


Abstract
Androgen receptor (AR) splice variants (ARVs) are implicated in development of castration-resistant prostate cancer (CRPC). Upregulation of ARVs often correlates with persistent AR activity after androgen deprivation therapy (ADT). However, the genomic and epigenomic characteristics of ARV-dependent cistrome and the disease relevance of ARV-mediated transcriptome remain elusive. Through integrated chromatin immunoprecipitation coupled sequencing (ChIP-seq) and RNA sequencing (RNA-seq) analysis, we identified ARV-preferential-binding sites (ARV-PBS) and a set of genes preferentially transactivated by ARVs in CRPC cells. ARVs preferentially bind to enhancers located in nucleosome-depleted regions harboring the full AR-response element (AREfull), while full-length AR (ARFL)-PBS are enhancers resided in closed chromatin regions containing the composite FOXA1-nnnn-AREhalf motif. ARV-PBS exclusively overlapped with AR binding sites in castration-resistant (CR) tumors in patients and ARV-preferentially activated genes were up-regulated in abiraterone-resistant patient specimens. Expression of ARV-PBS target genes, such as oncogene RAP2A and cell cycle gene E2F7, were significantly associated with castration resistance, poor survival and tumor progression. We uncover distinct genomic and epigenomic features of ARV-PBS, highlighting that ARVs are useful tools to depict AR-regulated oncogenic genome and epigenome landscapes in prostate cancer. Our data also suggest that the ARV-preferentially activated transcriptional program could be targeted for effective treatment of CRPC.
INTRODUCTION
ADT is the standard treatment for patients with advanced prostate cancer. Approximately 10–20% of these patients relapse into CRPC within 5 years, and the mean survival time is ∼14 months after CRPC diagnosis (1). Despite the depletion of circulating testicular androgen after ADT, sustained AR signaling remains the major molecular mechanism driving castration resistance (2,3). To re-target the persistent AR activity in CRPC, next-generation AR axis inhibitors have been developed, which include abiraterone acetate (an inhibitor of androgen synthesis) and enzalutamide (an AR antagonist). Although these new drugs significantly improve overall survival, resistance has continued to be a problem in a majority of patients (4,5).
Prostate specific antigen (PSA) often resurges in enzalutamide-resistant patients, suggesting the growth of the tumors is still driven by AR signaling (6). Persistent AR activity in CRPC can be mediated by several mechanisms including AR gene amplification and overexpression (7–9), AR gene mutation (10), intra-tumoral androgen synthesis (11), overexpression of AR coactivators (12), aberrant kinase pathway activation (13) and the constitutive expression of AR splice variants (ARVs) (14). ARVs are important in CRPC because most ARVs lack the ligand-binding domain (LBD), the intended therapeutic target of hormone therapy regimens including abiraterone acetate and enzalutamide.
Recent efforts to determine how ARVs drive prostate cancer survival and progression discovered that overexpression of AR splice variant-7 (ARV7) or ARv567es in LNCaP cells resulted in increased cell proliferation, and knocking down endogenous ARVs in 22Rv1 cells lead to attenuated cell growth in the androgen-deprived condition in vitro and in vivo (15–18). These findings highlight the role of ARVs in promoting cell proliferation and tumor progression. Overexpression of ARV7 in metastatic and circulating tumor cells is significantly associated with shorter survival and resistance to enzalutamide and abiraterone treatments (19,20). These data indicate that ARVs are valuable predictive biomarkers of antiandrogen resistance. Nonetheless, the genome, cistrome, and epigenome features of ARVs remain incompletely characterized, and especially the relevance of ARV-regulated transcription program to the castration-resistant progression of patients is poorly understood. More importantly, it remains unclear whether increased expression of ARVs is a driving force or merely the by-product of other molecular mechanisms such as AR amplification and rearrangement. Therefore, the identification and characterization of ARV-regulated transcription programs could potentially lead to novel targets for the development of more effective therapeutics for CPRC.
In this study, we characterized the genome, cistrome and epigenome landscapes of ARVs. Specifically, we discovered that ARV-preferentially targeted genes are associated exclusively with CRPC, but not with treatment-responsive or untreated prostate cancer in patients, highlighting the role of ARV in driving castration resistance. We also demonstrated that the expression of ARV-preferentially activated genes, but not those driven by ARFL or total AR (ARVs + ARFL), was significantly increased in the tumor metastases of abiraterone-resistant patients compared to those of abiraterone-responsive patients, suggesting that ARV-preferentially targeted genes are involved in the development of therapeutic resistance. The ARV-preferentially targeted genes identified in this study may serve as prognostic biomarkers for predicting abiraterone resistance and as potential targets for developing new therapeutics for CRPC patients.
MATERIALS AND METHODS
Clinical samples
The whole transcriptome sequencing (RNA-seq) of 77 CRPC patients is part of the ‘PROMOTE’ (Prostate Cancer Medically-Optimized Genome-Enhanced Therapy) study that was initiated in May 2013 after obtaining approval from Mayo Clinic Institutional Review Board (IRB) (21). All patients enrolled in the trial provided a written informed consent approved by the IRB. All patients had to have sub-castrate testosterone levels (less than 50 ng/dl) and a metastatic site for biopsy. Tumor tissue biopsies were collected from bone (n = 54) or soft tissue (n = 23) before initiation of abiraterone acetate and prednisone therapy (AAP). Progression status at 12 weeks after initiating AAP was determined per the recommendations of the Prostate Cancer Working Group-2 criteria (PCWG2) (22). For assessing progression at 12-weeks, serum PSA, bone and CT imaging and symptom assessments using the Functional Assessment of Cancer Therapy-Prostate (FACT-P) scale were performed at the same time (week 12) (22). Response to therapy was defined as the absence of PSA progression (as defined by the PSA Working Group Criteria); absence of any new bone lesion on bone scan and no radiological (RECIST 1.0) progression of nodal or soft tissue metastases. Bone scans at 12 weeks in which new lesions were detected were repeated with a follow-up bone scan at least six more weeks and if additional new bone lesions were observed at the second follow-up scan patient was deemed to have progression. Patients who met one of these criteria for the progressive disease were defined as ‘non-responders’ after 12 weeks of drug exposure. As a result, 45 and 32 patients were determined as responders and non-responders, respectively.
ChIP, ChIP-qPCR and ChIP-seq library preparation
ChIP experiments were performed as described previously (http://younglab.wi.mit.edu/hESRegulation/Young_Protocol.doc). In brief, chromatin was cross-linked for 10 min at room temperature with 11% formaldehyde/PBS solution added to cell culture medium. Cross-linked chromatin was then sonicated, diluted and immunoprecipitated with Protein G-plus Agarose beads (Bio-Rad®) prebound with antibody (anti-AR, N-20, SC-816; anti-AR, C-19, SC-815 from Santa Cruz; anti-H3K4me1, ab8895; anti-H3K4me2, ab7766; anti-H3K27Ac, ab4729; anti-H2A.Z, ab4174; Pk-tag antibody (V5-tag antibody), ab9116 from Abcam) at 4°C overnight. Precipitated protein-DNA complexes were eluted and cross-linking was reversed at 65°C for 16 h. DNA fragments were purified and analyzed by real-time PCR. ChIP-seq libraries were prepared using previously described methods (23). High-throughput sequencing (51 nt, pair-end) was performed using the Illumina HiSeq™2000 platforms at the Mayo Genome Core Facility. Real-time quantitative PCRs were carried out in a Bio-Rad CFX96™ Real-Time System, using SYBR green PCR master mix (Bio-Rad, Hercules, CA, USA). Reactions were carried out in triplicate and with biological replicates. Primers are shown in Supplementary Table S10. ChIP-qPCR data were analyzed as % input after normalizing each ChIP DNA fraction's Ct value to the Input DNA fraction's Ct value.
Estimate ARV7 expression using TCGA RNA-seq data
RNA-seq BAM files were downloaded from the NCI Genomic Data Commons (https://gdc.cancer.gov/). The expression level of ARV7 was measured by the number of splice reads that span exon-3 (chrX:66905852–66905968, genome coordinates are based on the human reference genome hg19/GRCh37) and the downstream cryptic exon (chrX:66914515–66915580) located in intron-3. The number of splice reads was then normalized by total splice reads to correct sequencing depth (i.e. SRPM, splice read per million).
Cell lines, cell culture and transfection
22Rv1 and LNCaP prostate cancer cell lines and 293T cell lines were purchased from ATCC. C4–2 cells were purchased from Uro Corporation (Oklahoma City, OK, USA). 22Rv1 and LNCaP cells were maintained at 37°C and 5% CO2 in RPMI 1640 containing 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic (Thermo Fisher Scientific). 293T cells were maintained in DMEM medium with 10% FBS and C4–2 cells were maintained in RPMI medium with 10% FBS. Transient transfections of 22Rv1 and LNCaP cells were performed by electroporation (350 V, 10 ms. BTX, Harvard Apparatus) and then cells were cultured in RPMI 1640 containing 10% FBS or charcoal stripped serum (CSS).
Knockdown of ARFL and ARVs by siRNAs
Small interference RNAs (siRNAs) specifically against human ARVs (Pools), ARFL and nonspecific controls were purchased from Dharmacon (Thermo Fisher Scientific) and their sequences are listed in Supplementary Table S10. 22Rv1 cells were seeded in a 10-cm dish and transfected with 50 nM siRNA. Transfection was performed using Lipofectamine 2000 (Invitrogen). The effect of siRNA-mediated AR silencing was examined using western blot 48 h after transfection. To knock down ARFL, we used siRNA targeting AR exon 7/8 junction (siAR Ex7/8, Dharmacon D-003400-18-0002 target sequence CGUGCAGCCUAUUGCGAGAUU). We knocked down AR variants with siRNAs targeting AR-V1/V3/V4/V7 (four siRNA mixture, siARV1 target sequence GAGGGUGUUUGGAGUCUCAUU, siARV3 target sequence AAGAGCCGCUGAAGGAUUUUU, siARV4 target sequence GAUGACUCUGGGAGGAUUUUU, siARV7 target sequence GCAAUUGCAAGCAUCUCAAUU).
Chromatin conformation capture (3C) assay
3C assays were performed as described previously (24). Briefly, cross-linking was performed by incubating 1 × 107 cells in PBS containing 1% formaldehyde. Cell pellets were lysed and suspended with restriction enzyme EcoRI-HF (400 units). Restriction digestion was stopped by 1.6% SDS (final concentration and samples were incubated at 65°C for 25 min). DNA ligation was performed with T4 DNA ligase (2000 units) at 16°C for 4 h. The chromatin samples were reverse cross-linked with proteinase K at 65°C overnight. 3C samples were then purified using phenol-chloroform extraction and purified DNA was used for qPCR analysis using the specific primers listed in Supplementary Table S10. All 3C PCR products were confirmed by Sanger sequencing.
Lentiviral shRNA infection and ARV7 overexpression
293T cells were co-transfected with control or FOXA1 or RAP2A-specific shRNA along with packing and envelop plasmids by Lipofectamine 2000 according to the manufacturer's instructions. At two days post-transfection, virus particles containing shRNA were used to infect prostate cancer cells according to the protocol provided by Sigma-Aldrich. Individual shRNAs specifically targeting human FOXA1 and RAP2A were obtained from Sigma-Aldrich and their sequences are listed in Supplementary Table S10. ARV7 construct was cloned into the lentiviral vector pTSiN, and virus production was performed as described previously (25). C4–2 cells were transduced by culturing with a 1:1 mixture of fresh medium and virus supernatant with Polybrene (4 μg/mg final concentration) for 24–72 h. The infected cells were screened by 1.5 μg/ml puromycin for two days and then cells were maintained in medium containing 0.5 μg/ml puromycin for further use.
Cell proliferation assays
22Rv1 cells infected with shRAP2A or control shRNA or C4–2 cells infected with pTSiN-ARV7 and/or shPAP2A were seeded in 96-well plates (3000 cells/well) and cultured in medium containing 10% CSS. Cells were fixed at different time points (day 0–5) and cell growth was measured using sulforhodamine B (SRB) assay (26).
Tumor xenograft studies in mice
Six-week-old NOD-SCID IL-2-receptor gamma null (NSG) mice were generated in-house and used for animal experiments. All mice were housed under standard conditions with a 12 h light/dark cycle and access to food and water ad libitum. The mouse studies were approved by the IACUC at Mayo Clinic. C4–2 cells transduced with pTSiN-ARV7 or double infected by pTSiN-ARV7 and shPAP2A cells and empty vector (3 × 106 cells in 100 μl matrigel) were injected s.c. (n = 5 experiments) into the left flank of each mouse. Tumor size was measured every three days and calculated using the formula: length × width2 × 0.5.
ChIP-seq data analysis
All short reads were mapped to the human reference genome (hg19/GRCh37) using bowtie2 (version 2.1.0) with default configurations (28). On average, we obtained 81.7 million reads that were uniquely mapped to the reference genome for each sample. These uniquely mapped reads were then used for peak calling. MACS2 (version 2.0.10) was used to identify peaks with input samples used as background and a qvalue cutoff 0.05 (macs2 callpeak –bdg –SPMR -f BAM) (29). Peaks located in satellite repeats and centromere regions were removed. ChIP-seq tag intensity tracks (bedGraph files) were generated by MACS2, and then were converted into bigWig files using UCSC ‘wigToBigWig’ tool. Genomic distribution of peaks with regard to transcription start sites (TSS), and the association of peaks to target genes was performed by Genomic Regions Enrichment of Annotations Tool (GREAT) (30). Histone modification profiles were generated by Epidaurus (31).
RNA seqeuncing data analysis
RNA-seq libraries were prepared using Illumina's TruSeq RNA prep kit and standard protocol. The RNA-seq libraries were sequenced as 51 nt pair-end reads at one sample per lane of an Illumina HiSeq 2500, generating an average of 265 million reads per sample. Fragment size was estimated by RSeQC using the first 1 000 000 read pairs that were uniquely mapped (32). All reads were aligned to the human reference genome (hg19/GRCh37) by TopHat 2.0.9 using these options: TopHat:–keep-fasta-order, –keep-tmp, –no-coverage-search, –bowtie1, –library-type fr-unstranded, –max-multihits 20, –solexa1.3-quals, –fusion-search, –fusion-ignore-chromosomes chrM, –fusion-min-dist 50 000 (33). Gene expression counts were generated using HTseq software (http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html) from Illumina gene annotation files (http://support.illumina.com/sequencing/sequencing_software/igenome.html). Gene expression analysis was conducted using edgeR (version 3.6.8) and the built-in ‘TMM’ (trimmed mean of M-values) normalization method was used (34). For 22Rv1 cell line RNA-seq data, differentially expressed genes were determined based on the false discovery rate (FDR) threshold 0.01 and log2 fold change (FC = siARV/siNT) threshold of 1. Specifically, up-regulated genes were defined as those with FDR ≤ 0.01 and log2 (FC) ≥ 1 and down-regulated genes were defined as those with FDR ≤ 0.01 and log2 (FC) ≤ –1.
ARFL and ARV activity score
For each of the 77 CRPC patients, AR activity score was calculated based on 20 AR target genes as described previously (35) including ABCC4, ACSL3, ADAM7, C1ORF116, CENPN, EAF2, ELL2, FKBP5, GNMT, HERC3, KLK2, KLK3, MAF, MED28, MPHOSPH9, NKX3–1, NNMT, PMEPA1, PTGER4 and ZBTB10. Cell cycle progression (CCP) activity was calculated based on 31 genes described in (36) including: FOXM1, ASPM, TK1, PRC1, CDC20, BUB1B, PBK, DTL, CDKN3, RRM2, ASF1B, CEP55, CDK1, DLGAP5, SKA1, RAD51, KIF11, BIRC5, RAD54L, CENPM, KIAA0101, KIF20A, PTTG1, CDCA8, NUSAP1, PLK1, CDCA3, ORC6, CENPF, TOP2A and MCM10. ARV activity was calculated based on 63 genes defined by these criteria: (i) must differentially expressed with FDR < 0.01 and log2 (FC) < –1 and ( ii) must be ARV-preferentially targeted genes, which gave rise to 66 genes: ANKRD32, ANO6, ASPM, ATL2, CA8, CDK1, CEP128, CHAC2, CPS1, CROT, DIAPH3, DSCAM, DTL, E2F7, ENDOG, GCNT1, GJA1, GMPR, GRIN3A, IL1R2, INMT, INMT-FAM188B, INSC, INSIG1, ISL1, KBTBD8, KCNC4, KCNMB2, KRT19, LPL, LRFN2, MAD2L1, MAF, MESP1, MESP2, MID1, NCAPG2, NDST4, NUF2, NUP93, PARL, PHTF2, PREX2, PRIM2, RAB33A, RARB, RDH10, RECQL, RGS1, RGS2, SGOL1, SLC31A2, SLCO2A1, SMC2, STOX1, TIFA, TMEM169, TMEM241, TMEM97, TMPO, TRIM36, TTK, UCHL5, WNT16, ZBTB10, ZNF367; (iii) three genes (ASPM, CDK1and DTL) were removed due to overlapping with CCP gene list. ARFL activity was calculated based on 33 genes including ADAM9, SLC50A1, RAB4A, PHF8, PARVA, NRARP, NFIX, MYO1B, MMP20, LRIG3, KLF5, KCNJ1, GLI3, G3BP2, FAM96B, ERN1, CUZD1, CLDN8, CDK2AP2, KNSTRN, TIGD6, GPR65, GALNT18, FOXD4, ELP5, DNM1L, ABCA1, CKB, CEACAM16, CAB39L, PXYLP1, ABCA5 and AGAP9. We used the same method to calculate the activity score as described in (37). In brief, gene expression values (log2(FPKM)) of each sample were converted to Z-scores by Z = (x – μ)/σ, where μ is the average Log2(FPKM) across all samples of a gene and σ is the standard deviation of the log2(FPKM) across all samples of a gene. The Z-scores were then summed across all genes for each sample. Finally, the summed Z-scores were converted to a percentile and normalized between 0 and 100 with 0 being the lowest and 100 being the highest.
Motif analysis
De novo motif discovery was performed using MEME-ChIP on 100 nucleotides DNA sequence centered around ChIP-seq peak summit (38). In brief, the most significant 500 peaks were selected and the corresponding genomic sequences (500 bp centered on the peak summit) were retrieved from human reference genome (hg19/GRCh37) and submitted to MEME-ChIP webserver (http://meme-suite.org/tools/meme-chip). For candidate motif search, we used C++/Python candidate motif finding package (motility, http://cartwheel.caltech.edu/motility/intro.html) to map motif with no mismatch allowed. When there were multiple motifs, pick the one that is most conserved (i.e the one with the highest PhastCon score).
Survival analysis
We downloaded the expression and the associated clinical outcome data of 390 TCGA prostate cancer patients from the TCGA cancer browser website (https://genome-cancer.ucsc.edu/). The gene expression values were measured experimentally using the IlluminaHiSeq_RNASeqV2 and then mean-normalized across all TCGA cohorts by the UCSC Cancer Browser team. The recurrence-free survival time and status were determined by the ‘_RFS’ and ‘_RFS_IND’ fields from the ‘clinical_data’ file, respectively. We first stratified the 390 prostate patients into two groups according to the median expression value of E2F7 (or RAP2A), and then performed the Kaplan-Meier survival analysis using the ‘survival’ R package available from http://cran.r-project.org. The Log-rank test was used to evaluate if the difference in recurrence-free survival between the above two subgroups was statistically significant.
Meta-analysis of AR ChIP-seq data derived from clinical tissues and cell lines
We analyzed AR ChIP-seq data from two cohorts of clinical samples. One cohort included 7 normal prostate epithelium tissues and 13 prostate tumor tissues reported previously (39). The original peaks and associated ChIP-seq reads intensity signals (bigWig files) were downloaded from the Gene Expression Omnibus (GEO) database (GSE56288). The other cohort included five castration-resistant prostate cancer tissues, three untreated prostate tumor tissues and two treatment responsive prostate tumor tissues reported previously (40). The original peaks were downloaded from GEO database (GSE28219) and lifted over to the hg19 assembly using CrossMap (41). We regenerated the ChIP-seq read intensity signals using MACS2 after aligning read to hg19 reference genome. AR ChIP-seq data generated from LNCaP cells were downloaded from GEO with accessions (GSM1328169 and GSM1328170, biological replicates), and AR ChIP-seq data generated from C4–2 cells were downloaded from GEO with accessions (GSM1328166, and GSM1328167, biological replicates) reported previously (42). Peak calling was performed as described previously. Only the top 50,000 peaks were used to compare to ARV-preferentially binding sites (ARV-PBS), ARFL-preferentially binding sites (ARFL-PBS), and the Common binding sites (Common-BS).
GC content and sequence conservation analysis
GC content (i.e. GC percent) was calculated for every five nucleotides window as (G+C)*100/(A+T+C+G). Precomputed GC content file downloaded from UCSC database (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/gc5BaseBw.txt.gz) and then converted into bigwig format to facilitate analysis. Similarly, we computed CpG dinucleotide content for every 50 nucleotides window. Precomputed sequence conservation (PhastCon) file was also downloaded from UCSC database (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/phastCons100way.txt.gz) and converted into bigwig format.
Peak overlapping coefficient
We used the overlapping coefficient (or Szymkiewicz-Simpson coefficient) to measure the degree of overlaps between two lists of peaks (X and Y). It is defined as the number of the overlapped peaks divided by the smaller size of the two lists of peaks. The overlapping coefficient equals 0 when no overlap exists, and 1 when one list is a subset of another list.
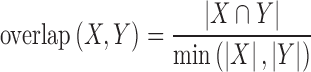 |
Pathway analysis
Gene Set Enrichment Analysis (GSEA) tool was used to analyze ARV activated and repressed genes (43).
Statistical test
In ARFL-PBS, out of 1089 FOXA1 and AREhalf motif pairs, 138 (or 12.67%) have the gap size of 4. To test if the composite motif with the gap size of 4 (i.e. FOXA1-nnnn-AREhalf) is significantly more frequent than other gap sizes, we performed Grubbs's outlier test using function ‘grubbs.test’ in R package ‘outliers’ (https://cran.r-project.org/web/packages/outliers/outliers.pdf). Similar analyses were performed for ARV-PBS and Common-BS. All data from cell culture studies were expressed as mean ± SD for experiments performed at least three times. The difference between two groups was analyzed using paired Student's t-test unless otherwise specified. A P value <0.05 is considered statistically significant.
RESULTS
Genomic features of ARV-PBS, ARFL-PBS and Common-BS
We effectively knocked down ARV and ARFL in 22Rv1 cells treated with or without the synthetic androgen R1881 and performed ChIP-seq in duplicates for ARFL (si-ARVs), ARV (si-ARFL), and total AR (si-Control) (Figure 1A). The high correlation between ChIP-seq replicates in each group was noticed (Supplementary Figure S1). We identified 5040 ARFL-preferential binding sites (ARFL-PBS), 1304 Common binding sties (Common-BS), and 946 ARV- preferential binding sites (ARV-PBS) by comparing the cistromes of ARFL and ARVs (Figure 1A and B and Supplementary Tables S1–S3). Since 22Rv1 is an atypical cell line in which ARFL has a duplicated exon-3 and ARVs are particularly highly expressed (44), we sought to determine if the cistrome of the ARFL isoform in 22Rv1 cells resembles that of the canonical ARFL in ARV-negative cells. To this end, we compared ARFL-PBS, Common-BS and ARV-PBS identified from 22Rv1 cells to AR binding sites (ARBS) identified from two other AR-positive prostate cancer cell lines including LNCaP and C4–2. We found that 91.6% and 93.7% of Common-BS in 22Rv1 overlapped with ARBS identified from LNCaP and C4–2 cells, respectively, suggesting that Common-BS are conserved among these cell lines. We found that 69.1% and 68.7% of ARFL-PBS overlapped with ARBS identified in LNCaP and C4–2 cells, respectively (Supplementary Table S4). Interestingly, these values are very close to the overlapping percentage between LNCaP-ARBS and C4–2-ARBS (69.7%), indicating that the cistrome of ARFL in 22Rv1 cells is highly similar to that of ARFL in LNCaP and C4–2 cells. We found that the overlapping percentage between 22Rv1-ARV-PBS and C4–2-ARBS (71.8%) was much higher (P = 1.86 × 10−9, Fisher's exact test) than that between 22Rv1-ARV-PBS and LNCaP-ARBS (58.7%), possibly because both 22Rv1 and C4–2 cell lines are CRPC in nature whereas LNCaP is androgen-dependent.
Figure 1.

Genome features of ARV-PBS, ARFL-PBS and Common-BS. (A) Western blots and diagram showing experiments performed to identify total AR binding stites (ARBS), ARFL-preferential binding sites (ARFL-PBS), ARV-preferential binding sites (ARV-PBS) and common binding sites (Common-BS). 22Rv1 cells transfected with Control siRNA (si-Con), siRNA targeting AR exon 7/8 to knock down ARFL (si-ARFL) or with siRNAs targeting AR-V1/V3/V4/V7 to knock down AR variants (si-ARVs) and 24 h later were treated with vehicle or 1 nM of R1881, a synthetic androgen. The effect of siRNA-mediated AR silencing was examined using western blot 48 h after transfection and indicated samples were utilized for RNA-seq or ChIP-seq analysis. (B) Heatmaps showing ChIP-seq read intensity of ARBS (si-Con, R1881+), ARFL (si-ARVs, R1881+) and ARVs (si-ARFL, R1881−) at ARFL-PBS, Common-BS and ARV-PBS loci. (C) Genomic distributions of ARFL-PBS (red), Common-BS (purple), ARV-PBS (blue) around transcription start site (TSS). (D) Sequence conservation (measured by PhastCon score) of ARFL-PBS (red), Common-BS (purple) and ARV-PBS (blue). Each peak was extended 1 kb to the up- and down-stream from the center. Wilcoxon rank sum test was used to compare conservation scores of ARFL-PBS and ARV-PBS (P = 0.22). (E–H) GC content profiles of ARFL-PBS (red), Common-BS (purple) and ARV-PBS (blue). Each peak was extended 1 kb to up- and down-stream from the center. Red, purple and blue lines represent the local regression (Loess) curves.
As expected, all three groups of ARBS exhibited similar patterns of genomic distribution with >60% located in distal enhancer regions that are at least 50 kb away from the transcription start site (TSS) (Figure 1C). Due to functional constraints, most transcription factor binding sites (TFBS) exhibited higher sequence-conservation than that of the flanking background regions (Supplementary Figure S2). Compared to the flanking background regions, the centers of all three groups of ARBS were highly conserved among 100 vertebrate genomes (Figure 1D), suggesting that they are genuine TFBS, which likely underwent purifying selection. When investigating the DNA GC content of the three groups of ARBS, we found that ARFL-PBS had higher GC content and that the GC content profile was relatively uniform across the entire binding region (Figure 1E and F). In contrast, ARV-PBS and Common-BS had lower GC content, and the GC content was dramatically reduced in the center of the binding region, forming V-shaped profiles (Figure 1E, G and H). The central dip in GC content profile has been linked frequently to nucleosome-depleted regions (NDRs) (45–47). This result suggests that, different from ARFL, ARVs bind primarily to NDRs. This hypothesis is further confirmed by our cistromic and epigenomic data described below.
Cistrome landscapes of ARV-PBS, ARFL-PBS and Common-BS
To further determine the role of ARVs and ARFL in regulating ARV-PBS and ARFL-PBS gene expression, we focused on two specific loci. The small G protein gene RAP2A, a member of RAS oncogene family, is an ARV-targeted gene with an ARV-PBS located 108 kb downstream (Figure 2A). IGF1 (insulin-like growth factor 1) is an ARFL-targeted gene with an ARFL-PBS located 53 kb downstream (Figure 2B). The long-range interaction between the ARV-PBS and the RAP2A promoter or the ARFL-PBS and the IGF1 promoter was confirmed by chromosome conformation capture (3C) assay (Figure 2C and D). Ectopic expression of ARV7 in ARV-negative LNCaP cells induced RAP2A expression in a dose-dependent manner, but had no overt effect on IGF1 expression (Figure 2E). Moreover, knocking down ARVs in 22Rv1 cells decreased RAP2A expression, but had no effect on IGF1 expression (Figure 2F). In contrast, knocking down ARFL increased the expression of IGF1 (Figure 2F). These results demonstrate that ARV and ARFL specifically regulate the expression of RAP2A and IGF1, respectively, and that ARV activates RAP2A expression while ARFL represses IGF1 expression.
Figure 2.

Assessment of characteristics of ARV-PBS and ARFL-PBS in the RAP2A and IGF1 loci. (A and B) UCSC genome browser screenshots showing ARV, ARFL and total AR (ARV+ARFL) ChIP-seq data at the RAP2A locus (A) and the IGF1 gene locus (B). Read intensities of ARV ChIP-seq (siARFL), ARFL ChIP-seq (siARV) and total AR ChIP-seq were indicated by the purple, green and red tracks, respectively. KD, knockdown. (C and D) LNCaP cells were transfected with empty vector (E.V.) or ARV7 and Chromosome Conformation Capture (3C) assays was performed to demonstrate the chromatin looping between the ARV binding site and the promoter of RAP2A (C) or IGF1 (D). (E) Different doses of ARV7 plasmid were transfected into LNCaP cells cultured in regular (androgen-undepleted) media and the effect of ARV7 overexpression on the RAP2A and IGF1 mRNA expression was detected by RT-qPCR. (F) Control siRNA (si-Con), specific siRNA of ARFL (si-ARFL) and ARVs (si-ARVs) were transfected into 22Rv1 cells cultured in regular (androgen-undepleted) media and the effect of knocking down ARFL or/and ARVs on RAP2A and IGF1 mRNA expression was measured by RT-qPCR. Knockdown effectiveness was measured by western blots. (G) LNCaP cells were transfected with empty vector (E.V.) or Pk tagged ARV7 (Pk-ARV7) and the effect of ARV7 overexpression on the occupancy of ARV7 and ARFL in RAP2A and IGF1 gene loci was detected by ChIP-qPCR. (H) Control siRNA (si-Con) and ARV-specific siRNA (si-ARVs) were transfected into 22Rv1 cells and the effect of knocking down ARVs on the occupancy of ARFL in RAP2A and IGF1 gene loci in 22Rv1 cells was detected by ChIP-qPCR.
Since ARV7 specific antibody is not suitable for ChIP assay, we used Pk (V5) tag antibody for ARV7 ChIP and AR-C19 antibody (recognized the C-terminal of AR) for ARFL ChIP. We showed the Pk tag antibody performed much better than ARV7 specific antibody for the ChIP-qPCR experiments (Supplementary Figure S3). Intriguingly, ChIP-qPCR data showed that ARFL could also bind to the ARV-PBS of RAP2A in LNCaP cells, which do not express endogenous ARVs (Figure 2G, middle). However, when exogenous Pk-tagged ARV7 was introduced, ARV7 preferentially bound to this locus in the presence of ARFL (Figure 2G, middle). Moreover, overexpression of ARV7 did not affect ARFL occupancy at IGF1, an ARFL-PBS (Figure 2G, right). Similar results were found in other ARV-PBS (Supplementary Figure S4) and ARFL-PBS genes (Supplementary Figure S5). In addition, knocking down ARVs significantly increased the binding of ARFL in the loci of ARV-PBS but not in the loci of ARFL-PBS in 22Rv1 cells (Figure 2H and Supplementary Figure S6). These data suggest that ARFL is able to bind to ARV-PBS in absence of ARVs and that ARVs can re-occupy these loci even in the presence of ARFL, recapitulating the definition of the sites preferentially bound by ARVs.
Next, we performed de novo motif search in these three groups of ARBS. We detected palindromic AR response element (AREfull) and FOXA1 motifs as the most enriched motifs in both Common-BS and ARV-PBS (Figure 3A). In contrast, the most enriched motif in ARFL-PBS was a composite motif consisting of FOXA1 and AREhalf with a 4-nt gap in between (i.e. FOXA1-nnnn-AREhalf) (Figure 3A). The 4-nt gap size between FOXA1 and AREhalf was significantly (P < 2.2 × 10−16, Grubbs’ outlier test) over-represented than other gap sizes (Figure 3A), and the 4-nt spacer between FOXA1 and AREhalf was recognizable through direct sequence pileup (Supplementary Figure S7). The preferred 4-nt gap between FOXA1 and AREhalf in ARFL-PBS prompted us to explore the spatial organizations between FOXA1 and AREfull in Common-BS and ARV-PBS, respectively. We found that FOXA1 and AREfull motifs were separated primarily by a 5-nt gap (P = 5.1 × 10−9, Grubbs’ outlier test) in Common-BS (Figure 3A), but there was no preferred gap size in ARV-PBS (Figure 3A). These data suggest that although FOXA1 motif is enriched in all three groups of AR bindings (Supplementary Figure S8A), the spatial organizations of AR and FOXA1 motifs in the three groups of AR bindings are different. It has been reported in LNCaP cells that 25% and 75% of ARBS are co-occupied by GATA2 and FOXA1, respectively (48). However, we did not detect the DNA motif of GATA2 from any of the three groups of ARBS, probably because of the intrinsic differences among different cell lines used.
Figure 3.

Cistrome characteristics of ARV-PBS, ARFL-PBS and Common-BS. (A) DNA motifs and frequency spectrum of gap sizes between AREfull and FOXA1 motif identified from ARFL-PBS, Common-BS and ARV-PBS loci. (B) FOXA1 ChIP-seq signal profiles for ARV-PBS and ARFL-PBS in 22Rv1 cells. 22Rv1 cells were treated with the synthetic androgen R1881 (methyltrienolone) for 24 h and samples were utilized for FOXA1 ChIP-seq analysis. FOXA1 binding signal at ARFL-PBS was significantly higher than that at ARV-PBS (P = 0.0087, two-tailed Wilcoxon rank sum test). (C and D) Human 293T cells were transfected with Pk-tagged (V5-Tag) ARFL (Pk-ARFL) or Pk-tagged AR splice variant 7 (Pk-ARV7) followed by cell lysis and Pk-tag antibody immunoprecipitation. Detected protein interactions were illustrated in both (C) and (D). (E) 22Rv1 cells transfected with siRNA targeting AR exon 7/8 to knock down ARFL or with siRNAs targeting AR-V1/V3/V4/V7 to knock down AR variants for 48 h followed by cell lysis and immunoprecipitation with AR-N20 or FOXA1 antibody. The effect of siRNA-mediated AR silencing and the protein interactions between FOXA1 and ARFL or ARVs were examined using western blots. (F and G) ARV-knockdown (F) or ARFL-knockdown 22Rv1 cells (G) were transfected with control siRNA (si-Con) or siRNA targeting ARFL or FOXA1 alone or in combination (F) or siRNAs for ARVs or FOXA1 alone or in combination (G), and then cells were seeded in 35-mm dishes (3,000 cells/dish). The ARFL knockdown 22Rv1 cells were cultured with charcoal stripped-FBS medium to further inactivate ARFL. After 10 days, cells were fixed and stained with crystal violet, and the colony number in every group was counted (n = 3). The colony numbers were normalized to the control siRNA group.
The different spatial organizations between FOXA1 and AR motifs in ARFL-PBS and ARV-PBS indicate that DNA binding of ARFL and ARV might be regulated through different mechanisms. The enrichment of the composite FOXA1-nnnn-AREhalf motif in ARFL-PBS suggests that ARFL and FOXA1 might form a protein complex and that DNA binding of ARFL is likely FOXA1 dependent. This hypothesis is supported by a large body of data gathered from androgen-sensitive prostate cancer LNCaP cells. LNCaP cells do not express ARVs, and the ARBSs identified in LNCaP cells are arguably bound by ARFL only. This notion is supported by previous findings including (i) AR-binding regions are enriched for FOXA1 motifs (49–51); (ii) more than half of the AR-binding sites in LNCaP cells overlap with FOXA1 binding sites (50,52); (iii) silencing of FOXA1 abolishes AR binding at AR target gene enhancers (52); and (iv) FOXA1 is instrumental in recruiting AR to low-affinity AREhalf motifs by opening local chromatin adjacent to FOXA1 sites (27). More importantly, the same composite motif was identified from the ‘lost AR binding program’ caused by siFOXA1 in LNCaP cells, further supporting the notion that DNA binding activity of ARFL is FOXA1-dependent (53) (Supplementary Figure S9). The enrichment of AREfull in ARV-PBS indicates that ARV homodimerization might be necessary for ARV to bind to these locations. This concept is supported by a recent report that dimerization is an essential requirement for DNA binding and transcriptional activation of constitutive ARV (54).While FOXA1 and AREfull co-exist in ARV-PBS, the presence of various gap sizes between these two motifs suggests that the dependency of ARV DNA binding on FOXA1 may vary among ARV-PBS (Figure 3A and Supplementary Figures S8B and S9C). In Common-BS, the identification of AREfull suggests that dimers (ARFL-ARFL, ARFL-ARV or ARV-ARV) might be a prerequisite for their binding to these loci. The observation that AREfull and FOXA1 motifs are separated by a 5-nt gap in Common-BS (Figure 3A) suggests that the pioneer factor FOXA1 might be needed to facilitate AR binding to these loci. However, it is still not clear why the gap size between AREfull and FOXA1 site (i.e. 5 nt) in Common-BS is different from that AREhalf and FOXA1 site (i.e. 4 nt) in ARFL-PBS.
To further assess the possible role of FOXA1 in DNA binding of ARVs, we performed FOXA1 ChIP-seq in 22Rv1 cells treated with androgen (R1881) and compared the FOXA1 binding signals in ARFL-PBS and ARV-PBS. We demonstrated that FOXA1 binding signal in ARV-PBS was significantly lower (P = 0.0087, two-tailed Wilcoxon rank sum test) compared to that in ARFL-PBS (Figure 3B and Supplementary Figure S10). These data further support the notion that chromatin binding of ARV and ARFL is likely regulated by FOXA1 through different mechanisms.
To interrogate the possible formation of protein complexes between ARFL, ARV, and FOXA1, we performed co-immunoprecipitation assays followed by western blots. We found that ARFL formed protein complexes with itself (ARFL-ARFL), ARV (ARFL-ARV) and FOXA1 (ARFL-FOXA1) (Figure 3C). However, ARV7 could only form protein complexes with itself and ARFL but not with FOXA1 (Figure 3D). This observation was also confirmed by the interaction of endogenous proteins in 22Rv1 cells (Figure 3E) and in previous reports (27,53,55,56). These data support our hypothesis that, unlike ARFL, ARV does not interact directly with FOXA1 and that its DNA binding is less likely affected by a physical interaction with FOXA1.
For the functional study, we performed cell proliferation analysis after FOXA1 knockdown in the condition of ARFL or ARV alone in 22Rv1 cells grown in regular (androgen-undepleted) media. We demonstrated that knockdown of ARVs largely decreased growth of 22Rv1 cells and this effect was much greater than that of ARFL knockdown (Supplementary Figure S11A), which is consistent with the previous report (57). In ARV-knockdown cells we demonstrated that knockdown of ARFL resulted in a moderate reduction (approximately 28%) in cell growth in FOXA1-proficient cells (Figure 3F). Importantly, the effect of ARFL knockdown was completely abolished by FOXA1 knockdown (Figure 3F), providing further support to the conclusion that FOXA1 is required for ARFL function. In contrast, in ARFL-knockdown cells we found that knockdown of ARVs resulted in a robust reduction (∼60%) in cell growth in FOXA1-proficient cells (Figure 3G). Most importantly, the effect of ARV knockdown was only partially diminished (∼31%) by FOXA1 knockdown (Figure 3G). These data suggest that different from ARFL, the pro-proliferation function of ARVs is only partially dependent on FOXA1. This result is not only in agreement with the findings reported previously by Jones et al. (58), but also consistent with our finding that FOXA1 binding motif is present in ARV-PBS (Figure 3A and B).
We also performed ChIP-qPCR to dissect the functionality of FOXA1 at the loci of ARFL-PBS and ARV-PBS. To characterize the binding sites of ARFL and ARV, we transfected Pk-tagged ARV7 into ARV-negative LNCaP cells and knocked down FOXA1 using small hairpin RNAs (shRNAs). FOXA1 knockdown had no overt effect on ARFL or ARV binding at the locus of RAP2A (ARV-PBS) (Supplementary Figure S11B, middle panel). In contrast, there was almost no ARV7 binding at the locus of IGF1 (ARFL-PBS) and the ARFL binding at this locus was obviously diminished after FOXA1 knockdown. This observation was confirmed in other ARFL-PBS loci (Supplementary Figure S11C). Furthermore, FOXA1 knockdown altered expression of IGF1 and other ARFL-PBS genes (G3BP2, ADAM9, AGAP9, RAB4A, MMP20 and LRIG3), suggesting these are FOXA1 dependent genes (Supplementary Figure S12A). Accordingly, their expression responded to androgen treatment (Supplementary Figure S12B). In contrast, we found that while FOXA1 knockdown failed to affect expression of majority of ARV-PBS genes examined, including RAP2A, E2F7, ZNF367, SLC31A2, RDH10 and RAB33A (Supplementary Figure S12C), it did decrease expression of a subset of ARV-PBS genes examined, including AUTS2, SLCO2A1, and LSG1 (Supplementary Figure S12D) and this result is consistent with the effect of FOXA1 knockdown on ARV binding at these loci (Supplementary Figure S12E). Thus, in agreement with the partial dependency of ARV-mediated cell growth on FOXA1 (Figure 3F and G), our data reveal that DNA binding of ARVs can be mediated through FOXA1-dependent or -independent mechanism, depending on the loci of ARV-PBS.
Collectively, these data indicate that although ARVs and ARFL preferentially utilized distinct cis-elements, ARFL can occupy ARV-PBS sites in cells without expression of ARVs since ARFL only require AREhalf. Notably, ARV could expel and re-occupy these loci when conditions became favorable (e.g. overexpression of ARV following ADT (18)). These findings indicate a potential role of ARVs in contributing to the persistent AR activity in CRPC where the ARFL’s activity is largely deactivated due to androgen deprivation.
Distinct epigenome landscapes at ARV-PBS, ARFL-PBS and Common-BS
It has been reported that the central nucleosome of active ARBS is dismissed, and replaced by two flanking nucleosomes marked by H3K4me2 (45). To interrogate the epigenome landscape of ARV-PBS, ARFL-PBS and Common-BS, we performed ChIP-seq experiments for histone markers including H3K4me1, H3K4me2, H3K27ac and histone H2A.Z variant in 22Rv1 cells treated with and without androgen. H3K4me1 and H3K27ac are the predominant histone modifications found at nucleosomes around enhancer regions (59,60). H3K4me2 is enriched at both promoters and distal enhancers of active genes.
Our histone ChIP-seq data revealed consistent bimodal distributions of H3K4me1, H3K4me2, H3K27ac and H2A.Z signal intensities at ARV-PBS and Common-BS in 22Rv1 cells regardless of androgen treatment (Figure 4A–E). Importantly, the status of these modifications remained unchanged even after knockdown of ARVs (Figures 1A and 4A), suggesting that chromatin is constitutively open at ARV-PBS and Common-BS. The open chromatin (or nucleosome depletion) status of ARV-PBS and Common-BS is consistent with the GC content dip at the centers of ARV-PBS and Common-BS as described above (Figure 1E, G and H). The constitutively open chromatin status is also consistent with our findings (Figure 3 and Supplementary Figure S12C–E) that there is only a small subset of ARV-PBS whose DNA binding is partially dependent on FOXA1, a pioneer factor with the capacity to open local chromatin (61). In contrast, we found that the chromatin states at ARFL-PBS depended on androgen treatment: the chromatin of ARFL-PBS was open only when cells were treated with androgen (Figure 4B–D). This result confirms that the transcription activity of ARFL is androgen dependent. Consistent with H3K4me1, H3K4me2 and H3K27ac data, we observed that H2A.Z-occupied central nucleosomes were undetectable at ARV-PBS and Common-BS in the presence or absence of androgen stimulation (Figure 4A and E). However, H2A.Z-occupied central nucleosomes were persistent at ARFL-PBS regardless of androgen treatment although the signal intensity was slightly lower after androgen treatment (Figure 4A and E). These data are in concordance with previous H2A.Z ChIP-qPCR data (45). They are also consistent with other genome-wide studies showing that H2A.Z-containing nucleosomes are intrinsically liable, and therefore likely instrumental in facilitating the binding of transcription factors (62).
Figure 4.

Epigenome landscapes of ARV-PBS, ARFL-PBS and Common-BS. (A) Heatmaps showing normalized read intensities for histone modification ChIP-seq and histone variant H2A.Z ChIP-seq at ARFL-PBS, Common-BS and ARV-PBS. Magenta and cyan indicate high and low read intensities, respectively. (B–E) H3K27ac, H3K4me2, H3K4me1 and histone variant H2A.Z ChIP-seq signal profiles for ARFL-PBS (red), Common-BS (purple) and ARV-PBS (blue) in 22Rv1 cells without (left) or with (right) R1881 (methyltrienolone) treatment. Zoom-in views of the y-axis for common-BS and ARV-PBS were displayed on the top-left corner of the panel (E).
ARV target genes associate with tumor progression and castration resistance
We next compared ARV-PBS, ARFL-PBS and Common-BS to the ARBS identified from 13 primary prostate tumors (tumor-ARBS) and seven histologically normal samples (normal-ARBS) published previously (39). We found that ARV-PBS was significantly associated with tumor-ARBS. Specifically, the overlapping coefficients (see Methods) between ARV-PBS and the tumor-ARBS were 3.79 times higher (P = 2.58 × 10−5, two-tailed Wilcoxon rank sum test) than that of ARV-PBS and the normal-ARBS. In contrast, the overlapping coefficient between ARFL-PBS and the tumor-ARBS was only 1.56 times higher (P = 0.0036) than that of ARFL-PBS and the normal-ARBS (Figure 5A). These results suggest that ARV-PBS is more tumor specific than ARFL-PBS. We also compared ARV-PBS, ARFL-PBS and Common-BS to ARBS identified from five castration resistant (CR), two treatment-responsive (TR) and three untreated prostate cancer samples reported previously (40). We found ARV-PBS, but not ARFL-PBS, was exclusively associated with ARBS identified from CR but not from TR and untreated prostate tumors (Figure 5B), suggesting ARV-PBS was strongly associated with castration resistance. It is worth noting that although ARV-PBS exhibits the highest tumor specificity in primary prostate cancers and the highest association with CR-ARBS, the binding affinity (measured by ChIP-seq signal intensity) of ARV-PBS is the lowest compared to those of ARFL-PBS and Common-BS (Supplementary Figure S13). Collectively, we found ARV-PBS were not only tumor-specific but more prone to CRPC, highlighting their potential role in prostate cancer development, progression, and castration resistance. Thus, our findings reinforce the importance of ARVs in mediating castration and therapeutic resistance (17–19,44).
Figure 5.

Analysis of ARFL-PBS, Common-PBS and ARV-PBS in prostate cancer patient specimens. (A) Overlap of ARFL-PBS, Common-PBS, and ARV-PBS with AR binding sites identified from seven normal prostate epithelium tissues (green) and 13 prostate tumor tissues (chocolate). The AR ChIP-seq data in 20 men were downloaded from GEO with accession number GSE56288. (B) Overlap of ARFL-PBS, Common-BS, and ARV-PBS with AR binding sites identified from 5 castration resistant (CR) prostate cancer tissues (deep pink), 2 treatment responsive (TR) prostate tumor tissues (cyan) and 3 untreated (UT) prostate tumor tissues (dark green). The AR ChIP-seq data in 10 men were downloaded from GEO with accession number GSE28219. The overlapping coefficient is defined as the number of the overlapped peaks divided by the smaller size of the two lists of peaks. (C–E) UCSC tracks showing AR ChIP-seq signal profiles in 7 normal (green), 13 prostate tumor tissues (chocolate), 22Rv1 (ARFL-specific KD, ARV-specific KD or control non-specific KD) and LNCaP cells at the RAP2A (C), E2F7 (D) and IGF1 loci (E). KD, knockdown. (F and G) Association of ARV7 with RAP2A (F) or E2F7 (G) expressions in 38 TCGA samples with detectable ARV7 expression. (H and I) Kaplan–Meier survival curves for RAP2A and E2F7 using 390 TCGA samples. Y-axis, the probability of recurrence free survival; X-axis, time in month. Red curves, overexpression; blue curves, low expression.
To further define ARV-regulated transcriptome, we performed RNA-seq in 22Rv1 cells. We identified a total of 7627 genes that were differentially expressed after knocking down ARVs (FDR < 0.01) (Supplementary Table S5). By overlapping with ARV-PBS identified from ChIP-Seq, we identified 213 genes activated by ARVs and 329 genes repressed by ARVs (Supplementary Table S6). The Gene Set Enrichment Analysis (GSEA) revealed that ‘cell cycle’ and ‘axon guidance’ were the top enriched pathways in the 213 ARV-activated genes and the 329 ARV-repressed genes, respectively (Supplementary Figures S14 and S15).
The RAP2A locus is an ARV-PBS (Figure 3A). This site was occupied by AR in 7 out of 13 primary tumor samples but in none of the 7 normal samples (P = 0.045, two-tailed Fisher's exact test) (Figure 5C). E2F7 was another ARV-target gene with ARV-PBS locating 122 kb downstream of its TSS. E2F7 was bound by AR in all of the tumor samples but none of the normal samples (P = 1.29 × 10−5, two-tailed Fisher's exact test) (Figure 5D). Both RT-qPCR and ChIP-qPCR analyses confirmed that E2F7 is an ARV-PBS (Supplementary Figures S4 and S6). In contrast, IGF1, a gene specifically targeted by ARFL (Figure 3B), was occupied by AR in both cancer and normal samples (Figure 5E). However, AR ChIP-seq generated from androgen-treated LNCaP cells indicated that ARFL could bind to all these three loci (Figure 5C–E, bottom). These results are not surprising because LNCaP cells do not express ARVs, but provide further support to the notion that ARFL is able to bind to ARV-PBS in cells lacking ARVs.
We next investigated whether expression of these genes is associated with ARV7 expression in clinical samples. Despite the low expression of ARV7 in primary prostate cancers, we successfully detected ARV7 expression in 38 TCGA RNA-seq samples (Supplementary Table S7). We found that the expression of RAP2A and E2F7 was significantly associated with ARV7 expression, with Pearson's correlation coefficients of 0.49 (P = 0.0019) and 0.63 (P = 2.57 × 10−5), respectively (Figure 5F and G). The expressions of these two genes were also significantly associated with ARFL (Supplementary Figure S16), probably because ARFL can still bind to ARV-PBS in primary prostate cancers similar to what we demonstrated in LNCaP cells (Figure 5C–E). In addition, survival analysis using 390 TCGA samples suggested that overexpression of RAP2A and E2F7 is significantly associated with shorter recurrence-free survival (RFS) (Figure 5H and I). These clinical data suggest that overexpression of ARV-preferentially bound genes such as E2F7 and RAP2A associates with a worse clinical outcome.
ARV activity associates with abiraterone resistance in CRPC patients
Expression of ARV7 in circulating tumor cells has been linked to resistance to enzalutamide and abiraterone treatments (19), but little is known if ARV-mediated transcription program drives therapy resistance. To explore whether ARV-targeted genes could predict potential resistance to abiraterone acetate, we performed RNA-seq analysis in 77 metastatic CRPC specimens including 54 bone and 23 lymph node biopsies acquired from patients in a clinical trial at Mayo Clinic (Supplementary Table S8) (21). All 77 patients were treated with abiraterone acetate initially, and the 3-month progression-free survival status of these patients was evaluated based on PSA level, bone and CT image scans, and symptom assessments. In total, 32 patients exhibited primary resistance (i.e. non-responders) to abiraterone acetate treatment (Supplementary Table S8). AR activity score was calculated using 20 AR target genes defined by previous studies (35,37). ARV activity score was calculated using 63 ARV-activated genes (See Methods). ARFL activity score was calculated using 33 genes (see Materials and Methods), which were specifically bound by ARFL in 22Rv1, LNCaP and C4–2 cells, but not by ARV in 22Rv1 cells (see Materials and Methods).
As we expected, ARV activity was significantly higher in the ARV7-positive group than that of the ARV7-negative group in both our CRPC cohort (n = 77, Supplementary Figure S17A) and the TCGA cohort (n = 498, Supplementary Figure S17B). The ARV7 detection in the 38 TCGA tumors was associated with increased total AR expression (Supplementary Figure S17C). ARV and ARFL activities were positively correlated with ARV and ARFL expression levels, respectively (Supplementary Figure S17D and E). However, no significant association was observed between ARV activity and the ratio of ARV to ARFL expression (Supplementary Figure S17F). We observed high pairwise correlations between AR, ARFL and ARV activities (Figure 6A–C). When comparing AR, ARFL and ARV activities between responders (n = 45) and non-responders (n = 32), we found no differences in the activity of total AR (P = 0.88, two-tailed Wilcoxon test) and ARFL (P = 0.64) between responders and non-responders (Figure 6D and E). In contrast, ARV activity was significantly higher in non-responders compared to responders (P = 0.0037), suggesting that the ARV-preferentially activated transcription program might play a causal role in the development of abiraterone resistance in CRPC patients (Figure 6F). Thus, our findings provide functional evidence for the previous report that overexpression of ARV7 in circulating tumor cells is associated with resistance to abiraterone in CRPC patients (19). Our data also indicate that overexpression of ARVs in CPRC serves as a surrogate of abiraterone resistance and that its high activity and its downstream pathways may also be viable targets to overcome drug resistance.
Figure 6.

ARV activity stratifies abiraterone-responders from non-responders of 77 metastatic CRPC patients. (A) Correlation between AR activity and ARFL activity. Purple dots indicate patients who were not responsive (non-responder, n = 32) to abiraterone acetate treatment at 12-week end point, and green dots indicate patients who were responsive (Responder, n = 45) to abiraterone acetate treatment. Purple and green dashed lines indicate linear regression lines calculated from non-responders and responders, respectively. The black solid line indicates linear regression line calculated from the whole cohort. r, Pearson correlation coefficient. (B) Correlation between AR activity and ARV activity. (C) Correlation between ARFL activity and ARV activity. (D–F) Comparing AR activity (D), ARFL activity (E) and ARV activity (F) between abiraterone responders and non-responders. (G) Associations between AR activity and cell cycle progression (CCP) activity. (H) Associations between ARFL activity and CCP activity. (I) Associations between ARV activity and CCP activity. CCP, cell cycle progression.
Since the cell cycle pathway (CCP) was significantly enriched in the ARV-activated transcription program (Supplementary Figure S14), we calculated CCP activity score using 31 cell cycle genes defined previously (36). As expected, the CCP score was significantly higher in non-responders compared to responders (P = 0.039, two-tailed Wilcoxon rank sum test) (Supplementary Figure S18). We detected positive, albeit weak, associations between total AR activity and the CCP score (r = 0.38, P = 0.00067), and between ARFL activity and the CCP score (r = 0.25, P = 0.025) (Figure 6G and H). In contrast, we detected strong positive correlation (r = 0.65, P = 2.4 × 10−10) between ARV activity and the CCP score (Figure 6I). More importantly, the correlation between ARV activity and CCP score is much higher in non-responders (r = 0.72) than that of responders (r = 0.51) (Figure 6I). It is also worth noting that there was no overlap of genes used to calculate ARV and CCP activities. Collectively, our data indicates that preferential activation of ARV-PBS targets such as cell cycle progression genes favors tumor progression and drug resistance.
RAP2A plays a key role in ARV7-mediated castration-resistant growth of prostate cancer cells in mice
Expression of RAP2A is important for the androgen-stimulated growth of LNCaP cells (63). Given that overexpression of RAP2A associates with shorter progression-free survival in the TCGA patients (Figure 5H) and the AR binding site of this gene is highly tumor specific (Figure 5C), we chose RAP2A to determine its role in ARV-mediated growth of CRPC cells. To this end, we focused on ARV7, the most abundant AR splice variant implicated in CRPC growth (16,64,65). We demonstrated that knocking down RAP2A using two independent shRNAs in 22Rv1 cells significantly (P < 0.01, two-sided t-test) inhibited androgen-independent growth (Figure 7A). Consistent with a previous report (63), the level of RAP2A is relatively low in ARV7-negative C4–2 cells (Figure 7B). Importantly, overexpression of ARV7 not only substantially elevated expression of RAP2A in C4–2 cells, but also significantly increased cell growth, and this effect was reversed by co-knockdown of RAP2A (P < 0.01, two-sided t-test) (Figure 7B). We found that tumor volumes were significantly decreased in RAP2A knockdown cells compared with cells with ARV7 overexpression alone in castrated male NOD-SCID IL-2-receptor gamma null (NSG) immune-deficient mice (Figure 7C). These data suggest that RAP2A is a key downstream mediator of ARV7 and plays a pivotal role in castration-resistant growth of prostate cancer cells in vitro and in vivo.
Figure 7.

The role of the ARV7 target gene RAP2A in the growth of CRPC cells in vitro and in mice. (A) 22Rv1 cells were infected with control shRNA (shCon), RAP2A-specific shRNA (shRAP2A#8 and shRAP2A#9) lentivirus and then seeded into 96-well plates with 3000 cells/well. Cells were harvested 5 days after infection and subjected to western blot analysis (top). Cell proliferation was measured using SRB assay (means ± S.D.; n = 6) at the indicated time points (bottom). **P < 0.01; ERK2, a loading control. (B) C4–2 cells stably expressing control empty vector (E.V.) or ARV7 were infected with lentivirus expressing control (shCon) or RAP2A-specific shRNAs. Cells were seeded to 96-well plates with 3000 cells/well. Cells were harvested 5 days after infection and subjected to western blotg analysis (top). Cell proliferation was measured using SRB assay (means ± S.D.; n = 6) at the indicated time points (bottom). ** P < 0.01; ERK2, a loading control. (C) C4–2 cells infected as in (B) were injected into the right flank of 6-month-old castrated male NSG mice. Tumor size in each individual mouse was measured every three days. At day 21, tumors were harvested and images was taken and shown (left). Tumor volumes (Y-axis) were measured as mean ± S.D. (n = 5) (right); **P < 0.01.
DISCUSSION
Increasing evidence from cell culture, xenografts in mice and patient samples suggests that expression of ARVs associates with castration resistance in prostate cancer. However, how transcription programs regulated by ARVs contribute to disease relapse remains undefined. In this study, we identified ARV-PBS from 22Rv1 CRPC cells and systematically delineated their genome, cistrome and epigenome features. Our motif analysis revealed that ARVs bind preferentially to canonical AREfull and that ARFL binds preferentially to composite FOXA1-nnnn-AREhalf. We found that ARVs bind to genomic regions where chromatin is constitutively open and central nucleosomes are depleted, as evident by the bimodal distribution of H3K4me1/2, H3K27ac, and H2A.Z ChIP-seq signals, as well as the GC content dip. We further demonstrated that there is only a small subset of ARV-PBS whose DNA binding is partially dependent on the pioneer factor FOXA1. The unique genomic and epigenomic characteristics of ARV-PBS, ARFL-PBS and Common-BS are summarized in Suplementary Table S9.
The distinct histone modification landscapes and nucleosome configurations of different forms of AR suggest that the DNA binding of ARVs and ARFL might be regulated by different molecular mechanisms. The dynamics of chromatin status at ARFL-PBS could be explained by the fact that ARFL is localized in the cytoplasm and its translocation into nucleus requires androgen activation. In contrast, ARVs, particularly ARV7, are localized in the nucleus in both prostate cancer cell lines and clinical tissues (17,66,67), and occupy open chromatin regions. Thus, our discovery of the constitutively open chromatin at ARV-PBS regardless of androgen treatment reveals a molecular basis underpinning the previous observation that ARVs are constitutively active. This also provides molecular insight into the persistent AR activity in CRPC patients after ADT.
Our data indicate that Common-BS are similar to ARV-PBS but different from ARFL-PBS in terms of genome, cistrome and epigenome characteristics. First, unlike ARFL-PBS that have relatively uniform GC content distribution, both Common-BS and ARV-PBS have a GC content dip at the center. Second, different from the composite FOXA1-nnnn-AREhalf motif identified from ARFL-PBS, independent/separated FOXA1 and AREfull motifs are found in both Common-BS and ARV-PBS. Third, while the open chromatin status at ARFL-PBS is dependent on androgen treatment, chromatin at both Common-BS and ARV-PBS is constitutively open. We demonstrated that the RAP2A locus, an ARV-PBS site, can be bound by ARFL in ARV-negative LNCaP cells, but ARVs such as ARV7 predominantly re-occupy this locus when introduced into LNCaP cells. In contrast, introducing ARV7 into LNCaP did not affect ARFL binding at the IGF1 locus, an ARFL-PBS site. This observation provides a plausible explanation as to why Common-BS are more similar to ARV-PBS in the genome, cistrome, and epigenome landscapes, and presumably, most Common-BS may actually be ARV-PBS that are temporarily bound by the remnant ARFL.
It is worth noting that a recent survey shows that the overlap of ARV unique target genes defined in six previous studies was very poor (68). Specifically, only one down-regulated gene (IGFBP3) was identified in 3 independent studies, and 14 up-regulated and 13 down-regulated genes were identified in two independent studies. When overlapping these 28 ARV-regulated genes with the ARV-PBS targets identified in the current study, we only found one gene (TLE1) is overlapped with previous studies (68). These findings are not surprising because there is little or no unique ARV-specific binding sites, but primarily are ARV-preferential binding sites (ARV-PBS). Most importantly, we provide evidence that ARV-PBS can be bound by ARFL in the absence or the presence of very low levels of ARVs.
We found that the FOXA1 binding motif and AREhalf are separated by 4 nt in ARFL-PBS, whereas FOXA1 motif and AREfull are mainly separated by 5 nt in Common-BS. AR binding to ARFL-PBS and Common-BS appears to be FOXA1-dependent. However, the molecular basis underlying their preference of different space sizes and FOXA1 dependency remains unclear. Moreover, we confirmed that expression of ARV-PBS genes examined was sensitive to ARV overexpression or knockdown. On the contrary, expression of ARFL-PBS genes was resistant to ARV overexpression or knockdown. In addition, using ChIP-qPCR, we demonstrated that ARV-PBS genes (e.g. RAP2A, E2F7, ZNF367 and SLC31A2) but not ARFL-PBS genes (IGF1, G3BP2, ADAM9, and AGAP9) can be occupied by ARV when it is expressed in ARV-negative LNCaP cells. Our data indicate ARFL still can bind to ARV-PBS if ARV are not expressed. In contrast, it seems that ARFL-PBS are very specific to ARFL. It can be postulated that AR monomer and dimer may require a different space size between ARE and FOXA1 motif at ARFL-PBS and Common-BS in order to work cooperatively with FOXA1 and/or other associated factors, and this concept warrants further investigation.
When overlapping ARV-PBS, ARFL-PBS and Common-BS with ARBS identified from 13 tumor and 7 normal prostate tissues (39), we found that sites in all three categories overlapped more significantly with tumor-ARBS than with normal-ARBS; however, ARV-PBS exhibit the highest degree of tumor specificity. Similarly, when examining the overlap of ARV-PBS, ARFL-PBS and Common-BS with ARBS identified from CR, TR and untreated patients (40), we found that ARV-PBS are almost exclusively associated with CR-ARBS. Notably, despite the fact that ARV-PBS exhibit the highest degree of tumor specificity in primary prostate cancers and the highest association with CR-ARBS, the binding affinity (measured by ChIP-seq signal intensity) of ARV-PBS is the lowest compared to those of ARFL-PBS and Common-BS. These data indicate that ARV-PBS are not only tumor-prone but also involved in castration resistance. However, it remains unclear whether those ARBS identified from clinical tissues were bound by ARV, ARFL or both. Presumably, ARBS identified from the primary tumors and normal prostate tissues are bound mainly by ARFL since ARVs are not expressed or expressed at very low levels in these tissues. In contrast, ARBS identified from CR patients might be mainly bound by ARVs since ARFL activity is presumably very low or largely deactivated due to androgen depletion. ARV- and ARFL-specific ChIP-seq data using ChIP-grade antibodies are warranted to fully understand the full spectrum and functional importance of ARV-PBS and ARFL-PBS in prostate tumors in patients. Addtionaly, expression of ARV7 is significantly associated with therapy resistance to enzalutamide and abiraterone. However, the low abundance of ARV7 makes it difficult to be detected and manipulated. For example, in the study reported by Antonarakis et al (19), only 39% enzalutamide-treated patients and 19% abiraterone-treated patients had detectable ARV7 in circulating tumor cells (CTCs). Therefore, alternative approaches in addition to ARV7 expression are warranted. The benefits of identifying ARV gene signature include: 1) Demonstrate ARV7 is not merely a biomarker, but it is also functionally important for the development of therapeutic resistance; 2) Identification of gene signature preferentially activated by ARVs could also offer new therapeutic targets.
CONCLUSIONS
In summary, we have identified a unique set of ARV-PBS with distinct genomic, cistromic and epigenomic features. Our integrated analysis of AR ChIP-seq and RNA-seq data defines a group of ARV-preferentially bound genes, activation of which not only associates with castration-resistant progression of prostate cancer, but also contributes to abiraterone therapy resistance in prostate cancer in the clinic. We also provide experimental evidence that ARV target genes such as RAP2A play a causal role in ARV7-induced castration-resistant growth of prostate tumors in vivo. Proteins encoded by aberrantly spliced mRNA could confer unique functions to promote cancer cell growth and survival (15,69). Our study reveals that the AR splice variants, although have the same DNA binding domain as the primary isoform (ARFL), could regulate a distinct class of genes that potentially drive castration and drug resistance in prostate cancer. Thus, identification of ARV-preferentially activated downstream pathways provides new targets for development of effective arsenal for CRPC therapy.
DATA AVAILABILITY
ChIP-seq and RNA-seq data have been deposited into NCBI Gene Expression Omnibus (GEO) with accession number GSE80743. RNA-seq of 77 CRPC metastasis tissues has been deposited into the database of Genotypes and Phenotypes (dbGaP) with accession number phs001141.v1.p1.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all patients who participated in the ‘PROMOTE’ study for their selfless contribution in bringing precision medicine to future advanced prostate cancer patients. We highly appreciate the support of family members as well. We gratefully acknowledge the helpful recruitment efforts of the following physicians who made patient referrals to the ‘PROMOTE’ program: Sandeep Basu (Mayo Clinic Health Systems), Daniel Burns (Mayo Clinic Health Systems), Kevin Cockerill (Mayo Clinic Health Systems), Sarah Kratz (Mayo Clinic Health Systems), Mohammad Ranginwala (Mayo Clinic Health Systems), Amrit Singh (Mayo Clinic Health Systems), Gautam Jha (University of Minnesota), Badrinath Konety (University of Minnesota), Mir Ali Khan (CGH Medical Center), Ferdinand Addo (Prairie Lakes Healthcare System), Kevin Panico (Altru Health System) and Laura Joque (Essentia Health Brainerd Clinic).
Author contributions: L.W., D.J.T., B.L., R.Z. and H.H. conceived the study. L.W. and Z.Y. performed bioinformatics analyses. Y.H., J.L. and S.H. performed the experiments. Z.Y. performed data analysis. M.K. oversaw the abiraterone-based clinical trial and M.K. and L.(Liewei)W. participated in study design and coordination in clinical sample collection. L.W. and H.H. wrote the manuscript. All authors read and approved the final manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [CA134514, CA130908, CA193239]; Department of Defense [W81XWH-14-1-0486, W81XWH-15-1-0634]; National Natural Science Foundation of China [31501052]; Mayo Clinic Center for Individualized Medicine [MC1351]; Minnesota Partnership for Biotechnology and Medical Genomics [MNP#14.37]; Prostate Cancer Foundation (2015 Prostate Cancer Foundation Challenge Award); A.T. Suharya and Ghan D.H.; Joseph and Gail Gassner; and Mayo Clinic Schulze Cancer for Novel Therapeutics in Cancer Research. Other contributing groups include the Mayo Clinic Cancer Center, the Pharmacogenomics Research Network (PGRN), and the Janssen Research & Development, LLC, which provided drug support from April 2014 onward for patient number 45 to 92, enrolled on the study and funding support for bioinformatics analysis. Funding for open access charge: National Institutes of Health [CA134514, CA130908, CA193239]; Department of Defense [W81XWH-14-1-0486, W81XWH-15-1-0634].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kirby M., Hirst C., Crawford E.D.. Characterising the castration-resistant prostate cancer population: a systematic review. Int. J. Clin. Pract. 2011; 65:1180–1192. [DOI] [PubMed] [Google Scholar]
- 2. Lonergan P.E., Tindall D.J.. Androgen receptor signaling in prostate cancer development and progression. J. Carcinogen. 2011; 10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scher H.I., Sawyers C.L.. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 2005; 23:8253–8261. [DOI] [PubMed] [Google Scholar]
- 4. Claessens F., Helsen C., Prekovic S., Van den Broeck T., Spans L., Van Poppel H., Joniau S.. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat. Rev. Urol. 2014; 11:712–716. [DOI] [PubMed] [Google Scholar]
- 5. Scher H.I., Fizazi K., Saad F., Taplin M.E., Sternberg C.N., Miller K., de Wit R., Mulders P., Chi K.N., Shore N.D. et al. . Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012; 367:1187–1197. [DOI] [PubMed] [Google Scholar]
- 6. Schrader A.J., Boegemann M., Ohlmann C.H., Schnoeller T.J., Krabbe L.M., Hajili T., Jentzmik F., Stoeckle M., Schrader M., Herrmann E. et al. . Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur. Urol. 2014; 65:30–36. [DOI] [PubMed] [Google Scholar]
- 7. Koivisto P., Kononen J., Palmberg C., Tammela T., Hyytinen E., Isola J., Trapman J., Cleutjens K., Noordzij A., Visakorpi T. et al. . Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997; 57:314–319. [PubMed] [Google Scholar]
- 8. Linja M.J., Savinainen K.J., Saramaki O.R., Tammela T.L., Vessella R.L., Visakorpi T.. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001; 61:3550–3555. [PubMed] [Google Scholar]
- 9. Visakorpi T., Hyytinen E., Koivisto P., Tanner M., Keinanen R., Palmberg C., Palotie A., Tammela T., Isola J., Kallioniemi O.P.. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995; 9:401–406. [DOI] [PubMed] [Google Scholar]
- 10. Waltering K.K., Urbanucci A., Visakorpi T.. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell. Endocrinol. 2012; 360:38–43. [DOI] [PubMed] [Google Scholar]
- 11. Cai C., Balk S.P.. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocrine-related Cancer. 2011; 18:R175–R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gregory C.W., He B., Johnson R.T., Ford O.H., Mohler J.L., French F.S., Wilson E.M.. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001; 61:4315–4319. [PubMed] [Google Scholar]
- 13. Gao H., Ouyang X., Banach-Petrosky W.A., Gerald W.L., Shen M.M., Abate-Shen C.. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:14477–14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dehm S.M., Tindall D.J.. Alternatively spliced androgen receptor variants. Endocrine-related Cancer. 2011; 18:R183–R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fackenthal J.D., Godley L.A.. Aberrant RNA splicing and its functional consequences in cancer cells. Dis. Models Mech. 2008; 1:37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo Z., Yang X., Sun F., Jiang R., Linn D.E., Chen H., Kong X., Melamed J., Tepper C.G., Kung H.J. et al. . A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009; 69:2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun S., Sprenger C.C., Vessella R.L., Haugk K., Soriano K., Mostaghel E.A., Page S.T., Coleman I.M., Nguyen H.M., Sun H. et al. . Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 2010; 120:2715–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watson P.A., Chen Y.F., Balbas M.D., Wongvipat J., Socci N.D., Viale A., Kim K., Sawyers C.L.. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:16759–16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Antonarakis E.S., Lu C., Wang H., Luber B., Nakazawa M., Roeser J.C., Chen Y., Mohammad T.A., Fedor H.L., Lotan T.L. et al. . AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014; 371:1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hornberg E., Ylitalo E.B., Crnalic S., Antti H., Stattin P., Widmark A., Bergh A., Wikstrom P.. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE. 2011; 6:e19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang L., Dehm S.M., Hillman D.W., Sicotte H., Tan W., Gormley M., Bhargava V., Jimenez R., Xie F., Yin P. et al. . A prospective genome-wide study of prostate cancer metastases reveals association of Wnt pathway activation and increased cell cycle proliferation with primary resistance to abiraterone acetate-prednisone. Ann. Oncol. 2017; doi: 10.1093/annonc/mdx689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scher H.I., Halabi S., Tannock I., Morris M., Sternberg C.N., Carducci M.A., Eisenberger M.A., Higano C., Bubley G.J., Dreicer R. et al. . Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J. Clin. Oncol. 2008; 26:1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peng J.C., Valouev A., Swigut T., Zhang J., Zhao Y., Sidow A., Wysocka J.. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell. 2009; 139:1290–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hagege H., Klous P., Braem C., Splinter E., Dekker J., Cathala G., de Laat W., Forne T.. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2007; 2:1722–1733. [DOI] [PubMed] [Google Scholar]
- 25. Miest T., Saenz D., Meehan A., Llano M., Poeschla E.M.. Intensive RNAi with lentiviral vectors in mammalian cells. Methods. 2009; 47:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He Y., Peng S., Wang J., Chen H., Cong X., Chen A., Hu M., Qin M., Wu H., Gao S. et al. . Ailanthone targets p23 to overcome MDV3100 resistance in castration-resistant prostate cancer. Nat. Commun. 2016; 7:13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin H.J., Zhao J.C., Wu L., Kim J., Yu J.. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 2014; 5:3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. . Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang L., Huang H., Dougherty G., Zhao Y., Hossain A., Kocher J.P.. Epidaurus: aggregation and integration analysis of prostate cancer epigenome. Nucleic Acids Res. 2015; 43:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L., Wang S., Li W.. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012; 28:2184–2185. [DOI] [PubMed] [Google Scholar]
- 33. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hieronymus H., Lamb J., Ross K.N., Peng X.P., Clement C., Rodina A., Nieto M., Du J., Stegmaier K., Raj S.M. et al. . Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006; 10:321–330. [DOI] [PubMed] [Google Scholar]
- 36. Cuzick J., Swanson G.P., Fisher G., Brothman A.R., Berney D.M., Reid J.E., Mesher D., Speights V.O., Stankiewicz E., Foster C.S. et al. . Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol. 2011; 12:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kumar A., Coleman I., Morrissey C., Zhang X., True L.D., Gulati R., Etzioni R., Bolouri H., Montgomery B., White T. et al. . Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016; 22:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Machanick P., Bailey T.L.. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011; 27:1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pomerantz M.M., Li F., Takeda D.Y., Lenci R., Chonkar A., Chabot M., Cejas P., Vazquez F., Cook J., Shivdasani R.A. et al. . The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015; 47:1346–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharma N.L., Massie C.E., Ramos-Montoya A., Zecchini V., Scott H.E., Lamb A.D., MacArthur S., Stark R., Warren A.Y., Mills I.G. et al. . The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013; 23:35–47. [DOI] [PubMed] [Google Scholar]
- 41. Zhao H., Sun Z., Wang J., Huang H., Kocher J.P., Wang L.. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics. 2014; 30:1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao Y., Wang L., Ren S., Blackburn P.R., McNulty M.S., Gao X., Qiao M., Vessella R.L., Kohli M., Zhang J. et al. . Activation of P-TEFb by androgen receptor-regulated enhancer RNAs in castration-resistant prostate cancer. Cell Rep. 2016; 15:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dehm S.M., Schmidt L.J., Heemers H.V., Vessella R.L., Tindall D.J.. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008; 68:5469–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. He H.H., Meyer C.A., Shin H., Bailey S.T., Wei G., Wang Q., Zhang Y., Xu K., Ni M., Lupien M. et al. . Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 2010; 42:343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peckham H.E., Thurman R.E., Fu Y., Stamatoyannopoulos J.A., Noble W.S., Struhl K., Weng Z.. Nucleosome positioning signals in genomic DNA. Genome Res. 2007; 17:1170–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yuan G.C., Liu J.S.. Genomic sequence is highly predictive of local nucleosome depletion. PLoS Comput. Biol. 2008; 4:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao J.C., Fong K.W., Jin H.J., Yang Y.A., Kim J., Yu J.. FOXA1 acts upstream of GATA2 and AR in hormonal regulation of gene expression. Oncogene. 2016; 35:4335–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jia L., Berman B.P., Jariwala U., Yan X., Cogan J.P., Walters A., Chen T., Buchanan G., Frenkel B., Coetzee G.A.. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS ONE. 2008; 3:e3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lupien M., Eeckhoute J., Meyer C.A., Wang Q., Zhang Y., Li W., Carroll J.S., Liu X.S., Brown M.. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008; 132:958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Q., Li W., Liu X.S., Carroll J.S., Janne O.A., Keeton E.K., Chinnaiyan A.M., Pienta K.J., Brown M.. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell. 2007; 27:380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Q., Li W., Zhang Y., Yuan X., Xu K., Yu J., Chen Z., Beroukhim R., Wang H., Lupien M. et al. . Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009; 138:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang D., Garcia-Bassets I., Benner C., Li W., Su X., Zhou Y., Qiu J., Liu W., Kaikkonen M.U., Ohgi K.A. et al. . Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011; 474:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan S.C., Selth L.A., Li Y., Nyquist M.D., Miao L., Bradner J.E., Raj G.V., Tilley W.D., Dehm S.M.. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015; 43:5880–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gao N., Zhang J., Rao M.A., Case T.C., Mirosevich J., Wang Y., Jin R., Gupta A., Rennie P.S., Matusik R.J.. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 2003; 17:1484–1507. [DOI] [PubMed] [Google Scholar]
- 56. Yu X., Gupta A., Wang Y., Suzuki K., Mirosevich J., Orgebin-Crist M.C., Matusik R.J.. Foxa1 and Foxa2 interact with the androgen receptor to regulate prostate and epididymal genes differentially. Ann. N. Y. Acad. Sci. 2005; 1061:77–93. [DOI] [PubMed] [Google Scholar]
- 57. Liu C., Lou W., Zhu Y., Nadiminty N., Schwartz C.T., Evans C.P., Gao A.C.. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014; 20:3198–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jones D., Wade M., Nakjang S., Chaytor L., Grey J., Robson C.N., Gaughan L.. FOXA1 regulates androgen receptor variant activity in models of castrate-resistant prostate cancer. Oncotarget. 2015; 6:29782–29794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al. . Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. et al. . Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007; 39:311–318. [DOI] [PubMed] [Google Scholar]
- 61. Cirillo L.A., Lin F.R., Cuesta I., Friedman D., Jarnik M., Zaret K.S.. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell. 2002; 9:279–289. [DOI] [PubMed] [Google Scholar]
- 62. Jin C., Zang C., Wei G., Cui K., Peng W., Zhao K., Felsenfeld G.. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009; 41:941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bigler D., Gioeli D., Conaway M.R., Weber M.J., Theodorescu D.. Rap2 regulates androgen sensitivity in human prostate cancer cells. Prostate. 2007; 67:1590–1599. [DOI] [PubMed] [Google Scholar]
- 64. Li Y., Chan S.C., Brand L.J., Hwang T.H., Silverstein K.A., Dehm S.M.. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013; 73:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yu Z., Chen S., Sowalsky A.G., Voznesensky O.S., Mostaghel E.A., Nelson P.S., Cai C., Balk S.P.. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin. Cancer Res. 2014; 20:1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bluemn E.G., Nelson P.S.. The androgen/androgen receptor axis in prostate cancer. Curr. Opin. Oncol. 2012; 24:251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hu R., Dunn T.A., Wei S., Isharwal S., Veltri R.W., Humphreys E., Han M., Partin A.W., Vessella R.L., Isaacs W.B. et al. . Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009; 69:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lu J., Van der Steen T., Tindall D.J.. Are androgen receptor variants a substitute for the full-length receptor?. Nat. Rev. Urol. 2015; 12:137–144. [DOI] [PubMed] [Google Scholar]
- 69. Skotheim R.I., Nees M.. Alternative splicing in cancer: noise, functional, or systematic?. Int. J. Biochem. Cell Biol. 2007; 39:1432–1449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq and RNA-seq data have been deposited into NCBI Gene Expression Omnibus (GEO) with accession number GSE80743. RNA-seq of 77 CRPC metastasis tissues has been deposited into the database of Genotypes and Phenotypes (dbGaP) with accession number phs001141.v1.p1.