 | 1916 | 3 | Yes | 17 | 3 | 17 kb | CP021388 |
| B. breve CNCM I-4321* | 2464852 | 8 | 98.4 | 1 | 339 | 2082 | 2 | Yes | 18 | 5 | No | CP021559 |
| B. breve DRBB30* | 2471118 | 8 | 98.4 | 1 | 434 | 2098 | 3 | No | - | 5 | No | CP023199 |
| B. breve DRBB26 | 2396387 | 15 | 98.6 | 1 | 335 | 1978 | 3 | Yes | 16 | 2 | No | CP021390 |
| B. breve NRBB01 | 2269404 | 17 | 98.6 | 1 | 536 | 1902 | 2 | Yes | 26 | 2 | No | CP021384 |
| B. breve NRBB04 | 2324647 | 18 | 98.7 | 1 | 168 | 1874 | 3 | Yes | 15 | 1 | 11 kb | CP021386 |
| B. breve 139W4–23 | 2411276 | 22 | 98.8 | 1 | 289 | 2013 | 3 | Yes | 45 | 3 | No | CP021556 |
| B. breve NRBB02* | 2289884 | 20 | 98.7 | 1 | 85 | 1872 | 2 | Yes | 37 | 1 | 12.5 kb | CP021385 |
| B. breve NRBB08* | 2289759 | 20 | 98.7 | 1 | 91 | 1875 | 2 | No | - | 1 | 12.5 kb | CP023192 |
| B. breve NRBB18* | 2289686 | 20 | 98.7 | 1 | 197 | 1874 | 2 | No | - | 1 | 12.5 kb | CP023193 |
| B. breve NRBB19* | 2289726 | 20 | 98.7 | 1 | 112 | 1875 | 2 | No | - | 1 | 12.5 kb | CP023194 |
| B. breve NRBB20* | 2289892 | 20 | 98.7 | 1 | 111 | 1875 | 2 | No | - | 1 | 12.5 kb | CP023195 |
| B. breve NRBB27* | 2289838 | 20 | 98.7 | 1 | 147 | 1871 | 2 | No | - | 1 | 12.5 kb | CP023196 |
| B. breve NRBB49* | 2289791 | 20 | 98.7 | 1 | 346 | 1869 | 2 | No | - | 1 | 12.5 kb | CP023197 |
| B. breve 215W4–47a | 2589602 | 9 | 98.7 | 1 | 274 | 2219 | 3 | Yes | 37 | 3 | No | CP021558 |
| B. breve 082W4–8 | 2286339 | 21 | 98.8 | 1 | 121 | 1885 | 3 | Yes | 8 | 2 | No | CP021555 |
| B. breve 180W8–3 | 2273173 | 23 | 98.6 | 1 | 309 | 1891 | 3 | Yes | 15 | 2 | No | CP021557 |
| B. breve 017W4–39 | 2301422 | 23 | 98.7 | 1 | 287 | 1923 | 2 | Yes | 9 | 2 | No | CP021554 |
| B. breve NRBB50 | 2409058 | 13 | 98.8 | 1 | 125 | 2021 | 3 | Yes | 22 | 2 | No | CP021391 |
| B. breve NRBB51 | 2402272 | 13 | 98.5 | 1 | 471 | 1960 | 3 | Yes | 14 | 2 | 13 kb | CP021392 |
Full length alignment of the generated genome sequences with the reference genome of B. breve UCC2003 returned an average nucleotide identity of 98.7% across all strains (Table 2), confirming their identity as members of the B. breve species. Dotplot alignments of each genome against that of B. breve UCC2003 revealed full length genome synteny with interruptions observed where (presumed) mobile genetic elements are located, in support of the notion that B. breve genomes appear more stable and do not (or very rarely) suffer from major chromosomal rearrangements (14) (Supplementary Figure S1). This observation differs from the situation in the closely related species B. longum, where such chromosomal rearrangements occur more frequently, suggesting that conservation of genome synteny is not a shared feature among related taxa (18). As apparent from dotplot alignments between representatives of B. longum species, such large chromosomal rearrangements are possibly promoted by insertion elements, as they involve regions that are commonly flanked by transposase-encoding genes (families IS30, IS21, ISL3) (Supplementary Figure S2).
The performed CGH analysis indicated that 11 out of 27 strains (isolated from the same source) were closely related. In order to determine the degree of similarity among these strains at nucleotide level and to verify if the observed differences were genuine, whole genome sequencing and nucleotide sequence alignments were performed. As the obtained result showed that these strains are clonal derivatives, we deduced that the observed differences were caused by hybridization bias (Table 2). For this reason only one representative per clonal strain group was selected for subsequent comparative purposes (NB. for the phylogenomic analysis we used all fully sequenced genomes, see below), reducing the dataset to 19 newly sequenced and distinct B. breve representatives (Table 2).
Comparative analysis was performed using all-against-all BLASTP alignments followed by clustering of (protein-encoding) gene families. These comparisons included the 19 newly sequenced strains, plus six previously completely sequenced B. breve genomes (14) which had been included in the CGH analysis, for a total of 25 representatives of this species. This analysis returned a total number of 3082 gene families, 27% higher than the previously computed B. breve pan-genome (14), indicating that the inclusion of 25 additionally sequenced strains substantially increases the B. breve pangenome. We furthermore confirmed our previous observations (14,15) that mobile genetic elements, CRISPR genes and R/M systems constitute a significant portion of the dispensable genome (i.e. gene functions not common to all members of the species).
Phylogenomics of B. breve
A phylogenomic investigation based on nucleotide alignments of the core genome of individual strains allowed us to deduce the evolutionary relationships between strains (as 16S rRNA sequence-based phylogeny does not provide sufficient resolution at the intraspecies level) (48). Unlike CGH and comparative genome-derived clustering (which are based on the presence-absence of genes), phylogenomic analysis is based on sequence alignment of core genes and for this reason more suitable for an in depth investigation of phylogenetic relationships between closely related taxa. For this analysis we used a total of 1031 core genes that are present in single copy, including a number of housekeeping (and thus essential) functions such as genes encoding DnaA, DNA gyrase, chaperone proteins DnaJ, GroEL and GroES, as well as functions involved in central carbon metabolism (e.g. transaldolase and transketolase) and amino acid transport and biosynthesis. The resulting phylogenomic tree (Figure 2) was computed using the 27 B. breve strains sequenced as part of this study as well as seven publicly available and fully sequenced B. breve representatives, while furthermore including B. longum subsp. longum NCIMB 8809 as an outgroup. The resulting tree clusters the B. breve strains in 12 phylogenetic groups where they appear to distribute evenly across the tree (with the exception of the B. breve NRBB02 clonal group which includes 7 representatives). It is worth noting that no strict correlation exists between the HCL clustering results derived from CGH analysis (Figure 1) and the phylogenomic tree (Figure 2). However, this may have been expected because the latter is based on sequence alignment of concatenated core genes, while the former is based on presence-absence of variably distributed gene families, and thus will not contain information on the relatedness of strains at phylogenetic level. In this regard it is worth mentioning that phylogenetic inference based on a selected set of concatenated genes may suffer from a number of inaccuracies (49).
Figure 2.

Phylogenomics of B. breve. Phylogenetic supertree computed on concatenated single-copy core genes and showing the existing relationship between 35 sequenced B. breve species. B. longum subsp. longum NCMIB 8809 was used as the outgroup and online available strains are also indicated in purple. B. breve clonal strains determined by our analysis are indicated in orange.
Methylated bases and recognition motifs in B. breve
PacBio SMRT sequencing also generates information on DNA methylation, and this data was used, in combination with BS-seq of selected strains, to determine the diversity and occurrence of methylated motifs that are recognized by R/M systems. Analysis of the sequencing kinetics indicated the presence of m6A, m5C and, though at a lower frequency, m4C across the investigated strains. Extraction of the sequence context of these modifications allowed the identification of nine motifs assignable to Type I R/Ms and thirteen to Type II/IIG R/Ms (Figure 3; Table 3 and Supplementary Table S2), constituting a total of 22 unique methylated motifs in the assessed B. breve strain collection.
Figure 3.

The B. breve methylome. (A) Circos plot representing the occurrence of detected motifs and methylated positions in B. breve. Methylated motifs are ordered as in the figure legend on the right (from outer to inner circle) and grouped based on their assignment to Type I (green), Type II (red) and Type IIG (blue) R/M system.The motif occurrence in genomes is also indicated. (B) Heatmap representing presence/absence of detected motifs across 20 B. breve strains. Motifs are grouped based on their assignment to Type I, Type II and Type IIG R/M system and the type of modification (m6A, m4C and m5C) is also indicated.
Table 3. Bifidobacterium breve RM-COGs locus tags with associated methylome.
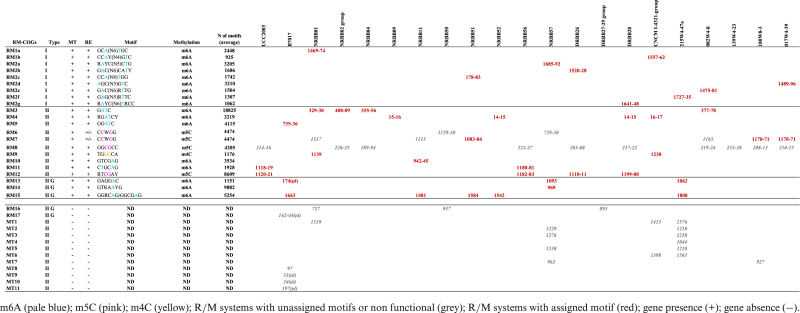
|
Adenine modification was detectable with high accuracy by PacBio SMRT sequencing (a given motif was detected as being methylated in >90% of the predicted positions in a genome) and m6A methylation was assigned to nine distinct Type II/IIG motifs. Of these, four were palindromic and thus methylated on both strands, while the remaining five do not constitute a perfect palindrome and are hemi-methylated (Figure 4). Interestingly, 5′-Gm6ATC-3′ represents the most abundant m6A modification across the investigated B. breve strains in terms of number of methylated sites, followed by 5′-GTRAm6AYG-3′ and 5′-GGRCm6AG-3′ (Figure 3, panel A). Regarding methylated consensus sequences that are commonly known to be associated to Type I R/Ms (50), our analysis identified nine motifs that were assigned to Type I R/Ms, where m6A modification is present in the top and bottom strand (Figure 3).
Figure 4.

B. breve R/M systems and corresponding methylation motifs. Locus maps showing the gene organization of each R/M system detected in B. breve. For each system the RM-COG number, assigned nomenclature, type of system (I, II or IIG) and type of modification (m6A, m5C or m4C) are indicated. Methylated motifs obtained by PacBio and BS-seq are also reported (blue for m6A, pink for m5C and yellow for m4C), with an indication of single-strand (*) and double strand (**) methylation.
Cytosine modification was detectable using PacBio SMRT sequencing only in the case of m4C, although with lower confidence compared to m6A (average motif detection rate of 52%), being an intrinsic feature of SMRT technology. However, this type of methylation seems to be rare in B. breve, being observed in just a single instance (5′-TGGm4CCA-3′) (Figure 3, panel B) (Table 3), and being assigned to a possible Type II alpha R/M encoded within a mobile genetic region (see below). Finally, m5C modifications were detected by BS-seq in eight strains that had been selected based on the presence of REBASE-predicted cytosine (or unknown orphan) methylases, confirming the presence of this methylation type in three cases (Figure 3, panel B). Of these motifs, 5′-RTm5CGAY-3′ seems to be the most abundant motif in B. breve in terms of the number of methylated sites, followed by 5′-Cm5CWGG-3′ and 5′-GGm5CGCC-3′ (Figure 3, panel A).
Matching methyltransferases to their cognate motif
To establish associations between predicted MTases and identified methylation motifs, an initial search for R/M systems across all 19 B. breve genomes was performed using the publicly available database (ftp://ftp.neb.com/pub/rebase) (21) and BLASTP alignments (37). All genes identified with this approach were organized into R/M systems when a predicted MTase-encoding gene was found in close proximity to a putative REase-specifying gene (or vice versa), or if both activities were predicted to be encoded by a single gene. In the case of genes encoding a predicted MTase or REase (with associated domains) without a closely positioned cognate partner, they were considered as orphan, possibly representing truncated, incomplete or non-functional systems (51).
Combining the SMRT/BS-seq-assisted methylome identification outputs (detecting methylated bases and the sequence context of occurrence) with comparative genomics (generating information on the presence or absence of R/M-encoding genes among strains) we were able to link the presence of specific methylated motifs with the presence of particular (predicted) R/M systems in corresponding B. breve strains (Supplementary Table S2). Comparative analysis assisted by MCL-clustering allowed us to group the identified R/M system genes into clusters of orthology (here designated as RM-COGs), reflecting the presence of homologous systems across strains. Our analysis associated 15 distinct RM-COGs with 22 methylated motifs showing a certain degree of overlap across strains (Tables 3 and 4). Furthermore, for each of the identified R/M systems we were able to assign a corresponding recognition sequence from the pool of detected methylated motifs (Table 4). Of these 22 motifs, nine were assigned to Type I with unique specificities for each strain. Type I systems belonging to the RM1-COGs are present in B. breve NRBB01 and B. breve CNCM I-4321, while those belonging to RM2-COGs are present in seven B. breve strains (Figure 5; Tables 3 and 4).
Table 4. Bifidobacterium breve MTases with assigned recognition sequence in REBASE.

|
Figure 5.

Diversity of Type I R/M systems in B. breve. (A) Locus map showing gene organization and comparison between all identified Type I R/M systems found in B. breve. Genes are indicated as arrows and are coloured based on their predicted function. Percentage of similarity in BLASTP alignment across homologous genes is indicated. (B) Phylogenetic tree obtained following the alignment of the concatenated M and R-encoding genes of Type I R/M systems detected in B. breve. The resulting Neighbour Joining tree shows the clustering of Type I R/M systems in two main phylogenetic groups (red boxes).
The remaining 13 motifs were predicted to correspond to: (i) five Type II R/M systems responsible for m6A modification, (ii) one Type II R/M system possibly responsible for m4C modification, (iii) three Type II R/M systems responsible for m5C modification, and (iv) four Type IIG R/M systems responsible for m6A modification (Figure 4; Tables 3 and 4).
In silico analysis of the distribution of methylated motifs across B. breve revealed that adenine modification (m6A) is the preferred methylation activity in R/Ms of this species, occurring in 10 out of 15 RM-COGs identified. Closer inspection of the genetic organization of each R/M system also revealed that members of the RM7-COG and RM8-COG appear to constitute non- or partially functional R/M systems, where methylome analysis was able to detect a methylated motif assignable to a particular MTase-encoding gene, although the corresponding RE-encoding gene appears to be truncated (Supplementary Figure S3, panels A and B). In the case of members of the RM7-COG we found either complete or truncated R/M systems, depending on the strain analysed (Supplementary Figure S3, panel B). Interestingly, these apparently partially functioning R/Ms are predicted to be responsible for cytosine modification (m5C) and are present in 15 out of the 21 strains investigated. It is worth mentioning that a recent study based on genome wide insertional mutagenesis in B. breve UCC2003 identified M.BbrI of the apparently truncated BbrI R/M system as essential for the survival of this strain, which suggests that this system (a member of the RM8-COG group) is at least partially functional (52).
Based on the occurrence of each motif across genomes and the type of modification observed (methylation of either both strands or one strand of a given recognition sequence), it is likely that not all of these systems impact equally on the genetic accessibility of strains or horizontal gene transfer in general. For example, the 5′-Gm6ATC-3′ palindromic recognition motif of the RM3-COG group, which is present in four B. breve strains, occurs at a high frequency in DNA and indeed may constitute a strong barrier to horizontal transfer and genetic accessibility. Other systems instead recognize larger, less frequently occurring and sometimes non-palindromic motifs (e.g. members of RM10-, RM13-, RM14- and RM15-COGs) (Figure 4).
Of note, our analysis also identified 12 presumed orphan MTases with no associated motif as based on methylome data. A large proportion of these are found within mobile genetic regions in B. breve NRBB57 and B. breve 215W4–47a, and they may constitute either non-functional or non-expressed methylases of degenerated R/M systems (Table 3).
Type I R/M systems in B. breve
Type I RM systems are commonly present in B. breve, as they are observed in nine out of the 20 examined strains (Table 3). Based on sequence similarity of M and R subunits the identified Type I systems are classified into two main phylogenetic groups (corresponding to the RM1- and RM2-COGs assignment as based on comparative genome analysis, see above) (Figure 5). Detailed inspection of the Type I R/M-encoding genomic region in individual B. breve strains revealed in each of the nine cases the presence of multiple copies of the S-encoding gene, which does not always represent a complete S-subunit (and are located adjacent to single R- and M-encoding genes). Alignment of Type I R/M system-encoding regions extracted from the nine B. breve strains showed that these loci differ mainly at the level of the partial/complete hsdS genes (Tables 3 and 4; Figure 5). In all cases we observed that these specificity determinants are located adjacent to a gene encoding a putative site-specific recombinase, which may catalyze hsdS reshuffling to generate diversity in recognition motifs, a phenomenon that has previously been described (26).
When comparing the observed Type I R/M systems with the phylogenetic relatedness of B. breve strains, it is worth noting that B. breve NRBB01 and B. breve CNCM I-4321 which exhibit a similar Type I R/M arrangement (Figure 5, panel B) also appear to be closely related at phylogenetic level (Figure 2).
It is also worth mentioning that (with the exception of B. breve NRBB51) the Type I R/M system is encoded within the same genomic region that also harbours a tRNA synthetase-encoding gene, a phenomenon that has previously been observed for other bacteria (53,54). Interestingly, of the nine identified motifs associated with B. breve Type I RMs, 7 represent novel motifs (http://rebase.neb.com/rebase/rebase.html), suggesting that hsdS reshuffling causes a high level of recognition motif diversity and dynamics.
Type II and IIG R/M systems in B. breve
Type II R/M-associated motifs (also including the Type IIG subgroup, in which MTase and REase activities are encoded by a single gene) are diverse in B. breve (13 out of the 22 identified motifs) (Table 3). Interestingly, nine of the 13 identified Type II R/Ms in B. breve are presumed to recognize palindromic sequences (Figure 4). The remaining four Type II R/Ms are deduced to recognize an asymmetric motif (which, when methylated result in single strand methylation within that recognition sequence). Interestingly, the genes encoding these four Type II R/M systems are located within putative mobile genetic elements, indicative of horizontal acquisition.
Notably, among the 13 identified Type II R/M systems M.Bbr11ORF942P is associated with a novel motif (according to http://rebase.neb.com/rebase/rebase.html; (21)). Furthermore, we observed similarity between Type II M.Bbr11ORF942P and Type IIG Bbr57III, where the individual MTase display ∼50% similarity with the first half of Bbr57III (Supplementary Figure S4), suggesting its origin from a fusion between MTase- and RE-encoding genes of a Type II system.
Genomic DNA digestion with commercially available enzymes
It was reasoned that the deduced methylated recognition sequences of these genomes provides protection against restriction enzymes that target such sequences (if they are sensitive to the corresponding methylation position). In order to verify this presumption, genomic DNA from a number of selected strains (B. breve NRBB01, B. breve NRBB02, B. breve NRBB57, B. breve NRBB04, B. breve 180W8–3 and B. breve NRBB52) was treated with selected restriction enzymes and analysed by agarose gel electrophoresis. Restriction of B. breve NRBB01 gDNA with DpnII, MscI or BamHI (control) demonstrated that NRBB01, NRBB02 and NRBB04 gDNA is protected from restriction by DpnII. This shows that, as expected from the methylome analysis, homologs of the predicted MTase M.Bbr01I member of the RM3-COG (Tables 3 and 4) are responsible for methylation at a 5′-Gm6ATC-3′ recognition sequence (Supplementary Figure S5). Digestion of B. breve NRBB02 genomic DNA with EheI confirmed that homologs of M.Bbr02II members of the RM8-COG (Tables 3 and 4) are responsible for the methylation of 5′-GG m5CGCC-3′ sites (Supplementary Figure S5). Indeed, the M.Bbr02II homolog in B. breve UCC2003 (constituting an R/M named BbrI), has previously been described to recognize a 5′-GGm5CGCC-3′ motif (19). Restriction of B. breve NRBB57 gDNA with EcoRII or BamHI (the latter being a positive control) demonstrated that B. breve NRBB57 gDNA is protected by the methylation activity provided by M.Bbr57ORF730P of RM6-COG (Tables 3 and 4; Supplementary Figure S5). Furthermore, restriction analysis of B. breve 180W8–3 gDNA with EcoRI, EcoRII or EheI demonstrated that gDNA of this strain is cleaved by EcoRI, but protected from restriction by EcoRII or EheI. This suggests that M.Bbr180I of RM7-COG in B. breve 180W8–3 is responsible for methylation at 5′-C m5CWGG-3′ sites, while M.Bbr180II of RM8-COG is responsible for methylation of 5′-GG m5CGCC-3′ recognition sequences (Supplementary Figure S5). Finally, restriction of B. breve NRBB52 gDNA with DpnII, PsuI and EcoRI (control) showed that gDNA of this strain is not protected from restriction with any of the enzymes tested (Supplementary Figure S5). As expected, M.Bbr52I which together with its homologs form RM4-COG, methylates 5′-RGm6ATCY-3′ sites (Tables 3 and 4), does not provide (full) protection against DpnII or PsuI digestion.
Methylase cloning and evaluation of genetic accessibility of B. breve strains
In order to obtain information on the genetic accessibility of B. breve strains, a set of seven plasmids (Table 1) was tested across a panel of six sequenced B. breve representatives in order to determine their transformation efficiencies (Table 5). As from the obtained results the transformation efficiencies were shown to be extremely variable across B. breve strains and plasmids, suggesting that certain strains impose barriers, such as R/M systems, that limit the introduction of foreign DNA (Table 5).
Table 5. Evaluation of genetic accessibility of B. breve strains with different plasmids (expressed as average of duplicate attempts) and impact of R/M systems. Results are expressed in CFUs/microgram of plasmid DNA.
| B. breve strain | E. coli strain | E. coli plasmid DNA | B. breve plasmid DNA | E. coli ER2796 (pNZ-M.Bbr52I) plasmid DNA | Plasmid | Active R/Ms |
|---|---|---|---|---|---|---|
| DRBB27 | ER2796 (Dam-) | 5.95 × 104 | 6.0 × 104 | - | pNZ8048 (Cm) | No |
| NRBB50 | ER2796 (Dam-) | 5.4 × 104 | 4.85 × 104 | - | pAM5 (Tet) | Partial R/M |
| NRBB01 | EC101 (Dam+) | 8.8 × 103 | 1.4 × 105 | - | pNZ8048 (Cm) | Yes |
| NRBB02 | EC101 (Dam+) | 2.01 × 105 | 2.8 × 105 | - | pAM5 (Tet) | Yes |
| NRBB02 | ER2796 (Dam-) | No transformants | - | - | pAM5 (Tet) | Yes |
| NRBB57 | EC101 (Dam+) | 5.37 × 104 | 1.03 × 106 | - | pDM1 (Spec) | Yes |
| NRBB52 | C29251 (Dam-/Dcm-) | 3.53 × 102 | 7.05 × 105 | 1.37 × 106 | pAM5 (Tet) | Yes |
In order to determine the impact of R/M systems on genetic accessibility, six B. breve representatives were examined (B. breve NRBB01, B. breve NRBB02, B. breve NRBB57, B. breve DRBB27, B. breve NRBB50 and B. breve NRBB52). For this purpose B. breve strains were first transformed with plasmids isolated from E. coli and the obtained bifidobacterial transformants were then used to recover these same plasmids, which would now be assumed to be methylated by native R/M systems. Interestingly and in line with our expectations, subsequent reintroduction of such methylated plasmid DNA into the same B. breve strains was shown to result in an increased transformation efficiency for those strains harbouring (functional) R/M systems. In contrast, the transformation efficacy of strains that did not encode, or were predicted to encode non-functional R/M systems was not increased when such methylated plasmid DNA was employed (Table 5). In particular, the absence of active R/M systems in B. breve DRBB27 and B. breve NRBB50 correlated with a high transformation efficiency of these strains.
A first indication that methylome information can be employed to circumvent the R/M barrier to make B. breve strains genetically more accessible was obtained by B. breve strain NRBB02 which harbours an active Bbr02I R/M system with a GATC-specific restriction endonuclease. For this strain no transformants were obtained when pAM5-tet plasmid DNA (containing twenty eight 5′-GATC-3′ sites) isolated from E. coli ER2796 (DAM– strain that does not methylate 5′-Gm6ATC-3′ sites) was used to transform this strain. Results from the transformation experiments show that M.Bbr02I is extremely active against 5′-GATC-3′ sites because a high transformation efficiency was obtained when plasmid DNA was used to transform B. breve NRBB02 that had been isolated from E. coli EC101 (DAM+ strain) (Table 5). This result confirms that a 5′-GATC-3′ targeting R/M system imposes a significant barrier to DNA acquisition.
In order to further substantiate our findings and to improve the genetic accessibility of this species, B. breve NRBB52 was selected as a target strain, based on the fact that it harbours a Type II R/M system (Bbr52I), which recognizes the relatively frequently occurring 5′-RGATCY-3′ sequence motif, and which thus is expected to imposes a substantial barrier to transformation. The M.Bbr52I methylase gene was cloned into pNZ8048 and introduced into E. coli ER2796 strain (see M&M), to generate strain E. coli ER2796.pNZ-M.NBB52. When the methylated pAM5 shuttle vector isolated from E. coli ER2796.pNZ-M.NBB52 harbouring the M.Bbr52I cloned MTase was used to transform B. breve NRBB52, a 4000-fold increase in the transformation efficiency was observed compared to the unmethylated pAM5 DNA from E. coli C29251 (Table 5). These results are not only consistent with the presence of an active Bbr52I system in B. breve NRBB52, but also confirm that the deduced methylome in this study can be succesfully used to improve the genetic accessibility of a given B. breve strain.
R/M systems and horizontal transfer in B. breve
R/M and CRISPR-Cas systems are believed to constitute the main barriers to horizontal transfer (HGT) in bacteria (16). To investigate this presumed barrier function, we searched (using PFAM and visual inspection) the 19 newly sequenced B. breve genomes for the presence of large chromosome-associated mobile regions in the form of prophages, integrated episomes and genomic regions (>20 kb) that contain a high density of hypothetical proteins and transposases possibly constituting a remnant of an acquired mobile genetic element. The analysis revealed the presence of such elements across B. breve ranging between a minimum of 1 to a maximum of 5 elements per genome. Interestingly, we found conserved prophages and Integrative Conjugative Elements (ICEs) always integrated in putative tRNA genes located in conserved positions along the chromosome, indicating the existence of integration hot spots in B. breve (Figure 6, panel A). Furthermore, it also appears that the occurrence and integration of these elements is more frequent around the Ter region of the B. breve chromosome (Figure 6, panel A).
Figure 6.

The B. breve mobilome and the impact of HGT on bacterial genome size. (A) Schematic representation of 20 B. breve strains and their predicted mobile genetic elements with genome size. Strains are divided in two groups based on the genome length and size of the cumulative mobilome. (B) Barplot showing differential distribution of mobilome DNA in the two size groups (orange and blue), ordered by genome length.
In order to determine if B. breve CRISPR-Cas and R/M systems impact on the frequency of (predicted) HGT, we searched for correlations between the presence/absence of such systems and the number of identified mobile genetic elements. Interestingly, 12 out of the 20 examined strains (19 B. breve representatives here sequenced plus the reference strain B. breve UCC2003) lack a CRISPR-Cas system, perhaps lowering the barrier to acquire exogenous genetic material (Table 2). In addition, our analysis revealed a correlation between the absence of CRISPR or short-target R/Ms (e.g. 5′-Gm6ATC-3′ associated with the RM3 cluster), and an increase of accessory gene families (variome) within the species, which possibly reflects the impact of the former systems on DNA acquisition by horizontal transfer means in B. breve (Figure 7, panel B).
Figure 7.

Horizontal Gene Transfer in B. breve strains. (A) Presence/absence of detected motifs in genomes ordered by frequency of occurrence. B. breve strains are grouped into two distinct clusters based on the genome length and the contribution of presence of CRISPR and R/M on HGT. Discriminant of the two groups at 2.4 Mb is also indicated. (B) Boxplot showing the inverse correlation between the presence of CRISPR and/or short-target R/M systems and variome size (expressed as number of accessory gene families [GF] in genomes). Median of each group and exact P-value are also indicated. Hypothesis tested with the Mann–Withney U test (significance P < 0.05).
In contrast, when neither a CRISPR locus nor a 5′-Gm6ATC-3′-targeting R/M system is present in a given B. breve strain, we observed a higher number of integrated mobile genetic elements in the corresponding genome. For example, the B. breve NRBB57 and B. breve 215W4-47a genomes contain three and five acquired regions, respectively, of which one is an ICE of ∼200 kb. The same can be said for B. breve CNCM I-4321 containing the highest number (five) of large mobile genetic elements so far observed in B. breve integrated in the chromosome (two prophages and three ICSs), accounting for a total of ∼300 kb of putatively acquired DNA (Figure 6).
CONCLUSIONS
The present study demonstrates the successful application of PacBio SMRT RSII sequencing to a large B. breve collection for the purpose of methylome analysis, in selected cases supplemented with BS-seq analysis and MTase cloning. These efforts generated a comprehensive overview of the R/M systems present in this bifidobacterial species.
The methylome definition of B. breve strains showed a high prevalence of m6A and m5C methylation across strains, while m4C methylation appears to be a rare occurrence. Accurate inspection of the methylation context of each modification allowed the identification of 15 distinct R/M systems assignable to Type I and Type II/IIG systems. As the occurrence of their recognized motifs also appears to be quite variable, it follows that they variably affect genetic accessibility of strains or horizontal gene transfer in general.
Among the various differences observed across the genomes analyzed, the considerable variability in genome size of strains was a rather striking feature. Moreover, the occurrence of additional DNA was limited to certain chromosomal locations (in particular the Ter region) and allowed the identification of tRNA genes as possible hot-spots for insertion of not only prophages, but also mobile genetic elements in general.
The frequent presence of mobile elements in those genomes lacking a CRISPR locus and/or R/M systems also suggests considerable impact of bacterial defense mechanisms on acquisition of exogenous DNA in B. breve and bifidobacteria in general (with possible advantages for the host which still needs to be further elucidated).
In conclusion, the present work demonstrates that representatives of B. breve exhibit considerable genomic diversity data, though such diversity seems to be balanced by the need of the host to protect itself against genetic invasions. The large diversity of defence systems, in particular R/M systems, may pose a substantial barrier to studies that want to investigate the genetic traits associated with these gut commensals, in particular if such traits are considered beneficial to its host. The findings of our methylome analysis coupled with the most complete identification of R/M systems in B. breve, represents an important advance in making bifidobacteria more genetically accessible. Ultimately, this will result in an improved understanding of the genetics and health-promoting potential of members of this species.
ACCESSION NUMBERS
The sequence here generated have been submitted to GenBank database under the following accession numbers: CP021394, CP021393, CP021553, CP021389, CP021552, CP023198, CP021387, CP021388, CP021559, CP023199, CP021390, CP021384, CP021386, CP021556, CP021385, CP023192, CP023193, CP023194, CP023195, CP023196, CP023197, CP021558, CP021555, CP021557, CP021554, CP021391, CP021392. CGH raw data are available in GEO database under the following accession numbers: GSE104927 and GSE27491.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Department of Agriculture Food and Marine (DAFM) for supporting the INFANTMET (Infant Nutrition for Programming the Gut Microbiota in Neonates) project, which allowed the isolation of some of the B. breve strains; we acknowledge all students and co-workers for their contribution and enthusiasm.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Nutricia Research, Utrecht, The Netherlands; D.vS., F.B., R.M., M.O.M., K.J., M.E. and J.vB. are members of The Alimentary Pharmabiotic Centre, which is a research centre funded by Science Foundation Ireland (SFI), through the Irish Government's National Development Plan; SFI [SFI/12/RC/2273]; FEMS Research Grant [FEMS-RG-2016-0103]; HRB [513 PDTM/20011/9]. Funding for open access charge: SFI [SFI/12/RC/2273]; FEMS Research Grant [FEMS-RG-2016-0103]; HRB [513 PDTM/20011/9].
Conflict of interest statement. R.J.R. works for New England Biolabs, a company that sells research reagents, including restriction enzymes and DNA methylases to the scientific community. J.L., K.vL. and J.K. are employees of Nutricia Research.
REFERENCES
- 1. Ursell L.K., Clemente J.C., Rideout J.R., Gevers D., Caporaso J.G., Knight R.. The interpersonal and intrapersonal diversity of human-associated microbiota in key body sites. J. Allergy. Clin. Immunol. 2012; 129:1204–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Turroni F., Peano C., Pass D.A., Foroni E., Severgnini M., Claesson M.J., Kerr C., Hourihane J., Murray D., Fuligni F. et al. . Diversity of bifidobacteria within the infant gut microbiota. PloS One. 2012; 7:e36957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O’Callaghan A., van Sinderen D.. Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 2016; 7:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tojo R., Suarez A., Clemente M.G., de los Reyes-Gavilan C.G., Margolles A., Gueimonde M., Ruas-Madiedo P.. Intestinal microbiota in health and disease: role of bifidobacteria in gut homeostasis. World J. Gastroenterol. 2014; 20:15163–15176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Duranti S., Gaiani F., Mancabelli L., Milani C., Grandi A., Bolchi A., Santoni A., Lugli G.A., Ferrario C., Mangifesta M. et al. . Elucidating the gut microbiome of ulcerative colitis: bifidobacteria as novel microbial biomarkers. FEMS Microbiol. Ecol. 2016; 92:fiw191. [DOI] [PubMed] [Google Scholar]
- 6. Boesten R., Schuren F., Ben Amor K., Haarman M., Knol J., de Vos W.M.. Bifidobacterium population analysis in the infant gut by direct mapping of genomic hybridization patterns: potential for monitoring temporal development and effects of dietary regimens. Microb. Biotechnol. 2011; 4:417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garrido D., Dallas D.C., Mills D.A.. Consumption of human milk glycoconjugates by infant-associated bifidobacteria: mechanisms and implications. Microbiology. 2013; 159:649–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yatsunenko T., Rey F.E., Manary M.J., Trehan I., Dominguez-Bello M.G., Contreras M., Magris M., Hidalgo G., Baldassano R.N., Anokhin A.P. et al. . Human gut microbiome viewed across age and geography. Nature. 2012; 486:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Avershina E., Storro O., Oien T., Johnsen R., Wilson R., Egeland T., Rudi K.. Bifidobacterial succession and correlation networks in a large unselected cohort of mothers and their children. Appl. Environ. Microbiol. 2013; 79:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arboleya S., Watkins C., Stanton C., Ross R.P.. Gut bifidobacteria populations in human health and aging. Front. Microbiol. 2016; 7:1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruiz L., O’Connell Motherway M., Lanigan N., van Sinderen D.. Transposon mutagenesis in Bifidobacterium breve: construction and characterization of a Tn5 transposon mutant library for Bifidobacterium breve UCC2003. PloS One. 2013; 8:e64699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Egan M., O’Connell Motherway M., Ventura M., van Sinderen D.. Metabolism of sialic acid by Bifidobacterium breve UCC2003. Appl. Environ. Microbiol. 2014; 80:4414–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. James K., O’Connell Motherway M., Bottacini F., van Sinderen D.. Bifidobacterium breve UCC2003 metabolises the human milk oligosaccharides lacto-N-tetraose and lacto-N-neo-tetraose through overlapping, yet distinct pathways. Sci. Rep. 2016; 6:38560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bottacini F., O’Connell Motherway M., Kuczynski J., O’Connell K.J., Serafini F., Duranti S., Milani C., Turroni F., Lugli G.A., Zomer A. et al. . Comparative genomics of the Bifidobacterium breve taxon. BMC Genomics. 2014; 15:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O’Connell Motherway M., Zomer A., Leahy S.C., Reunanen J., Bottacini F., Claesson M.J., O’Brien F., Flynn K., Casey P.G., Munoz J.A. et al. . Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:11217–11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Makarova K.S., Wolf Y.I., Koonin E.V.. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013; 41:4360–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oliveira P.H., Touchon M., Rocha E.P.. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014; 42:10618–10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O’Callaghan A., Bottacini F., O’Connell Motherway M., van Sinderen D.. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. BMC Genomics. 2015; 16:832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O’Connell Motherway M., O’Driscoll J., Fitzgerald G.F., Van Sinderen D.. Overcoming the restriction barrier to plasmid transformation and targeted mutagenesis in Bifidobacterium breve UCC2003. Microb. Biotechnol. 2009; 2:321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. O’Connell Motherway M., Watson D., Bottacini F., Clark T.A., Roberts R.J., Korlach J., Garault P., Chervaux C., van Hylckama Vlieg J.E., Smokvina T. et al. . Identification of restriction-modification systems of Bifidobacterium animalis subsp. lactis CNCM I-2494 by SMRT sequencing and associated methylome analysis. PLoS One. 2014; 9:e94875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015; 43:D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flusberg B.A., Webster D.R., Lee J.H., Travers K.J., Olivares E.C., Clark T.A., Korlach J., Turner S.W.. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods. 2010; 7:461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krueger F., Kreck B., Franke A., Andrews S.R.. DNA methylome analysis using short bisulfite sequencing data. Nat. Methods. 2012; 9:145–151. [DOI] [PubMed] [Google Scholar]
- 24. Murray N.E. 2001 Fred Griffith review lecture. Immigration control of DNA in bacteria: self versus non-self. Microbiology. 2002; 148:3–20. [DOI] [PubMed] [Google Scholar]
- 25. Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev S., Dryden D.T., Dybvig K. et al. . A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003; 31:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murray N.E. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev. 2000; 64:412–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khosaka T., Kiwaki M., Rak B.. Two site-specific endonucleases BinSI and BinSII from Bifidobacterium infantis. FEBS Lett. 1983; 163:170–174. [DOI] [PubMed] [Google Scholar]
- 28. Khosaka T., Sakurai T., Takahashi H., Saito H.. A new site-specific endonuclease BbeI from Bifidobacterium breve. Gene. 1982; 17:117–122. [DOI] [PubMed] [Google Scholar]
- 29. Hartke A., Benachour A., Boutibonnes P., Auffray Y.. Characterization of a complex restriction/modification system detected in a Bifidobacterium longum strain. Appl. Microbiol. Biotechnol. 1995; 45:132. [Google Scholar]
- 30. Skrypina N.A., Kramarov V.M., Liannaia A.M., Smolianinov V.V.. Restriction endonucleases from Bifidobacteria. Molekuliarnaia genetika, mikrobiologiia i virusologiia. 1988; 15–16. [PubMed] [Google Scholar]
- 31. Yasui K., Kano Y., Tanaka K., Watanabe K., Shimizu-Kadota M., Yoshikawa H., Suzuki T.. Improvement of bacterial transformation efficiency using plasmid artificial modification. Nucleic Acids Res. 2009; 37:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O’Riordan K., Fitzgerald G.F.. Evaluation of bifidobacteria for the production of antimicrobial compounds and assessment of performance in cottage cheese at refrigeration temperature. J. Appl. Microbiol. 1998; 85:103–114. [DOI] [PubMed] [Google Scholar]
- 33. Garcia De La Nava J., Santaella D.F., Alba J.C., Carazo J.M., Trelles O., Pascual-Montano A.. Engene: the processing and exploratory analysis of gene expression data. Bioinformatics. 2003; 19:657–658. [DOI] [PubMed] [Google Scholar]
- 34. van Hijum S.A.F.T., Garcia De La Nava J., Trelles O., Kok J., Kuipers O.P.. MicroPreP: a cDNA microarray data pre-processing framework. Appl. Bioinformatics. 2003; 2:241–244. [PubMed] [Google Scholar]
- 35. van Hijum S.A.F.T., De J.A., Baerends R.J., Karsens H.A., Kramer N.E., Larsen R., den Hengst C.D., Albers C.J., Kok J., Kuipers O.P.. A generally applicable validation scheme for the assessment of factors involved in reproducibility and quality of DNA-microarray data. BMC Genomics. 2005; 6:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Long A.D., Mangalam H.J., Chan B.Y., Tolleri L., Hatfield G.W., Baldi P.. Improved statistical inference from DNA microarray data using analysis of variance and a Bayesian statistical framework. Analysis of global gene expression in Escherichia coli K12. J. Biol. Chem. 2001; 276:19937–19944. [DOI] [PubMed] [Google Scholar]
- 37. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 38. Lagesen K., Hallin P., Rodland E.A., Staerfeldt H.H., Rognes T., Ussery D.W.. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007; 35:3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L.. Versatile and open software for comparing large genomes. Genome Biol. 2004; 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Enright A.J., Van Dongen S., Ouzounis C.A.. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002; 30:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gutierrez J., Maere S.. Modeling the evolution of molecular systems from a mechanistic perspective. Trends Plant Sci. 2014; 19:292–303. [DOI] [PubMed] [Google Scholar]
- 42. Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guindon S., Gascuel O.. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003; 52:696–704. [DOI] [PubMed] [Google Scholar]
- 44. De Man J.C., Rogosa M., Elisabeth Sharpe M.. A medium for the cultivation of lactobacilli. Appl. Microbiol. 1960; 130–135. [Google Scholar]
- 45. Law J., Buist G., Haandrikman A., Kok J., Venema G., Leenhouts K.. A system to generate chromosomal mutations in Lactococcus lactis which allows fast analysis of targeted genes. J. Bacteriol. 1995; 177:7011–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anton B.P., Mongodin E.F., Agrawal S., Fomenkov A., Byrd D.R., Roberts R.J., Raleigh E.A.. Complete genome sequence of ER2796, a DNA methyltransferase-deficient strain of Escherichia coli K-12. PLoS One. 2015; 10:e0127446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Milani C., Lugli G.A., Duranti S., Turroni F., Bottacini F., Mangifesta M., Sanchez B., Viappiani A., Mancabelli L., Taminiau B. et al. . Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl. Environ. Microbiol. 2014; 80:6290–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ventura M., Canchaya C., Del Casale A., Dellaglio F., Neviani E., Fitzgerald G.F., van Sinderen D.. Analysis of bifidobacterial evolution using a multilocus approach. Int. J. Syst. Evol. Microbiol. 2006; 56:2783–2792. [DOI] [PubMed] [Google Scholar]
- 49. Molloy E.K., Warnow T.. To include or not to include: the impact of gene filtering on species tree estimation methods. Syst. Bio. 2017; doi:10.1093/sysbio/syx077. [DOI] [PubMed] [Google Scholar]
- 50. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE: restriction enzymes and methyltransferases. Nucleic Acids Res. 2003; 31:418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murphy J., Mahony J., Ainsworth S., Nauta A., van Sinderen D.. Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 2013; 79:7547–7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ruiz L., Bottacini F., Boinett C.J., Cain A.K., O’Connell Motherway M., Lawley T.D., van Sinderen D.. The essential genomic landscape of the commensal Bifidobacterium breve UCC2003. Sci. Rep. 2017; 7:5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Claus H., Friedrich A., Frosch M., Vogel U.. Differential distribution of novel restriction-modification systems in clonal lineages of Neisseria meningitidis. J. Bacteriol. 2000; 182:1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Naderer M., Brust J.R., Knowle D., Blumenthal R.M.. Mobility of a restriction-modification system revealed by its genetic contexts in three hosts. J. Bacteriol. 2002; 184:2411–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.