


Abstract
The mouse is widely used as system to study human genetic mechanisms. However, extensive rewiring of transcriptional regulatory networks often confounds translation of findings between human and mouse. Site-specific gain and loss of individual transcription factor binding sites (TFBS) has caused functional divergence of orthologous regulatory loci, and so we must look beyond this positional conservation to understand common themes of regulatory control. Fortunately, transcription factor co-binding patterns shared across species often perform conserved regulatory functions. These can be compared to ‘regulatory sentences’ that retain the same meanings regardless of sequence and species context. By analyzing TFBS co-occupancy patterns observed in four human and mouse cell types, we learned a regulatory grammar: the rules by which TFBS are combined into meaningful regulatory sentences. Different parts of this grammar associate with specific sets of functional annotations regardless of sequence conservation and predict functional signatures more accurately than positional conservation. We further show that both species-specific and conserved portions of this grammar are involved in gene expression divergence and human disease risk. These findings expand our understanding of transcriptional regulatory mechanisms, suggesting that phenotypic divergence and disease risk are driven by a complex interplay between deeply conserved and species-specific transcriptional regulatory pathways.
INTRODUCTION
The mouse is a powerful and flexible model system for exploring human genetic diseases and regulatory mechanisms. Many studies have used comparisons between the human and mouse genomes to gain insight into transcriptional regulatory mechanisms and evolution. Most of these have relied on overlapping functional annotations across species at orthologous loci, and the term ‘positional conservation’ was recently coined to describe this strategy (1). Different studies have employed one or more of positional conservation of phylogenetic sequence conservation, open chromatin, or histone modifications, to infer regulatory conservation. However, while most protein coding genes, developmental and physiological pathways remain highly conserved between these two species, only ∼40% of their DNA sequences are directly alignable (2). The majority of regulatory elements reside in the non-alignable fraction, with some estimates placing up to 60% of regulatory features within non-orthologous sequence (1,3). Although the pool of transcription factors (TFs) and their binding specificities have changed little among vertebrates, the majority of occupied TF binding sites (TFBSs) are species specific (4), and even positionally conserved sequences sharing similar epigenetic marks often do not contain the same set of bound TFs (1,3). At the evolutionary distance between human and mouse, mutational processes frequently change the combinations and spatial arrangements of TFBSs within regulatory loci even when target gene expression patterns are conserved (5). The functional consequences of such changes are unclear, and, notably, they are not always associated with decreased phylogenetic sequence conservation. Indeed, phylogenetically conserved sequences often contain nucleotide level changes that quantitatively affecting TF binding. Furthermore, the majority of positionally conserved loci do not share the same set of bound TFs, and often drive divergent regulatory outcomes (1). This may explain why many clinical and mechanistic findings in mouse fail to translate back to human (6–8).
Positional-conservation methods rely on sharing of various annotations at the same genomic locus across species in order to infer conserved or divergent regulatory function. As a result, the utility of these methods is restricted to the 40% human and mouse sequences that can be aligned. This is problematic given that the majority of regulatory elements reside in non-orthologous sequences. These features are known to contribute to species-specific functional and structural properties of the human and mouse genomes (9–15). Without looking beyond positional conservation, we cannot understand how these features fit into the broader regulatory and evolutionary landscapes. While open chromatin and epigenetic modifications can give us clues to their functions, conserved chromatin signatures do not always equate to conserved regulatory functions (1,16,17).
An alternative approach is to focus not on the underlying sequence conservation of a given locus, but rather the combination of co-bound TFs occupying the locus across species. Indeed, there is considerable interest in combinations of TFBSs often found in combination with each other, or cis-regulatory modules (CRMs), as drivers of adaptive evolution (18–21). Studies have shown that gene expression is mediated by combinations of TFs acting cooperatively to regulate their target genes (22), that distinct CRMs elicit specific and reproducible gene expression patterns (23–26), and that these regulatory outcomes often directly translate between species (27). CRMs can be thought of as ‘regulatory sentences’ with meanings that are retained across tissues and species. Much like in natural language, the meaning of a regulatory sentence depends primarily on the combination of TFBS ‘words’ it contains, and these words can be combined in many ways to produce diverse regulatory instructions. Intuitively, adding, removing or exchanging TFBSs words may dramatically change the meaning of a regulatory sentence at a given locus, regardless of its underlying phylogenetic conservation. Furthermore, in much the same way that many natural language sentences can share equivalent meanings, it seems likely that many regulatory sentences can encode equivalent instructions. Hence, there must exist a set of rules that dictates how TFBS words can be combined into valid regulatory instructions. In natural language, the set of rules describing how words are combined into meaningful sentences is called a grammar; these grammars can be learned by observing the sentences that make up a language (28). We believe that a similar set of rules—a regulatory grammar, must exist to direct how TFBS are combined into meaningful regulatory sentences. We sought to learn this grammar by analyzing the regulatory sentences used in humans and mice.
We analyzed CRMs composed of 27 TFs, in four human and mouse cell types, independent of their sequence, cellular and species context. Investigating all observed co-binding patterns individually would have been intractable, and so we first sought to group regulatory sentences with equivalent meanings into discrete sets, which we call grammatical patterns. We applied a self-organizing map (SOM) algorithm to partition the regulatory sentences observed among all CRMs into a set of grammatical patterns. The SOM algorithm projects these grammatical patterns to a two-dimensional grid topology, upon which we can map covariates to explore the underlying properties of each grammatical pattern. SOMs have been previously used to describe CRM data in humans (22) and across distantly related species (29) but, as far as we are aware, this is the first application of an SOM to infer a mammalian regulatory grammar.
We found that the SOM partitions human and mouse regulatory sentences into both conserved and species-specific grammatical patterns. In contrast to the widespread divergence observed when positional conservation is used to partition the regulatory space, a majority of grammatical patterns in our study are shared between human and mouse, with most regulatory loci harboring conserved grammatical patterns. These grammatical patterns carry stable functional signatures and are enriched for functional GWAS variants relevant to human immune disorders. Importantly, these observations held true for both orthologous and non-orthologous regulatory loci, underscoring the advantages of our approach in predicting regulatory function genome-wide. Although none of the TFs in our dataset are tissue-specific and the SOM was trained without species and cell-type labels, we observed many grammatical patterns specific to single cell types, tissues and species. We coin the term ‘grammatical class’ to describe the cell specificity of a grammatical pattern, and show that grammatical classes are correlated with matching tissue-specific expression profiles of nearby genes. Surprisingly, we also found a significant correlation between non-orthologous sequences carrying conserved grammatical patterns and species-specific gene expression, suggesting that recruitment of existing regulatory pathways to novel target genes is a prevalent mechanism in regulatory evolution. Applying this regulatory grammar can facilitate reliable genome-wide prediction of regulatory function across species, regardless of underlying sequence conservation, allowing us to determine which genetic pathways have diverged significantly between species and which remain under similar control. This may highlight which pathways hold the most promise for translational research using mouse model systems.
MATERIALS AND METHODS
Preparation of the dataset
ChIP-seq datasets used in this manuscript are listed in Supplementary Table S1. All were retrieved from the ENCODE DCC portal (https://www.encodeproject.org/), and follow ENCODE standards for preprocessing, quality control and uniform peak calling. Human data map to hg19 and mouse data to mm9. We collected data for 27 TFs for which data were available in human K562 and GM12878, and in mouse MEL and CH12 cell types. All peaks in each species were merged using BedTools mergeBed (30) to create a base set of CRMs in each species. These regions were labeled with TF binding data specific to each cell type and all regions with at least two TFs bound were retained for further analysis. The SOM analysis was performed using methods defined by us previously. Briefly, we train a 47 × 34 toroidal SOM with hexagonal neurons with an update radius of one-third map size and alpha of 0.05 for 100 epochs. We repeat this for 1000 trials to minimize the quantization error. We performed this analysis with a modified version of the kohonen R package available through our github site here: https://github.com/Boyle-Lab/kohonen2.
In contrast to our previous methods, we also performed 10 000 random permutations of the SOM, in which we shuffled TF labels within each region while maintaining the length, cell type and organism associations. We used these permutations to generate empirical distributions for co-binding pattern occurrence, which allowed for us to correct for patterns that occur at low frequency by chance and for factors with many apparent associations due to their high frequency in the datasets. We considered a neuron significant if it had an empirical P-value ≤ 0.0001 (i.e. has not appeared in any random permutations). Grammatical pattern labels were assigned to each significant neuron by enumerating occurrences of each of the 27 TFs in all its constituent CRMs, retaining all labels present in ≥ 90% of CRMs. Neurons were assigned to grammatical classes based on the empirical P-values observed for each cell. We required an empirical P-value ≤ 0.0001 to include a cell type in a pattern’s grammatical class and a P-value > 0.95 to exclude a cell from a pattern’s grammatical class. If any cell types did not pass either criterion, resulting in an ambiguous grammatical class assignment, the pattern was discarded.
Preparation of background sequences
To serve as a baseline for our statistical tests, we selected background regions, matched in length, GC content and distance to the nearest transcription start site (TSS), from portions of the human and mouse genomes not occupied by CRMs in our dataset. These were analyzed following the same procedures as CRMs whenever possible. Briefly, CRMs were randomly drawn from the human or mouse dataset, without replacement. The length, GC content and distance from the midpoint to the nearest TSS were calculated and used to assign the element to one of five distance bins: 0–500, 501–2000, 2001–5000, 5001–10 000 and >10 000 bp. No significant differences were found within the same distance bin and sequences of matching length were randomly drawn until one with GC content ±10% was found. The coordinates for this sequence were stored, along with the metadata from its CRM counterpart, and then masked to prevent the same sequence from being drawn multiple times. This was repeated until a matched sequence for each CRM was found, all potential background windows were exhausted or 100 000 background sequences were produced, whichever came first. Visual inspection of density plots of length, GC content and TSS distance for CRMs and matched background sequences revealed good agreement in all three parameters, and no significant differences were found between any based on individual t-tests performed in R.
Database and browser preparation
All data and annotations pertaining to the grammatical patterns and individual CRMs and background sequences, were loaded into a MySQL database to facilitate further analysis and to serve as the repository for the SOM browser. Data for various analyses were retrieved directly in MySQL, through the MySQL PERL API and through the RMySQL R package. The SOM Browser web application is based on the open source Catalyst framework (http://www.catalystframework.org/) and interfaces directly with the database through Catalyst’s MCV model.
Analysis of orthologous and non-orthologous sequences
In order to investigate the cross-species relationships between human and mouse CRMs, we used bnMapper (17) to map CRMs and background sequences across genomes using the settings described in (31). Human-to-mouse and mouse-to-human liftover chains were obtained from the UCSC download portal (http://hgdownload.soe.ucsc.edu/downloads.html) (32). We call these chains ‘one-to-many’ chains because one-to-one orthology is not enforced when they are prepared. We also produced reciprocal-best chains, representing the most likely set of one-to-one orthologs between both species, following procedures at (http://genomewiki.ucsc.edu/index.php/HowTo:_Syntenic_Net_or_Reciprocal_Best). Reciprocal-best chains were used to map each CRM definitively to its orthologous location across species. We used these as our benchmark ‘orthologs’ dataset. We identified a set of strict non-orthologs by exclusion of all CRMs mappable using the one-to-many chains from the original dataset. We further excluded any CRMs that mapped using the one-to-many chains but not the reciprocal-best chains from further analysis, as these were ambiguous in their orthology status. Based on these mappings, loci were assigned to positional classes by observing the set of cell(s) in which a CRM was present in either species.
Analysis of module gain and loss
Starting with the set of strict non-ortholog CRMs, we employed a phylogenetic maximum parsimony method to infer whether non-orthologous elements represented sequence gains or losses in human or mouse. We first retrieved the 46-species placental mammal phylogenetic model from UCSC (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/phyloP46way/placentalMammals.mod) (32), and used tree_doctor (33) to prune all branches but human, mouse, dog, horse and elephant (Supplementary Figure S3). We obtained liftover chains for human and mouse to each of the outgroup species from UCSC (32) and used bnMapper (17) to cross-map human and mouse sequences between each outgroup, using the same methods and settings described in the ‘Analysis of orthologous and non-orthologous sequences’ section. Given these mappings, the ancestral state at the most recent common ancestor was inferred by maximum parsimony using a custom perl script. In ∼12% of cases, the ancestral state was ambiguous based on the whole phylogeny. Dropping elephant from the phylogeny allowed us to unambiguously assign these to gain and loss groups. Database records for non-orthologous elements were update in the database to reflect these assignments.
Analysis of intersection with DNaseI hypersensitive sites
DNaseI hypersensitive sites (DHS) were retrieved from the ENCODE DCC portal in bed format (Supplementary Table S3). All datasets within each cell type were combined, sorted and merged with bedtools merge prior to calculation of intersections and coverages with bedtools intersect and bedtools coverage (30). Cell-wise coverages for all CRMs, background sequences and their orthologous locations were compiled into a table with a custom perl script, and loaded into the database. Bulk enrichments over background were tested in R using the fisher.test function with default options and P-values were adjusted using the holm method. Data were loaded into the database and SQL queries were used to extract intersections of CRMs and matched background regions with DHS elements for individual grammatical patterns and six aggregate grammatical classes (Supplementary Figure S4), within orthologous, non-orthologous, gain and loss fractions for both human and mouse. These were stored in text files and loaded into R, for subsequent enrichment testing using the fisher.test function with alternative ‘greater’. Multiple testing correction was performed with the holm method and results are presented in Supplementary Table S6. Percent intersections for each subset of the data were visualized using the ggplot2 R package (Figure 3A). Plots were reproduced after excluding the two largest cohesin-related patterns from the dataset (Supplementary Figure S5A).
Figure 3.

Grammatical patterns associate stably with genomic annotations associated with regulatory function, regardless of underlying sequence conservation. Regulatory sequences in six aggregate grammatical classes were divided into orthologous, and species-specific loss and gain fractions, based on sequence mappings between human, mouse and three outgroup species. Within each subset, we calculated the fraction of sequences overlapping functional annotations. (A) Overlaps with DHS, which indicate open chromatin regions characteristic of active regulatory sequences, were remarkably stable among all data subsets. (B) ACS from ChromHMM were also relatively stable among all subsets of the data, although more variable than DHS. (C) Intersections with PhastCons elements, which specifically measure phylogenetic conservation, decrease in direct correlation with the evolutionary ages of orthologous, loss and gain sequences, and are poorly correlated with DHS and ACS in non-orthologous sequences.
Analysis of intersection with active chromatin states (ACS)
ChromHMM annotations were obtained from the ENCODE DCC portal for all four cell types in bigBed format (Supplementary Table S2). A custom perl script was used to extract chromatin state intersections for all CRMs and background sequences and their orthologous locations in human or mouse were extracted from the bigBed files and load them into the database. We focused on four chromatin states annotated in the chromHMM data: promoter (H3K4me3), strong enhancer (H3K4me1+H3K4me3), weak enhancer (H3K4me1). We refer to the these as active chromatin states (ACS). Enrichments of ACS were tested across the entire dataset and in six aggregate grammatical classes (Supplementary Figure S4), relative to matched background sequences as described in the ‘DNaseI Hypersensitivity Analysis’ section. Results are presented in Supplementary Table S6, and percent intersections were plotted with the ggplot2 R package for the entire dataset (Figure 3B) and after excluding the two largest cohesin-related grammatical patterns excluded (Supplementary Figure S5B).
Analysis of phylogenetic conservation
PhastCons elements (PEs) and base-wise phastCons conservation scores for the human (46-way placental mammals) and mouse (30-way placental mammals) genomes were retrieved from the UCSC download portal in bed format (34). Intersections and coverages of CRMs and background sequences by phastCons elements were extracted with bedtools intersect and bedtools coverage (30) and the coverage, average, maximum and minimum scores for each module were extracted using a custom perl script. Bulk enrichment tests and tests within aggregate grammatical patterns were performed as described in the ‘DNaseI Hypersensitivity Analysis’ section. Count matrices were visualized using the ggplot2 R package for the complete dataset (Figure 3C), and after excluding the two largest cohesin-related patterns (Supplementary Figure S5C). PhastCons conservation scores plotted in Supplementary Figure S6 were extracted from bigWig files for hg19 phastCons46way, and mm9 phastCons30way tracks, which were retrieved from the UCSC download portal (35). Score vectors for 400 bp windows centered around 1000 randomly selected ChIP-seq peaks within our dataset were retrieved from the bigWig files using a custom R script based on the RTrackLayer package. These were assembled into a matrix, which was then sorted by descending row sum and plotted using a custom plotting function.
Analysis of correlation between grammatical patterns and ACS
We quantified the correlation between grammatical patterns and ACS by comparing the ACS annotations for all pairs of loci within each grammatical pattern (Figure 4A and Supplementary Figure S7B). Similarly, we quantified the extent to which positional conservation of ACS could predict conservation of grammatical patterns across cells and species by counting the fraction of sequence pairs with matched ACS at each positionally conserved locus in the dataset that also match in grammatical pattern (Figure 4B and Supplementary Figure S7A). In both analyses, the two largest cohesin-related grammatical patterns were excluded (Figure 4B and Supplementary Figure S5A). Euler diagrams in Figure 4A–B and Supplementary Figure S7A-B were generated using the draw.pairwise.venn function from the VennDiagram R package.
Figure 4.

Grammatical patterns predict ACS better than positional conservation of ACS predicts positional conservation of grammatical patterns. We intersected all CRMs with ACS from ChromHMM. (A) To evaluate the predictive value of positional conservation of ACS for underlying grammatical patterns, we compared the grammatical patterns present at positionally conserved pairs of loci with matched ACS. The Euler diagram shows the proportion of such pairs that also have matched grammatical patterns. (B) To assess the degree to which matched grammatical patterns predict matched ACS, we calculated the fraction of CRM pairs within each grammatical pattern that also matched in ACS. The Euler diagram shows the proportion of sequence pairs among all grammatical pattern carry matched ACS. (C and D) Heat maps describe the extent to which grammatical classes (C) and positional classes (D) predict the cell-specificity of underlying ACS. Shading densities reflect the fraction of loci or grammatical patterns that carry a given chromatin state in the expected cell type(s) based on their assigned positional or grammatical class. (C) All grammatical patterns in which at least 50% of CRMs intersect a given chromatin state were counted (‘total’ column). The ‘in class’ column describes the number of patterns in each row in which at least 50% of CRMs from cell types belonging to its grammatical class carry the same mark (see Supplementary Figure S3B). The ‘outside class’ column shows the number of patterns in each row in which the given chromatin mark is present in cell types not belonging to its grammatical class. (D) For each locus in the dataset, chromatin states were gathered for all four cell types. We used these to evaluate how well positional class labels agreed with the observed cell specificity of ACS associations. All loci carrying a given ACS in at least one cell type were counted (‘total’ column). The match column describes the number of loci where the observed cell specificity of ACS annotations matched what we would expect based on its positional class. The mismatch 1 column gives the number of loci at which the given state was observed in one or more ‘outside class’ cells. The mismatch 2 column gives the number of loci at which one or more ‘in-class’ cell types lacks the given ACS.
Procedures used to evaluate cell specificity of ACS within grammatical patterns are described in Supplementary Figure S8A and C. Briefly, grammatical patterns for which at least 50% of the CRMs, either from all cells (Supplementary Figure S8A) or each individual cell (Supplementary Figure S8C) were counted toward the ‘total’ column. For the pooled analysis (Figure 4C and Supplementary Figure S8A), we counted the number of occurrences where the same chromatin state appeared in ‘in-class’ cells (i.e. those that belong to a pattern’s grammatical class) and ‘outside-class’ cells (i.e. those that do not belong to a pattern’s grammatical class) to fill in the corresponding matrix cells. For the cell-wise analysis (Supplementary Figure S8C), we counted the number of times we observed the same chromatin state in each cell type, adding the counts to the corresponding matrix cell. Count matrices were visualized using the geom_tile function from the ggplot2 R package with a fixed shading scale to enable direct comparison of maps. We repeated these procedures using thresholds of 75, 90 and 100% to evaluate whether the match rates we observed were robust to our choice of threshold (Supplementary Figure S10A–C).
Procedures used to evaluate cell-specificity of ACS within positional classes are illustrated in Supplementary Figure S6B. For the summary matrix (Figure 4D) each ACS (P, SE, WE), we looped over all regulatory loci in the dataset that contained the given ACS. The ‘total’ column contains the count of total loci carrying each ACS in at least one occupied cell. Three categories of loci were defined: matches, in which the specificity of ACS matched that expected based on the positional class; mismatch 1, in which one or more ‘outside class’ cell carries the given ACS; and mismatch 2, in which one or more ‘in-class’ cells lacks the given ACS. For the full matrix (Supplementary Figure S9B), we looped over all combinations of chromatin state, cell, and positional class, and counted the number of CRMs where the reference cell carried a given ACS. For each of those loci, we then counted the number of times the same ACS was observed in each of the other cells. Count matrices were visualized using geom_tile with a fixed shading scale.
Graphical representations of chromatin states observed within grammatical pattern 821 (Supplementary Figure S7C) were prepared by extracting chromatin state intersections from the database directly into R, using the RMySQL package, for all member CRMs. Chromatin state annotations were converted to count vectors, which were normalized to a uniform length and sorted by ascending numerical order. These were assembled into a matrix and the rows were sorted by descending row sums. The matrix was then visualized with a custom plotting function. Plots of chromatin states for CG positional class loci in Supplementary Figure S7D were produced in similar manner, except that state intersections for all occupied and unoccupied cells in each of the 10 loci shown were extracted from the database, followed by within-row sorting and plotting of locus-wise groups using a custom plotting function.
Analysis of tissue-specific target gene expression
Single-ended RNA-seq data for all four cell types were obtained from the ENCODE portal as raw sequencing reads in fastq format (Supplementary Table S1). In order to mitigate the impact of sequencing batch effects, we restricted our choice of datasets to those produced within the same laboratory using the same protocols and sequencing equipment whenever possible. We used Kallisto version 0.42.4 (36) to quantify expression levels directly from the fastq files. Fasta sequences for all UCSC ‘knownGene’ transcripts (32), were prepared for hg19 and mm9 genomes using gffread 0.9.5 (https://github.com/gpertea/gffread). These were used to prepare transcript indexes with ‘kallisto index’. We then ran ‘kallisto quant -b 100 –single -l 200 -s 80 -t 8’, to calculate raw expression values and bootstrap data for each replicate individually. The sleuth R package (http://pachterlab.github.io/sleuth/) was then used to combine replicates and calculate normalized expression values for each species and cell. R analysis scripts are available through our github repository (https://github.com/Boyle-Lab/mouse-human-SOM). UCSC transcript IDs were converted to gene names with a custom perl script based on data obtained from the UCSC Table Browser (37) and transcripts per kilobase million (TPM) was calculated for each gene by totaling the expression over all isoforms. Gene ortholog relationships were established between human and mouse genes using the modEncode common orthologs list (http://compbio.mit.edu/modencode/orthologs/modencode.common.orth.txt.gz) (38). These were used to assemble TPM values into a 6-column table, with gene symbols for human and mouse in columns 1 and 2, using a placeholder value where no orthologous gene existed, and expression values for each cell in columns 3–6. This table was read into R and normalized with the normalize.quantiles function from the preprocessCore package.
As an alternative method to normalizing gene expression levels, we obtained the bam alignment files corresponding to the same fastq datasets described previously and performed a detailed process to correct for sequencing batch effects, as described in (39). Briefly, all individual isoforms from UCSC knownGenes transcripts were collapsed into gff file with one record per gene with mergeOverlappingExons.py (39). Gene-wise GC content was computed using bedtools nuc (30) and raw fragment counts for each gene were extracted from the bam files with featureCounts (40). Counts for each cell type were totaled over replicates and assembled into a 4xN matrix, with expression values for each cell in the columns and one row for each gene. Counts were read into R and normalized following the exact process described in (39), with one modification: we did not filter out the bottom 30% of genes by expression value. This modification had a very small quantitative effect on observed expression levels: density plots were visually indistinguishable and, although a paired t-test showed a significant difference (P = 5.99e-15), the average difference of 0.016 is unlikely to be meaningful. Finally, we converted the normalized counts to TPM to facilitate direct comparison to the Kallisto expression values, using the weighted average of the all the transcript-wise effective lengths for each gene, as reported by Kallisto, as the effective length of the gene.
Differential gene expression analysis was performed with DESeq (41). Raw counts for all RNA-seq replicates were first normalized for batch effects following the procedures described above. DESeq size factors were fixed at 1 to disable internal normalization and differential expression predictions were performed using the nbinomGLMTest procedure using a full model including both species and cell effects and a reduced model including only species. Predictions with multiple-testing-corrected P-values ≤ 0.05 were retained as differentially expressed. From these, we selected gene sets with expression specific to a given species or cell type based on the predicted partial slope coefficients in the full model. For example, myeloid specific genes were selected by setting a lower-bound of four on the ‘cellTypeMyeloid’ coefficient and upper, indicating a positive correlation with expression in myeloid cells, and lower bounds of 0.05 and −0.05 on the ‘speciesMm9’ coefficient, indicating little or no correlation with species. Similarly, a set of stably expressed genes was selected from among all predictions by setting the upper and lower bounds for both coefficients at 0.065 and −0.065, yielding a set of 724 stably expressed genes.
Gene assignments for CRMs were made based on the nearest TSS, based on the list of genes used by the Chip-Enrich R package (42). CRM counts from each grammatical class nearest to genes in each set were extracted from the databse in R using the RMySQL package. Within each aggregate grammatical class (Supplementary Figure S4), we calculated fractions of CRMs nearest to genes with stable expression (πS) and CRMs nearest to in-class genes (πD) and calculated a tissue-specificity coefficient, 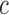 , as
, as 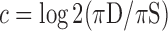 , where
, where 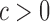 indicates tissue-specific enrichment and
indicates tissue-specific enrichment and 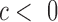 indicates tissue-specific depletion. We also calculated a corresponding coefficient,
indicates tissue-specific depletion. We also calculated a corresponding coefficient, 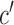 , as
, as 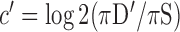 , where (πD′
, where (πD′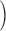 represents the fraction of CRMs nearest to cross-class genes. We interpret positive values of these ratios as evidence for enrichment, and negative values as evidence for depletion. We visualized the values of
represents the fraction of CRMs nearest to cross-class genes. We interpret positive values of these ratios as evidence for enrichment, and negative values as evidence for depletion. We visualized the values of 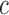 and
and 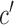 as paired bars in Figure 5 using the geom_bar function from ggplot2. Pairwise significance tests were performed on the count matrices using the fisher.test function in R with alternative ‘greater’, for tests of enrichment among CRMs nearest genes with matching specificity, or ‘less’, for tests of depletion of CRMs nearest genes with mismatched specificity.
as paired bars in Figure 5 using the geom_bar function from ggplot2. Pairwise significance tests were performed on the count matrices using the fisher.test function in R with alternative ‘greater’, for tests of enrichment among CRMs nearest genes with matching specificity, or ‘less’, for tests of depletion of CRMs nearest genes with mismatched specificity.
Figure 5.
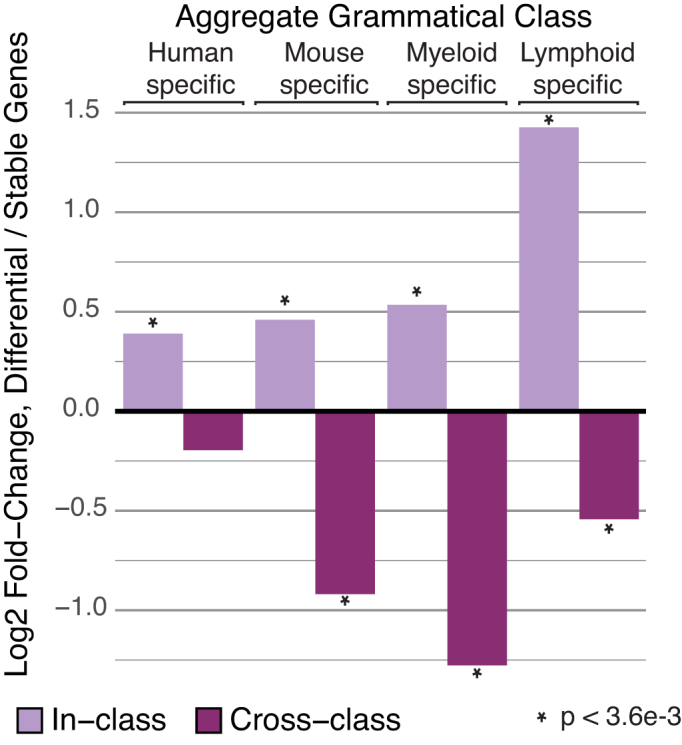
Gene expression patterns are correlated with cell- and species-specific grammatical patterns. To evaluate a potential causal relationship between CRMs within tissue- and species-specific grammatical classes and corresponding gene expression patterns, we calculated log-ratios of the proportion of tissue/species-specific CRMs targeting differentially expressed genes compared to stably expressed genes for each aggregate grammatical class. Light bars represent the log-ratio observed for tissue/species-specific CRMs targeting genes with the matching tissue/species-specific expression profile. Dark bars indicate log-ratios observed for tissue/species-specific CRMs targeting genes with mismatched tissue/species-specific expression profile. Positive values indicate enrichments while negative values indicate depletions. Statistical significance of enrichments/depletions was evaluated using Fisher's exact test, with significant P-values indicated by an asterisk. Notably, significant enrichment among matching tissue/species-specific genes was seen for all aggregate grammatical classes, along with corresponding depletions among non-matching genes.
Analysis of correlation with disease-associated variants in human
We retrieved a list of human GWAS lead SNPs and associated GWAS Ontology terms (43) from the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/) in hg38 coordinates. Coordinates were converted to hg19 by matching RSIDs to dbSNP build 142 (44). A total of 61 records that had been merged into newer dbSNP records were successfully mapped using the active RSID after initial failure. A total of 46 SNPs could not be mapped using the RSID; their coordinates were converted to hg19 frame using liftOver (45). Four records failed to map due to anomalies in the RSID field. One, ‘rs1083 2417’, contained a space character, removal of which resolved the issue. Three had letters appended to RSID: rs4460079b, rs7658266b, rs6917824L. None of these could be found in the cited references under the given RSID, nor under any other associated RSIDs in dbSNP. These were excluded from further analysis.
We next used a custom perl script to merge in all SNPs in linkage disequilibrium (R2 ≥ 0.8) with GWAS Catalog lead SNPs based on data from HapMap (46). The unified list was intersected with CRMs and background sequences with bedtools intersect (30). Intersections and associated GWAS Ontology terms were counted using a combination of command-line utilities and custom perl scripts, counting duplicate terms within a single sequence only once. GWAS Ontology terms were categorized into 29 groups (Supplementary Table S5) based on the EBI Experimental Factor Ontology and primary literature sources, describing known associations with specific tissues, disease states, biological functions and processes similar to (47). Single-tailed Fisher’s Exact tests were used to assess bulk enrichment of GWAS SNPs and for enrichments of 27 functional ontology categories (48) (Supplementary Table S8). Binomial tests were used to test for enrichment of individual ontology terms with the observed frequency of each term in the GWAS Catalog used as the expected binomial probability (Supplementary Table S9). This procedure was repeated for CRMs in each aggregate grammatical class (Supplementary Table S10) and across individual grammatical classes (Supplementary Table S11). All P-values were corrected for multiple testing using the Benjamini–Hochberg FDR method.
RegulomeDB scores (49) for CRM-associated and background-associated SNPs were retrieved with a custom Python script, and read into R. Significance of the trend toward lower scores in CRM-associated SNPs was assessed by comparing score vectors using the wilcox.test function with alternative = ‘l’. A table of normalized frequencies of CRM-associated and background-associated SNPs within each RegulomeDB score was prepared and visualized as a stacked bar graph with geom_bar from the ggplot2 R package (Figure 6B).
Figure 6.

Conserved and species-specific grammatical patterns are enriched for causal SNPs for relevant human GWAS phenotypes. (A) CRM-associated GWAS SNPs were tested for enrichment of 27 functional ontology categories. Only categories in which at least one individual ontology term was enriched among CRM-associated SNPs in (C) or (D) are shown. Terms with adjusted P-values ≤ 0.05 (Fisher's exact tests) are marked with an asterisk. (B) RegulomeDB scores were retrieved for all GWAS SNPs in CRMs and matched background regions. The fraction of observations contributed by CRMs and background sequences is plotted for each RegulomeDB score. There is a significant enrichment of lower scores, corresponding to greater functional evidence, among CRMs (Wilcoxon P-value < 3.0e-20). (C) We tested CRM-associated SNPs for enrichment of individual GWAS ontology terms using individual binomial tests. Adjusted P-values are shown as shading densities on the heat map, with values ≤0.05 starred. (D) We further broke down the dataset among six aggregate grammatical classes and tested for enrichment of GWAS ontology terms within each class. Binomial P-values were adjusted for multiple testing and are presented as a heat map. All terms significant at ≤0.05 are marked with an asterisk.
RESULTS
Grammatical patterns reflect known qualitative properties of transcription factors
We used an SOM to learn significantly occurring co-binding patterns of TFs for 27 TFs among two pairs of biologically matched cell types from human and mouse: K562 (K) and MEL (M) myeloid cells, and GM12878 (G) and CH12 (C) lymphoid cells (Figure 1A–C and see ‘Materials and Methods’ section) (22). ChIP-seq peak data for each of the factors in all four cell types were obtained from the ENCODE portal (3). These were aggregated into CRMs based on mutual overlap of peaks and encoded as binary occupancy vectors, which were assigned to nodes in a 47 × 34 map grid by the SOM algorithm (Figure 1C and see ‘Materials and Methods’ section). We wanted to focus only on the regulatory effects of different TF combinations, and so we ignored factors such as spacing, order and orientation of individual TFBSs. Each of these nodes represents a set of regulatory loci, all of which share a similar TF co-binding profile. We call these co-binding profiles ‘grammatical patterns.’ All grammatical patterns occurring at a frequency exceeding that observed in the actual dataset in any of 10 000 random permutations were excluded from further analysis. We grouped grammatical patterns into sets, which we call ‘grammatical classes,’ based on their observed cell specificities (Figures 1D and 2A). All patterns that could not be unambiguously assigned to a grammatical class (see ‘Materials and Methods’ section) were discarded, leaving 780 distinct grammatical patterns (Figure 1C). This filtration process had minimal effects on the length distribution of the grammatical patterns and did not qualitatively affect their partitioning into grammatical classes (Supplementary Figure S1).
Figure 1.

The SOM, grammatical classes and positional classes. (A) ChIP-seq datasets from human and mouse myeloid (K562 and MEL) and lymphoid (GM12878 and CH12) immune cells were obtained from the ENCODE DCC portal. (B) Overlapping ChIP-seq peaks were assembled into CRMs, which can be thought of as individual regulatory sentences. Seven actual CRMs observed in human and/or mouse are presented, with human and mouse coordinates given in the left-most column, cells of origin in the center column and schematic diagrams showing the arrangement of ChIP-seq peaks for seven different TFs, each shown as a color-coded ‘hill’ within the diagram. Orthologous loci are grouped together in dark gray boxes. (C) All CRMs with two or more overlapping ChIP-seq peaks were encoded as binary occupancy vectors. These were analyzed with an SOM algorithm in order to cluster CRMs into sets of similar regulatory sentences, which we believe are likely to share equivalent regulatory meanings. We call these sets of regulatory sentences grammatical patterns, and each hexagonal cell on the map represents a discrete grammatical pattern, which are depicted as hexagonal cells on the map. Color-coded pie charts within each hexagon represent the cell specificity of each grammatical pattern, or its grammatical class. (D) Positional class assignments describing the set of cells in which we observed positional conservation of TF occupancy (for any TF) at each locus. Positional conservation is defined as overlapping presence of functional annotations at orthologous loci across cells and/or species. Note that positional classes are defined at the locus level, and so every CRM at a given locus belongs to the same positional class. (E) Grammatical pattern assignments for each CRM in (B). Grammatical patterns can be represented as the string of TFs that make up its core regulatory sentence. Note that individual CRMs at a given locus often carry different grammatical patterns. (F) Grammatical class assignments for each CRM in (B). Grammatical classes describe the cell specificity of each grammatical pattern (also see panel D). Note that grammatical classes are a property of the grammatical pattern to which they are assigned, and so individual CRMs at a given locus need not belong to the same grammatical class.
Figure 2.

Grammatical patterns capture regulatory conservation better than positional conservation. (A and B) Venn diagrams show the segmentation of regulatory space into 15 possible grammatical and positional classes. The first letter of each cell type was used to construct a class label for each cell in the diagrams. These labels describe the cell-specificity of the corresponding grammatical patterns and positionally conserved loci. (A) Grammatical classes represent collections of grammatical patterns that share the same observed cell specificity. Each segment in the Venn diagram is labeled with its grammatical class, the number of grammatical patterns assigned to the class (first number), and the total number of CRMs contributing to those patterns (number in parentheses). Overall, the SOM partitions the dataset into 780 grammatical patterns, 593 of which are used in both species and 187 that are species-specific (103 mouse and 84 human). (B) Positional classes describe regulatory conservation in terms of shared sequence occupancy, or positional conservation. Regulatory loci were assigned to positional classes based on the cell(s) in which we observed TF occupancy, regardless of the specific TFs present. Each segment in the Venn diagram is labeled with its positional class, the total number of loci within the class (first number), and the total number of CRMs assigned to the class (number in parentheses). In all, 209 768 CRMs were observed at 128 380 distinct loci, 54 491 of which are positionally conserved. (C and D) Pie charts show the fraction of CRMs in each cell type assigned to seven aggregate grammatical classes (C) or positional classes (D). The regulatory landscape appears highly conserved when defined as a set of grammatical patterns, with the bulk of CRMs in all cell types falling into grammatical patterns shared between human and mouse (C). By contrast, the vast majority of regulatory loci are not positionally conserved across human and mouse (D)—i.e. the orthologous locus in the other species is not bound by any TFs.
The grammatical patterns we observed reflected many known associations between TFs (22,29,31), and generally reflect previously reported spatial biases for their member TFs (31), with most patterns showing a preference for either promoters or enhancers. Overall, 76% of grammatical patterns showed stable spatial preferences between human and mouse, and of the remaining 164 patterns, 159 contained ETS1, which is known to be promoter-biased in humans but enhancer-biased in mouse (31). Given the consistency of our dataset with previously reported qualitative properties of the constituent TFs, we wondered how well our data captured the evolutionary properties of these elements. Specifically, we wanted to see if grammatical patterns presented a more-conserved picture of regulatory conservation than positional conservation.
Grammatical patterns are more evolutionarily stable than positionally conserved regulatory loci
Recent studies have shown positive correlations between positionally conserved TF occupancy and functional outcomes, including tissue-specific enhancer activity (31) and tissue-specific expression of nearby genes (31,47). However, while these studies and others (50,51) suggest a positive correlation between deeply shared orthologous TF binding and conserved regulatory outcomes, they focus on tissue-specific TFs and their findings may not generalize to broader tissue sets. By contrast, there is substantial evidence for extensive tissue-specific repurposing (i.e. alterations in tissue-specific activity patterns between human and mouse) of positionally conserved regulatory elements, with up to 69% of DHS shared between human and mouse repurposed across species (1). It seems likely that this repurposing results from changes in TF occupancy at positionally conserved loci between species. Consistent with this possibility, several studies have shown that a minority of CRMs and DHS shared between human and mouse share the same sets of TFs (1,5,17,50–52). One of these showed that these changes in TF occupancy are indeed correlated with divergent expression of liver-specific genes (1). We speculate that the site-specific gain and loss (turnover) of liver-specific TFBS observed at these loci directly altered the meaning of their attendant regulatory sentences in human and/or mouse. In other words, TFBS turnover caused a switch from one grammatical pattern to another, leading to divergent regulatory outcomes. We wondered if we would see evidence for grammatical pattern turnover (i.e. changes in the grammatical pattern observed at a locus across cells due to differential TF occupancy and/or TFBS turnover) in our dataset.
To evaluate the prevalence of grammatical pattern turnover in positionally conserved sequences, we quantified the fraction of our CRMs that could be mapped between human and mouse and analyzed the extent to which grammatical patterns differed between cell types. Human and mouse CRMs were mapped across species with bnMapper (17) and grouped into orthologous and non-orthologous loci. Consistent with previous studies (1,5,47,51), we found that only 30–50% of CRMs could be aligned between human and mouse (Figure 2D). Orthologous loci were assigned to ‘positional classes’, representing the set of cells in which TF binding was observed at each locus (Figure 2B). These can be compared to grammatical classes, in that they express the observed cell-specificity of a positionally conserved locus. At each positionally conserved locus, we calculated the fraction of cases in which grammatical patterns matched across the represented cell types, finding that 74% housed different grammatical patterns in at least one cell type (Supplementary Figure S2). Interestingly, the fraction of mismatches was positively correlated with the number of cells sharing occupancy at each locus, and exceeded 90% in the deeply conserved MCKG positional class. These findings corroborate previous observations of extensive TFBS turnover between orthologous regulatory loci at all evolutionary distances (5,50,51,53–55), demonstrating the limitations inherent to positional conservation in predicting regulatory output. This led us to ask whether grammatical patterns would present a more conserved regulatory picture between human and mouse compared to positionally conserved loci.
In order to investigate conservation of grammatical patterns themselves, we counted how many loci fell into each grammatical class (Figure 2A) and aggregated those counts into species-specific and tissue-specific classes (Figure 2C). For comparison, we repeated this procedure for regulatory loci in each positional class (Figure 2B and D). We found that 89% of regulatory loci carried grammatical patterns shared between human and mouse (Figure 2A and C) while, in contrast, 82% of positionally conserved loci were species specific (Figure 2B and D). We next compared grammatical class representation between orthologous and non-orthologous regulatory loci. Importantly, we observed no significant differences, with 88% of orthologous loci and 92% of non-orthologous loci carrying conserved grammatical patterns. Therefore, although grammatical patterns at positionally conserved loci change frequently, most of these turnover events involve switching between conserved grammatical patterns rather than creation of species-specific regulatory logic. We find that this observation applies equally to orthologous and non-orthologous regulatory loci. This suggests that regulatory grammar is deeply conserved, leading to broad sharing of grammatical patterns between human and mouse. We conclude that grammatical patterns are more informative than positional conservation in predicting the regulatory output of a locus, especially when phylogenetic methods cannot be applied.
Grammatical patterns have stable functional signatures across orthologous and species-specific loci despite differential sequence conservation
Given how strongly conserved grammatical patterns are between human and mouse, we wondered if they would also associate with conserved functional signatures across species and evolutionary contexts. Toward this end, we assigned non-orthologous loci as human or mouse gains and losses, and compared their functional annotation content with orthologous elements. The functional annotations we used were DHS, ACS from ChromHMM (56) and PhastCons elements (PE) (57). We first mapped each non-orthologous element to three outgroup species (dog, horse and elephant) and applied a phylogenetic maximum parsimony algorithm to determine the most likely branch along which a sequence was gained or lost (Supplementary Figure S3). We then measured overlaps with DHS, ACS and PE across six aggregate grammatical classes: human-specific, mouse-specific, lineage-specific, two-cell-mixed, three-cell and MCKG (Supplementary Figure S4).
DHS, which are commonly used to indicate noncoding regulatory function (58), had remarkably consistent intersections between orthologs, gains and losses (Figure 3A and Supplementary Table S3). All aggregate classes were enriched for DHS relative to matched background sequences (all P-values < 2.1e-24, Supplementary Table S3) and, in total, 88% of regulatory loci overlapped a DHS. Similarly, the percentage of CRMs labeled with ACS remained relatively stable between orthologous, gain and loss fractions in all aggregate classes (Figure 3B), although the effect was not as uniform as with DHS. Orthologs, gains, and losses were significantly enriched for ACS in all aggregate classes except one when compared to matched background sequences (all P-values < 2.6e-3, Supplementary Table S4). Similar to previous observations (31), ACS in our dataset were present in 58% of all CRMs. For both DHS and ACS, we see a decreased intersection in the MCKG class in orthologs, gains and losses. This effect was largely explained by grammatical patterns composed primarily of the cohesin subunits, CTCF, RAD21 and SMC3. Removing the two most prevalent cohesin-related patterns markedly reduces its magnitude (Supplementary Figure S5, increasing intersection with DHS to 96% and ACS to 79%, but the underlying reasons for this effect are not clear. Regardless, the stability we see in both DHS and ACS across orthologs, gains and losses, is consistent with our hypothesis that grammatical patterns retain conserved functions across all contexts.
In contrast, intersections between regulatory loci and PE, which reflect phylogenetic conservation, declined steadily between orthologous, loss and gain fractions (Figure 3C). Overall, 65% of regulatory loci (58% mouse, 75% human) contain PE, and CRMs were 3.3 times more likely to contain PE than matched background sequences (P < 6.2e-258). Similar to our observations among DHS and ACS, we saw weaker enrichment in the MCKG class, but excluding the two largest cohesin grammatical patterns only modestly increased PE intersection, to 68%. Significant enrichments over background were observed in all six aggregate classes among orthologs, gains and losses, except in three subsets with insufficient data (Supplementary Table S2). As expected, orthologous sequences contained more PE than both losses and gains, and sequence conservation was correlated with the evolutionary age of the regulatory loci. Species-specific gains showed the least overlap with PE, while PE intersection in losses occurred at an intermediate level relative to gains and orthologs (P-values < 2.2e-240). Notably, evolutionary conservation in many regulatory loci appears to arise largely from direct TF binding, as shown by clustering of phastCons scores approaching 1 within 50 bp of ChIP-seq peak summits (Supplementary Figure S6). As in Figure 3C, we saw a trend toward declining phastCons scores progressing from highest in orthologs, to intermediate in losses, and lowest in gains (Supplementary Figure S5). These results contrast with the relatively stable associations we see with DHS and ACS, and we conclude that grammatical patterns are more informative regarding regulatory function than underlying phylogenetic sequence conservation.
Grammatical patterns predict underlying chromatin states and are correlated with cell-specific chromatin state conservation patterns
Based on a previous study showing a correlation between cell-specific sets of histone modifications and corresponding gene expression patterns (16), we wondered if a deeper relationship exists between ACS and regulatory grammar. Specifically, we wanted to know if positionally conserved loci that share the same ACS signature also tend to carry matched grammatical patterns. To investigate this possibility, we examined all sequence pairs with matched ACS at each positionally conserved locus to determine the fraction that share the same grammatical pattern. Surprisingly, the overwhelming majority of matched-ACS pairs carried different grammatical patterns, with only 1.5% matching (Figure 4A). For comparison, we counted the fraction of sequence pairs within each grammatical pattern for which underlying ACS match. We observed a 29% match rate – a ∼19-fold increase over positionally conserved loci with matched ACS (Figure 4B). This fraction was relatively stable across promoters, strong enhancers, and weak enhancers (Supplementary Figure S7A and B). These results show that there is a stronger, potentially causal, link between grammatical patterns and underlying ACS than vice versa. This is consistent with our hypothesis that these patterns carry the same meanings regardless of where they are found in the genome.
We next asked whether the grammatical class a pattern belongs to corresponds to its functional specificity or simply reflects the cells in which it has been observed so far. One possible scenario is that cell-specific grammatical patterns will carry the same ACS associations when placed in a non-native cell type, suggesting functional stability regardless of cellular context. Another is that cell-specific grammatical patterns will associate with different ACS when placed in a non-native cell type, suggesting that proper function requires proper cellular context. To differentiate between these possibilities, we counted grammatical patterns in which at least 50% of member sequences were annotated with promoter, strong enhancer or weak enhancer ACS. Among these sets, we counted the number of patterns where at least 50% of sequences from ‘in-class’ cell types (those included in its grammatical class label) carried the matched ACS, and those in which 50% or more of sequences from ‘outside-class’ cells (those not included in its grammatical class label) carried the matched ACS (Supplementary Figure S8A). In all, 694 of the 780 grammatical patterns showed evidence of ACS specificity and, in keeping with our previous observations, we observed a 99.8% match rate among ‘in-class’ cells (Figure 4C, second column and Supplementary Figure S9A). Interestingly, only 15–28% of patterns had matched ACS in sequences from ‘outside-class’ cells (Figure 4C, third column), demonstrating that associations between grammatical patterns and underlying ACS match the cell-specificity of their grammatical classes in a majority of cases. However, when comparing ACS content between individual cell types, we rarely observed chromatin states in ‘outside-class’ cells that were not observed in a subset of ‘in-class’ cells (Supplementary Figure S9A). Therefore, although grammatical classes appear to correlate with ACS in matching cell-specific patterns, ACS associations in ‘outside-class’ cells still reflect those found in the grammatical pattern at large. As a result, although we do observe differences in ACS profiles among ‘outside-class’ elements, it is impossible to say without follow up experiments whether these represent functionally divergent states in their native cellular context. These results were robust to our choice of inclusion threshold (Supplementary Figure S10A–C), indicating that our observations were unlikely to be an artifact of our thresholding procedure. A plot of chromatin state annotations from a representative CG-class grammatical pattern also shows remarkable consistency with our expectations (Supplementary Figure S7C).
We also wondered to what extent observing a given ACS in one cell type at a positionally conserved locus could predict the ACS in other occupied cell types. For example, if a locus carries an enhancer mark in one occupied cell type, how likely is it that all occupied cell types are also marked as enhancers? We would expect this rate to be high if positional conservation is a good predictor of stable regulatory function. To investigate this possibility, within each positional class, we counted the proportion of loci for which all ‘in-class’ cells and no ‘outside-class’ cells, carried a given ACS annotation (Supplementary Figure S8B). Loci that did not satisfy both criteria were classified as ‘mismatch 1’ loci, at which a given ACS was found in ‘outside-class’ cells, and ‘mismatch 2’ loci, at which one or more ‘in-class’ cells lacked the ACS. Notably, ‘match’ rates at positionally conserved loci (Figure 4D and Supplementary Figure S9B) were consistent with previous observations (3), and much lower than those we saw for grammatical patterns. These observations were robust to the choice of inclusion thresholds (Supplementary Figure S10D and E). Most interestingly, we found that up to 42% of positionally conserved loci harbored mismatched ACS in at least one occupied cell type. These differences in ACS may correspond to functional repurposing across species and/or cell types, possibly caused by underlying grammatical pattern changes.
Tissue and species specificity of CRMs and grammatical classes correlate with gene expression patterns
We wanted to determine if there was an association between grammatical classes and cell-specific gene expression patterns. Under this hypothesis, we would expect to see a positive correlation between presence of tissue and species-specific CRMs and matching tissue and species-specific expression profiles in nearby genes. To test this theory, we looked for enrichments of tissue and species-specific grammatical patterns among CRMs nearest to matching tissue/species-specific genes (in-class genes), and depletion of the same patterns among CRMs near genes with non-matching tissue/species-specific expression (cross-class genes), relative to stably expressed genes.
We generated lists of stably expressed genes, and genes specific to human, mouse, lymphoid cells and myeloid cells, using DESeq (41). Within each of the corresponding aggregate grammatical classes, we calculated the fractions of CRMs nearest to genes with stable expression (πS) and CRMs nearest to in-class genes (πD) and calculated a tissue-specificity coefficient, c, as 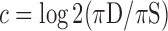 , where
, where 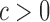 indicates tissue-specific enrichment and
indicates tissue-specific enrichment and 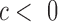 indicates tissue-specific depletion. We also calculated a corresponding coefficient,
indicates tissue-specific depletion. We also calculated a corresponding coefficient, 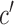 , as
, as 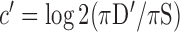 , where (πD′) represents the fraction of CRMs nearest to cross-class genes.
, where (πD′) represents the fraction of CRMs nearest to cross-class genes.
Consistent with our hypothesis, we observed significant enrichment of tissue and species-specific CRMs near in-class genes in all aggregate grammatical classes (Figure 5, light purple bars). Likewise, we saw a consistent trend toward depletion of species-specific CRMs near cross-class genes, which was significant in all but one aggregate grammatical class (Figure 5, dark purple bars). We also observed a modest but significant depletion of tissue-specific and species-specific CRMs near stably expressed genes (P = 3.4e-4). Notably, MCKG patterns were neither enriched nor depleted in CRMs near any category of genes. This is consistent with our theory that MCKG grammatical patterns serve broad regulatory roles spanning both tissues and species, while tissue-specific and species-specific grammatical patterns are important in producing corresponding tissue and species-specific gene expression profiles.
Based on the previous observation that mouse regulatory sequences without human orthologs are enriched near immune-specific genes (3), we wondered if we would see enrichments of species-specific, non-orthologous CRMs near genes with corresponding species-specific expression. Using our lists of species-specific and stably expressed genes, we counted species-specific CRMs and CRMs with 1:1 orthologs nearest to species-specific and stably expressed genes and performed Fisher’s exact tests to identify significant departures. As expected, we found strong enrichments of species-specific CRMs near genes with expression patterns specific to the same species (Mouse: OR 2.1, p 3.3e-38; Human: OR 2.9, p 1.2e-69). Surprisingly, 89% of these CRMs carry grammatical patterns shared between human and mouse suggesting that associated expression changes were caused by recruitment of common regulatory pathways to novel target genes.
Taking these results into account, there are two primary ways in which regulatory grammar extends our understanding of gene regulation. First, it allows us to identify positionally conserved loci that have diverged sufficiently in their TF content as to produce a divergent regulatory outcome. Second, it broadens our understanding of how regulatory networks evolve. We observe that species-specific gene expression patterns correlate with both species-specific grammatical patterns and non-orthologous CRMs holding conserved grammatical patterns, and this suggests two parallel processes driving regulatory divergence. When TFBS turnover at positionally conserved loci causes a switch from a conserved grammatical pattern to a tissue or species-specific pattern, divergent gene expression may result. Likewise, an insertion or deletion event that creates a non-orthologous regulatory locus may create a species-specific association between a conserved grammatical pattern and a target gene, leading to divergent gene expression. By applying regulatory grammar, we can to quantify the relative effects of these processes and gain further insight into how they relate to phenotypic divergence.
Immune-related GWAS variants are enriched in both conserved and species-specific grammatical patterns
Given the complex interplay between conserved and tissue-specific grammatical patterns we observed in our differential gene expression analysis, we wanted to explore how conserved and species-specific grammatical patterns contribute to human genetic disease. Previous work has shown a significant association between tissue-specific GWAS variants and corresponding tissue-specific epigenetic signatures (16). We wondered if we would see a similar association in our dataset, with immune-related human GWAS variants overrepresented among human-specific grammatical patterns, or if enrichments would also appear among conserved grammatical patterns. We tested this by intersecting human CRMs with variants from the NHGRI-EBI GWAS Catalog (43) and looking for significant enrichments of GWAS SNPs associated with sets of previously published ontology terms (48) across the dataset as a whole and among aggregate grammatical classes.
Consistent with their functions in immune-cell regulation, we found a 1.5-fold enrichment for immune-specific GWAS SNPs (P < 5.9e-8) in CRMs pooled across all grammatical patterns relative to matched background sequences. We wanted to know how many of these SNPs were potentially causal, and so we used RegulomeDB, a SNP annotation tool which evaluates noncoding variants for potential regulatory activity using a heuristic scoring system (49), to score each variant in CRMs and background datasets. We observed strong evidence for enrichment of causal SNPs among CRM-associated variants based on systematically lower RegulomeDB scores, which are inversely correlated with the amount of functional evidence overlapping a SNP (49) (Figure 6B, Wilcoxon P-value < 3.0e-20). To further explore the physiological pathways affected by these SNPs, we separated the data among 27 categories of ontology terms, testing for enrichments across each category and for each individual gwas ontology term (Supplementary Table S5). Among the 27 categories, we found only two significant enrichments: immune system (1.4-fold, P < 2.3e-90) and cardiovascular (1.2-fold, P < 2.8e-3) (Figure 6A). We also found enrichments for 33 individual ontology terms (Figure 6C). In all, 12 immune-related terms were enriched, 10 of which ranked among the top fifteen terms (Supplementary Table S6). Interestingly, several enriched non-immune terms relate to disorders with recent evidence for immune involvement, including schizophrenia (59) and Parkinson’s disease (60). Furthermore, cardiovascular, blood-related and cancer-related GWAS SNPs appearing in our results hint at emerging relationships between the immune system and the pathology of cardiovascular disease (reviewed in (61) and (62)) and cancer (63).
We next asked if conserved and human-specific grammatical patterns contribute equally to human disease risk. After further dividing the GWAS associations into the same six aggregate grammatical classes we used in conservation and gene expression analyses, we retested for enrichments of all individual GWAS ontology terms. We found that 16 of the original 33 terms, plus three more, were enriched in at least one aggregate grammatical pattern (Figure 6D and Supplementary Table S6). In addition, four more terms were enriched in single grammatical classes (Supplementary Table S6). All novel terms related to the same ontology categories observed in the pooled data. Surprisingly, we found that 78% of the GWAS enrichments affected conserved grammatical patterns (i.e. those in MCKG, Three-cell and two-cell mixed grammatical classes) (Figure 6D). Five individual terms were enriched in human-specific grammatical patterns; however, only two of these were enriched solely in human-specific classes.
Encouragingly, we noted substantial overlaps between our results and those reported in a previous study using the same cell types (31). This included enrichment of our top term, ‘Inflamatory Bowel Disease’, among occupancy conserved TFBSs in GM12878 cells (31). We extend their observations, and those reported in (16), by noting that immune-related GWAS SNPs affect both conserved and human-specific grammatical patterns. This suggests that, as we saw in our differential expression analysis, genetic disease risk stems from an interplay between conserved and divergent regulatory grammar. Grammatical patterns, therefore, may add context by which GWAS SNPs can be more effectively functionally classified, allowing deeper insight into the regulatory conservation of their associated pathways. For instance, pathways in which GWAS SNPs are concentrated among conserved grammatical patterns may represent promising targets for translational research. Conversely, a preponderance of GWAS SNPs in human-specific grammatical patterns may warrant caution in using the mouse as a model system for a given pathway. This may partially explain, for example, mixed results seen in clinical trials of investigational lupus treatments following success in mouse model systems (64).
Human–mouse SOM data browser
To facilitate data analysis and visualization, the SOM data are presented in a public browser with extensive search and visualization capabilities (https://boylelab.med.umich.edu/SOMbrowser/). Interactive density maps are supplied for visual comparison of annotations across all grammatical patterns. Clicking a pattern within these maps reveals pattern-wise summary data and CRM-level annotations in a tabbed browsing pane. Links to external resources are provided throughout to facilitate deeper analysis. Furthermore, the ‘compare maps’ utility gives the ability to compare multiple maps side-by-side, and a flexible search tool allows users to construct arbitrarily complex queries directly against the browser database. Search results are presented as an interactive density map showing how search results are divided among all grammatical patterns, accompanied by a tabular section containing detailed results. As in the main browser, clicking a pattern within the results map reveals summary data related to the selected pattern, along with a link to the primary browser record. Throughout the browser, integrated help links provide instructions on how to use various feature and information on data sources and interpretation. A separate detailed help section is also available with more detailed information on browser features and analysis procedures.
DISCUSSION
These results build upon previous significant studies in this area (1,5,17,22,29,31,47,50–52) by expanding our understanding of the relationship between positional conservation and underlying gene regulatory logic. SOMs have been used previously to study gene regulation, in a study of TFBS co-binding profiles across multiple human cell types (22), across distant species (29), and, most recently, to investigate combinatorial regulatory logic in the mouse liver (65), but this is, as far as we know, the first application of an SOM to identify a mammalian regulatory grammar. Our results recapitulate several observations from these studies, supporting the utility of SOMs to illuminate underlying properties of the regulatory landscape. Namely, the existence of conserved, tissue-specific, and species-specific regulatory compartments, the ability of the SOM to cluster regulatory patterns into sets with stable chromatin signatures, and the association of distinct TF co-binding patterns with expression (and dysregulation) of tissue-specific genes.
We extend the body of knowledge regarding human and mouse gene regulation by showing widespread turnover of grammatical patterns at positionally conserved regulatory loci, frequently accompanied by differences in associated chromatin states. In most cases, we saw a stronger correlation between functional markers among different loci with matched grammatical patterns than between different cell types at positionally conserved loci. Notably, positionally conserved loci with matched chromatin states were no more likely to carry matched grammatical patterns than positionally conserved loci in general. We conclude that positional conservation alone has limited predictive value for underlying regulatory logic, even when augmented by chromatin state data. By contrast, grammatical patterns have stable functional signatures that span both species and tissue, and these signatures remain relatively constant regardless of the evolutionary history of an individual regulatory locus. This gives grammatical patterns a distinct advantage for predicting the function of regulatory sequences genome-wide compared to methods relying on positional conservation, especially considering that such methods are uninformative for roughly 60% of regulatory loci. Regulatory grammar enables reliable functional prediction within non-orthologous regions, substantially advancing our ability to interrogate regulatory mechanisms across genomes and species.
We find evidence that divergent gene regulation results from a complex interplay between changes in grammatical patterns at positionally conserved loci as a result of TFBS turnover, and physical gain and loss of regulatory sequences, which carry mostly conserved grammatical patterns. This offers new understanding of the mechanisms by which gene expression programs evolve. We show a clear correlation between the tissue and species specificity of grammatical patterns (i.e. their grammatical class) with matching tissue and species-specific gene expression patterns. This shows that grammatical patterns capture the functional conservation of regulatory logic and can predict tissue and species-specific regulatory activities. As such, tissue-specific grammatical patterns may offer new understanding of the regulatory basis of relevant diseases, especially if CRMs within a given pattern are enriched for related GWAS SNPs. We speculate that, by combining grammatical conservation and GWAS data, it may be possible to identify disease pathways that are most promising for translational research based on conservation of their associated regulatory circuitry. Similarly, species-specific grammatical patterns may represent emergent portions of the regulatory language with potentially causal roles in evolutionary divergence and speciation. These portions of regulatory grammar may help us understand why mouse models often fail to translate to the human system.
Our choice of cell types in this study was a direct reflection of data availability from ENCODE at the time of the analysis, and we acknowledge that all four are cancer-derived and/or immortalized. Although this may restrict how broadly our results can be interpreted, the strong presence of conserved grammatical patterns in all cell types shows that regulatory mechanisms have remained fairly stable between these cell types. Therefore, despite some apparent regulatory divergence, especially in K562, it is probably safe to assume that the bulk of our observations will also apply to normal cell types. As sufficient data become available for various primary and immortalized cell types, we believe these methods can yield deep insights into how conserved and divergent grammatical patterns participate in normal biology and pathological states. In summary, we believe that grammatical patterns concisely capture the meanings associated with different regulatory sentences, regardless of their evolutionary history, and that these methods can allows us to globally predict functional conservation and divergence. Applying these methods across may differentiate between genetic pathways that are conserved in their regulation, and those that have diverged significantly between species. This may highlight the pathways that hold the most promise for translational approaches using mouse models.
DATA AVAILABILITY
All analyses available through https://boylelab.med.umich.edu/SOMbrowser/ and https://github.com/Boyle-Lab/mouse-human-SOM. All datasets used in this analysis are documented in Supplementary Tables S1-S4, and are available through the ENCODE project at encodeproject.org.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the ENCODE project for making well-documented data freely available for use in this project. We also thank Haley Amemiya, Christopher Castro, Melissa Englund, Torrin McDonald, Sierra Nishizaki, Marcus Sherman and Jessica Switzenberg for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Alfred P. Sloan Foundation [FG-2015-65465]; National Human Genome Research Institute Grant [R00HG007356]; NSF CAREER Award [DBI-1651614 to A.B.]. Funding for open access charge: NSF CAREER Award [DBI-1651614].
Conflict of interest statement. None declared.
REFERENCES
- 1. Vierstra J., Rynes E., Sandstrom R., Zhang M., Canfield T., Hansen R.S., Stehling-Sun S., Sabo P.J., Byron R., Humbert R. et al. . Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science. 2014; 346:1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Consortium M.G.S., Waterston R.H., Lindblad-Toh K., Birney E., Rogers J., Abril J.F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M. et al. . Initial sequencing and comparative analysis of the mouse genome. Nature. 2002; 420:520–562. [DOI] [PubMed] [Google Scholar]
- 3. Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D. et al. . A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014; 515:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shibata Y., Sheffield N.C., Fedrigo O., Babbitt C.C., Wortham M., Tewari A.K., London D., Song L., Lee B.-K., Iyer V.R. et al. . Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLOS Genet. 2012; 8:e1002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Odom D.T., Dowell R.D., Jacobsen E.S., Gordon W., Danford T.W., MacIsaac K.D., Rolfe P.A., Conboy C.M., Gifford D.K., Fraenkel E.. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet. 2007; 39:730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seok J., Warren H.S., Cuenca A.G., Mindrinos M.N., Baker H.V., Xu W., Richards D.R., McDonald-Smith G.P., Gao H., Hennessy L. et al. . Genomic responses in mouse models poorly mimic human inflammatory diseases. PNAS. 2013; 110:3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mestas J., Hughes C.C.W.. Of mice and not men: differences between mouse and human immunology. J. Immunol. 2004; 172:2731–2738. [DOI] [PubMed] [Google Scholar]
- 8. Shay T., Jojic V., Zuk O., Rothamel K., Puyraimond-Zemmour D., Feng T., Wakamatsu E., Benoist C., Koller D., Regev A. et al. . Conservation and divergence in the transcriptional programs of the human and mouse immune systems. PNAS. 2013; 110:2946–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidt D., Schwalie P.C., Wilson M.D., Ballester B., Goncalves A., Kutter C., Brown G.D., Marshall A., Flicek P., Odom D.T.. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012; 148:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. . A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kunarso G., Chia N.-Y., Jeyakani J., Hwang C., Lu X., Chan Y.-S., Ng H.H., Bourque G.. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010; 42:631–634. [DOI] [PubMed] [Google Scholar]
- 12. Bourque G., Leong B., Vega V.B., Chen X., Lee Y.L., Srinivasan K.G., Chew J.-L., Ruan Y., Wei C.-L., Ng H.H. et al. . Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008; 18:1752–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du J., Leung A., Trac C., Lee M., Parks B.W., Lusis A.J., Natarajan R., Schones D.E.. Chromatin variation associated with liver metabolism is mediated by transposable elements. Epigenet. Chromatin. 2016; 9:28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chuong E.B., Elde N.C., Feschotte C.. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016; 351:1083–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lowe C.B., H. D.. 29 mammalian genomes reveal novel exaptations of mobile elements for likely regulatory functions in the human genome. PLoS One. 2012; 7:e43128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mortazavi A., Pepke S., Jansen C., Marinov G.K., Ernst J., Kellis M., Hardison R.C., Myers R.M., Wold B.J.. Integrating and mining the chromatin landscape of cell-type specificity using self-organizing maps. Genome Res. 2013; 23:2136–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Denas O., Sandstrom R., Cheng Y., Beal K., Herrero J., Hardison R.C., Taylor J.. Genome-wide comparative analysis reveals human-mouse regulatory landscape and evolution. BMC Genomics. 2015; 16:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoekstra H.E., Coyne J.A.. The locus of evolution: evo devo and the genetics of adaptation. Evolution. 2007; 61:995–1016. [DOI] [PubMed] [Google Scholar]
- 19. Romero I.G., Ruvinsky I., Gilad Y.. Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 2012; 13:505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wray G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007; 8:206–216. [DOI] [PubMed] [Google Scholar]
- 21. Carroll S.B. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008; 134:25–36. [DOI] [PubMed] [Google Scholar]
- 22. Xie D., Boyle A.P., Wu L., Zhai J., Kawli T., Snyder M.. Dynamic trans-acting factor colocalization in human cells. Cell. 2013; 155:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oliveri P., Oliveri P., Tu Q., Tu Q., Davidson E.H., Davidson E.H.. Global regulatory logic for specification of an embryonic cell lineage. PNAS. 2008; 105:5955–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davidson E.H. Emerging properties of animal gene regulatory networks. Nature. 2010; 468:911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coulombe-Huntington J., Xia Y.. Regulatory network structure as a dominant determinant of transcription factor evolutionary rate. PLoS Comput. Biol. 2012; 8:e1002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007; 8:450–461. [DOI] [PubMed] [Google Scholar]
- 27. Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.-K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R. et al. . Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chomsky N. On certain formal properties of grammars. Inf. Control. 1959; 2:137–167. [Google Scholar]
- 29. Boyle A.P., Araya C.L., Brdlik C., Cayting P., Cheng C., Cheng Y., Gardner K., Hillier L.W., Janette J., Jiang L. et al. . Comparative analysis of regulatory information and circuits across distant species. Nature. 2014; 512:453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng Y., Ma Z., Kim B.-H., Wu W., Cayting P., Boyle A.P., Sundaram V., Xing X., Dogan N., Li J. et al. . Principles of regulatory information conservation between mouse and human. Nature. 2014; 515:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M. et al. . The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015; 43:D670–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hubisz M.J., Pollard K.S., Siepel A.. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief. Bioinform. 2011; 12:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rosenbloom K.R., Dreszer T.R., Pheasant M., Barber G.P., Meyer L.R., Pohl A., Raney B.J., Wang T., Hinrichs A.S., Zweig A.S. et al. . ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 2010; 38:D620–D625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bray N.L., Pimentel H., Melsted P., Pachter L.. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016; 34:525–527. [DOI] [PubMed] [Google Scholar]
- 37. Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J.. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004; 32:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu Y.C., Bansal M.S., Rasmussen M.D., Herrero J., Kellis M.. Phylogenetic identification and functional characterization of orthologs and paralogs across human, mouse, fly, and worm. bioRxiv. 2014; doi:10.1101/005736. [Google Scholar]
- 39. Gilad Y., Mizrahi-Man O.. A reanalysis of mouse ENCODE comparative gene expression data. F1000Res. 2015; 4:121–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 41. Anders S., Huber W.. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:R106–R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Welch R.P., Lee C., Imbriano P.M., Patil S., Weymouth T.E., Smith R.A., Scott L.J., Sartor M.A.. ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic Acids Res. 2014; 42:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L. et al. . The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014; 42:D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kuhn R.M., Haussler D., Kent W.J.. The UCSC genome browser and associated tools. Brief. Bioinform. 2013; 14:144–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. International HapMap Consortium The International HapMap Project. Nature. 2003; 426:789–796. [DOI] [PubMed] [Google Scholar]
- 47. Ballester B., Medina-Rivera A., Schmidt D., Gonzàlez-Porta M., Carlucci M., Chen X., Chessman K., Faure A.J., Funnell A.P.W., Goncalves A. et al. . Multi-species, multi-transcription factor binding highlights conserved control of tissue-specific biological pathways. Elife Sci. 2014; 3:e02626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Malone J., Holloway E., Adamusiak T., Kapushesky M., Zheng J., Kolesnikov N., Zhukova A., Brazma A., Parkinson H.. Modeling sample variables with an Experimental Factor Ontology. Bioinformatics. 2010; 26:1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S. et al. . Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012; 22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stefflova K., Thybert D., Wilson M.D., Streeter I., Aleksic J., Karagianni P., Brazma A., Adams D.J., Talianidis I., Marioni J.C. et al. . Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell. 2013; 154:530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schmidt D., Wilson M.D., Ballester B., Schwalie P.C., Brown G.D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C.P., Mackay S. et al. . Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010; 328:1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stergachis A.B., Neph S., Sandstrom R., Haugen E., Reynolds A.P., Zhang M., Byron R., Canfield T., Stelhing-Sun S., Lee K. et al. . Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature. 2014; 515:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Borneman A.R., Gianoulis T.A., Zhang Z.D., Yu H., Rozowsky J., Seringhaus M.R., Wang L.Y., Gerstein M., Snyder M.. Divergence of transcription factor binding sites across related yeast species. Science. 2007; 317:815–819. [DOI] [PubMed] [Google Scholar]
- 54. Jones F.C., Grabherr M.G., Chan Y.F., Russell P., Mauceli E., Johnson J., Swofford R., Pirun M., Zody M.C., White S. et al. . The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012; 484:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kvon E.Z., Stampfel G., Yáñez-Cuna J.O., Dickson B.J., Stark A.. HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev. 2012; 26:908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. et al. . Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E.. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008; 132:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Strous R.D., Shoenfeld Y.. Schizophrenia, autoimmunity and immune system dysregulation: a comprehensive model updated and revisited. J. Autoimmun. 2006; 27:71–80. [DOI] [PubMed] [Google Scholar]
- 60. Panaro M.A., Cianciulli A.. Current opinions and perspectives on the role of immune system in the pathogenesis of Parkinson's disease. Curr. Pharm. Des. 2012; 18:200–208. [DOI] [PubMed] [Google Scholar]
- 61. Frostegård J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013; 11:117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Danesh J., Collins R., Peto R.. Chronic infections and coronary heart disease: is there a link?. Lancet. 1997; 350:430–436. [DOI] [PubMed] [Google Scholar]
- 63. de Visser K.E., Eichten A., Coussens L.M.. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer. 2006; 6:24–37. [DOI] [PubMed] [Google Scholar]
- 64. Perry D., Sang A., Yin Y., Zheng Y.-Y., Morel L.. Murine models of systemic lupus erythematosus. J. Biomed. Biotechnol. 2011; 2011:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dubois-Chevalier J., Dubois V., Dehondt H., Mazrooei P., Mazuy C., Sérandour A.A., Gheeraert C., Guillaume P., Baugé E., Derudas B. et al. . The logic of transcriptional regulator recruitment architecture at cis-regulatory modules controlling liver functions. Genome Res. 2017; 27:985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All analyses available through https://boylelab.med.umich.edu/SOMbrowser/ and https://github.com/Boyle-Lab/mouse-human-SOM. All datasets used in this analysis are documented in Supplementary Tables S1-S4, and are available through the ENCODE project at encodeproject.org.