

AAV-mediated CRISPR screens empower autochthonous functional cancer genomics in the mouse liver.
Abstract
Cancer genomics consortia have charted the landscapes of numerous human cancers. Whereas some mutations were found in classical oncogenes and tumor suppressors, others have not yet been functionally studied in vivo. To date, a comprehensive assessment of how these genes influence oncogenesis is lacking. We performed direct high-throughput in vivo mapping of functional variants in an autochthonous mouse model of cancer. Using adeno-associated viruses (AAVs) carrying a single-guide RNA (sgRNA) library targeting putative tumor suppressor genes significantly mutated in human cancers, we directly pool-mutagenized the livers of Cre-inducible CRISPR (clustered regularly interspaced short palindromic repeats)–associated protein 9 (Cas9) mice. All mice that received the AAV-mTSG library developed liver cancer and died within 4 months. We used molecular inversion probe sequencing of the sgRNA target sites to chart the mutational landscape of these tumors, revealing the functional consequence of multiple variants in driving liver tumorigenesis in immunocompetent mice. AAV-mediated autochthonous CRISPR screens provide a powerful means for mapping a provisional functional cancer genome atlas of tumor suppressors in vivo.
INTRODUCTION
Large-scale molecular profiling of patient samples has tremendously improved our understanding of human cancers (1–6). The multidimensional landscapes produced by international consortia such as The Cancer Genome Atlas (TCGA) and Catalogue of Somatic Mutations in Cancer (COSMIC), encompassing key data sets such as somatic mutations, copy number variants, epigenetic marks, mRNA and microRNA transcriptomes, as well as protein levels, have illuminated the molecular underpinnings of cancer at an unprecedented resolution and scale (7–9). Consequently, we now have an extensive catalog of genes that are recurrently mutated across different patients, both within and across histological subtypes (1, 2, 10, 11). Whereas some of these recurrently mutated genes (RMGs) are well-known tumor suppressors or oncogenes, many other RMGs still have an unclear role in cancer. Although the identification of RMGs is an important first step toward the development of new therapeutic avenues, functional evidence is required to definitively determine which genomic alterations are essential for the growth of an individual cancer (4, 6, 12, 13). A number of statistical algorithms, which aim to distinguish RMGs that are “drivers” of cancer growth from those that are mere “passengers,” have been developed (2, 12, 14, 15). These RMGs are represented by thousands of mutant variants; however, the functional roles of many of these mutants remain to be explicitly tested in controlled experimental settings.
Genetically engineered mouse models (GEMMs) have been instrumental for studying the mechanisms of oncogenes and tumor suppressors in vivo (16). Conditional or germline knockout alleles enable in vivo modeling of diverse diseases, including a wide variety of cancer types. Because the microenvironment is increasingly recognized to have a critical influence on cancer progression, GEMMs enable autochthonous modeling of cancer, that is, in the native tissue of origin (17), which provides a higher degree of precision for cancer modeling and preclinical testing. However, the production of GEMMs is time-consuming and requires a complex multistep process, involving embryonic stem cell modifications, the generation of chimeras, germline transmission, and mouse colony expansion (18). Owing to the technical difficulties of this process and the complexity of breeding with large numbers of genetic modifications, GEMMs have largely been limited to the study of only a handful of genes at a time. Thus, a systematic characterization of the hundreds of RMGs identified through tumor sequencing studies is impractical using regular GEMMs.
One promising approach for high-throughput assessment of cancer RMGs is through the use of clustered regularly interspaced short palindromic repeats (CRISPR)–mediated genome engineering in mammalian species (19–22). Hydrodynamic injection of plasmids encoding single-guide RNAs (sgRNAs) and CRISPR-associated protein 9 (Cas9) (23, 24) has been used to directly mutate several tumor suppressor genes (TSGs) in the mouse liver (23, 25). Viruses have also been used to generate loss-of-function or gain-of-function mutations in tumor suppressors and oncogenes in vivo (26–30). In addition, the CRISPR system has been used to perform genome-scale knockout screens in vitro and in transplant models (31–34). However, current screens rely on the sequencing of sgRNAs, which is an indirect measurement of the selective forces acting on specific gene perturbations.
To directly interrogate the comparative selective advantage of mutants in the tumor-initiating organs, it is necessary to first generate and then subsequently sequence pools of mutant cells within the native tumor environment. Adeno-associated viruses (AAVs) are powerful carriers of transgenes and have been shown to mediate efficient genome editing in various organs in mice (29, 35). Given that AAVs can efficiently infect the liver after intravenous injection (36), we reasoned that liver hepatocellular carcinoma (LIHC; also known as HCC), a deadly cancer with poor 5-year survival (37), would be a suitable and relevant model.
Here, we directly mapped functional cancer genome variants of tumor suppressors in the autochthonous mouse liver using massively parallel CRISPR/Cas9 genome editing. We performed a direct in vivo CRISPR screen by intravenously injecting AAV pools carrying a library of 278 sgRNAs that target a set of the most frequently mutated, known, or putative TSGs into Rosa-LSL-Cas9-EGFP knock-in mice (LSL-Cas9 mice) to generate highly complex autochthonous liver tumors, followed by direct readout of the Cas9-generated variants at predicted sgRNA cut sites using molecular inversion probe (MIP) sequencing. This combination of direct mutagenesis and pooled variant readout illuminated the mutational landscape of the tumors. Mutagenesis of individual or combinations of the top genes represented by high-frequency variants led to liver tumorigenesis in fully immunocompetent mice.
RESULTS
We first sought to compile a list of the top RMGs in the pan-cancer TCGA data sets. Applying a similar approach, as in previous studies (1–3), we identified the top 50 RMGs after excluding known oncogenes (Fig. 1A). Of the top 50 putative TSGs, 49 genes had mouse orthologs (mouse TSGs, hereafter referred to as mTSG). We also selected seven housekeeping genes to serve as controls. Then, we designed a library of sgRNAs targeting these 56 different genes, with 5 sgRNAs for each gene, totaling 280 sgRNAs (hereafter referred to as the mTSG library; Fig. 1A and table S1). For Cdkn2a and Rpl22, only four unique sgRNAs were synthesized, with the fifth sgRNA being a duplicate. The duplicates were treated as identical in downstream analyses. After oligo synthesis, we cloned the mTSG library into a base vector containing a U6 promoter driving the expression of the sgRNA cassette, as well as a Cre expression cassette (Fig. 1A). Because mutation of a single TSG rarely leads to rapid tumorigenesis in humans or autochthonous mouse models, we included an sgRNA targeting Trp53 in the base vector with the initial hypothesis that concomitant Trp53 loss of function might facilitate tumorigenesis. Sequencing of the plasmid pool revealed a complete coverage of the 278 unique sgRNAs represented in the mTSG library (table S2). After generating AAVs (serotype AAV9) containing the base vector or the mTSG library, we then intravenously injected phosphate-buffered saline (PBS), vector AAVs, or mTSG AAVs into fully immunocompetent LSL-Cas9 mice (Fig. 1A). Upon AAV infection, Cre is expressed and excises the stop codon, activating Cas9 and enhanced green fluorescent protein (EGFP) expression.
Fig. 1. AAV-CRISPR mTSG library rapidly induces robust liver tumorigenesis in LSL-Cas9 mice.
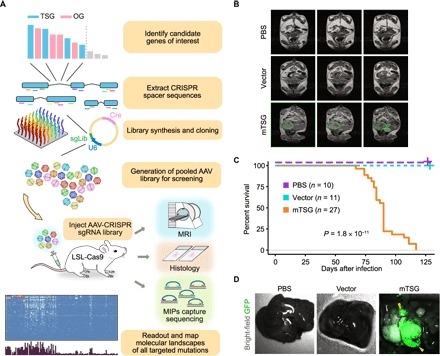
(A) Schematics of the overall design and experimental outline. First, the top MGs were identified from pan-cancer TCGA data sets. After removing known oncogenes and genes without mouse orthologs, a set of 49 most recurrently mutated putative TSGs were chosen (mTSG). Seven additional genes with housekeeping functions were spiked-in, leading to a final set of 56 genes. sgRNAs targeting these genes were then identified computationally, and five were chosen for each gene. Two hundred eighty sgRNAs plus 8 NTC sgRNAs were synthesized, and the sgRNA library (mTSG; 288 sgRNAs) was cloned into an expression vector that also contained Cre recombinase and a Trp53 sgRNA. AAVs carrying the mTSG library were produced and injected into the tail veins of LSL-Cas9 mice. After a specified time period, the mice were subjected to MRI, histology, and MIP capture sequencing for readout and deep variant analysis of molecular landscape of all targeted genes and mutations. (B) MRI of abdomens of mice treated with PBS, vector, or mTSG library. Detectable tumors are circled with green dashed lines. PBS-treated mice (n = 3) did not have any detectable tumors, whereas vector-treated mice (n = 3) occasionally had small nodules. In contrast, mTSG-treated mice (n = 4) often had multiple detectable tumors. (C) Kaplan-Meier survival curves for PBS-treated (purple, n = 10), vector-treated (teal, n = 11), and mTSG-treated (orange, n = 27) mice. No mTSG-treated mice survived longer than 4 months after treatment, whereas all PBS- and vector-treated animals survived the duration of the experiment. Statistical significance was assessed by the log-rank test (P = 1.8 × 10−11). (D) Bright-field images with GFP fluorescence overlay (green) of livers from representative PBS-, vector-, and mTSG-treated mice 4 months after treatment. Large GFP+ tumors are marked with yellow arrowheads. In contrast to PBS- or vector-treated mice, mTSG-treated mice had numerous detectable GFP+ nodules.
Live magnetic resonance imaging (MRI) of mice 3 months after treatment revealed large nodules in mTSG-treated animals (n = 4), whereas vector-treated animals (n = 3) only occasionally had small nodules and PBS-treated animals (n = 3) were devoid of detectable nodules (Fig. 1B; fig. S1, A and B; and table S3). The total tumor volume in each mouse was significantly larger in mTSG samples compared to PBS and vector samples (one-sided Mann-Whitney test, P = 0.0286 and P = 0.0286, respectively; fig. S1B). These data suggest that the AAV-CRISPR mTSG library is sufficient to induce rapid tumorigenesis in the livers of LSL-Cas9 transgenic mice.
Mice that received the AAV-CRISPR mTSG library (n = 27) did not survive more than 4 months (median survival, 90 days; 95% confidence interval, 84 to 90 days), whereas mice that were treated with PBS (n = 10) or vector control (n = 11) all survived the duration of the experiment (log-rank test, P = 1.8 × 10−11; Fig. 1C and table S4). By gross examination under a fluorescent dissecting scope, detectable GFP+ nodules were observed in mTSG-treated livers but not in PBS or vector samples (Fig. 1D and fig. S2). Notably, in mTSG-treated mice, we occasionally observed tumors that were not primarily located in the liver. Chief among these were several big abdominal tumors (BATs; n = 6), as well as a few sarcomas (n = 4) and ear tumors (n = 2), although BATs were later found to be of liver origin on the basis of histological analysis.
We analyzed endpoint histological sections from PBS-treated (n = 7), vector-treated (n = 5), and mTSG-treated mice (n = 13), sacrificed 3 to 4 months after treatment (Fig. 2A and figs. S3 and S4). No tumors were found in PBS-treated mice, whereas rare small tumors were found in vector-treated mice (total tumor area = 5.96 ± 3.27 mm2; Fig. 2B). Consistent with the MRI results, mice that received the mTSG library had significantly larger liver tumors, with the pathology of LIHC (total tumor area = 100.6 ± 47.19 mm2; one-sided Welch’s t test, P = 0.027 compared to PBS and P = 0.034 compared to vector; Fig. 2, A and B, and table S5). Because these mice were found to have multiple liver tumors, we also compared the size of each individual tumor across the three treatment groups (Fig. 2C). The mTSG-treated mice collectively had tumors that were significantly larger (26.69 ± 6.18 mm2) than those found in vector-treated animals (3.31 ± 1.55 mm2; P = 0.0003), although the latter were too small to be detected by gross examination under a GFP dissecting scope. We assessed the proliferation of liver samples from PBS-, vector-, and mTSG-treated mice by Ki67 expression and found that rapid proliferation was restricted to tumor cells (fig. S4B). In addition, we found that the tumors in mTSG-treated mice, but not in vector-treated mice, were largely positive for AE1/AE3 (pan-cytokeratin), which is a marker of LIHC (Fig. 2D and fig. S4C). These data collectively indicate that the AAV-CRISPR mTSG library directly promotes aggressive liver tumorigenesis in otherwise wild-type LSL-Cas9 mice.
Fig. 2. Histology analysis of autochthonous tumors generated by AAV-CRISPR mTSG library.
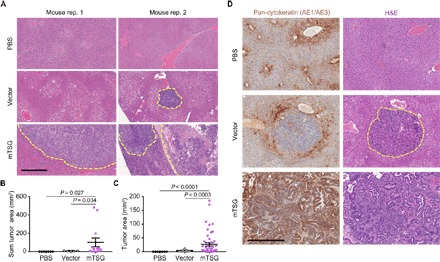
(A) Hematoxylin and eosin (H&E) staining of liver sections from mice treated with PBS (n = 7), vector (n = 5), or mTSG library (n = 13). Tumor-normal boundaries are demarcated with yellow dashed lines. No tumors were found in PBS samples, whereas small nodules were found, although rare, in vector samples. On the other hand, mTSG-treated livers were replete with tumors [statistics in (B) and (C)]. (B) Dot plot of the total tumor area per mouse (mm2) in liver sections from mice treated with PBS (black, n = 7), vector (gray, n = 5), or mTSG library (purple, n = 13). mTSG-treated mice had a significantly higher total tumor burden than PBS-treated (one-sided Welch’s t test, P = 0.027) or vector-treated mice (P = 0.034). (C) Dot plot of the individual tumor area (mm2) in liver sections from mice treated with PBS (black, n = 7), vector (gray, n = 9), or mTSG library (purple, n = 49). mTSG-treated mice had significantly larger tumors than PBS-treated (one-sided Welch’s t test, P < 0.0001) or vector-treated mice (P = 0.0003). (D) Representative immunohistochemical staining of an LIHC marker, pan-cytokeratin (AE1/AE3), from mice treated with PBS, vector, or mTSG library. The tumors from mTSG-treated samples shown revealed positive staining for AE1/AE3, consistent with LIHC pathology. Certain mTSG tumors were partially positive for cytokeratin, revealing tumor heterogeneity. The tumors from vector-treated samples were relatively small and almost always negative or slightly positive for cytokeratin. Scale bar, 0.5 mm.
To understand the molecular alterations driving the development of tumors in mTSG-treated mice, we designed MIPs to enable capture sequencing of the ±70–base pair (bp) regions surrounding the predicted cut site of each sgRNA in the mTSG library (namely, the +17 position of each 20-bp spacer sequence; Materials and Methods). As opposed to simply sequencing the sgRNA cassettes to find the relative enrichment of each sgRNA within the cell population, MIP capture sequencing enables a direct quantitative analysis of the mutations induced by the Cas9-sgRNA complex. To generate this pool of MIPs (termed mTSG-MIPs; table S6), we synthesized a total of 266 extension and ligation probes targeting 266 genomic loci with an average size of 158 ± 8 (SEM) bp, covering 278 unique sgRNA sites. Liver genomic DNA was extracted from PBS-treated (n = 8 mice), vector-treated (n = 8 mice), and mTSG-treated mice (n = 27 mice; 37 liver lobes in total). To assess the potential for AAV-CRISPR–mediated mutagenesis of other organs, we also collected DNA from all observed non-liver tumors (n = 23), as well as a wide variety of tissues (such as brain, lung, colon, spleen, and kidney) without detectable tumors under a fluorescent dissecting scope (n = 57 samples) from all three groups. We performed MIP capture sequencing on all genomic DNA samples (total n = 133; table S7). Sequencing depth of the sgRNA target regions was sufficiently powerful to detect variants at <0.01% frequency, with a mean read depth of 13,286 ± 1033 (SEM) across all MIPs after mapping to the mouse genome (table S8). Median read depth across all MIPs approximated a log-normal distribution, indicating relatively even capture of the target loci (fig. S5A). Insertions and deletions (indels) were then called across all samples to reveal detectable indel variants at each sgRNA cut site (Materials and Methods; table S9). We excluded single-nucleotide variants from the analysis because indels are the dominant variants generated by nonhomologous end-joining (NHEJ) following Cas9-mediated double-strand breaks (DSBs) in vivo (38, 39). For downstream analysis, we only considered indels that overlapped the ±3-bp flanks around each of the predicted sgRNA cut sites because Cas9 tends to create DSBs within a tight window near the predicted sgRNA cut site in mammalian cells (39). A representative example of the genotypes observed by MIP capture sequencing is shown at the Setd2 sgRNA 1 cut site for PBS-, vector-, or mTSG-treated samples (Fig. 3A), illustrating the diversity of Cas9-induced indels in mTSG-treated mice.
Fig. 3. Mutational variant-level mutational landscape of mouse AAV-mTSG–induced LIHC.
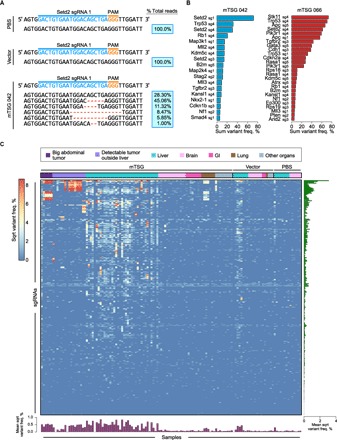
(A) Unique variants observed at the genomic region targeted by Setd2 sg1 in representative PBS-, vector-, and mTSG-treated liver samples. The percentage of total reads that correspond to each genotype is indicated on the right in the blue boxes. No indels were found in the PBS- or vector-treated samples, whereas several unique variants were identified in the mTSG-treated sample (mTSG 042). (B) Waterfall plots of two mTSG-treated samples (042 and 066) detailing sum variant frequencies in mutated sgRNA sites (MSs). Individual mice presented with distinct mutational signatures, suggesting that a wide variety of mutations induced by the mTSG library had undergone positive selection. (C) Global heat map detailing the square root of sum variant frequency across all sequenced samples (n = 133) from mTSG-treated (n = 98 samples), vector-treated (n = 21 samples), or PBS-treated mice (n = 14 samples) in terms of sgRNAs. Square root transformation was used to even out the distribution of variant frequencies for visualization. Each row represents one sgRNA, whereas each column represents one sample. Treatment conditions and tissue type are annotated at the top of the heat map: BAT (dark purple), detectable tumor outside liver (light purple), liver (teal), brain (light pink), gastrointestinal (GI; dark pink), lung (brown), and other organs (gray). Bar plots of the mean square root variant frequencies for each sgRNA (right, green bars) and each sample (bottom, purple bars) are also shown. mTSG-treated organs without visible tumors (0.148 ± 0.037 SEM) had significantly lower mean variant frequencies compared to mTSG-treated tumors and livers (BATs, 3.098 ± 0.600; two-sided unpaired t test, P < 0.0001), non-liver tumors (1.919 ± 0.338; P < 0.0001), and livers (1.451 ± 0.203; P < 0.0001). Livers and other organs from vector-treated animals (0.398 ± 0.179 and 0.054 ± 0.004, respectively) and PBS-treated animals (0.140 ± 0.067 and 0.063 ± 0.021, respectively) all had significantly lower variant frequencies than mTSG-treated livers (P < 0.0001 for all comparisons).
After collapsing each of the filtered indel calls to the closest sgRNA by summing their constituent variant frequencies (table S10), we plotted the overall spectrum of variant frequencies across all sequenced samples (Fig. 3C). We then calculated the mean variant frequency for each sgRNA and each sample (Fig. 3C, right and bottom panels, respectively). The mTSG-treated organs without visible tumors (0.148 ± 0.037 SEM) had significantly lower mean variant frequencies compared to mTSG-treated tumors and livers (BATs, 3.098 ± 0.600; unpaired t test, P < 0.0001), non-liver tumors (1.919 ± 0.338; P < 0.0001), and livers (1.451 ± 0.203; P < 0.0001). Livers and other organs from vector-treated animals (0.398 ± 0.179 and 0.054 ± 0.004, respectively) and PBS-treated animals (0.140 ± 0.067 and 0.063 ± 0.021, respectively) all had significantly lower variant frequencies than mTSG-treated livers (P < 0.0001 for all comparisons). The low background variant frequencies observed in vector- and PBS-treated samples may be due to noise that was generated during sequencing, as well as stochastic or germline mutations. Notably, the vector contains a Trp53 sgRNA that potentially contributes to higher variant frequencies in vector-treated livers due to genome instability of Trp53-deficient cells.
We identified MSs in the mTSG-treated liver samples using the false discovery rate (FDR) method as compared to PBS- and vector-treated liver samples such that no control sample would have any called MSs (Materials and Methods). Because we were most interested in analyzing dominant clones that had undergone strong positive selection in the tumor, we further required that at least 5% of the reads must have an indel in that region to call an MS (table S11). Different mTSG-treated liver samples presented with highly heterogeneous mutational signatures, indicating that a diverse array of mutations had undergone positive selection in different samples (Fig. 3B and fig. S6).
We then collapsed MSs in each sample to the gene level to find MGs within individual tumors (table S12). Analysis of all mTSG liver samples revealed a full mutational landscape of the entire cohort, unfolded as a binary mutation spectrum (Fig. 4) and a quantitative spectrum with sum allele frequencies of each gene in a tumor (fig. S7). Of the 37 mTSG-treated liver samples, 33 (89%) were found to have major indels (≥5% sum variant frequency and FDR < 0.0625; Materials and Methods) in one or more of the 56 genes in the mTSG library (average number of MGs per sample, 11.7 ± 1.53). Trp53, Setd2, Cic, and Pik3r1 were the top MGs in the cohort (mutated in 24 of 37, 18 of 37, 17 of 37, and 17 of 37 samples, respectively). Trp53 is a well-known tumor suppressor that has been found to directly induce liver tumors upon loss of function in hepatocytes (40); Setd2 is an epigenetic modifier that has been implicated in clear cell renal carcinoma (41) but is not yet functionally characterized in liver cancer; Cic is a transcriptional repressor that has been shown to be a negative regulator of epidermal growth factor receptor (EGFR) signaling (42); Pik3r1 is a modulator of phosphatidylinositol 3-kinase (PI3K) signaling, and loss-of-function mutations of this gene have been found to induce liver tumorigenesis in mice (43). In terms of cellular pathways, epigenetic modifiers and cell death/cell cycle regulators were frequently mutated, with multiple genes that were mutated in more than 20% of samples (Fig. 4). Although the importance of epigenetic modifiers in cancer is now accepted (44), direct functional testing of groups of epigenetic regulators in an autochthonous model of tumorigenesis has not yet been shown in a systems manner.
Fig. 4. Mouse gene-level mutational landscape of LIHC.
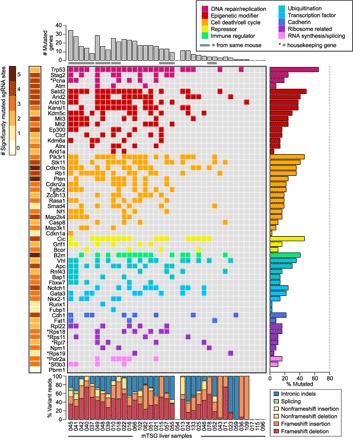
Each row in the figure corresponds to one gene in the mTSG library, whereas each column corresponds to one mTSG-treated liver sample. (Top) Bar plots of the total number of MGs identified in each mTSG-treated liver sample (n = 37). Samples originating from the same mouse are grouped together and denoted with a gray bar underneath. (Middle) Tile chart depicting the mutational landscape of primary liver samples infected with the mTSG library. Genes are grouped and colored according to their functional classifications (DNA repair/replication, epigenetic modifier, cell death/cycle, repressor, immune regulator, ubiquitination, transcription factor, cadherin, ribosome-related, and RNA synthesis/splicing), as noted in the legend in the top-right corner. Colored boxes indicate that the gene was mutated in a given sample, whereas a gray box indicates no significant mutation. Asterisks denote several preselected genes that were generally considered housekeeping genes. (Right) Bar plots of the percentage of liver samples that had a mutation in each of the genes in the mTSG library. Trp53, Setd2, Pik3r1, Cic, B2m, Vhl, Notch1, Cdh1, Rpl22, and Polr2a were the top MGs in each of the 10 functional classifications, respectively. (Bottom) Stacked bar plots describing the type of indels observed in each sample, color-coded according to the legend in the bottom-right corner. Frameshift insertions or deletions comprised the majority of variant reads (median, 59.2% across all samples). (Left) Heat map of the number of mutated sgRNA sites (0 to 5 MSs) for each gene. Multiple mutated sgRNA sites for a given gene are indicative of a strong selective force for loss-of-function mutations in that gene.
Of the genes that were mutated in at least one sample, the vast majority (91%, or 50 of 55) had multiple MSs (median, 3 MSs of 5 total sgRNAs per gene), suggesting that these genes are functional tumor suppressors (Fig. 4). ANNOVAR analysis of the indels present in the mTSG liver cohort revealed that frameshift insertions and frameshift deletions comprised the majority of total variant reads (median, 59.2% across all samples; Fig. 4 and fig. S5B), consistent with the notion that frameshift mutations are expected to cause loss of function in genes. Intronic, splice site, and non-frameshift mutations nevertheless comprised a sizeable proportion of total variant reads (Fig. 4).
To explore synergistic effects between different genes in the mTSG library, we performed co-mutation analysis. For each pair of genes, by tabulating the number of samples that were double mutant, single mutant, or double wild-type, we determined the strength of mutational co-occurrence (Materials and Methods; Fig. 5A). Of all 1540 possible gene pairs, we found that a total of 226 pairs were significantly enriched beyond what would be expected by chance (hypergeometric test, Benjamini-Hochberg–adjusted, P < 0.05), with highly significant pairs such as Cdkn2a + Pten (co-occurrence rate = 7/10 = 70%; hypergeometric test, P = 2.63 × 10−5), Cdkn2a + Rasa1 (co-occurrence rate = 6/9 = 67%; P = 7.96 × 10−5), Arid2 + Cdkn1b (co-occurrence rate = 11/17 = 65%; P = 9.13 × 10−5), and B2m + Kansl1 (co-occurrence rate = 11/18 = 61%; P = 3.6 × 10−4; Fig. 5, B and C, and table S13). Loss-of-function mutations in both genes of these combinations might synergistically enhance tumor progression.
Fig. 5. Co-mutation analysis of liver samples from mTSG-treated mice reveals potential synergistic combinations of driver mutations.
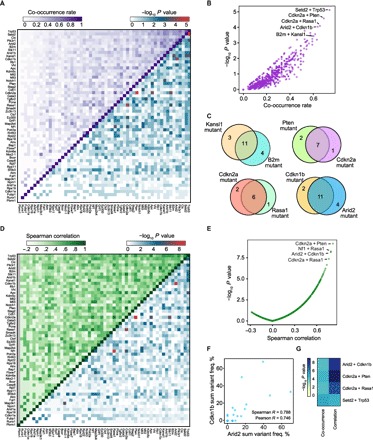
(A) Upper-left triangle: Heat map of the co-occurrence rates for each gene pair. To calculate co-occurrence rates, the intersection is defined as the number of double-mutant samples, and the union is defined as the number of samples with a mutation in either of the two genes. The co-occurrence rate was then calculated as the intersection divided by the union. Lower-right triangle: Heat map of −log10 P values by hypergeometric test to evaluate whether specific pairs of genes are statistically significantly co-mutated. (B) Scatterplot of the co-occurrence rates for each gene pair, plotted against −log10 P values by hypergeometric test. Highly co-occurring pairs include Cdkn2a + Pten (co-occurrence rate = 7/10 = 70%; hypergeometric test, P = 2.63 × 10−5), Cdkn2a + Rasa1 (co-occurrence rate = 6/9 = 67%; P = 7.96 × 10−5), Arid2 + Cdkn1b (co-occurrence rate = 11/17 = 65%; P = 9.13 × 10−5), and Kansl1 + B2m (co-occurrence rate = 11/18 = 61%; P = 3.6 × 10−4). (C) Venn diagrams showing the strong co-occurrence of mutations in B2m + Kansl1 (top left), Cdkn2a + Pten (top right), Cdkn2a + Rasa1 (bottom left), and Arid2 + Cdkn1b (bottom right). Numbers shown correspond to the number of mTSG-treated liver samples with a given mutation profile. (D) Upper-left triangle: Heat map of the pairwise Spearman correlation of sum % variant frequency for each gene, summed across sgRNAs. Lower-right triangle: Heat map of −log10 P values by t distribution to evaluate the statistical significance of the pairwise correlations. (E) Scatterplot of pairwise Spearman correlations plotted against −log10 P values. The top four correlated pairs were Cdkn2a + Pten (R = 0.817, P = 6.97 × 10−10), Nf1 + Rasa1 (R = 0.791, P = 5.86 × 10−9), Arid2 + Cdkn1b (R = 0.788, P = 7.16 × 10−9), and Cdkn2a + Rasa1 (R = 0.761, P = 4.45 × 10−8). (F) Scatterplot comparing sum level % variant frequency for Arid2 versus Cdkn1b across all mTSG-treated liver samples. Spearman and Pearson correlation coefficients are noted on the plot (Spearman R = 0.788; Pearson R = 0.746). (G) Heat map of the P values associated with the top mutation pairs that were found to be statistically significant (Benjamini-Hochberg–adjusted, P < 0.05) in both co-occurrence (left) and correlation (right) analyses.
We then investigated whether genes correlated with each other in terms of mutation frequencies within individual tumors. Because the variant frequency is a metric for the positive selection that acts on a given mutation, genes whose variant frequencies are highly correlated across samples could also be synergistic in driving tumorigenesis. A caveat is that some passenger mutations could be hitchhiking on strong drivers within a given tumor; however, the probability of finding a co-occurring passenger-driver mutation pair is vanishingly small across increasing numbers of mice. We calculated the total variant frequency for each gene by summing all the values from all five sgRNAs, used these summed gene-level variant frequencies across each sample to calculate the Spearman correlation between all 1540 possible gene pairs, and assessed whether the correlations were statistically significant (Materials and Methods; Fig. 5D and table S14). A total of 128 gene pairs were significantly correlated (Spearman correlation, Benjamini-Hochberg–adjusted, P < 0.05). The top four correlated pairs were Cdkn2a + Pten (Spearman R = 0.817, P = 6.97 × 10−10), Nf1 + Rasa1 (R = 0.791, P = 5.86 × 10−9), Arid2 + Cdkn1b (R = 0.788, P = 7.16 × 10−9), and Cdkn2a + Rasa1 (R = 0.761, P = 4.45 × 10−8; Fig. 5, E and F). We performed the same analysis using Pearson correlation, finding extensive similarities in the identified pairs (fig. S8, A and B, and table S14). Because the base vector contained a Trp53 sgRNA, we also performed the co-mutation analyses excluding all pairs involving Trp53 (fig. S8, C and D). The correlation analysis thus revealed a number of highly significant associations in specific pairs of genes. Four gene pairs were statistically significant at Benjamini-Hochberg–adjusted P < 0.05 in both the co-occurrence and correlation analyses (Fig. 5G). One of the top gene pairs was Arid2 + Cdkn1b, representing a previously unreported synergistic interaction between an epigenetic regulator and a cell cycle regulator.
To examine the mutational landscape of the liver tumors induced by the AAV-CRISPR mTSG library at a finer resolution, we reframed our analysis to the level of specific indel variants. Across all 37 mTSG-treated liver samples, we identified 593 unique variants that had a variant frequency of ≥1% in at least one sample (table S15). The majority of these variants (80.94%) were deletions rather than insertions (table S15). Hierarchical clustering of the variant-level data across all mTSG-treated liver samples revealed the existence of sample-specific variants: 70.15% (416 of 593) of the variants were sample-specific (private variants), whereas 29.85% (177 of 593) of the variants were found across multiple samples (shared variants; fig. S9). Shared variants could originate from convergent processes of NHEJ following Cas9/sgRNA-mediated DSBs, leading to the same indel pattern. Alternatively, shared variants in different liver lobes from the same mouse could also arise from clonal expansion or metastasis.
To deepen our understanding of the clonal architecture in this genetically complex, highly heterogeneous yet fully gene-targeted autochthonous tumor model, we focused on a single mTSG-treated mouse that had presented with multiple visible tumors in several liver lobes, five of which had been harvested for MIP capture sequencing (Fig. 6A). Analysis of the sgRNA-level variant frequencies in the five lobes revealed strong pairwise correlations between multiple lobes (Fig. 6B and table S16). For instance, lobes 3 and 5 were significantly correlated [Spearman rank correlation (R) = 0.700, P < 2.2 × 10−16]. Lobes 2 and 4 were also significantly correlated, though to a lesser extent (R = 0.207, P = 5.08 × 10−4). Furthermore, lobes 1, 2, and 4 were also significantly correlated with lobe 5 (R = 0.248, P = 2.99 × 10−5; R = 0.146, P = 0.0146; and R = 0.243, P = 4.31 × 10−5, respectively). The interlobe correlations are suggestive of similar variant compositions within these liver lobes.
Fig. 6. Systematic dissection of variant compositions across individual liver lobes within a single mTSG-treated mouse reveals substantial clonal mixture between lobes.
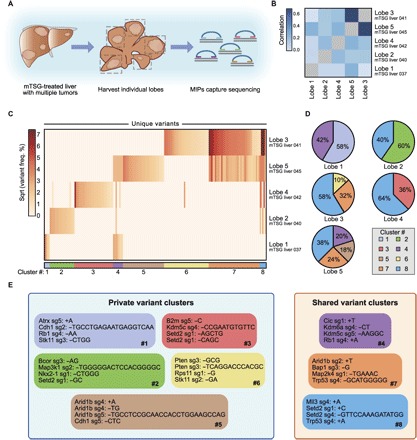
(A) Schematic of the experimental workflow for analysis of multiple liver lobes (n = 5) from a single mTSG-treated mouse. (B) Heat map of Spearman rank correlation coefficients among five liver samples from a single mTSG-treated mouse, calculated on the basis of variant frequency for all unique variants present within the five samples. Notably, lobes 1 to 4 are all significantly correlated with lobe 5, with lobe 3 having the strongest correlation to lobe 5. (C) Heat map of variant frequencies for each unique variant identified across the five individual liver lobes after square root transformation. Rows correspond to different liver lobes, whereas columns denote unique variants. Eight clusters were identified based on binary mutation calls and are indicated on the bottom of the heat map. (D) Pie charts depicting the proportional contribution of each cluster to the five liver lobes. In order for a cluster to be considered, at least half of the variants within the cluster must be present in that particular sample. For each lobe, variant frequencies within a cluster were averaged and converted to relative proportions, as shown in the pie charts. The pie charts accurately recapture the correlation analysis in (B) while additionally providing quantitative insight into the shared variants between the five liver lobes. (E) Each box corresponds to one cluster, color-coded as in (C) and (D), showing the top four variants in each cluster. On the basis of whether a variant cluster was present in multiple liver lobes, each box is also classified as either a private or a shared variant cluster. Clusters 1, 2, 3, 5, and 6 are largely unique to individual lobes (private variant clusters), whereas clusters 4, 7, and 8 are present in multiple lobes (shared variant clusters). Cluster 8 was found in four of five lobes and is characterized by mutations in Mll3, Setd2, and Trp53.
To delineate any potential clonal mixtures among the five lobes, we next examined the unique variant patterns across these samples. We identified 178 unique variants (≥1% variant frequency threshold) represented within the five liver lobes (table S17). Using binary variant calls (that is, whether a given variant is present or absent in a sample), we clustered these 178 variants into eight groups (Fig. 6C and table S17). Variants in clusters 1, 2, 3, 5, and 6 were specific to a single lobe (private variant clusters), whereas variants in clusters 4, 7, and 8 were present across multiple lobes (shared variant clusters). By averaging the variant frequencies within each cluster for a given sample, we then analyzed the relative contribution of each cluster to the overall composition of the five lobes (Fig. 6, D and E). The degree of correlation between lobes (Fig. 6B) is echoed by their degree of variant cluster sharing (lobe 1 shares cluster 4 with lobe 5, lobes 2 and 4 share variant cluster 8 with lobe 5, and lobe 3 shares clusters 7 and 8 with lobe 5; Fig. 6, D and E). The presence of cluster 8 in four of five lobes is especially notable, because it comprised a large percentage of the mutational burden in these four lobes (Fig. 6, D and E). Cluster 8 was defined by mutations in Mll3 (also known as Kmt2c), Setd2, and Trp53 (Fig. 6E). Variant-level analyses therefore recaptured the pairwise correlations identified on the sgRNA level, suggesting clonal mixture between individual liver lobes within a single mouse.
We individually tested the functional roles of mutations in several of the top genes in a Trp53-sensitized background. We chose gene pairs based on their ranking in the screen, potential biological function, and literature. We generated an AAV vector for liver-specific CRISPR knockout that expressed Cre recombinase under a TBG promoter, together with a Trp53-targeting sgRNA cassette and an open (GeneX-targeting) sgRNA cassette (fig. S10A). The vector also contained a firefly luciferase gene (FLuc) co-cistronic with Cre under the TBG promoter for live imaging of tumorigenesis in mice. We cloned either a nontargeting control (NTC) sgRNA (thus only mutating Trp53) or a top candidate GeneX-targeting sgRNA (GTS; thus mutating both GeneX and Trp53) into the second sgRNA expression cassette. After AAV packaging, we injected NTC + Trp53 or GTS + Trp53 AAVs into LSL-Cas9 mice (fig. S10A). We assessed the growth of potential liver tumors by monitoring their luciferase activities using a bioluminescent in vivo imaging system (IVIS; fig. S10B). Compared to mice treated with NTC AAVs (n = 8), sgRNAs targeting multiple candidates identified in the screen, including Cic (n = 4), Pik3r1 (n = 7), Pten (n = 4), Stk11 (n = 8), Arid2 (n = 3), and Kdm5c (n = 3), had significantly stronger luciferase activity (two-sided unpaired t test, P < 0.05 for all groups; fig. S10, B to D), suggesting that knocking out these genes accelerated liver tumorigenesis at high penetrance in a Trp53-sensitized background. Double knockouts such as Pik3r1 + Pten (n = 3) and Arid2 + Kdm5c (n = 4) also had significantly stronger luciferase activity compared to NTC (two-sided unpaired t test, P < 0.001) but not significant compared to respective single knockouts (fig. S10, C and D), suggesting that these genes are strong drivers alone but do not have synergistic effect with each other. B2m + Kansl1 is one of the top co-occurring gene pairs identified in the screen (co-occurrence rate = 11/18 = 61%, P = 3.6 × 10−4). Whereas LSL-Cas9 mice injected with AAVs for individual knockout of B2m or Kansl1 alone did not show significantly stronger luminescence intensities compared to the NTC group, AAVs targeting the B2m + Kansl1 combination showed significantly higher luminescence intensities as compared to NTC (two-sided unpaired t test, P < 0.01), B2m alone (P < 0.01), and Kansl1 alone (P < 0.05; fig. S10, C and D). These results suggested that combinatorial knockout of B2m and Kansl1 had a synergistic effect in accelerating liver tumor development, whereas the single knockouts of B2m or Kansl1 were not sufficient to induce liver tumorigenesis in a Trp53-sensitized background. In summary, the single and combinatorial AAV-CRISPR knockout experiments further confirmed the phenotypes of several top-ranked genes and co-occurring gene pairs in liver tumorigenesis. Our study demonstrates a powerful strategy for quantitatively mapping functional suppressors in the cancer genome and their synergistic relationships directly in vivo in a fully immunocompetent setting.
DISCUSSION
We developed an approach for direct in vivo CRISPR screens to directly map functional variants of tumor suppressors in mice in an autochthonous cancer model. Using an AAV library carrying 278 different CRISPR sgRNAs, we generated hundreds of variants in the top 49 known or putative TSGs and assayed their ability to promote tumorigenesis in the mouse liver upon loss of function by Cas9 mutagenesis. Capture sequencing of the resultant liver tumors revealed a heterogeneous mutational landscape, indicating that several of the genes in the mTSG library function as tumor suppressors. Notably, our experiments were conducted in immunocompetent mice; considering the critical role of the tumor microenvironment and associated immune context in human disease progression, the use of autochthonous tumor models in immunocompetent animals is especially important to enhance clinical relevance (17).
Compared to conventional sgRNA sequencing, MIP capture sequencing enabled direct, multiplexed analysis of the indels induced by Cas9 mutagenesis. By refocusing our analysis to the variant level, we systematically dissected the variant compositions across multiple liver lobes from a single mouse, uncovering evidence of clonal mixture between lobes. We thus demonstrate that massively parallel autochthonous in vivo CRISPR screens can be achieved through the use of pooled AAVs in conjunction with MIP capture sequencing in the mouse liver. To date, library-scale CRISPR screens have largely been demonstrated in in vitro or cellular transplant studies; however, it has remained a challenge to perform such screens in a direct in vivo manner. Because AAVs do not typically integrate into the genome, direct sgRNA cassette readout is rendered infeasible. Instead, we read out a high-throughput in vivo CRISPR experiment by targeted capture sequencing, demonstrating a new approach for in vivo CRISPR screens. Whereas traditional sgRNA sequencing can only provide information about the relative abundances of each sgRNA, capture sequencing enables high-resolution analysis of individual indel variants for clonal analysis of tumor heterogeneity.
Co-mutation analysis identified several pairs of significantly co-occurring mutations. A binary metric, such as mutation occurrence (that is, number of mice or patient with a gene mutated), tends to reflect the prevalence of a driver. On the other hand, a quantitative metric such as allele frequency of a mutant or a gene in a complex tumor tends to reflect the strength of in vivo selection, where dominant mutants with strong effects may outcompete other mutant cells in the same mouse. To gain a more comprehensive picture of both processes, we had leveraged both binary metrics and allele frequency as surrogate statistics (tables S10 and S11), each of which has distinct advantages and limitations. We picked a top co-occurring pair (B2m + Kansl1), two moderate co-occurring pairs (Pik3r1 + Pten and Pik3r1 + Stk11), and a nonsignificantly co-occurring pair (Arid2 + Kdm5c). We then knocked out these genes, individually and combinatorially, to study their independent and synergistic roles in liver tumorigenesis along with Trp53 loss in immunocompetent mice. According to the IVIS data (fig. S10), combinatorial knockout of the top co-occurring pair B2m + Kansl1 led to significantly faster liver tumorigenesis than knocking out either B2m or Kansl1 individually, whereas the combinatorial knockout of the gene pair Arid2 + Kdm5c that was not found to significantly co-occur did not show any synergistic effect. The validation results were consistent with our analysis of the screening data, indicating that the in vivo AAV-CRISPR screen is an effective approach to identify potent TSGs and potential synergistic drivers of liver tumorigenesis in immunocompetent mice.
In the pooled AAV-CRISPR tumorigenesis system, some passenger mutations could be hitchhiking on advantageous mutated clones, thus showing relatively high mutation frequencies. Nevertheless, because neutral mutations hitchhike by chance, the probability for a passenger mutation to be recurrently co-mutated with a strong driver across multiple samples is low compared to co-mutation of two true drivers. Here, we adopted both binary mutant calls and allele frequency analysis in exploring potentially synergistic interactions to minimize the possibility of identifying selectively neutral passenger-driver pairs.
As an approximation to the clonality of these tumors, we also calculated the number of major clusters (fig. S11), in which each major cluster has one or more mutations at similar frequencies as compared to other mutants. From this analysis, we found that 6 of 30 mTSG livers had single-cluster tumors, with the majority (24 of 30) being composed of multiple clusters (fig. S11). Given the nature of pooled mutagenesis, the detected mutations comprising co-occurring gene pairs can be either in the same clone or in different clones within the same tumor. On the basis of allele frequency analysis, we would expect that most of the significantly correlated gene pairs had coevolved in the same clone. To definitively distinguish the monoclonal or multiclonal synergistic effects of gene pairs in vitro and in vivo, double knockouts must be introduced simultaneously into the same cell using a dual-sgRNA expression system and compare them to single knockout cells.
The AAV-CRISPR screen approach can potentially be extended to identify genetic factors with a significant impact on various cancer types and other human diseases. Our strategy for selecting genes to target in the mTSG library was based on pan-cancer TCGA data sets, with an initial aim of identifying genes that are more likely to function as tumor suppressors in a wide variety of tissues such that the same AAV-CRISPR mTSG library could potentially be used in other organs. We recently applied this approach to map functional tumor suppressors in glioblastoma (45), finding that the strongest single and co-occurring driver mutations differ between mouse liver and brain. A technical distinction is that the present study used MIPs for reading out variants, whereas the glioblastoma study used Roche probes. MIPs provide more flexibility and are more cost-efficient as compared to Roche probes, making it simple for utilization of the AAV-CRISPR–MIP screening approach. In light of the previous success of in vivo transposon-based, short hairpin RNA (shRNA), or lentiviral screens in other cancers (46–49), we anticipate that our approach (AAV-CRISPR mutagenesis followed by MIP capture sequencing) can be readily expanded to other organ systems, potentially enabling the construction of a multiorgan functional variant mapping of tumor suppressors.
One limitation of this study is the exclusion of oncogenes. Because advantageous alterations in proto-oncogenes are generally thought to occur through several different mechanisms, such as mutations to specific amino acid residues, increased expression by transcriptional amplification, or copy number amplification (3), modeling proto-oncogenes in a massively parallel manner requires more sophisticated methods. Several in vitro gene activation screens using CRISPR have been described, opening the possibility for overexpression screens of proto-oncogenes (50, 51). CRISPR has also recently been engineered to mutate specific DNA nucleotides (52, 53). Adapting these new tools for use in vivo would allow high-throughput oncogene screens, thereby enabling functional variant mapping of oncogenes. Although we focused on a set of highly mutated pan-cancer tumor suppressors in mouse liver in this study, given the immense programmability of CRISPR-mediated genome editing, it is feasible to apply this AAV-CRISPR screen approach to target different sets of genes or noncoding elements, potentially at genome scale, to functionally assess phenotypes in an unbiased fashion. The versatility of this new platform provides a powerful means for mapping a provisional functional cancer genome atlas of tumor suppressors, oncogenes, and other types of genetic events of tumor evolution, in isolation or as combinations and larger pools, in virtually any cancer type. Application of this approach in conjunction with therapeutic agents will enable precision preclinical testing for rapid identification of effective compounds against specific mutant genotypes, paving new ways for cancer target discovery.
MATERIALS AND METHODS
Design, synthesis, and cloning of the mTSG library
Pan-cancer mutation data from 15 cancer types were retrieved from the TCGA portal via cBioPortal (54) and Synapse (www.synapse.org). RMGs were calculated similarly to previously described methods (1–3, 15). Known oncogenes were excluded, and only known or predicted TSGs were included. The top 50 TSGs were chosen, and their mouse homologs (mTSG) were retrieved from mouse genome informatics (www.informatics.jax.org). A total of 49 mTSGs were found. A total of seven known housekeeping genes were chosen as internal controls. sgRNAs against these 56 genes were designed using a previously described method (31, 32) with our custom scripts. Five sgRNAs were chosen for each gene, plus eight NTCs, making a total of 288 sgRNAs in the mTSG library. NTCs do not target any predicted sites in the genome; thus, they were not included in subsequent MIPs analysis. Notably, two sgRNA pairs happened to be identical by design, namely, Rpl22_sg4/sg5 and Cdkn2a_sg2/sg5. These sgRNAs were treated as the same in subsequent analyses.
Design, cloning of AAV-CRISPR vectors, and mTSG sgRNA library cloning
AAV-CRISPR vectors were designed to express Cre recombinase for the induction of Cas9 expression using constitutive or conditional promoters when delivered to LSL-Cas9 mice (29). Two sgRNA cassettes were built in these vectors, one encoding an sgRNA targeting Trp53, with the other being an open sgRNA cassette (double Sap I sites for sgRNA cloning). The vector was generated by gBlocks Gene Fragment synthesis [Integrated DNA Technologies (IDT)] followed by Gibson assembly [New England Biolabs (NEB)]. The mTSG library was generated by oligo synthesis, pooled, and cloned into the double Sap I sites of the AAV-CRISPR vectors. Library cloning was done at over 100× coverage to ensure proper representation. Representation of plasmid libraries was read out by barcoded Illumina sequencing as described previously (33) with customized primers.
AAV-mTSG viral library production
The AAV-CRISPR plasmid vector (AAV-vector) and library (AAV-mTSG) were subjected to AAV9 production and chemical purification. Briefly, human embryonic kidney (HEK) 293FT cells (Thermo Fisher Scientific) were transiently transfected with AAV-vector or AAV-mTSG, AAV9 serotype plasmid, and pDF6 using polyethyleneimine. Each replicate consisted of five 80% confluent HEK 293FT cells in 15-cm tissue culture dishes or T-175 flasks (Corning). Multiple replicates were pooled to enhance production yield. Approximately 72 hours after transfection, cells were dislodged and transferred to a conical tube in sterile PBS. Pure chloroform (1/10 volume) was added, and the mixture was incubated at 37°C and vigorously shaken for 1 hour. NaCl was added to a final concentration of 1 M, and the mixture was shaken until dissolved and then pelleted at 20,000g at 4°C for 15 min. The aqueous layer was discarded, whereas the chloroform layer was transferred to another tube. PEG8000 was added to 10% (w/v) and shaken until dissolved. The mixture was incubated at 4°C for 1 hour and then spun at 20,000g at 4°C for 15 min. The supernatant was discarded, and the pellet was resuspended in Dulbecco’s PBS plus MgCl2 and treated with Benzonase (Sigma) and incubated at 37°C for 30 min. Chloroform (1:1 volume) was then added, shaken, and spun down at 12,000g at 4°C for 15 min. The aqueous layer was isolated and passed through a 100-kDa molecular weight cutoff (Millipore). The concentrated solution was washed with PBS, and the filtration process was repeated. Genomic copy number (GC) of AAV was titrated by real-time quantitative polymerase chain reaction (qPCR) using custom TaqMan assays (Thermo Fisher Scientific) targeted to Cre.
Animal work statements
All animal work was performed under the guidelines of Yale University Institutional Animal Care and Use Committee and Massachusetts Institute of Technology Committee for Animal Care, with approved protocols (Chen-2015-20068 and Sharp-0914-091-17), and were consistent with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) (institutional animal welfare assurance no. A-3125-01).
Intravenous virus injection for liver transduction
Conditional LSL-Cas9 knock-in mice were bred in a mixed 129/C57BL/6 background. Mixed-gender (randomized males and females) 8- to 14-week-old mice were used in experiments. Mice were maintained and bred in standard individualized cages with a maximum of five mice per cage, with regular room temperature (65° to 75°F, or 18° to 23°C), 40 to 60% humidity, and a 12-hour–12-hour light-dark cycle. To intravenously inject AAVs, mice were restrained in a rodent restrainer (Braintree Scientific), their tails were dilated using a heat lamp or warm water and sterilized by 70% ethanol, and 200 μl of concentrated AAV (~1 × 1010 GC/μl, 2 × 1012 GC per mouse) was injected into the tail vein of each mouse. One hundred percent of the mice survived the procedure. Animals that failed injections (<70% of total volume injected into the tail vein after multiple attempts) were excluded from the study. No specific methods were implemented to choose sample sizes.
Magnetic resonance imaging
MRI was performed using standard imaging protocol with MRI machines (Varian 7T/310/ASR-whole mouse MRI system or Bruker 9.4T horizontal small animal systems). Briefly, animals were anesthetized using isoflurane and positioned in the imaging bed with a nose cone providing constant isoflurane. A total of 20 to 30 frontal views were acquired for each mouse using a custom setting: echo time (TE) = 20, repetition time (TR) = 2000, slicing = 1.0 mm. Raw image stacks were processed using OsiriX or Slicer tools. Rendering and quantification were performed using Slicer (www.slicer.org). Tumor size was calculated with the following formula: volume (mm3) = 1/6 × 3.14 × length (mm) × height (mm) × depth (mm). Statistical significance was assessed by nonparametric Mann-Whitney test, as sample numbers and sample distributions varied across treatment conditions.
Survival analysis
We observed that LSL-Cas9 mice receiving AAV-mTSG intravenous injections rapidly deteriorated in their body condition scores (BSCs) (due to tumor development in most cases). Mice with BSC < 2 were euthanized, and the euthanasia date was recorded as the last survival date. Occasionally, mice bearing tumors died unexpectedly early, and the date of death was recorded as the last survival date. Cohorts of mice intravenously injected with PBS, AAV-vector, or AAV-mTSG virus were monitored for their survival. Survival analysis was analyzed by standard Kaplan-Meier method, using the survival and survminer R packages. Differences among the three treatment groups were assessed by the log-rank test. Notably, several AAV-vector– or PBS-injected mice were sacrificed at time points earlier than the last day of survival analysis (at times when certain AAV-mTSG mice were found dead or euthanized due to poor body conditions) to provide time-matched histology, although those mice presented with good body condition (BSC ≥ 4). Mice euthanized early in a healthy state were excluded from calculation of survival percentages.
Mouse organ dissection, fluorescent imaging, and histology
Mice were sacrificed by carbon dioxide asphyxiation or deep anesthesia with isoflurane followed by cervical dislocation. Mouse livers and other organs were manually dissected and examined under a fluorescent stereoscope (Zeiss, Olympus, or Leica). Bright-field and/or GFP fluorescent images were taken for the dissected organs and overlaid using ImageJ. Organs were then fixed in 4% formaldehyde or 10% formalin for 48 to 96 hours, embedded in paraffin, sectioned at 6 μm, and stained with H&E for pathology. For tumor size quantification, H&E slides were scanned using an Aperio digital slide scanner (Leica). Tumors were manually outlined as region of interest (ROI) and subsequently quantified using ImageScope (Leica). Statistical significance was assessed by Welch’s t test, given the unequal sample numbers and variances for each treatment condition.
Mouse tissue collection for molecular biology
Mouse livers and various other organs (Supplementary Materials) were dissected and collected manually. For molecular biology, tissues were flash-frozen with liquid nitrogen and ground in 24-well polyethylene vials with metal beads in a GenoGrinder machine (OPS Diagnostics). Homogenized tissues were used for DNA/RNA/protein extractions using standard molecular biology protocols.
Genomic DNA extraction from cells and mouse tissues
For genomic DNA extraction, 50 to 200 mg of frozen ground tissue were resuspended in 6 ml of lysis buffer [50 mM tris, 50 mM EDTA, 1% SDS (pH 8)] in a 15-ml conical tube, and 30 μl of proteinase K (20 mg/ml; Qiagen) was added to the tissue/cell sample and incubated at 55°C overnight. The next day, 30 μl of ribonuclease A (10 mg/ml; Qiagen) was added to the lysed sample, which was then inverted 25 times and incubated at 37°C for 30 min. Samples were cooled on ice before the addition of 2 ml of prechilled 7.5 M ammonium acetate (Sigma) to precipitate proteins. The samples were vortexed at high speed for >20 s and then centrifuged at ≥4000g for 10 min. A tight pellet was visible in each tube, and the supernatant was carefully decanted into a new 15-ml conical tube. Then, 6 ml of 99% isopropanol was added to the tube, inverted 50 times, and centrifuged at ≥4000g for 10 min. Genomic DNA was visible as a small white pellet in each tube. The supernatant was discarded, 6 ml of freshly prepared 70% ethanol was added, and the tube was inverted 10 times and then centrifuged at ≥4000g for 5 min. The supernatant was discarded, and the remaining ethanol was removed using a P200 pipette. After air drying for 10 to 30 min, the DNA changed appearance from a milky white pellet to slightly translucent. Then, ~500 μl of double-distilled water was added, and the tube was incubated at 65°C for 1 hour and at room temperature overnight to fully resuspend the DNA. The next day, the genomic DNA samples were vortexed briefly. The concentration of genomic DNA was measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific).
MIP design and synthesis
MIPs were designed according to previously published protocols (55, 56). Briefly, the 70 bp flanking the predicted cut site of each sgRNA of all 278 unique sgRNA were chosen as targeting regions, and the bed file with these coordinates was used as an input. Because Trp53 sg4 targets a similar region as the Trp53 sgRNA within the base vector, the same MIP was used to sequence both of these loci.
These coordinates contained overlapping regions, which were subsequently merged into 173 unique regions. Each probe contains an extension probe sequence, a ligation probe sequence, and a 7-bp degenerate barcode (NNNNNNN) for PCR duplicate removal. A total of 266 MIP probes were designed, covering a total amplicon of 42,478 bp. The statistics for the MIP target size were as follows: minimum, 155 bp; maximum, 190 bp; mean, 159.7 bp; median, 156.0 bp (Supplementary Materials). Each of the mTSG-MIPs was synthesized using standard oligo synthesis (IDT), normalized, and pooled.
MIP capture sequencing
Genomic DNA sample (150 ng) from each mouse organ was used as input. MIP capture sequencing was done according to previously published protocols (55, 56) with some slight modifications. The multiplexed library was then quality controlled using qPCR and subjected to high-throughput sequencing using the HiSeq 2500 or HiSeq 4000 platforms (Illumina) at the Yale Center for Genome Analysis. Targeted sgRNAs (280 of 281, 99.6%) were captured for all samples from this experiment, with the missing one being Arid1a sg5.
Illumina sequencing data preprocessing
FASTQ reads were mapped to the mm10 genome using the bwa mem function in BWA v0.7.13 (57). Bam files were sorted and indexed using SAMtools v1.3 (58).
Variant calling
For each sample, indel variants were called using SAMtools and VarScan v2.3.9 (59). Specifically, we used SAMtools mpileup (−d 1000000000 –B –q 10) and piped the output to VarScan pileup2indel (−−min-coverage 1 --min-reads2 1 --min-var-freq 0.001 --p-value 0.05). To link each indel to the sgRNA that most likely caused the mutation, we took the center position of each indel and mapped it to the closest sgRNA cut site.
Calling MSs and MGs
We further filtered all detected indels by requiring that each indel must overlap the ±3-bp flank of the closest sgRNA cut site, because Cas9-induced DSBs are expected to occur within a narrow window of the predicted cut site. To exclude any possible germline mutations, we also removed any sgRNAs with indels present in more than half of the control samples with greater than 5% variant frequency. In particular, high variant frequencies were observed across all samples at the Rps19 sg5 cut site, suggesting that these were germline variants; thus, we excluded Rps19 sg5 from all analyses.
To determine MSs in each liver sample, we used a false discovery approach based on the PBS and vector control samples. For each sgRNA, we first took the highest % variant read frequency across all control liver samples; for a mutation to be called in an mTSG sample, the % variant read frequency had to exceed the control sample cutoff. However, because the base vector contained a Trp53 sgRNA (p53 sg8) whose cut site was only 1 bp away from the target site of Trp53 sg4 (from the mTSG library), we only considered PBS samples when calculating the false discovery cutoff for Trp53 sg4. Finally, because we were most interested in identifying the dominant clones in each sample, we further set a 5% variant frequency cutoff on top of the false discovery cutoff. These criteria gave us a binary table (that is, not mutated versus mutated) detailing each sgRNA and whether its target site was mutated in each sample. To convert mutated sgRNA sites into MGs, we simply collapsed the binary sgRNA scores by gene such that, if any of the five sgRNAs for a gene were found to be strongly cutting, the entire gene would be called as mutated.
Coding frame analysis
For coding frame and exonic/intronic analysis, we only considered the indels that were associated with an sgRNA, which had been considered mutated in that particular sample. This final set of significant indels was converted to .avinput format and subsequently annotated using ANNOVAR v. 2016Feb01, using default settings (60).
Co-occurrence and correlation analysis
Co-occurrence analysis was performed by first generating a double-mutant count table for each pairwise combination of genes in the mTSG library. Statistical significance of the co-occurrence was assessed by two-sided hypergeometric test. To calculate co-occurrence rates, we defined the “intersection” as the number of double-mutant samples, and the “union” as the number of samples with a mutation in either (or both) of the two genes, and then divided the intersection by the union. For correlation analysis, we first collapsed the table of variant frequencies to the gene level (that is, summing the variant frequencies for all five of the targeting sgRNAs for each gene). Using these summed variant frequency values, we calculated the Spearman or Pearson correlation between all gene pairs across each mTSG sample. Statistical significance of the correlation was determined by converting the correlation coefficient to a t statistic and then using the t distribution to find the associated probability. For both co-occurrence and correlation analyses, P values were adjusted for multiple hypothesis testing by the Benjamini-Hochberg method to obtain q values.
Unique variant analysis
Instead of first collapsing variant calls to the sgRNA level as above, unique variants and their associated mutant frequencies were compiled across all sequenced samples. To be considered present in a given sample, a particular variant must have a mutant frequency of ≥1%. Heat maps of the unique variant landscape were created in R using the NMF package, with average linkage and Euclidean distance. We also performed a focused analysis on the unique variant landscape within a single mouse, as presented in Fig. 5. For the correlation heat map in Fig. 5B, we used Spearman rank correlation to assess the pairwise correlation between different liver lobes. In Fig. 5C, clusters of variants were defined on the basis of binary mutation calls (that is, whether a given variant is present or not within each sample). To determine the proportional contribution of each cluster, for each sample, we only included the clusters in which at least half of the variants in the cluster are present in that sample. We then took the average mutant frequency across the variants within each cluster and used these values to determine the relative contribution of each cluster to the overall sample. To identify the top four variants in each cluster, we ranked all the variants by the average variant frequency across all lobes in which the variant cluster was considered present.
Clustering of variant frequencies to infer clonality of tumors
For each mTSG liver sample, we extracted the individual variants that comprised the MS calls in that sample, with a cutoff of 5% variant frequency to eliminate low-abundance variants. To identify clusters of variant frequencies in an unbiased manner, we modeled the variant frequency distribution with a Gaussian kernel density estimate, using the Sheather-Jones method to select the smoothing bandwidth. From the kernel density estimate, we then identified the number of local maxima (that is, “peaks”) within the density function. The number of peaks thus represents the number of variant frequency clusters for an individual sample, which is an approximation for the clonality of the tumors.
Direct in vivo validation of drivers or combinations
Liver-specific AAV-CRISPR vectors were designed to co-cistronically express FLuc and Cre recombinase for induction of Cas9 expression under a TBG promoter when delivered to LSL-Cas9 mice (plasmids available at Addgene). Two sgRNA expression cassettes were built in these vectors, one encoding an sgRNA targeting Trp53, with the other being an open sgRNA cassette (double Sap I sites for GTS cloning). The vector was generated by gBlocks Gene Fragment synthesis (IDT) followed by Gibson assembly (NEB). Each specific sgRNA targeting a driver gene was cloned separately into this vector. AAV9 virus was produced and qPCR-titrated, as described above. Total viral particles (1 × 1011) were introduced by intravenous injection into LSL-Cas9 mice. For combinations of two AAVs, 5 × 1010 viral particles were used from each AAV to generate equal titer mixtures and injected. Four to six mice were injected per group. Starting 1 month after injection, mice were imaged by IVIS each month. Briefly, mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) and imaged for in vivo tumor growth using an IVIS machine (PerkinElmer) with firefly d-luciferin potassium salt (150 mg/kg body weight) injected intraperitoneally. Relative luciferase activity was quantified using Living Image software (PerkinElmer).
Blinding statement
Investigators were blinded for histology scoring and MIP sequencing but were not blinded for dissection, MRI, or survival analysis.
Supplementary Material
Acknowledgments
We thank all members in the Chen, Sharp, Zhang, and Platt laboratories, as well as various colleagues in the Yale Department of Genetics, Systems Biology Institute, Yale Cancer Center and Stem Cell Center, Koch Institute, and Broad Institute at Massachusetts Institute of Technology (MIT) for assistance and/or discussion. We thank Yale Center for Genome Analysis, Yale Center for Molecular Discovery, Yale High Performance Computing Center, Yale West Campus Analytical Chemistry Core, Yale West Campus Imaging Core, and Yale Keck Biotechnology Resource Laboratory, as well as MIT Swanson Biotechnology Center, for technical support. Funding: S.C. is supported by the Yale SBI/Genetics Startup Fund, Damon Runyon Cancer Research Foundation (DRG-2117-12; DFS-13-15), Melanoma Research Foundation (412806, 16-003524), St. Baldrick’s Foundation (426685), American Cancer Society (IRG 58-012-54), Breast Cancer Alliance, Cancer Research Institute (CRI) (Clinic and Laboratory Integration Program), American Association for Cancer Research (499395), U.S. Department of Defense (W81XWH-17-1-0235), and NIH/National Cancer Institute (1U54CA209992, 5P50CA196530-A10805, and 4P50CA121974-A08306). P.A.S. is supported by the NIH (R01-CA133404, R01-GM034277, CCNE), Skoltech Center, and the Casimir-Lambert Fund. F.Z. is supported by the NIH (grants 1R01-HG009761, 1R01-MH110049, and 1DP1-HL141201); Howard Hughes Medical Institute; New York Stem Cell, Simons, Paul G. Allen Family, Vallee Foundations; Poitras Center for Affective Disorders Research; Hock E. Tan and K. Lisa Yang Center for Autism Research; and J. and P. Poitras, R. Metcalfe, and D. Cheng. F.Z. is a New York Stem Cell Foundation–Robertson Investigator. R.D.C. and M.B.D. are supported by an NIH Medical Scientist Training Program training grant (T32GM007205). C.D.G. is supported by an NIH PhD training grant (T32 GM007499). G.W. is supported by R. J. Anderson and CRI Irvington Postdoctoral Fellowships. Author contributions: S.C. and R.J.P. planned, designed, and initiated the experiments. G.W. and S.C. performed the majority of molecular, cellular, and animal work. S.C. conceived the functional genome atlas concept. R.D.C. designed the functional genome variant mapping approach, analyzed all data, and prepared the first manuscript draft. G.W. performed all validation experiments. L.Y., C.D.G., X.D., and M.B.D. contributed to parts of the experiments. F.Z., P.A.S., R.J.P., and S.C. jointly supervised the work. R.D.C., G.W., and S.C. wrote the paper with inputs from all authors. Competing interests: F.Z. is a co-founder of Editas Medicine and a scientific advisor for Editas Medicine and Horizon Discovery. P.A.S., F.Z., R.J.P., and S.C. are inventors on two patent applications related to this work (application nos. US2015/051446 and 15/467888). S.C. and R.D.C. are also inventors on an additional patent application related to this work (application no. 62/600,802). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. CRISPR reagents (plasmids and libraries) will be deposited to Addgene to share with the academic community. All custom scripts used to process and analyze the data in this study will be available upon reasonable requests. Genomic sequencing data are deposited in the National Center for Biotechnology Information Sequence Read Archive under accession number PRJNA416821.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaao5508/DC1
fig. S1. Representative full-spectrum MRI series of livers from PBS-, vector-, and mTSG-treated mice.
fig. S2. Additional bright-field images of mTSG-treated livers with GFP overlay.
fig. S3. Representative full-slide scanning images of mouse liver sections in PBS, vector, and mTSG treatment groups.
fig. S4. Representative histology and immunohistochemistry images of mouse liver sections in PBS, vector, and mTSG groups.
fig. S5. MIP capture sequencing statistics and indel size distribution of mTSG livers.
fig. S6. Mutated sgRNA sites across all liver samples from mice treated with AAV-mTSG library.
fig. S7. Heat map of gene-level sum variant frequency across all mTSG liver samples.
fig. S8. Additional co-mutation analysis.
fig. S9. Heat map of all unique variants across all mTSG liver samples.
fig. S10. Investigation and comparison of single or combinatorial knockout of screened TSGs in liver tumorigenesis.
fig. S11. Mutant clonality and clustering analysis.
table S1. DNA sequences of sgRNA spacers in mTSG library.
table S2. Raw read counts of mTSG plasmid library.
table S3. Tumor volume data as measured by MRI.
table S4. Survival data for PBS-, vector-, or mTSG-treated animals.
table S5. Tumor area data as measured by tissue histology.
table S6. Sequence information and annotation for all MIPs used in the study.
table S7. Metadata for all of the 133 sequenced samples.
table S8. MIP capture sequencing coverage statistics across all predicted cutting sites of sgRNAs in AAV-mTSG library.
table S9. Raw indel variant calls of all samples with targeted capture sequencing before filtering.
table S10. sgRNA-level sum indel frequency table for all samples with targeted capture sequencing.
table S11. sgRNA-level binary MS calls in livers from mice treated with AAV-mTSG library.
table S12. Gene-level binary MG calls in livers from mice treated with AAV-mTSG library.
table S13. Co-occurrence analysis of MG pairs in livers from mice treated with AAV-mTSG library.
table S14. Correlation analysis of gene-level sum indel frequency in livers from mice treated with AAV-mTSG library.
table S15. Mutant frequencies for all unique variants present across all mTSG liver samples.
table S16. Spearman rank correlation matrix for five individual liver lobes within a single mouse.
table S17. Mutant frequencies for all unique variants present in five individual liver lobes from a single mouse.
REFERENCES AND NOTES
- 1.Kandoth C., McLellan M. D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J. F., Wyczalkowski M. A., Leiserson M. D. M., Miller C. A., Welch J. S., Walter M. J., Wendl M. C., Ley T. J., Wilson R. K., Raphael B. J., Ding L., Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawrence M. S., Stojanov P., Polak P., Kryukov G. V., Cibulskis K., Sivachenko A., Carter S. L., Stewart C., Mermel C. H., Roberts S. A., Kiezun A., Hammerman P. S., McKenna A., Drier Y., Zou L., Ramos A. H., Pugh T. J., Stransky N., Helman E., Kim J., Sougnez C., Ambrogio L., Nickerson E., Shefler E., Cortés M. L., Auclair D., Saksena G., Voet D., Noble M., DiCara D., Lin P., Lichtenstein L., Heiman D. I., Fennell T., Imielinski M., Hernandez B., Hodis E., Baca S., Dulak A. M., Lohr J., Landau D.-A., Wu C. J., Melendez-Zajgla J., Hidalgo-Miranda A., Koren A., McCarroll S. A., Mora J., Lee R. S., Crompton B., Onofrio R., Parkin M., Winckler W., Ardlie K., Gabriel S. B., Roberts C. W. M., Biegel J. A., Stegmaier K., Bass A. J., Garraway L. A., Meyerson M., Golub T. R., Gordenin D. A., Sunyaev S., Lander E. S., Getz G., Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davoli T., Xu A. W., Mengwasser K. E., Sack L. M., Yoon J. C., Park P. J., Elledge S. J., Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garraway L. A., Lander E. S., Lessons from the Cancer Genome. Cell 153, 17–37 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Stratton M. R., Campbell P. J., Futreal P. A., The cancer genome. Nature 458, 719–724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A. Jr, Kinzler K. W., Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forbes S. A., Bindal N., Bamford S., Cole C., Kok C. Y., Beare D., Jia M., Shepherd R., Leung K., Teague A. M. J. W., Campbell P. J., Stratton M. R., Futreal P. A., COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R. M., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., Stuart J. M., The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zack T. I., Schumacher S. E., Carter S. L., Cherniack A. D., Saksena G., Tabak B., Lawrence M. S., Zhang C.-Z., Wala J., Mermel C. H., Sougnez C., Gabriel S. B., Hernandez B., Shen H., Laird P. W., Getz G., Meyerson M., Beroukhim R., Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciriello G., Miller M. L., Aksoy B. A., Senbabaoglu Y., Schultz N., Sander C., Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 45, 1127–1133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoadley K. A., Yau C., Wolf D. M., Cherniack A. D., Tamborero D., Ng S., Leiserson M. D. M., Niu B., McLellan M. D., Uzunangelov V., Zhang J., Kandoth C., Akbani R., Shen H., Omberg L., Chu A., Margolin A. A., van’t Veer L. J., Lopez-Bigas N., Laird P. W., Raphael B. J., Ding L., Robertson A. G., Byers L. A., Mills G. B., Weinstein J. N., Van Waes C., Chen Z., Collisson E. A. The Cancer Genome Atlas Research Network, Benz C. C., Perou C. M., Stuart J. M., Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin L., Hahn W. C., Getz G., Meyerson M., Making sense of cancer genomic data. Genes Dev. 25, 534–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogelstein B., Kinzler K. W., Cancer genes and the pathways they control. Nat. Med. 10, 789–799 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez-Perez A., Perez-Llamas C., Deu-Pons J., Tamborero D., Schroeder M. P., Jene-Sanz A., Santos A., Lopez-Bigas N., IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 10, 1081–1082 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrence M. S., Stojanov P., Mermel C. H., Robinson J. T., Garraway L. A., Golub T. R., Meyerson M., Gabriel S. B., Lander E. S., Getz G., Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frese K. K., Tuveson D. A., Maximizing mouse cancer models. Nat. Rev. Cancer 7, 654–658 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Quail D. F., Joyce J. A., Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heyer J., Kwong L. N., Lowe S. W., Chin L., Non-germline genetically engineered mouse models for translational cancer research. Nat. Rev. Cancer 10, 470–480 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D. A., Horvath P., CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E., A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M., RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue W., Chen S., Yin H., Tammela T., Papagiannakopoulos T., Joshi N. S., Cai W., Yang G., Bronson R., Crowley D. G., Zhang F., Anderson D. G., Sharp P. A., Jacks T., CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin H., Xue W., Chen S., Bogorad R. L., Benedetti E., Grompe M., Koteliansky V., Sharp P. A., Jacks T., Anderson D. G., Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 32, 551–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber J., Öllinger R., Friedrich M., Ehmer U., Barenboim M., Steiger K., Heid I., Mueller S., Maresch R., Engleitner T., Gross N., Geumann U., Fu B., Segler A., Yuan D., Lange S., Strong A., de la Rosa J., Esposito I., Liu P., Cadiñanos J., Vassiliou G. S., Schmid R. M., Schneider G., Unger K., Yang F., Braren R., Heikenwälder M., Varela I., Saur D., Bradley A., Rad R., CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc. Natl. Acad. Sci. U.S.A. 112, 13982–13987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiou S.-H., Winters I. P., Wang J., Naranjo S., Dudgeon C., Tamburini F. B., Brady J. J., Yang D., Grüner B. M., Chuang C.-H., Caswell D. R., Zeng H., Chu P., Kim G. E., Carpizo D. R., Kim S. K., Winslow M. M., Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 29, 1576–1585 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heckl D., Kowalczyk M. S., Yudovich D., Belizaire R., Puram R. V., McConkey M. E., Thielke A., Aster J. C., Regev A., Ebert B. L., Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 32, 941–946 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maddalo D., Manchado E., Concepcion C. P., Bonetti C., Vidigal J. A., Han Y.-C., Ogrodowski P., Crippa A., Rekhtman N., de Stanchina E., Lowe S. W., Ventura A., In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Platt R. J., Chen S., Zhou Y., Yim M. J., Swiech L., Kempton H. R., Dahlman J. E., Parnas O., Eisenhaure T. M., Jovanovic M., Graham D. B., Jhunjhunwala S., Heidenreich M., Xavier R. J., Langer R., Anderson D. G., Hacohen N., Regev A., Feng G., Sharp P. A., Zhang F., CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sánchez-Rivera F. J., Papagiannakopoulos T., Romero R., Tammela T., Bauer M. R., Bhutkar A., Joshi N. S., Subbaraj L., Bronson R. T., Xue W., Jacks T., Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelsen T. S., Heckl D., Ebert B. L., Root D. E., Doench J. G., Zhang F., Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang T., Wei J. J., Sabatini D. M., Lander E. S., Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S., Sanjana N. E., Zheng K., Shalem O., Lee K., Shi X., Scott D. A., Song J., Pan J. Q., Weissleder R., Lee H., Zhang F., Sharp P. A., Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song C.-Q., Li Y., Mou H., Moore J., Park A., Pomyen Y., Hough S., Kennedy Z., Fischer A., Yin H., Anderson D. G., Conte D. Jr, Zender L., Wang X. W., Thorgeirsson S., Weng Z., Xue W., Genome-wide CRISPR screen identifies regulators of mitogen-activated protein kinase as suppressors of liver tumors in mice. Gastroenterology 152, 1161–1173.e1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ran F. A., Cong L., Yan W. X., Scott D. A., Gootenberg J. S., Kriz A. J., Zetsche B., Shalem O., Wu X., Makarova K. S., Koonin E. V., Sharp P. A., Zhang F., In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zincarelli C., Soltys S., Rengo G., Rabinowitz J. E., Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 16, 1073–1080 (2008). [DOI] [PubMed] [Google Scholar]
- 37.El–Serag H. B., Rudolph K. L., Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F., Jaenisch R., One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu P. D., Lander E. S., Zhang F., Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katz S.-F., Lechel A., Obenauf A. C., Begus-Nahrmann Y., Kraus J. M., Hoffmann E. M., Duda J., Eshraghi P., Hartmann D., Liss B., Schirmacher P., Kestler H. A., Speicher M. R., Rudolph K. L., Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology 142, 1229–1239.e3 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Duns G., van den Berg E., van Duivenbode I., Osinga J., Hollema H., Hofstra R. M. W., Kok K., Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 70, 4287–4291 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Tseng A.-S. K., Tapon N., Kanda H., Cigizoglu S., Edelmann L., Pellock B., White K., Hariharan I. K., Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Curr. Biol. 17, 728–733 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taniguchi C. M., Winnay J., Kondo T., Bronson R. T., Guimaraes A. R., Alemán J. O., Luo J., Stephanopoulos G., Weissleder R., Cantley L. C., Kahn C. R., The phosphoinositide 3-kinase regulatory subunit p85α can exert tumor suppressor properties through negative regulation of growth factor signalling. Cancer Res. 70, 5305–5315 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dawson M. A., Kouzarides T., Cancer epigenetics: From mechanism to therapy. Cell 150, 12–27 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Chow R. D., Guzman C. D., Wang G., Schmidt F., Youngblood M. W., Ye L., Errami Y., Dong M. B., Martinez M. A., Zhang S., Renauer P., Bilguvar K., Gunel M., Sharp P. A., Zhang F., Platt R. J., Chen S., AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 20, 1329–1341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zender L., Xue W., Zuber J., Semighini C. P., Krasnitz A., Ma B., Zender P., Kubicka S., Luk J. M., Schirmacher P., McCombie W. R., Wigler M., Hicks J., Hannon G. J., Powers S., Lowe S. W., An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 135, 852–864 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starr T. K., Allaei R., Silverstein K. A. T., Staggs R. A., Sarver A. L., Bergemann T. L., Gupta M., O’Sullivan M. G., Matise I., Dupuy A. J., Collier L. S., Powers S., Oberg A. L., Asmann Y. W., Thibodeau S. N., Tessarollo L., Copeland N. G., Jenkins N. A., Cormier R. T., Largaespada D. A., A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 323, 1747–1750 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mann K. M., Ward J. M., Yew C. C. K., Kovochich A., Dawson D. W., Black M. A., Brett B. T., Sheetz T. E., Dupuy A. J. Australian Pancreatic Cancer Genome Initiative, Chang D. K., Biankin A. V., Waddell N., Kassahn K. S., Grimmond S. M., Rust A. G., Adams D. J., Jenkins N. A., Copeland N. G., Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. U.S.A. 109, 5934–5941 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beronja S., Janki P., Heller E., Lien W.-H., Keyes B. E., Oshimori N., Fuchs E., RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 501, 185–190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qi L. S., Larson M. H., Gilbert L. A., Doudna J. A., Weissman J. S., Arkin A. P., Lim W. A., Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Konermann S., Brigham M. D., Trevino A. E., Joung J., Abudayyeh O. O., Barcena C., Hsu P. D., Habib N., Gootenberg J. S., Nishimasu H., Nureki O., Zhang F., Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim Y. B., Komor A. C., Levy J. M., Packer M. S., Zhao K. T., Liu D. R., Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Komor A. C., Kim Y. B., Packer M. S., Zuris J. A., Liu D. R., Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., Schultz N., The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hardenbol P., Banér J., Jain M., Nilsson M., Namsaraev E. A., Karlin-Neumann G. A., Fakhrai-Rad H., Ronaghi M., Willis T. D., Landegren U., Davis R. W., Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat. Biotechnol. 21, 673–678 (2003). [DOI] [PubMed] [Google Scholar]
- 56.O’Roak B. J., Vives L., Fu W., Egertson J. D., Stanaway I. B., Phelps I. G., Carvill G., Kumar A., Lee C., Ankenman K., Munson J., Hiatt J. B., Turner E. H., Levy R., O’Day D. R., Krumm N., Coe B. P., Martin B. K., Borenstein E., Nickerson D. A., Mefford H. C., Doherty D., Akey J. M., Bernier R., Eichler E. E., Shendure J., Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koboldt D. C., Zhang Q., Larson D. E., Shen D., McLellan M. D., Lin L., Miller C. A., Mardis E. R., Ding L., Wilson R. K., VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang K., Li M., Hakonarson H., ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaao5508/DC1
fig. S1. Representative full-spectrum MRI series of livers from PBS-, vector-, and mTSG-treated mice.
fig. S2. Additional bright-field images of mTSG-treated livers with GFP overlay.
fig. S3. Representative full-slide scanning images of mouse liver sections in PBS, vector, and mTSG treatment groups.
fig. S4. Representative histology and immunohistochemistry images of mouse liver sections in PBS, vector, and mTSG groups.
fig. S5. MIP capture sequencing statistics and indel size distribution of mTSG livers.
fig. S6. Mutated sgRNA sites across all liver samples from mice treated with AAV-mTSG library.
fig. S7. Heat map of gene-level sum variant frequency across all mTSG liver samples.
fig. S8. Additional co-mutation analysis.
fig. S9. Heat map of all unique variants across all mTSG liver samples.
fig. S10. Investigation and comparison of single or combinatorial knockout of screened TSGs in liver tumorigenesis.
fig. S11. Mutant clonality and clustering analysis.
table S1. DNA sequences of sgRNA spacers in mTSG library.
table S2. Raw read counts of mTSG plasmid library.
table S3. Tumor volume data as measured by MRI.
table S4. Survival data for PBS-, vector-, or mTSG-treated animals.
table S5. Tumor area data as measured by tissue histology.
table S6. Sequence information and annotation for all MIPs used in the study.
table S7. Metadata for all of the 133 sequenced samples.
table S8. MIP capture sequencing coverage statistics across all predicted cutting sites of sgRNAs in AAV-mTSG library.
table S9. Raw indel variant calls of all samples with targeted capture sequencing before filtering.
table S10. sgRNA-level sum indel frequency table for all samples with targeted capture sequencing.
table S11. sgRNA-level binary MS calls in livers from mice treated with AAV-mTSG library.
table S12. Gene-level binary MG calls in livers from mice treated with AAV-mTSG library.
table S13. Co-occurrence analysis of MG pairs in livers from mice treated with AAV-mTSG library.
table S14. Correlation analysis of gene-level sum indel frequency in livers from mice treated with AAV-mTSG library.
table S15. Mutant frequencies for all unique variants present across all mTSG liver samples.
table S16. Spearman rank correlation matrix for five individual liver lobes within a single mouse.
table S17. Mutant frequencies for all unique variants present in five individual liver lobes from a single mouse.