


Abstract
RNA plays a central role in the expression of all genes. Because any sequence within RNA can be recognized by complementary base pairing, synthetic oligonucleotides and oligonucleotide mimics offer a general strategy for controlling processes that affect disease. The two primary antisense approaches for regulating expression through recognition of cellular RNAs are single-stranded antisense oligonucleotides and duplex RNAs. This review will discuss the chemical modifications and molecular mechanisms that make synthetic nucleic acid drugs possible. Lessons learned from recent clinical trials will be summarized. Ongoing clinical trials are likely to decisively test the adequacy of our current generation of antisense nucleic acid technologies and highlight areas where more basic research is needed.
INTRODUCTION
DNA makes RNA and RNA makes protein. The universal model for gene expression suggests that synthetic compounds that bind to RNA should modulate protein production. Because protein expression affects disease, recognition of RNA has the potential to be a broadly valuable approach to drug development. To make the concept even more inviting, RNA can be recognized by complementary base pairing. Identification of a lead compound that can bind to an RNA target with high affinity can be as simple as designing a complementary oligonucleotide followed by routine synthesis and testing.
The concept of using synthetic oligonucleotides to control the expression of genes that impact disease has been pursued for 40 years (1). Until recently, clinical success was elusive—for good reason. Synthetic nucleic acids are large, highly negatively charged molecules. They have fundamentally different chemical properties relative to the typical small molecule drugs that have been the backbone of the pharmaceutical industry. Unlike antibodies, oligonucleotides that aim to regulate gene expression must cross cell membranes to reach their molecular targets.
These obstacles to success required a new science of drug development (2). In some cases, the lessons from small molecule drugs needed to be ignored. For example, the dogma that drugs needed to be non-polar small molecules to be active inside cells was incompatible with the fact that oligonucleotides are large, highly charged molecules (Figure 1).
Figure 1.

Properties of small molecule, antibody and oligonucleotide drugs.
In other cases, lessons from traditional pharmaceutical development proved invaluable. Even though oligonucleotides have atypical chemical properties, they remain synthetic molecules. Like other drugs, they must reach disease-associated tissues to function. By manipulating their chemical structure, it is possible to enhance potency and limit toxicities. While development of large negatively charged drugs presented novel challenges, oligonucleotides are not some strange alien drug development platform that fall outside of known models for pharmaceutical development.
After many years of slow progress, the pace of development for nucleic acid therapeutics is quickening with several clinical trials reaching decisive phases during 2016–2019. The purpose of this review is to introduce the mechanisms of action used by approved nucleic acid therapeutics and those in advanced clinical trials. The impact of chemical modifications on drug design will be discussed and the lessons learned from key clinical trials will be described.
CHEMICAL MODIFICATIONS
Background
Single-stranded DNA and RNA oligonucleotides have properties that complicate drug development. Unfavorable properties include: (i) degradation by nucleases when introduced into biological systems, (ii) poor uptake through cell membranes, (iii) unfavorable biodistribution and pharmacokinetic properties and (iv) sub-optimal binding affinity for complementary sequences (3,4).
To improve these properties, oligonucleotides must be chemically modified by changing the phosphodiester linkages, the ribose backbone or the nucleobases (Figure 2). The chemical modification of oligonucleotides for therapy has been the subject of recent authoritative reviews (5,6) and this section will be restricted to the factors that mostly influence drug development. Methods for large-scale nucleic acid synthesis are well developed (7) and will not be covered here.
Figure 2.

Chemical modifications of oligonucleotides used in major clinical trials. (X = OH or H).
Phosphorothioate modifications
Phosphorothioate (PS) (replacement of a non-bridging phosphodiester oxygen by sulfur) modification is the most widely used single alteration in nucleic acid drug development (8). PS linkages serve two purposes. The first role is to increase stability toward digestion to nucleases. The modification transforms DNA or RNA sequences with half-lives of minutes to half-lives of days. The second role is to increase binding to proteins, especially serum proteins. Serum interactions include high affinity interactions with heparin binding proteins and lower affinity interactions with albumin (9). Increased binding to serum proteins preserves oligonucleotide in circulation, slows removal by the liver and boosts the time available for uptake into target tissues.
PS linkages have two stereoisomers, while phosphodiester linkages are prochiral. PS antisense oligonucleotides (ASOs) synthesized by standard methods will be a collection of diastereomers, some of which may be more active than others. A recent report has suggested that PS stereochemistry affects the pharmacological properties and efficacy of ASOs and that it is possible to identify stereopure ASOs that are more active (10). It will be interesting to follow the development of stereopure ASOs and published data defining their potential as antisense agents.
2′-Ribose modifications
Modifications to the ribose are a common class of alterations. The ribose can be modified through replacement of the 2′-hydroxyl by many different groups but most commonly by 2′-O-methyl, 2′-O-methoxyethyl and 2′-fluoro (11). 2′-Methoxyethyl has probably been the most widely used ribose modification employed in the clinic to date. The 2′ modifications increase stability toward digestion by nucleases by blocking the nucleophilic 2′ hydroxyl moiety. The modifications also increase the thermal stability of complementary hybridization, encouraging tighter binding and allowing use of shorter oligonucleotides.
Because 2′ modified nucleotides are more similar to RNA than DNA, hybrids formed between 2′-modified sequences and cellular RNA are not substrates for RNase H and will not be cleaved. As will be seen below, the avoidance of RNase H cleavage can be exploited for ‘steric blocking’ mechanisms used to preserve the mRNA target and is currently used in the design of oligomers that modulate alternative splicing.
Bridged nucleic acids
The 2′-oxygen can also be linked through bridging carbons to the 4′ carbon of the ribose to form a bridged nucleic acid (BNA). Perhaps the most commonly used BNA in laboratories is locked nucleic acid (LNA) characterized by a 2′,4′-methylene linkage (12–14). A second BNA, 2′,4′-constrained ethyl nucleic acid ((S)-cET) (15) is now being widely tested in the clinic (16).
BNA is an especially useful type of modification for increasing the strength of hybridization. The 2′,4′-constraint locks the ribose in a conformation that is ideal for binding complementary sequences, reducing the entropic price paid during Watson–Crick base-pairing. Introducing a single BNA substitution can enhance a melting temperature by as much as 5–10°C, allowing the affinity of complementary hybridization to be tailored for clinical applications. High affinity binding allows clinical candidates to be as short as thirteen nucleotides in length (17).
Cellular delivery of ASOs and dsRNAs
The current suite of chemical modifications provides good stability toward digestion by nucleases and achieves favorable binding to complementary sequences. Therefore, rather than achieving greater affinity or stability, the primary current need for innovation is the development of improved methods for the delivery of oligonucleotides to disease-related tissues (3).
In the laboratory, single-stranded PS-containing ASOs can be introduced into cells by gymnotic (‘naked’) uptake in the absence of lipid (18,19). This approach was shown to be general by Stein et al. (18), leading to the widespread application of gymnosis to large scale laboratory testing to facilitate identification of candidate ASOs for drug development. Advantages of gymnotic delivery include: (i) avoiding use of transfection agent and the associated potential for toxicity, (ii) application to cell lines that are difficult to transfect; and (iii) greater convenience, especially when large number of experiments are necessary for screening. The disadvantage is that the method requires relatively high concentrations (0.1–10 μM versus 1–10 nM when carrier is used) of ASO and is often cost-prohibitive for many laboratories.
In the clinic, single-stranded ASOs can be formulated in simple saline solutions and administered to patients. When administered systemically, ASOs accumulate in the liver and are a general approach to controlling gene expression (20). Although 90% of ASO in plasma binds serum albumin and are not subject to glomerular filtration, in mice and non-human primates, ASOs do accumulate in the kidneys and can be used to knock down gene expression (21–23). Intrathecal delivery of saline-formulated ASO’s has also been successful for targets in the central nervous system (CNS) related to neurodegenerative disease. Uptake in other tissues is less effective and achieving favorable biodistribution in a wider range of normal and diseased tissue remains a major challenge for research.
By contrast to ASOs, duplex RNAs (dsRNAs) do not efficiently enter tissues when administered in saline. One strategy for overcoming the roadblock posed by the need for duplex strands is to use single-stranded RNA that can function through the RNAi pathway. When suitably modified with PS and other modifications described above, single-stranded silencing RNAs (ss-siRNAs) can be stabilized while retaining the ability to function through the RNAi pathway (24,25).
In theory, the ss-siRNA approach offers the potential to combine the strengths of dsRNA and ASOs, but more research is required to determine that those theoretical strengths can be translated into improved pre-clinical or clinical outcomes. ‘Self-delivering’ dsRNAs are being developed and tested in vitro and in vivo that avoid the need for a delivery vehicle or conjugation to receptor target agents like GalNAc (26). These dsRNAs use relatively simple hydrophobic modifications like cholesterol, increasing the potential that they can help drive down the material cost of therapeutic RNAs.
Nanoparticle assisted delivery
To achieve uptake in vivo, dsRNAs can be complexed with nanoparticles (3). Nanoparticles are typically lipid-based formulations and their advantages include protection of RNA, increased half-life in serum and improved passage into cells. Disadvantages include increasing the complexity of the formulation and the potential to induce toxicity separate from the active dsRNA. Nanoparticles can alter the pharmacokinetics and the biodistribution of oligonucleotides and these new properties should be characterized with care. Patisiran, a dsRNA encapsulated in a nanoparticle for delivery to the liver is likely to be the first approved dsRNA drug (27) (see below). It remains to be seen whether nanoparticles will enable clinical success for dsRNA in non-hepatic organs where other delivery options are unavailable.
Targeting ligands
Another option is conjugation to targeting ligands. To date, the most widely used modification has been the addition of N-acetylgalactosamine (GalNAc) to the passenger strand of dsRNA (28) (Figure 3). This modification enhances delivery to hepatocytes through binding to the asialoglycoprotein receptor, increasing internalization and permitting potent gene knock down in vivo without the need for nanoparticle formulation. GalNAc delivery is now contributing to a wide spectrum of clinical and pre-clinical programs (29), and is demonstrating sustained efficacy with relatively low doses. The application of GalNAc to the delivery of dsRNAs is an exciting frontier and clinical data will be discussed in detail below.
Figure 3.

Conjugation of GalNAc to passenger strand of dsRNAs or ASOs to enhance delivery to the liver. ASGPR: asialoglycoprotein receptor.
GalNAc conjugation can also be used to enhance delivery of ASOs (30–32) and a key question for future research will be whether GalNAc will have as major impact on the delivery of ASOs as the modification has had for dsRNA. For ASOs, the modification is not necessary for potent hepatic gene knockdown but may widen the therapeutic window by increasing potency. A GalNAc-conjugated ASO, IONIS-APO(a)-LRX has been tested in a Phase 2 trial and produced lower lipoprotein(a) levels. Effects were long-lasting and suggest that GalNAc is a promising clinical option for ASOs (33).
Another key question is whether the strategy can be extended to other ligand–receptor pairs and therefore to other organs or tumors. The asialoglycoprotein receptor is expressed at an unusually high level on the surface of hepatocytes (34). It is not clear that the success of the approach can be widely replicated by ligand–receptor pairs that are less well expressed. Rates of receptor internalization and re-cycling will also be critical factors determining successful delivery.
MECHANISMS OF OLIGONUCLEOTIDE DRUGS
Background
There are many different classes of nucleic acids inside cells that might be targets for therapeutic oligonucleotides (Figure 4). These targets have different structures and functions and present different challenges during drug discovery and development. To achieve potent recognition of diverse targets, synthetic oligonucleotides can be designed to function by several different mechanisms (Figure 5).
Figure 4.

Types of target for therapeutic oligonucleotides.
Figure 5.
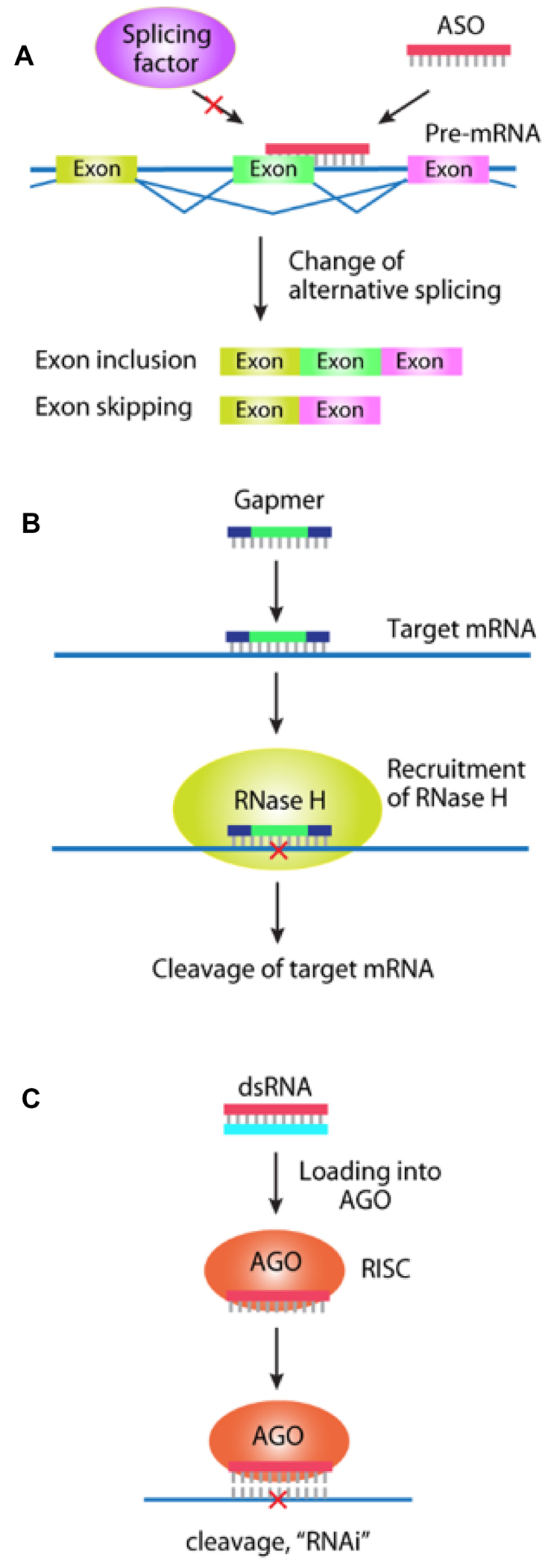
Mechanisms of action for therapeutic oligonucleotides. (A) control of splicing by ASOs and alternative splicing. In contrast to gapmers, ASOs that control splicing possess ribose or morpholino modifications throughout the oligomer; (B) block of translation by antisense gapmers (oligonucleotide that contain 2′-modified RNA flanking a central DNA region) targeting mRNA; (C) block of translation by a fully complementary dsRNA and RNA interference.
Regulating splicing with ASOs
Pre-mRNA is spliced into mature mRNA in the nucleus prior to translation in the cytoplasm. Alternative splicing produces different protein variants and changing the prevalence of one variant can offer therapeutic benefits. ASOs can modulate alternative splicing by binding to pre-mRNA and disrupting recognition by splicing factors (35,36) (Figures 5A and 6).
Figure 6.

Design and clinical status of oligonucleotides drugs discussed in this review. Source material for this figure is cited as Supplementary Data. For Inclisiran, Fitusiran, and Givosiran, we could not locate publically available information and the structures shown below are based on the cited descriptions of this class of compound.
The first step in developing an ASO to affect splicing is to identify a disease that might be alleviated by changing the expression of a splice variant. For example, it might be possible to use ASOs to increase expression of a protein variant and restore a therapeutic level of protein function. Active ASOs are identified empirically by testing multiple ASOs that are complementary to sequences near intron/exon junctions. Testing multiple ASOs is necessary because activities sometimes can differ dramatically, even when ASOs target closely related sequences (37).
ASOs affect splicing by blocking the binding of splicing factors. They are designed to contain modified ribose or morpholino modifications to remove any potential to form RNA–DNA hybrids that allow recruitment of RNase H. ‘Steric blocking’ ASOs do not promote cleavage of the target RNA and function by a different mechanism than RNase H-active ASOs (see below). Two splice modulating ASOs, nusinersen and eteplirsen, have recently been approved by the Food and Drug Administration (FDA) (Figure 6).
Regulating translation with ASOs by recruiting RNase H
DNA:RNA hybrids are substrates for the enzyme RNase H. While the textbook role of RNase H is degradation of the RNA–DNA hybrids produced by lagging strand synthesis in the nucleus, RNase H also exists in the cytoplasm and can degrade mature mRNA (38,39). When an oligonucleotide containing DNA bases binds to mRNA, it can induce cleavage of the mRNA. (Figures 5B and 6).
ASO ‘gapmers’ contain chemically modified RNA bases flanking both sides of a central eight to ten base DNA ‘gap’ (40). The RNA bases contain modifications that enhance affinity to complementary sequences, while the DNA bases serve as a substrate for RNase H. RNase-induced cleavage amplifies the potency of synthetic ASOs by destroying the target mRNA and is a key feature of therapeutic oligonucleotides designed to inhibit translation (41,42).
Regulating translation with duplex RNAs and RNA interference
In 2001, Tuschl and Elbashir demonstrated that RNA interference (RNAi) could function through synthetic 21 base RNA duplexes (dsRNAs) in human cells (43). These dsRNAs could be readily synthesized, transfected into cultured cells and used to control gene expression.
dsRNAs are composed of two complementary strands, the passenger strand and the guide strand. After cellular uptake, the dsRNAs associate with the RNA-induced silencing complex, which normally exists to facilitate the action of endogenous micro RNAs (miRNAs) (44) (Figure 5C). The passenger strand protects the guide strand from hydrolysis by nucleases. The guide strand binds to argonaute 2 (AGO2) protein (45,46) to form a ribonucleoprotein complex and provides the specificity domain that programs the complex for recognition of complementary sequences. AGO2 protects the guide strand and facilitates efficient location of target sequences. When sequences are fully complementary, AGO2 cleaves the target RNA.
Cleavage of target mRNA by AGO2 enhances the potency of RNAi, producing an outcome like that observed for RNase H-mediated cleavage by gapmer ASOs. When dsRNAs contain centrally-located mismatches relative to their target sequences they more closely resemble endogenous miRNAs. These mismatches disrupt cleavage by AGO2, permitting binding but leaving the target mRNA intact (47). Therefore, there are two mechanisms for dsRNA activity—cleavage by fully complementary dsRNA or steric blocking by mismatch containing (miRNA-like) RNA. Examples of fully complementary dsRNAs being tested in advanced clinical trials are shown in Figure 6.
ASOs versus dsRNAs
The discovery that dsRNAs could act through RNAi in mammalian cells rapidly led to a drug discovery technology capable of competing with gapmer ASOs (48) (Figure 5C). Both approaches are antisense technologies because they both involve recognition of mRNA by an antisense strand. Both use synthetic oligonucleotides and benefit from many of the same chemical modifications. Both aim to reduce RNA levels, and most lessons about the design of control experiments or dangers from off-target effects apply equally (49). Like ASOs, dsRNAs can also redirect alternative splicing (50) but this strategy has not been used for clinical development.
Key differences include the obvious fact that dsRNAs have two strands and ASOs have just one. The difference in strand number has several implications. The cost of goods for dsRNA will likely be more than for an ASO designed to modulate the same target. A second difference is the requirement for two strands reduces the cellular uptake in vivo relative to ASOs. While ASOs can be delivered to some tissues after formulation in saline, delivery of dsRNAs requires conjugation to GalNAc or other targeting agents or complexation with nanoparticles (3). As noted above, the mechanisms of action differ. ASO gapmers function by inducing RNase H cleavage of their targets. dsRNAs, by contrast, co-opt the endogenous RNAi machinery to facilitate efficient recognition and cleavage of their targets.
The exploitation of a natural mechanism led to robust results in cell culture and rapid adoption of RNAi and synthetic RNA as a widely-used laboratory tool for controlling gene expression. Another reason why dsRNAs have been adopted widely as experimental tools is that the chemical modifications necessary for efficient gene knock down by ASOs are either expensive or unavailable commercially. dsRNAs, by contrast, do not need to be chemically modified for laboratory use. Finally, there are useful algorithms for selecting good candidate dsRNAs, less so for ASOs.
Applications for dsRNAs in the laboratory can often be straightforward, but it is important to treat their use with caution. For example, the use of large panels of dsRNAs for screening can yield many off target effects (51). Because of this danger of off target effects, large-scale target screens should also be evaluated critically and candidate silencing agents validated individually.
While dsRNAs are often preferred relative to ASOs for use in the laboratory, this preference in cell culture applications may not parallel success in vivo. Seventeen years after the discovery of mammalian RNAi, dsRNAs and ASOs remain in close competition as development technologies for nucleic acid therapeutics. A mature understanding of the merits of each approach will await head to head comparisons of success with different targets from multiple clinical programs over the next 3–5 years.
Recognition of miRNAs by anti-miRNAs
microRNAs are endogenously expressed small RNAs that help regulate gene expression. Oligonucleotides that target miRNAs are known as anti-miRs. This topic has been recently reviewed (52) and will only be discussed briefly here. The design of anti-miRs draws from the same pool of chemical modifications as ASOs or dsRNAs. Unlike gapmers, anti-miRs do not recruit RNase H and act by directly binding to the miRNA, blocking it, and preventing recognition of target. Duplex oligonucleotides can also be designed as synthetic miRNAs to mimic function, providing another strategy for drug development.
In the clinic, RG-012 (Regulus/Sanofi) is being tested in Phase II trials. RG-012 is an anti-miR that inhibits the function of miR-21 for the treatment of Alport syndrome (53). Alport Syndrome is a severe genetic kidney disease and it was hypothesized that inhibition of miR-21 would block pathways that contribute to fibrogenesis. Pre-clinical data showed potent inhibition of miR-21 and increase in the lifespan of the mice by up to 50%. Results from a renal biopsy study are due in 2018 and may provide insights into target engagement and tissue biodistribution. Miravirsen (SPC3649, Santaris/Roche) (Figure 6) is an anti-miR with LNA, DNA and PS modifications that inhibits miR-122 for the treatment of Hepatitis C. Miravirsen-reduced HCV RNA levels without viral resistance in Phase II trials (54–57).
Recognition of proteins by aptamer oligonucleotides
Antibodies have emerged as a successful class of pharmaceutical. Therefore, while the focus of this review is therapeutic oligonucleotides that target cellular RNA, it is useful to note that oligonucleotides can also bind with high selectivity to proteins. Nucleic acids that recognize the shape and overall chemical structure of macromolecules are termed ‘aptamers’ and are selected by PCR-based methods. A library of oligonucleotides is synthesized, and applied to a support-bound target (usually a protein). Compounds that bound the target are then amplified through repeated rounds of purification. The development of aptamers recently been thoroughly reviewed (58).
The first commercially successful therapeutic oligonucleotide was an aptamer, pegaptanib (Macugen, Pfizer/Eyetech). Pegaptanib was selected for binding to vascular endothelial growth factor (VEGF) and approved by the FDA for the treatment of macular degeneration in 2004. While initially successful, pegaptanib soon succumbed to competition from VEGF-specific monoclonal antibodies bevacizumab (Avastin, Genentech) and ranibizumab (Lucentis, Genetech/Novartis) (58).
This outcome offers general lessons for oligonucleotide therapeutics. Pegaptanib was administered directly into the eye (intravitreal administration), revealing a potential strength and opportunity for oligonucleotide therapeutics. Because less material is needed and the dosing is local, costs and the potential for side effects are reduced. Administration was well tolerated. A general strength of aptamers relative to ASOs or dsRNAs is that they do not need to cross cell membranes to be functional, removing a critical barrier to efficacy. This strength, however, is shared by antibodies creating a direct head to head competition that, at least in the case of pegaptanib, was won by the antibodies. Competition with antibodies and other drug development strategies will be a recurrent theme when considering the prospects for clinical success of ASOs and dsRNAs.
Off-target effects: a threat that cannot be over emphasized
No drug is perfect. For every drug, there is a concentration required to achieve therapeutic benefit. There is another concentration at which toxicity is observed. The difference between these points is the ‘therapeutic window’ governing successful application. Like any other drugs, ASOs will always have associated toxicities and the key to success will be designing nucleic acid therapeutics to achieve a useful therapeutic window.
Off-target effects are physiologic changes that are unrelated to binding of a drug to its intended target. While all drugs have the potential to suffer from off-target effects, oligonucleotides are prone to a diverse array of off-target interactions because of their size, negative charge, and potential to bind RNA and DNA by complementary base-pairing (59–66). Potential off-target mechanisms include:
Binding of nucleic acid to cell surface proteins or to proteins inside cells. Any protein that has the potential to bind to a polyanionic macromolecule has the potential to bind to an oligonucleotide. Many proteins naturally interact with RNA or DNA. While the interactions with synthetic oligonucleotides may not be as strong as with their native partners, some interactions will occur. Because there are many negative charges on an oligonucleotide, even weak interactions can add up to significant overall binding affinities. One outcome is that synthetic oligonucleotides can bind serine/threonine protein kinase PKR or Toll-like receptors (TLRs) and activate the innate immune response. (67)
Binding to complementary nucleic acids. All ASOs and dsRNAs will be at least partially complementarity to DNA or RNA sequences inside cells that are not their intended targets. Such inappropriate binding might directly modulate the expression of other genes and yield confounding physiological effects. Because DNA sequences are bound in the double helix and are associated with chromatin proteins, they are a lesser concern.
For dsRNAs, interference with normal pathways that involve miRNAs has the potential to be problematic. In theory, introducing sufficient artificial RNA into cells may co-opt the endogenous protein machinery for RNAi and block the normal function of miRNAs (68). This phenomenon has not yet been observed to be a problem clinically.
It is impossible to eliminate these interactions; they will always contribute to some level of off-target effects. The challenge is to understand the origins of off-target effects well enough to design ASOs and dsRNAs to have the best likelihood of achieving acceptable toxicity profiles. For example, one recent report suggested that the propensity of gapmer ASOs to cleave their mRNA targets contributes to toxicity relative to steric blocking ASOs that leave mRNA intact (69). These data have significant implications for clinical trials using ASOs that function by these two mechanisms and provides guidance for identifying candidate gapmer oligonucleotides.
In the clinic, thrombocytopenia has attracted attention as a side effect with the potential to have a major impact on ASO development. Thrombocytopenia is a condition characterized by low platelet counts that decrease clot formation and increase the risk of bleeding. While thrombocytopenia had long been a concern for ASOs (70), it became the focus of renewed attention after severe adverse events due to thrombocytopenia were observed during trials of two different ASOs, volanesoren and IONIS-TTRrx (71). These events provoked serious concern that thrombocytopenia might be a general roadblock to clinical progress for ASOs as a class.
Each ASO, however, is different. The level of toxicity will be related to the sequence of the ASO and careful selection of clinical candidates may mitigate risk (72). While it is hoped that unsatisfactory candidates can be eliminated in pre-clinical testing or during Phase 1 trials, careful monitoring for thrombocytopenia may be useful during clinical trials using ASOs and after drug approval. Thrombocytopenia has not been noted to be a major adverse event associated with use of dsRNAs.
KEY CLINICAL CANDIDATES AND APPROVED DRUGS
Fomivirsen
Commercial development of therapeutic ASOs began in the late 1980’s when the tool box of chemical modifications was limited and knowledge of biodistribution and pharmacology was primitive. Given the challenge of developing new technology for drug development, it is not surprising that the undeveloped understanding of key obstacles led to a series of failed early clinical trials. The one exception was FDA approval of fomivirsen (Vitravene, Ionis Pharmaceuticals).
Fomivirsen was a 21 base PS-modified DNA oligonucleotide complementary to the critical UL123 gene within cytomegalovirus (CMV) for treatment of blindness caused by CMV retinitis (73,74) (Figure 6). Like the aptamer pegaptanib, fomivirsen was administered directly into the eye by intravitreal injection, taking advantage of local administration to simplify dosing and reduce cost. Clinical trials showed benefit to patients and the drug was approved by the FDA in 1998.
Fomivirsen’s time as a clinically used drug was short-lived because advances in anti-retroviral therapy were reducing the number of AIDS patients subject to opportunistic infections like CMV retinitis. The declining clinical need could not have been predicted when fomivirsen’s development began at the height of the AIDS crisis in western countries. The outcome reinforces the general lesson for future programs that clinical need must be a primary consideration when initiating programs but that subsequent periodic reconsideration of the need for the drug is necessary as competing technologies advance.
Despite the approval of fomivirsen, the clinical potential of gene silencing in the eye remains undetermined and the basic science of ocular activity of nucleic acid therapeutics remains an important area of research. For example, we lack an understanding of the fine detail characterizing ASO or dsRNA uptake in the different parts of the eye. There is one report that ASOs can be delivered by eye drops (75) but more studies are needed to explore the potential efficacy of topical delivery.
Oblimersen
Oblimersen (Genesense, Genta) was an 18 base PS DNA ASO-developed to target the initiation codon of bcl-2 mRNA (76) (Figure 6). The goal was to downregulate Bcl-2 expression and sensitize cancer cells to chemotherapy. Clinical trials proceeded for more than a decade including multiple phase III trials. Despite occasional tantalizing results, efficacy remained unproven and development was terminated in 2012 (77).
One important lesson from the oblimersen experience is that the mechanism of drug action matters. Oligonucleotides are large molecules that can have complex anti-proliferative effects that may not necessarily be related to recognition of the intended mRNA target (62). For ASOs that target bcl-2, Stein and co-workers (78,79) demonstrated that pro-apoptotic effects were independent of sequence. Equally troubling, an siRNA that did sequence-specifically downregulate bcl-2 did not have anti-proliferative effects.
There is no requirement that the mechanism of action for a drug must be understood for it to be successful (80). However, given the many unknowns involved in developing nucleic acid therapeutics, a failure to establish that a gene knockdown is sequence-specific and related to a therapeutic effect will increase the likelihood of poor clinical performance and eventual failure in Phase III trials for efficacy.
A second important lesson is that oblimersen was intended to target gene expression in non-hepatic cancerous cells, a difficult task. There was little precedent for believing that those cells could be targeted effectively when oblimersen was developed in the 1990’s and targeting cancer cells remains problematic today. Choice of drug target for a nucleic acid therapeutic should follow the demonstrated pharmacology and biodistribution of this class of drugs. Well characterized demonstrations of delivery of ASOs and dsRNAs to tumors followed by potent knockdown of the intended target gene’s expression in well controlled experiments remain important landmarks for research.
GRN163L
GRN163L (Imetelstat, Geron) (Figure 6) is a special case ASO. Rather than targeting mRNA, it targets the RNA component of the ribonucleoprotein telomerase (81,82). Telomerase is the enzyme responsible for maintaining telomere length and consists of a protein domain that polymerizes nucleotide addition and a template RNA domain. Telomerase activity is absent in most somatic cells but is reactivated in most cancers, leading to the hypotheses that (i) telomerase is necessary for maintain telomeres in proliferating cancer cells and (ii) inhibiting telomerase activity could curb cell proliferation. In cell culture, blocking telomerase with ASOs complementary to the RNA template leads to decreased cell proliferation over a period of weeks (83).
Anti-telomerase ASOs present an unusually promising therapeutic opportunity because a single ASO would have the potential to treat many different types of cancer where telomere renewal might be rate limiting. Despite this promise, progress has been slow and clinical trials inconclusive. Like oblimersen, an anti-telomerase ASO would need to enter cancerous tissue where uptake of ASOs is much less efficient than in the liver.
GRN163L is a thirteen base phosphoramidate oligonucleotide conjugated to a 5′-palmitoyl (C16) lipid group (Figure 6). The scientific basis for choosing the phosphoramidate modification rather than more commonly used 2′ derivatized RNA was not clear, and the need to develop protocols for large scale phosphoramidate synthesis delayed development. The 5′-palmitoyl group was intended to improve cell permeability, thereby improving uptake and potency.
Cancer cell telomeres are relatively long and inhibition of telomerase activity would be expected to cause a slow erosion that would take weeks before leading to reduced proliferation. Such slow effects complicate clinical trials and led to additional delays. While clinical trials have not yielded decisive results, they are ongoing have produced promising findings for essential thrombocytothemia (84), myelofibrosis (85) and myelodysplastic syndromes (86). Current clinical trials are directed toward myelofibrosis (Phase 2) and myelodysplastic syndromes (Phase 2/3).
Even if GRN163L fails in the clinic, it is possible that a redesigned ASO that is more potent or has better uptake properties may one day exploit the promise of telomerase as a target. If GRN163L does succeed as a drug, it is possible that it may not be functioning by inhibiting telomerase and careful study would be needed to definitively investigate mechanism. The story of ASOs as telomerase inhibitors should remain a productive area for research and clinical development.
Mipomersen
Mipomersen (Kynamro, Ionis Pharmaceuticals) (Figure 6) is a gapmer ASO designed to decrease expression of ApoB protein, reduce cholesterol levels and treat familial hypercholesterolemia. (87–89). Mipomersen was a landmark antisense drug because pre-clinical and clinical data convincingly demonstrated efficacy in vivo (87–91). While some patients had adverse reactions, the overall pre-clinical data demonstrated that ASOs can be administered systemically and reduce expression of the target gene in animals through the predicted antisense mechanism. Analysis of serum was used to confirm that both apoB and low density lipoprotein (LDL)-cholesterol were lowered. This outcome was ground breaking because it showed that ASOs could be potent enough to produce a physiologically relevant effect on protein expression.
From a practical standpoint, the results demonstrated the value of possessing an easily measured outcome that could be evaluated in Phase I trials. Phase I trials are relatively inexpensive, and having clear molecular markers of compound efficacy increases the likelihood that correct decisions will be made regarding whether to proceed with costly Phase II/III trials by definitively demonstrating that the appropriate target is being engaged and silenced.
Mipomersen was approved by the FDA under the trade name Kynamro, becoming the first FDA-approved systemically-delivered ASO in 2013. To date, however, mipomersen has not been a commercial success. Mipomersen was approved at the same time as a second drug, lomitapide (Juxtapid, Novelion). Lomitipide is a small molecule designed to bind microsomal triglyceride transfer protein, block lipid transfer and lower apoB secretion. Both drugs lower cholesterol levels, run the risk of hepatotoxicity and require strict treatment guidelines and physician training.
Lomitapide has been a commercial success even though mipomersen was offered at a lower price. Why the divergence? At least initially, concern regarding the toxicity of mipomersen was greater than lomitipide. Lomitipide is administered once a day orally, rather than by injection once a week, a convenience for patients. Business and marketing strategies may also have played a role. As with macugen, an effective nucleic acid drug was confronted with a competing agent and, in this case, the outcome of the competition was not favorable for mipomersen.
Eteplirsen and drisapersen
Duchenne muscular dystrophy (DMD) is a progressive neuromuscular disease with high unmet clinical need. Patients lose mobility and become wheel chair bound by their teens, require assisted ventilation by their 20’s and experience premature death in their 30’s or 40’s. DMD is caused by mutations within the dystrophin gene that disrupt the reading frame or cause premature termination of protein synthesis. It is difficult to imagine how a small molecule or antibody might restore dystrophin function, creating a need for alternative treatment strategies.
It is possible to use ASOs to alter splicing of dystrophin to generate a protein variant that is found in the less severe Becker muscular dystrophy, leading to the hypothesis that ASOs could be a treatment for DMD (92,93). To be successful, ASOs would need to be delivered into muscle at concentrations high enough to create the functional dystrophin needed to improve disease prognosis. The challenge is to achieve adequate delivery to muscle with ASOs that are best known for uptake by the liver when introduced systemically—an example of the interface between high unmet need and a technology pushed to its limits.
Two ASOs were developed to induce alternative splicing of mutant dystrophin and restore partial activity. Drisapersen (BioMarin) was a 2′-O-methyl PS oligonucleotide (94), while eteplirsen (Exondys 51, Sarepta) (95) was an uncharged morpholino oligomer (Figure 6). The histories of the two drugs and the dramatic events surrounding their clinical trials has been elegantly reviewed elsewhere (96–98).
Drisapersen was tested in Phase 2 and Phase 3 clinical trials that were placebo controlled and examined outcomes in more than 300 patients. It failed to show evidence of restoring dystrophin production in patients in these trials. While some differences in the primary endpoint—the 6 min walk test—were noted, these differences were not clinically significant. Thrombocytopenia was noted in some patients raising the risk that the treatment could cause harm. Drisapersen was not approved for marketing.
By contrast to the relative extensive testing of drisaperson, eteplirsen was tested in a Phase 3 trial that examined only twelve patients and was not placebo controlled (95). While there appeared to be some improvement in patients relative to historical controls, the size of the trial prevented the evaluation of statistical significance. Biopsies revealed a small, 0.28%, increase in dystrophin levels, a result that was difficult to reconcile with a favorable physiologic outcome.
While the clinical evidence for efficacy was underwhelming, there were also no reports of severe side effects. After contentious debate, the FDA approved eteplirsen in 2016 with the caveat that a larger placebo-controlled trial must be run to provide more conclusive evidence for efficacy. In a follow-up study, after 3 years of treatment patients treated with eteplirsen showed a slower rate of decline in the 6-min walk test than historical controls (99). Only 13% of DMD patients may be amenable by eteplirsen through exon 51 skipping. In addition, it is suggested that 80% of patients have genotypes amenable to exon-skipping and SRP-4053 (exon 53 skipping) and SRP-4045 (exon 45 skipping) are actively ongoing in Phase III trials by Sarepta (100). The debate over eteplirsen is ongoing (99,101–103).
The two drugs teach important lessons. The disease target, correction of dystrophin for treatment of muscular dystrophy, was an excellent one. The unmet need was so high and the absence of competing therapies so severe that a relatively unconvincing dataset was sufficient for FDA approval. More problematic, the choice to target dystrophin meant that access to muscle at an effective concentration was necessary. Muscle uptake is not a strong point for oligonucleotide therapeutics, and inefficient delivery likely contributed to the modest results. Another drawback was the need to biopsy tissue to observe molecular evidence for efficacy. Biopsies are uncomfortable for patients and it is difficult to provide a window into the effects within a tiny fraction of tissue.
Both 2′-O-methyl RNA and morpholino oligonucleotides are relatively old chemistries and their application in these trials is a legacy of the fact that the development programs began over a decade ago. It is possible that more efficient delivery and better effects might be achieved using more modern chemical modifications that allow stronger hybridization or more efficient uptake by muscle. For example, it is reported that peptide-PMO conjugate showed good exon skipping activity in skeletal and cardiac muscle cells from DMD mouse model (104,105). Like GRN163L, even if the eventual verdict for eteplirsen is negative or only shows modest benefit, the concept may still be a good one to revisit and the need for effective ASOs to help patients remains high.
Nusinersen
Spinal muscular atrophy (SMA) refers to several different motor neuron diseases and is most commonly associated with mutations in the survival motor neuron 1 (SMN1) gene (106). Mutations in SMN1 comprise the most common genetic cause of death in children and effective treatments have been a priority for research. Humans also possess a second SMN variant, SMN2. SMN2 is less active and cannot compensate for the loss-of functional SMN1, but oligonucleotides that modulate splicing can promote expression of an alternatively spliced variant (exon 7 inclusion) to compensate for the loss of active SMN1 (107–109).
An ASO, nusinersen (Spinraza, Ionis Pharmaceuticals and Biogen) (Figure 6) was developed to treat SMA and was administered by intrathecal injection into the CNS (110). Nusinersen demonstrated promise in multiple clinical trials, extending lifespan in treated children and increasing mobility. Autopsy specimens revealed enhanced expression of the alternatively spliced product in the CNS. Placebo-controlled trials were so successful that they were terminated early followed by rapid FDA approval in 2016.
As of this writing, nusinersen has been the biggest nucleic acid therapeutics success to date for both patients and commercially (111,112). Clinical data offer several important lessons. ASOs can enter tissues within the CNS at concentrations that are adequate to positively affect the course of disease. This result offers reason to hope that the expression of other disease-associated genes can be modulated in the CNS. ASO uptake and activity were not associated with undue toxicity and seen well tolerated by patients. Effects are long-lasting, with dosing required only three or four times per year. It is worth noting that that the high level of success achieved to date would probably not have been viewed as a likely outcome when development started, a reminder that risks are sometimes rewarded in the clinic.
The nusinersen story is ongoing. Gene therapy is being used to replace SMN1 while small molecules are being studied for their ability to enhance expression of the more active SMN2 splice variant (113–116). The long-term competitiveness of nusinersen against these other approaches remains undetermined. A key factor determining nusinersen's competitiveness will be its long-term toxicity in patients, a feature that will be revealed as patients are exposed for longer and longer periods. The potential for synergy between nusinersen and alternative approaches should also be defined. Finally, early clinical success was achieved in children. Some forms of SMA also afflict adults and ongoing trials will reveal whether nusinersen can also be a model for oligonucleotide therapeutics in the adult CNS.
Revusiran, patisiran and inotersen
ASOs and dsRNAs are competing antisense technologies for silencing gene expression by targeting mRNA. Both strategies silence their target genes in the liver in clinical trials. Until recently there has been little data directly comparing the two approaches and their relative strengths and weaknesses. That gap in our knowledge may be filled as dsRNAs and ASOs are developed for the same target and tested in clinical trials for the same disease (117).
Transthyretin (TTR) amyloidosis is a genetic disease in which misfolded mutant TTR builds up, leading to heart dysfunction and failure. Inhibition of mutant TTR would be predicted to alleviate the disease, leading to the development and testing of one ASO (inotersen, Ionis Pharmaceuticals) and two dsRNAs (revusiran and patisiran, Alnylam Pharmaceuticals) (27) (Figure 6).
Inotersen has been tested in a Phase III trial after being shown to reduce TTR production in preclinical and Phase 1/2 trials (118). The drug met both primary endpoints and showed benefit relative to placebo and validated TTR as a target for gene knockdown strategies (http://ir.ionispharma.com/news-releases/news-release-details/ionis-pharmaceuticals-announces-phase-3-neuro-ttr-study; http://ir.ionispharma.com/news-releases/news-release-details/new-data-presented-peripheral-nerve-society-meeting-further). Balanced against this success, three cases of thrombocytopenia were noted and ongoing clinical trials were modified to enhance monitoring of platelets and renal function in hopes of improving the management of side-effects.
Revusiran and patisiran are dsRNAs that differ in their in vivo delivery mechanisms. Revusiran is conjugated with GalNAc and enters cells through the asialoglycoprotein receptor as described above. Revusiran demonstrated potent reduction of TTR (119) and appeared to be progressing smoothly through clinical trials until analysis of Phase III data revealed excess mortality. While it remains unclear whether the excess mortality was due to the chemical composition of Revusiran or simply bad luck treating patients whose health was already fragile, the trial was halted and development abandoned (120). Other dsRNA–GalNAc conjugates (see below) have different chemical modifications that allow dosing at lower concentrations and might avoid any toxicities that contributed to failure of Revusiran.
Patisiran is delivered in complex with a lipid nanoparticle (121) and is being tested in clinical trials (122; http://investors.alnylam.com/releasedetail.cfm?ReleaseID=1022960). On September 20, 2017, Alnylam Pharmaceuticals announced that patisiran met its primary endpoint and all secondary endpoints in Phase III clinical trials. Clinical data have shown a potent and sustained knockdown of TTR expression and, while there have been side effects, there has been little evidence of safety concerns about platelets, renal function or liver enzyme elevations. The successful Phase III clinical trials may lead to patisiran being the first FDA-approved dsRNA drug, probably in 2018. (http://investors.alnylam.com/releasedetail.cfm?ReleaseID=1041081)
Both Ionis and Alnylam submitted U.S. marketing application for inotersen and patisiran respectively in November, 2017. It is possible that patisiran and inotersen will both be approved and will compete directly. This outcome would provide an informative clinical comparison between a dsRNA and an ASO. Every ASO is unique, as is every dsRNA. It is impossible to generalize with certainty based on the experience with a single compound. Nevertheless, the outcome of that competition will be a useful case study for the relative merits of ASOs and dsRNAs. Inotersen has the disadvantage of requiring platelet monitoring and the advantage of being administered subcutaneously. Patisiran has the advantage of not requiring monitoring but requires treatment with steroids to minimize injection site reactions and administration by intravenous infusion.
Fitusiran, inclisiran and givosiran
Fitusiran (Alnylam Pharmaceuticals) (123), inclisiran (Alnylam Pharmaceuticals, The Medicines Company) (124,125) and givosiran (Alnylam Pharmaceuticals) (126,127) (Figure 6) are dsRNAs that target antithrombin for treatment of hemophilia, proprotein convertase subtilisin kexin type 9 (PCSK9) to reduce elevated cholesterol, and hepatic 5-aminolevulinic acid synthase 1 (ALAS1) to treat acute hepatic porphyrias, respectively. All three are currently being tested in Phase II clinical trials and are GalNAc conjugates with enhanced stabilization chemistry that requires lower dosing than was used for Revusiran.
These programs took advantage of analysis of serum markers to increase the power of Phase I and Phase II clinical trials. Fitusiran-reduced anti-thrombin levels by 75% while inclisiran reduced LDL cholesterol levels by as much as 50%, and givosiran reduced ALAS1 mRNA by as much as 64%. For fitusiran, one death was reported, causing a halt in Phase II studies and delaying initiation of Phase III investigation. It is not clear that the death was due to fitusiran and trials are likely to restart after modification.
Some conclusions can be drawn from the published data from Phase I and Phase II trials. Fitusiran, inclisiran and givosiran offer three more examples supporting the conclusion that GalNAc conjugates can effectively knock down gene expression in humans and produce potent physiologic effects. A second important finding is that the effects are remarkably long lasting. Depending on the dose, fitursiran, inclisiran and givosiran show effects 2 to 6 months after initial dosing. Infrequent dosing suggests that these drugs might plausibly be able to compete with orally bioavailable small molecules even if they must be delivered by subcutaneous injection. Convenient delivery, coupled with a good safety profile might lead to larger markets and a breakthrough beyond treatment of rare diseases.
Lengthy duration of effects was also observed for nusinersen and appears to be a general characteristic of nucleic acid therapeutics. How can a nucleic acid have effects that last months? We normally think of DNA and RNA as being labile molecules and assume that cells have multiple pathways for turning over macromolecules. The nucleic acids used in these trials, however, are heavily chemically modified. The simplest explanation that fits the data is that the chemical modifications stabilize the ASOs and dsRNAs and that cells have no mechanism to remove them. The nucleic acids move from one target mRNA to another over a period of months. More research will be needed to resolve these issues and explain the remarkable half-lives being observed in the clinic.
Ionis-HTTRx
Huntington disease (HD) is a progressive neurological disorder caused by a CAG trinucleotide expansion within the coding region of the huntingtin (HTT) gene (128). There is currently no curative treatment for HD and developing a drug to slow or halt the course of the disease would be a major advance. HTT protein is a large scaffolding protein and developing a small molecule drug to affect the action of the mutant protein will not be straightforward (129). Because a path toward small molecule drugs is not obvious, ASOs or dsRNAs to inhibit expression of HTT are leading treatment options.
HD carriers and patients are heterozygous for the mutation. It is possible that silencing approaches that inhibit both the wild-type and mutant allele might cause toxicity by reducing levels of wild-type HTT. To counter this possibility, allele-selective dsRNAs and ASOs have been developed that primarily block expression of mutant HTT (130–135). However, animal studies have shown that non-allele selective ASOs and virally delivered dsRNAs can reverse HD phenotypes in mouse models and reduce HTT levels in primates without obvious toxicities related to decreased levels of HD (136–138). Specifically, data from Cleveland and co-workers were a remarkable demonstration of the potential of potential for ASOs in the CNS (138). Because of this favorable data, the clinical testing is focusing on non-allele selective ASOs, with allele selective approaches being held in reserve in case unexpected toxicities are observed.
The leading clinical candidate is HTTRx (Ionis Pharmaceuticals (Figure 6), a gapmer ASO complementary to the coding region of HTT outside the expanded repeat. This compound is being tested in a multicenter Phase 1/2a clinical trial in adults with early HD (https://clinicaltrials.gov/ct2/show/NCT02519036). Like nusinersen, the drug is administered intrathecally. While primarily designed to test safety, cognitive measures will be evaluated and results are expected by late 2017 or early 2018. While progress is being made using innovative chemical strategies to improve the distribution of dsRNAs in the CNS (139), more work needs to be done before dsRNAs reaches the confidence level achieved with ASOs.
The HTTRx clinical experience is significant because it further tests the hypothesis that ASOs can be generally useful drugs in the CNS. The trial is a major test of ASOs in adults. HD is a devastating disease and it is difficult to imagine any other treatment curative strategy being successful within the next decade. HTTRx has the potential to provide a major benefit to HD patients and the experience with HTTRx will have a large impact on other CNS programs.
Volanesorsen
Volanesorsen (Ionis Pharmaceuticals and Akcea) (Figure 6) is a 2′-MOE-modified gapmer ASO that targets the coding region of apolipoprotein C3 (apo-CIII) mRNA. ApoC-III plays an important role regulating the clearance of triglyceride-containing lipoproteins (140). Reducing its expression would be expected to decrease triglyceride levels and might be useful in patients with hypertriglyceridaemia. Clinical trials have revealed decreased ApoC-III levels, reducing cardiovascular risk and improving insulin sensitivity (141,142).
Phase III trials have examined volanesorsen for treatment of familial chylomicronemia and familial partial lipiddystrophy (http://ir.akceatx.com/news-releases/news-release-details/akcea-and-ionis-announce-positive-results-compass-phase-3-study; http://ir.akceatx.com/node/6121). These trials confirm reduction of apo-CIII and reduced triglycerides. These reductions met the primary endpoint for the study. Thrombocytopenia was observed in several patients and was reversed after dosing was halted. Volanesorsen appears likely to benefit some patients but platelet counts will require careful monitoring. The observed thrombocytopenia complicates application of volanesoren to a broader patient population that might benefit from reduced triglyceride levels. Clinical data were strong enough to support a marketing application to the FDA on 15 November 2017.
Summary
ASOs and dsRNAs have been the subject of research and clinical testing for many years. Until recently, success has been elusive. Progress over the past year, however, has begun to bring clinical potential into focus. This progress is the result of hard work by chemists, biologists, clinicians and project managers over decades to overcome obstacles and create drugs that fill unmet needs.
Chemical modifications are now powerful enough to permit potent gene knockdown months after drug administration. Conjugation to GalNAc appears to be a breakthrough for hepatic delivery, while the success of nusinersen suggests that ASOs have a promising future treating disease in the CNS. Decades of data are permitting more effective choice of target and better design of clinical trials.
Balanced against these advances, adverse events continue to be observed and must be better understood and carefully managed. The development of nucleic acid therapeutics is not occurring in a vacuum. The developers of other drug modalities recognize the same unmet needs, and antibodies and small molecules will continue to provide difficult competition. Gene therapy is likely to have a growing impact. With three marketing applications pending (Volanesorsen, Inotersen and Patisiran) and many clinical trials underway, the next 5 years will reveal whether ASOs and dsRNAs can become a major class of drugs capable of benefiting large numbers of patients across a range of diseases.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [GM118103 to D.R.C.]; Robert Welch Foundation [I-1244 to D.R.C.]; Rusty Kelley Professorship in Medical Science (to D.R.C.). Funding for open access charge: NIH [GM118103].
Conflict of interest statement. D.R.C. is an Executive Editor of Nucleic Acids Research.
REFERENCES
- 1. Zamecnik P.C., Stephenson M.L.. Inhibition of Rous Sarcoma virus replication and cell transformation by a specific oligonucleotide. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crooke S.T. Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Ther. 2017; 27:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Juliano R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016; 44:6518–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H.. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:230–237. [DOI] [PubMed] [Google Scholar]
- 5. Deleavey G.F., Damha M.J.. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012; 19:937–954. [DOI] [PubMed] [Google Scholar]
- 6. Sharma V.K., Watts J.K.. Oligonucleotide therapeutics: chemistry, delivery and clinical progress. Future Med. Chem. 2015; 7:22221–2242. [DOI] [PubMed] [Google Scholar]
- 7. Abramova T. Frontiers and approaches to chemical synthesis of oligodeoxyribonucleotides. Molecules. 2013; 18:1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014; 24:374–387. [DOI] [PubMed] [Google Scholar]
- 9. Dowdy S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017; 35:222–229. [DOI] [PubMed] [Google Scholar]
- 10. Iwamoto N., Butler D.C., Svrzikapa N., Mohapatra S., Zlatev I., Sah D.W., Standley S.M., Lu G., Apponi L.H., Frank-Kamenetsky M. et al. . Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:845–851. [DOI] [PubMed] [Google Scholar]
- 11. Manoharan M. 2′-carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim. Biophys. Acta. 1999; 1489:117–130. [DOI] [PubMed] [Google Scholar]
- 12. Koshkin A.A., Singh S.K., Nielsen P., Rajwanshi V.K., Kumar R., Meldgaard M., Olsen C.E., Wengel J.. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, 38ligomerization, and unprecedented nucleic acid recognition. Tetrahedron. 1998; 54:3607–3630. [Google Scholar]
- 13. Obika S., Nanbu D., Hari Y., Andoh J.I., Morio K.I., Doi T., Imanishi T.. Stability and structural features of the duplexes containing nucleoside analogues with a fixed N-type conformation, 2′-O,4′-C-methyleneribonucleosides. Tetrahedron Lett. 1998; 39:5401–5404. [Google Scholar]
- 14. Braasch D.A., Corey D.R.. Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem. Biol. 2001; 8:1–7. [DOI] [PubMed] [Google Scholar]
- 15. Seth P.P., Vasquez G., Allerson C.A., Berdeja A., Gaus H., Kinberger G.A., Prakash T.P., Migawa M.T., Bhat B., Swayze E.E.. Synthesis and biophysical evaluation of 2′4′-constrained 2′O-methoxyethyl and 2′4′;-constrained 2′O-ethyl nucleic acid analogues. J. Org. Chem. 2010; 75:1569–1581. [DOI] [PubMed] [Google Scholar]
- 16. Hong D., Kurzrock R., Kim Y., Woessner R., Younes A., Nemunaitis J., Fowler N., Zhou T., Schmidt J., Jo M. et al. . AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015; 7:314ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Staarup E.M., Fisker P.N., Hedtjarn M., Lindholm M.W., Rosenbohm C., Aarup V., Hansen H.F., Orum H., Hansen J.B., Koch T.. Short locked nucleic acid antisense oligonucleotides potently reduce apoliprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010; 38:7100–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stein C.A., Hansen J.B., Lai J., Wu S., Voskresenskiy A., Høg A., Worm J., Hedtjärn M., Souleimanian N., Miller P. et al. . Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2009; 38:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Z., Monia B.P., Corey D.R.. Telomerase inhibition, telomere shortening, and decreased cell proliferation by cell permeable 2 ‘-O-methoxyethyl oligonucleotides. J. Med. Chem. 2002; 45:5423–5425. [DOI] [PubMed] [Google Scholar]
- 20. Sehgal A., Vainshaw A., Fitzgerald K.. Liver as a therapeutic target for oligonucleotide therapeutics. J. Hepatol. 2013; 59:1354–1359. [DOI] [PubMed] [Google Scholar]
- 21. Srinivasan S. K., Tewary H. K., Iversen P.L.. Characterization of binding sites, extent of binding, and drug interactions of oligonucleotides with albumin. Antisense Res. Dev. 1995; 5:131–139. [DOI] [PubMed] [Google Scholar]
- 22. Henry S.P., Johnson M., Zanardi T.A., Fey R., Auyeung D., Lappin P.B., Levin A.A.. Renal uptake and tolerability of a 2′-O-methoxyethyl modified antisense oligonucleotide (ISIS 113715) in monkey. Toxicology. 2012; 301:13–20. [DOI] [PubMed] [Google Scholar]
- 23. Donner A.J., Yeh S.T., Hung G., Graham M.J., Crooke R.M., Mullick A.E.. CD40 generation 2.5 antisense oligonucleotide treatment attenuates doxorubicin-induced nephropathy and kidney inflammation. Mol. Ther. Nucleic Acids. 2015; 4:e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lima W.F., Prakash T.P., Murray H.M., Kinberger G.A., Li W., Chappell A.E., Li C.S., Murray S.F., Gaus H., Seth P.P. et al. . Single-stranded siRNAs activate RNAi in animals. Cell. 2012; 150:883–894. [DOI] [PubMed] [Google Scholar]
- 25. Yu D., Pendergraff H., Liu J., Kordasiewicz H.B., Cleveland D.W., Swayze E.E., Lima W.F., Crooke S.T., Prakash T.P., Corey D.R.. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell. 2012; 150:895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ly S., Navaroli D.M., Didiot M.C., Cardia J., Pandarinathan L., Alterman J.F., Fogarty K., Standley C., Lifshitz L.M., Bellve K.D. et al. . Visualization of self-delivering hydrophobically modified siRNA cellular internalization. Nucleic Acids Res. 2017; 45:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheridan C. With Alnylam's amyloidosis success, RNAi approval hopes soar. Nat. Biotechnol. 2017; 35:995–997. [DOI] [PubMed] [Google Scholar]
- 28. Nair J.K., Willoughby J.L., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Eil’in A.V., Milstein S. et al. . Multivalent N-aceylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014; 136:16958–16961. [DOI] [PubMed] [Google Scholar]
- 29. Tanowitz M., Hettrick L., Revenko A., Kinberger G.A., Prakash T.P., Seth P.P.. Asialoglycoproteinreceptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res. 2017; 45:12388–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol. Ther. Nucleic Acids. 2017; 6:116–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prakash T.P., Graham M.J., Yu J., Carty R., Low A., Chappell A., Schmidt K., Zhao C., Aghajan M., Murray H.F. et al. . Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014; 42:8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt K., Prakash T.P., Donner A.J., Kinberger G.A., Low A., Ostergaard M.E., Bell M., Swayze E.E., Seth P.P.. Characterizing the effect of GalNAc and phosphorothioate backbone on binding of antisense oligonucleotides to the asialoglycoprotein receptor. Nucleic Acids Res. 2017; 45:2294–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Viney N.J., Van Capelleveen J.C., Geary R.S., Xia S., Tami J.A., Rosie Z.Y., Marcovina S.M., Hughes S.G., Graham M.J., Crooke R.M. et al. . Antisense oligonucleotides targeting apolipoprotein (a) in people with raised lipoprotein (a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016; 388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 34. D’Souza A.A., Devarajan P.V.. Asialoglycoprotein receptor mediated hepatocyte targeting-strategies and applications. J. Control. Release. 2015; 203:126–139. [DOI] [PubMed] [Google Scholar]
- 35. Dominski Z., Kole R.. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Havens M.A., Hastings M.L.. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016; 44:6549–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Z., Jeon H.Y., Rigo F., Bennett C.F., Krainer A.R.. Manipulation of PK-M mutally exclusive alterative splicing by antisense oligonucleotides. Open Biol. 2012; 2:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vickers T.A., Crooke S.T.. The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res. 2015; 43:8955–8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lennox K.A., Behlke M.A.. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016; 44:863–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Monia B.P., Lesnik E.A., Gonzalez C., Lima W.F., McGee D., Guinosso C.J., Kawasaki A.M., Cook P.D., Freier S.M.. Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene expression. J. Biol. Chem. 1993; 268:14514–14522. [PubMed] [Google Scholar]
- 41. Minshull J., Hunt T.. The use of single-stranded DNA and RNase H to promote quantitative ‘hybrid arrest of translation’ of mRNA/DNA Hybrids in reticulocyte lysate cell-free translations. Nucleic Acids Res. 1986; 14:6433–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakamura H., Oda Y., Iwai S., Inoue H., Ohtsuka E., Kanaya S., Kimura S., Katsuda C., Katayanagi K., Morikawa K. How does RNase H recognize a DNA.RNA hybrid. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:11535–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elbashir S.M., Harborth J., Lendeckel W., Yalcin A.. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001; 411:494–498. [DOI] [PubMed] [Google Scholar]
- 44. Pratt A.J., MacRae I.J.. The RNA-induced silencing complex: a versatile gene-silencing machine. J. Biol. Chem. 2009; 284:17897–17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu J., Carmell M.A., Rivas F.V., Marsden C.G., Thomson J.M., Song J.J., Hammond S.M., Joshua-Tor L., Hannon G.J.. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004; 305:1437–1441. [DOI] [PubMed] [Google Scholar]
- 46. Meister G. Argonaute proteins: functional insights and emerging roles. Nat. Rev. Genet. 2013; 14:447–459. [DOI] [PubMed] [Google Scholar]
- 47. Wang Y., Juranek S., Li H., Sheng G., Tuschl T., Patel D.J.. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature. 2008; 456:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khvorova A., Watts J.K.. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017; 35:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Corey D.R. RNA learns from antisense. Nat. Chem. Biol. 2007; 3:8–11. [DOI] [PubMed] [Google Scholar]
- 50. Liu J., Hu J., Corey D.R.. Expanding the action of duplex RNAs into the nucleus: redirecting alternative splicing. Nucleic Acids Res. 2012; 40:1240–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adams R., Nicke B., Pohlenz H.D., Sohler F.. Deciphering seed sequence based off-target effects in a large-scale RNAi reporter screen for E-cadherin expression. PLoS One. 2015; 10:e0137640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li Z., Rana T.M.. Therapeutic targeting of miRNAs: current status and future challenges. Nat. Rev. Drug Discov. 2014; 13:622–638. [DOI] [PubMed] [Google Scholar]
- 53. Gomez I.G., MacKenna D.A., Johnson B.G., Kaimal V., Roach A.M., Ren S., Nakagawa N., Xin C., Newitt R., Pandya S. et al. . Anti–microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Invest. 2015; 125:141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Janssen H.L., Reesink H.W., Lawitz E.J., Zeuzem S., Rodriguez-Torres M., Patel K., van der Meer A.J., Patick A.K., Chen A., Zhou Y. et al. . Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013; 368:1685–1694. [DOI] [PubMed] [Google Scholar]
- 55. Gebert L.F., Rebhan M.A., Crivelli S.E., Denzler R., Stoffel M., Hall J.. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014; 42:609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ottosen S., Parsley T.B., Yang L., Zeh K., van Doorn L.J., van der Veer E., Raney A.K., Hodges M.R., Patick A.K.. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob. Agents Chemother. 2015; 59:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ree M.H., Meer A.J., Nuenen A.C., Bruijne J., Ottosen S., Janssen H.L., Kootstra N.A., Reesink H.W.. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment. Pharmacol. Ther. 2016; 43:102–113. [DOI] [PubMed] [Google Scholar]
- 58. Zhou J., Rossi J.. Aptamers as targeted therapeutics: current potential and challenges. Nat. Biotechnol. 2017; 16:181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stein C.A., Krieg A.M.. Problems in interpretation of data derived from in vitro and in vivo use of antisense oligonucleotides. Antisense Res. Dev. 1994; 4:67–69. [DOI] [PubMed] [Google Scholar]
- 60. Crooke S.T. Proof of mechanism of antisense drugs. Antisense Nucleic Acid Drug Dev. 1996; 6:145–147. [DOI] [PubMed] [Google Scholar]
- 61. Stein C.A. Keeping the biotechnology of antisense in context. Nat. Biotechnol. 1999; 17:209. [DOI] [PubMed] [Google Scholar]
- 62. Crooke S.T. Evaluating the mechanism of action of antiproliferative antisense drugs. Antisense Nucleic Acid Drug Dev. 2000; 10:123–126. [DOI] [PubMed] [Google Scholar]
- 63. Myers K.J., Dean N.M.. Sensible use of antisense: how to use oligonucleotides as research tools. Trends Pharm. Sci. 2000; 21:19–23. [DOI] [PubMed] [Google Scholar]
- 64. Stein C.A. The experimental use of antisense oligonucleotides: a guide for the perplexed. J. Clin. Invest. 2001; 108:641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Editorial Wither RNAi. Nat. Cell Biol. 2003; 5:489–490. [DOI] [PubMed] [Google Scholar]
- 66. Winkler J., Stessl M., Amartey J., Noe C.R.. Off-target effects related to the phosphorothioate modification of nucleic acids. Chemmedchem. 2010; 5:1344–1352. [DOI] [PubMed] [Google Scholar]
- 67. de Fougerolles A., Vornlocher H. P., Maraganore J., Lieberman J.. Interfering with disease: a progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007; 6:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Grimm D., Streetz K.L., Jopling C.L., Storm T.A., Pandey K., Davis C.R., Marion P., Salazar F., Kay M.A.. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006; 441:537–541. [DOI] [PubMed] [Google Scholar]
- 69. Burel S.A., Hart C.E., Cauntay P., Hsiao J., Machemer T., Katz M., Watt A., Bui H.H., Younis H., Sabripour M. et al. . Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acid Res. 2016; 44:2093–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Frazier K.S. Antisense oligonucleotide therapies: The promise and the challenges from a toxicologic pathologists perspective. Toxicol. Pathol. 2015; 43:78–89. [DOI] [PubMed] [Google Scholar]
- 71. Chi X., Gatti P., Papoian T.. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today. 2017; 22:823–833. [DOI] [PubMed] [Google Scholar]
- 72. Crooke S.T., Baker B.F., Witzum J.L., Kwoh J., Pham N.C., Salgado N., McEvoy B.W., Cheng W., Hughes S.G., Bhanot S. et al. . The effects of 2′-O-methoxyethyl containing antisense oligonucleotides on platelets in clinical trials. Nucleic Acid Ther. 2017; 27:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Geary R.S., Henry S.P., Grillone L.R.. Fomivirsen: clinical Pharmacology and potential drug interactions. Clin. Pharmacokinet. 2002; 41:255–260. [DOI] [PubMed] [Google Scholar]
- 74. Jabs D.A., Griffiths P.D.. Fomivirsen for the treatment of cytomegalovirus retinitis. Am. J. Opthamol. 2002; 133:552–556. [DOI] [PubMed] [Google Scholar]
- 75. Cursiefen C., Viaud E., Bock F., Geudelin B., Ferry A., Kadlecová P., Lévy M., Al Mahmood S., Colin S., Thorin E. et al. . Aganirsen antisense oligonucleotide eye drops inhibit Kaeratitis-induced corneal neovascularization and reduced need for tranplanation. The I-CAN study. Ophthamology. 2014; 121:1683–1692. [DOI] [PubMed] [Google Scholar]
- 76. Kim R., Emi M., Tababe K., Toge T.. Therapeutic potential of antisense Bcl-2 as a chemosensitizer for cancer therapy. Cancer. 2004; 101:2491–2501. [DOI] [PubMed] [Google Scholar]
- 77. Matsui M., Corey D.R.. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017; 27:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Benimetskaya L., Lai J.C., Khvorova A., Wu S., Hua E., Miller P., Zhang L.M., Stein C.A.. Relative Bcl-2 independence of drug-induced cytotoxicity and resistance in 518A2 melanoma cells. Clin. Cancer Res. 2004; 10:8371–8379. [DOI] [PubMed] [Google Scholar]
- 79. Anderson E.M., Miller P., Ilsley D., Marshall W., Khvorova A., Stein C.A., Benimetskaya L.. Gene profiling study of G3139- and Bcl-2 targeting siRNAs identifies a unique G3139 molecular signature. Cancer Gene Ther. 2006; 13:406–414. [DOI] [PubMed] [Google Scholar]
- 80. Westphal S.P. Behind the mask. New Scientist. 2003; 179:32–35. [Google Scholar]
- 81. Corey D.R. Telomeres and telomerase: from discovery to clinical trials. Chem. Biol. 2009; 16:1219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ouellette M.M., Wright W.E., Shay J.W.. Targeting telomerase-expressing cancer cells. J. Cell. Mol. Med. 2011; 15:1433–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Herbert B., Pitts A.E., Baker S.I., Hamilton S.E., Wright W.E., Shay J.W., Corey D.R.. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:14276–14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Baerlocher G.M., Leibundgut E.O., Ottmann O.G., Spitzer G., Odenike O., McDevitt M.A., Roth A., Daskalakiz M., Burington B., Stuart M. et al. . Telmoerase inhibitor imetelstat in patients with essential thrombocythemia. N. Eng. J. Med. 2015; 373:920–928. [DOI] [PubMed] [Google Scholar]
- 85. Tefferi A., Lasho T.L., Begna K.H., Patnaik M.M., Zblewski D.L., Finke C.M., Laborde R.R., Wassie E., Schimek L., Hanson C.A. et al. . A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N. Eng. J. Med. 2015; 373:908–919. [DOI] [PubMed] [Google Scholar]
- 86. Tefferi A., Al-Kali A., Begna K.H., Patnaik M.M., Lasho T.L., Rizo A., Wan Y., Hansen C.A.. Imetelstat therapy in refractory anemia with ring sideroblasts with or without thrombocytosis. Blood Cancer J. 2016; 6:e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Duell P.B., Santos R.D., Kirwan B-A., Witzum J.L., Tsimikas S., Kastelein J.J.P.. Long term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J. Clin. Lipidol. 2016; 10:1011–1021. [DOI] [PubMed] [Google Scholar]
- 88. Rader D.J., Kastelein J.J.. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014; 129:1022–1032. [DOI] [PubMed] [Google Scholar]
- 89. Won J.I., Zhang J., Tecson K.M., McCullough P.A.. Balancing low-density lipoprotein cholesterol reduction and hepatotoxicity with lomitapide mesylate and mipomersen in patients with homozygous familial hypercholesterolemia. Rev. Cardiovasc. Med. 2017; 18:21–28. [DOI] [PubMed] [Google Scholar]
- 90. Crooke R.M., Graham M.J., Lemonidis K.M., Whipple C.P., Ko S., Perera R.J.. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J. Lipid Res. 2005; 46:872–884. [DOI] [PubMed] [Google Scholar]
- 91. Kastelein J.J.P., Wedel M.K., Baker B.F., Su J., Bradley J.D., Yu R.Z., Chuang E., Graham M.J., Crooke R.M.. Potent reduction of apolipoprotein B and low-density cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006; 114:1729–1735. [DOI] [PubMed] [Google Scholar]
- 92. Mann C.J., Honeyman K., Cheng A.J., Ly T., Lloyd F., Fletcher S., Morgan J.E., Partridge T.A., Wilton S.D.. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kole R., Krieg A. M.. Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug. Deliv. Rev. 2015; 87:104–107. [DOI] [PubMed] [Google Scholar]
- 94. Goemans N., Tulinius M., Kroksmark A-K., Wilson R., van den Hauwe M., Campion G.. Comparison of ambulatory capacity and disease progression of Duchenne muscular dystrophy subjects enrolled in the drisapersen DMD114673 study with a matched natural history cohart of subjects on daily corticosteroids. Neuromuscul. Disord. 2017; 27:203–213. [DOI] [PubMed] [Google Scholar]
- 95. Mendell J.R., Goemans N., Lowes L.P., Alfano L.N., Berry K., Shao L.P., Kaye E.M., Mercuri E.. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016; 79:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Stein C.A., Castanotto D.. FDA-approved oligonucleotide therapies in 2017. Mol. Ther. 2017; 25:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kesselheim A.S., Avorn J.. Appoving a problematic muscular dystrophy drug Implications for FDA policy. J. Am. Med. Assoc. 2016; 316:2357–2358. [DOI] [PubMed] [Google Scholar]
- 98. Aartsma-Rus A., Krieg A.M.. FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017; 27:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mendell J.R., Goemans N., Lowes L.P., Alfano L.N., Berry K., Shao J., Kaye E.M., Mercuri E.. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016; 79:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Aartsma‐Rus A., Fokkema I., Verschuuren J., Ginjaar I., Van Deutekom J., van Ommen G.J., Den Dunnen J.T.. Theoretic applicability of antisense‐mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009; 30:293–299. [DOI] [PubMed] [Google Scholar]
- 101. Mendell J.R., Rodino‐Klapac L.R., Sahenk Z., Roush K., Bird L., Lowes L.P., Alfano L., Gomez A.M., Lewis S., Kota J. et al. . Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013; 74:637–647. [DOI] [PubMed] [Google Scholar]
- 102. Unger E.F., Califf R.M.. Regarding “Eteplirsen for the treatment of Duchenne muscular dystrophy”. Ann. Neurol. 2017; 81:162–164. [DOI] [PubMed] [Google Scholar]
- 103. Irwin A.N., Herink M.C.. Eteplirsen for the treatment of Duchenne muscular dystrophy: Quality of evidence concerns—an alternative viewpoint. Pharmacotherapy. 2017; 37:e109–e111. [DOI] [PubMed] [Google Scholar]
- 104. Betts C., Saleh A.F., Arzumanov A.A., Hammond S.M., Godfrey C., Coursindel T., Gait M.J., Wood M.J.. Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther. Nucleic Acids. 2012; 1:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lehto T., Castillo Alvarez A., Gauck S., Gait M.J., Coursindel T., Wood M.J., Lebleu B., Boisguerin P.. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2013; 42:3207–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Faravelli I., Nizzardo M., Comi G.P., Corti S.. Spinal muscular atrophy – recent therapeutic advances for an old challenge. Nat. Rev. Neurosci. 2015; 11:351–359. [DOI] [PubMed] [Google Scholar]
- 107. Lim S.R., Hertel K.J.. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J. Biol. Chem. 2001; 48:45476–45483. [DOI] [PubMed] [Google Scholar]
- 108. Cartegni L., Krainer A.R.. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002; 30:377–384. [DOI] [PubMed] [Google Scholar]
- 109. Hua Y., Vickers T.A., Baker B.F., Bennett C.F., Krainer A.R.. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007; 5:729–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Haché M., Swoboda K.J., Sethna N., Farrow-Gillespie A., Khandji A., Xia S., Bishop K.M.. Intrathecal injections in children with spinal muscular atrophy: Nusinersen clinical trial experience. J. Child. Neurol. 2016; 31:899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Corey D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017; 20:497–499. [DOI] [PubMed] [Google Scholar]
- 112. Aartsma-Rus A. FDA approval of nusinersen for spinal muscular atrophy makes 2016 the year of splice modulating oligonucleotides. Nucleic Acid Ther. 2017; 27:67–69. [DOI] [PubMed] [Google Scholar]
- 113. Scoto M., Finkel R.S., Mercuri E., Muntoni F.. Therapeutic approaches for spinal muscular atrophy (SMA). Gene Ther. 2017; 24:514–519. [DOI] [PubMed] [Google Scholar]
- 114. Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E. et al. . Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017; 377:1723–1732. [DOI] [PubMed] [Google Scholar]
- 115. Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. et al. . Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017; 377:1713–1722. [DOI] [PubMed] [Google Scholar]
- 116. van der Ploeg A.T. The dilemma of two innovative therapies for spinal muscular atrophy. N. Engl. J. Med. 2017; 377:1786–1787. [DOI] [PubMed] [Google Scholar]
- 117. Niemietz C., Chandhok G., Schmidt H.. Therapeutic oligonucleotides targeting liver disease: TTR amyloidosis. Molecules. 2015; 20:17944–17975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ackermann E.J., Guo S., Benson M.D., Booten S., Freier S., Hughes S.G., Kim T.W., Kwoh J.T., Matson J., Norris D. et al. . Suppressing transthyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid. 2016; 23:148–157. [DOI] [PubMed] [Google Scholar]
- 119. Zimmerman T.S., Karsten V., Chan A., Chiesa J., Boyce M., Bettencourt B.R., Huabarat R., Nochur S., Vaishnaw A., Gollob J.. Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol. Ther. 2017; 25:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Garber K. Alnylam terminates revusiran program, stock plummets. Nat. Biotechnol. 2016; 34:1213–1214. [DOI] [PubMed] [Google Scholar]
- 121. Tam Y.K., Madden T.D., Hope M.J.. Pieter Cullis’ quest for a lipid-based, fusogenic delivery system for nucleic acid therapeutics: success with siRNA so what about mRNA. J. Drug Target. 2016; 24:774–779. [DOI] [PubMed] [Google Scholar]
- 122. Suhr O.B., Coelho T., Buades J., Pouget J., Conceicao I., Berk J., Schmidt H., Waddington-Cruz M., Campistol J.M., Bettencourt B.R. et al. . Efficacy and safety of patisiran for famial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J. Rare Dis. 2015; 10:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Pasi K.J., Rangarajan S., Georgiev P., Mant T., Creagh M.D., Lissitchkov T., Bevan D., Austin S., Hay C.R., Hegemann I. et al. . Targeting antithrombin in hemophilia A or B with RNAi therapy. N. Eng. J. Med. 2017; 377:819–828. [DOI] [PubMed] [Google Scholar]
- 124. Ray K.K., Landmesser U., Leiter L.A., Kallend D., Dufour R., Karakas M., Hall T., Troquay R.P., Turner T., Visseren F.L. et al. . Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N. Eng. J. Med. 2017; 376:1430–1440. [DOI] [PubMed] [Google Scholar]
- 125. Fitzgerald K., White S., Borodovsky A., Bettencourt B.R., Strahs A., Clausen V., Wijngaard P., Horton J.D., Taubel J., Brooks A. et al. . A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 2017; 376:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yasuda M., Gan L., Chen B., Kadirvel S., Yu C., Phillips J.D., New M.I., Liebow A., Fitzgerald K., Querbes W. et al. . RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:7777–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chan A., Liebow A., Yasuda M., Gan L., Racie T., Maier M., Kuchimanchi S., Foster D., Milstein S., Charisse K. et al. . Preclinical development of a subcutaneous ALAS1 RNAi therapeutic for treatment of hepatic porphyrias using circulating RNA quantification. Mol. Ther. Nucleic Acids. 2015; 4:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jimenez-Sanchez M., Licitra F., Underwood B.R., Rubinsztein D.C.. Huntington's disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 2017; 7:a024240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. De Souuza E.B., Cload S.T., Pendergrast P.S., Sah D.W.. Novel therapeutic modalities to address nondruggable protein interaction targets. Neuropsychopharmacology. 2009; 34:142–158. [DOI] [PubMed] [Google Scholar]
- 130. Pfister E.L., Kennington L., Straubhaar J., Wagh S., Liu W., Difiglia M., Landwhrmeyer B., Vonsattel J-P., Zamore P.D., Aronin N.. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington's Disease patients. Curr. Biol. 2009; 19:774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hu J., Corey D.R.. Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem. Biol. 2010; 17:1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Gagnon K.T., Pendergraff H.M., deleavey G.F., Swayze E.E., Potier P., Randolph J., Roesch E.B., Chattopadhyaya J., Damha M.J., Bennett C.F. et al. . Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanding CAG repeat. Biochemistry. 2010; 49:10166–10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Skotte N.H., Southwell A.L., Ostergaard M.E., Carroll J.B., Warby S.C., Doty C.N., Petoukhov E., Vaid K., Kordasiewicz H., Watt A.T. et al. . Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: providing a therapeutic option for all huntingtin disease patients. PLoS One. 2014; 9:e107434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Miller J.R.C., Pfister E.L., Liu W., Andre R., Trager U., Kennington L.A., Lo K., Dijkstra S., Macdonald D., Ostroff G. et al. . Allele-selective suppression of mutant huntingtin in primary human blood cells. Sci. Rep. 2017; 7:46740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wild E. J., Tabrizi S. J.. Therapies targeting DNA and RNA in Huntington's disease. Lancet Neurol. 2017; 16:837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Boudreau R.L., McBride J.L., Martins I., Shen S., Xing Y., Carter B.J., Davidson B.L.. Nonallele-specific silencing of mutant and wild-type hunting demonstrates therapeutic efficacy in Huntington's Disease mice. Mol. Ther. 2009; 17:1053–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Drouet V., Perrin V., Hassig R., Dufour N., Auregan G., Alves S., Bonvento G., Brouileet E., Luthi-Cater R., Hantraye P. et al. . Sustained effects of nonallele-specific huntingtin silencing. Ann. Neurol. 2009; 65:276–285. [DOI] [PubMed] [Google Scholar]
- 138. Kordasiewicz H.B., Stanek L.M., Wancewicz E.V., Mazur C., McAlonis M.A., Pytel K.A., Artates J.W., Weiss A., Cheng S.H., Shihabuddin L.S. et al. . Sustained therapeutic reversal of Huntington's Disease by transient repression of huntingtin synthesis. Neuron. 2012; 74:1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Nikan M., Osborn M.F., Coles A.H., Godinho B.M., Hall L.M., Haraszti M.R., Echeverria D., Aronin N., Khvorova A.. Docosahexaenoic acid conjugation enhances distribution and safety of siRNA upon local administration in mouse brain. Mol. Ther. Nucleic Acids. 2016; 5:e344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Reiner Z. Hypertriglyceridaemia and risk of coronary artery disease. Nat. Rev. Cardiol. 2017; 14:401–411. [DOI] [PubMed] [Google Scholar]
- 141. Digenio A., Dunbar R.L., Alexander V.J., Hompesch M., Morrow L., Lee R.G., Graham M.J., Hughes S.G., Yu R., Singleton W. et al. . Antisense-mediated lowering of plasma apolipoprotein C-III by volanesorsen improves dyslipidemia and insulin sensitivity in type 2 diabetes. Diabetes Care. 2016; 39:1408–1415. [DOI] [PubMed] [Google Scholar]
- 142. Yang X., Liee S-R., Choi Y-S., Alexander V.J., Digenio A., Yang Q., Miller Y.I., Witzum J.L., Tsimikas S.. Reduction in lipoprotein-associated apoC-III levels following volanesorsen therapy: phase 2 randomized trials results. J. Lipid Res. 2016; 57:706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.