

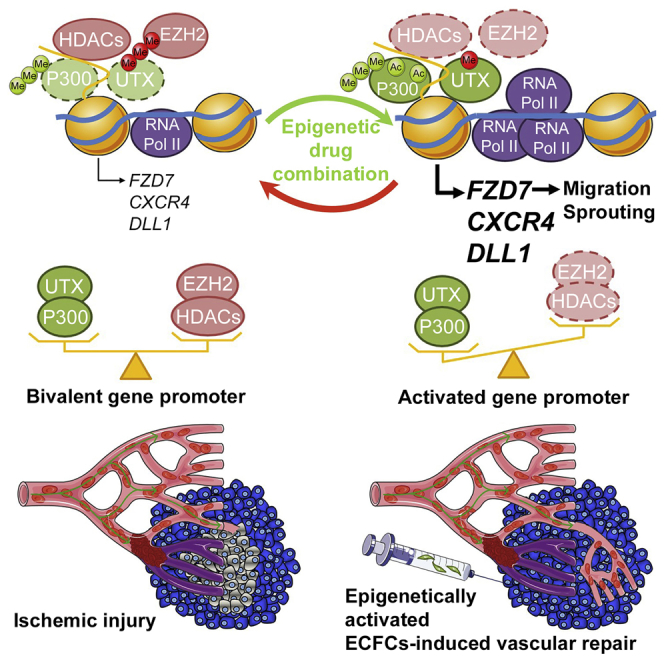
Summary
Human endothelial colony-forming cells (ECFCs) represent a promising source of adult stem cells for vascular repair, yet their regenerative capacity is limited. Here, we set out to understand the molecular mechanism restricting the repair function of ECFCs. We found that key pro-angiogenic pathways are repressed in ECFCs due to the presence of bivalent (H3K27me3/H3K4me3) epigenetic marks, which decreases the cells' regenerative potential. Importantly, ex vivo treatment with a combination of epigenetic drugs that resolves bivalent marks toward the transcriptionally active H3K4me3 state leads to the simultaneous activation of multiple pro-angiogenic signaling pathways (VEGFR, CXCR4, WNT, NOTCH, SHH). This in turn results in improved capacity of ECFCs to form capillary-like networks in vitro and in vivo. Furthermore, restoration of perfusion is accelerated upon transplantation of drug-treated ECFCs in a model of hindlimb ischemia. Thus, ex vivo treatment with epigenetic drugs increases the vascular repair properties of ECFCs through transient activation of pro-angiogenic signaling pathways.
Keywords: pro-angiogenic pathway, ECFCs, epigenetics, bivalent genes, human endothelial progenitors, vasculogenesis, angiogenesis, hindlimb ischemia, EZH2, UTX
Graphical Abstract
Highlights
-
•
Pro-angiogenic pathways are maintained in a poised state in ECFCs
-
•
Epigenetic drugs resolve bivalently marked genes toward an active state in ECFCs
-
•
Treatment with epigenetic drugs activates multiple pro-angiogenic pathways in ECFCs
-
•
Ex vivo treatment with epigenetic drugs increases ECFC-mediated vasculogenesis
Endothelial colony-forming cells (ECFCs) have the unique capability to form blood vessels in vivo. Here, Brand and colleagues show that the regenerative function of ECFCs is restricted by the presence of bivalent histone marks on pro-angiogenic genes. This poised status can be overcome through the combined action of epigenetic drugs that simultaneously activate multiple pro-angiogenic pathways to increase ECFC-mediated vasculogenesis.
Introduction
Human endothelial colony-forming cells (ECFCs) are endothelial progenitors that can be isolated from umbilical cord blood and peripheral blood (Ingram et al., 2004, Lin et al., 2000, Medina et al., 2010) or derived from induced pluripotent stem cells (iPSCs) (Prasain et al., 2014). These highly proliferative cells possess the unique property of forming functional blood vessels in vivo upon transplantation (Au et al., 2008, Melero-Martin et al., 2007, Reinisch et al., 2009, Yoder et al., 2007). This property distinguishes ECFCs from more mature endothelial cells and makes them particularly attractive as a cell therapy for enhanced vascular repair in ischemic diseases such as peripheral arterial disease, ischemic retinopathy, myocardial infarction, or stroke (Cooke and Losordo, 2015, Fadini et al., 2012, Fraineau et al., 2015, Park and Gerecht, 2014, Shantsila et al., 2007). Upon transplantation at sites of ischemic injury, ECFCs incorporate into damaged blood vessels and release pro-angiogenic growth factors to facilitate the repair process, ultimately improving blood perfusion and organ function (Alphonse et al., 2014, Bouvard et al., 2010, Palii et al., 2014, Saif et al., 2010, Schwarz et al., 2012). Furthermore, ECFCs can be used to provide trophic support to other therapeutically relevant cells (Lin et al., 2014). While ECFCs represent promising candidates for cell therapy, it is critical to improve their efficacy in vivo through promoting cell survival and increasing the kinetics of cell migration, homing, differentiation, and secretion of pro-angiogenic factors (Chavakis et al., 2008, Cooke and Losordo, 2015).
Studies in endothelial cells have identified several signal transduction pathways (e.g., CXCR4, VEGF, NOTCH, SHH, and WNT) that coordinate survival, differentiation, arterial/venous specification, and blood-vessel morphogenesis (Carmeliet and Jain, 2011, Herbert and Stainier, 2011, Le Bras et al., 2010). Therefore, a major question is how to activate pro-angiogenic signaling pathways in ECFCs such that we can boost the regenerative function of these cells and increase their survival in vivo post transplantation. Promising results have been obtained through pre-activation of the NOTCH pathway where it was shown that the vascular repair potential of transplanted murine endothelial progenitors is significantly increased if the cells are cultured ex vivo with stromal cells overexpressing the Notch ligand Jag-1 prior to transplantation (Kwon et al., 2008). Furthermore, it was recently found that co-implantation of stromal cells expressing the Notch ligand Dll1 decreases apoptosis and enhances the post-natal vasculogenic capacity of human ECFCs in murine subcutaneous implants (Kim et al., 2015). While activation of the Notch pathway through external stimulation of Notch receptors may provide a valuable strategy to enhance vascular repair properties of endothelial progenitors, several other signaling pathways (e.g., VEGFR, CXCR4, SHH, WNT) are also essential for angiogenesis and post-natal vasculogenesis. However, an approach that simultaneously activates multiple pro-angiogenic pathways in ECFCs is currently lacking.
We previously demonstrated that ex vivo treatment with the histone deacetylase (HDAC) inhibitor trichostatin A (TSA) increases ECFC-mediated vascular repair in hindlimb ischemia (Palii et al., 2014), providing proof of principle that the priming of ECFCs with epigenetic drugs prior to transplantation is a valuable strategy to improve their regenerative function (Fraineau et al., 2015). Stem and progenitor cells are characterized by a high prevalence of transcriptionally competent but repressed (i.e., “poised”) genes that are marked by both active (histone H3 lysine 4 trimethylation [H3K4me3]) and repressive (histone H3 lysine 27 trimethylation [H3K27me3]) histone modifications that play important roles in differentiation and development (Benyoucef and Brand, 2015, Voigt et al., 2013). While those bivalent genes are expressed at extremely low levels, it has been proposed that the presence of H3K4me3 may prime them for rapid activation upon removal of H3K27me3. Here, we set out to test the hypothesis that decreasing the levels of the repressive mark H3K27me3 with epigenetic drugs could improve the vascular repair properties of ECFC through activation of pro-angiogenic signaling pathways.
Results
A Combination of Epigenetic Drugs Increases ECFC Migration, Capillary Network Formation, and Resistance to Serum Starvation-Induced Apoptosis
Repressive histone marks H3K27me2/3 are established on chromatin by the polycomb group repressive complex 2 (PRC2), a protein complex comprised of three core subunits: methyltransferase EZH2 and two other proteins, EED and SUZ12, both of which are required for PRC2 catalytic activity (Di Croce and Helin, 2013). To decrease H3K27me2/3 levels, we treated human umbilical cord blood-derived ECFCs with GSK-343, a potent and selective inhibitor of EZH2 (Verma et al., 2012). As expected, GSK-343 exposure led to a global decrease of H3K27me2/3 levels in a time- and dose-dependent manner (Figures 1A, S1A, and S1B). Importantly, other histone marks, such as H3K27me1, H3K4me3, H3K9me3, H4K20me3, or histone acetylation, are not affected (Figures 1A and S1B). Furthermore, GSK-343 does not lead to a decrease of PRC2 subunits nor does it alter the level of other chromatin-modifying enzymes, including the acetyltransferase P300, the deacetylase HDAC1, the H3K27 demethylase UTX, the H3K9 methyltransferase G9a, or the DNA methyltransferases DNMT1 and DNMT3A (Figure 1B). This is in contrast to the widely used S-adenosylhomocysteine hydrolase inhibitor 3-deazaneplanocin (DZNep), which depletes PRC2 subunits and leads to a global decrease of multiple histone marks, including H3K27me2/3 but also H4K20me3, H3K9me3, H3K4me3, and others (Helin and Dhanak, 2013, Miranda et al., 2009, Ohtani et al., 2011, Tan et al., 2007). Thus, in contrast to DZNep, GSK-343 is a potent and selective inhibitor of EZH2 in ECFCs.
Figure 1.

Pharmacologic Treatment with a Combination of EZH2 and HDAC Inhibitors Enhances ECFC Migration, Capillary Network Formation, and Resistance to Serum Starvation-Induced Apoptosis
(A) GSK-343 and panobinostat lead to a global decrease in H3K27me2/3 levels and an increase in H3/H4-acetyl levels, respectively, as analyzed by western blot. Molecular masses are indicated in kDa. Total histone H3 and H4 serve as loading controls.
(B) GSK-343 and panobinostat do not affect the overall level of indicated proteins, as analyzed by western blot. Molecular masses are indicated in kDa. Tubulin serves as a loading control.
(C) GSK-343 and panobinostat increase the kinetics of ECFC migration as measured by gap closure assay. Left: representative pictures of ECFCs stained with crystal violet after 10 hr migration (10× magnification; scale bar, 250 μm). Dashed circles represent the gap area prior to cell migration. Right: fraction of the gap area invaded by migrating cells at the indicated times. Average from thee independent experiments (n = 3) each performed in duplicate are shown ±SEM.
(D) GSK-343 and panobinostat increase ECFC-mediated capillary-like structure formation on Matrigel. Left: representative pictures of capillary-like structures stained with calcein (2.5× magnification; scale bar, 1 mm). Right: total capillary length of the network is expressed as the mean percentage of control values corresponding to cells treated with vehicle ±SEM. Data shown from three independent experiments (n = 3) each performed in duplicate.
(E) GSK-343 and panobinostat do not induce apoptosis in ECFCs as measured by fluorescence-activated cell sorting (FACS) after 7-AAD and annexin V staining.
(F) GSK-343 and panobinostat reduce serum starvation-induced apoptosis in ECFCs as measured by FACS after 7-AAD and annexin V staining. ECFCs were grown in serum-containing EGM-2 medium or serum-depleted EBM-2 medium.
(E and F) Data expressed as mean percentage of control values corresponding to cells treated with vehicle ±SEM from three independent experiments (n = 3), each performed in duplicate.
(G) GSK-343 and panobinostat decrease ECFC proliferation as analyzed by bromodeoxyuridine (BrdU) incorporation. The percentage of cells incorporating BrdU is indicated ±SEM from four independent experiments (n = 4), each performed in duplicate.
∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, non-significant. See also Figures S1 and S2.
Next, we examined the effects of GSK-343 on the capacity of ECFCs to migrate (using a gap closure assay) and to form a capillary-like network on Matrigel. These experiments revealed that GSK-343 significantly improves the pro-angiogenic properties of ECFCs, increasing both the kinetics of cell migration (Figure S1C) and the capacity of ECFCs to form a capillary-like network (Figure S1D) in a dose-dependent manner. Consistent with this, we found that knocking down EZH2 also increases ECFC-mediated capillary network formation (data not shown). Thus, inhibition of EZH2 is an efficient strategy to improve the angiogenic properties of ECFCs in vitro.
We showed previously that pre-treatment with the histone deacetylase (HDAC) inhibitor TSA increases ECFC-mediated capillary network formation (Palii et al., 2014). Since GSK-343 has no effect on histone acetylation (Figures 1A and S1B), we hypothesized that combining EZH2 and HDAC inhibitors may further improve ECFC function by acting on complementary epigenetic pathways. To test this hypothesis, we sought to use the highly potent class I, II, and IV HDAC inhibitor panobinostat (LBH589), which is less toxic than TSA and is already clinically approved for cancer treatment (Andreu-Vieyra and Berenson, 2014, Dokmanovic et al., 2007). First, we confirmed that panobinostat used as a single agent is at least as efficient as TSA for improving the pro-angiogenic capacity of ECFCs, increasing both cell migration (Figure S2D) and capillary network formation (Figure S2E). Furthermore, we confirmed that, at the established optimal conditions (i.e., 10 nM for 2 hr), panobinostat leads to a significant increase in the global levels of histone H3 and H4 acetylation without affecting H3K27me3 or other histone modifications (Figures S2A–S2C and 1A). Having established optimal conditions for GSK-343 and panobinostat treatments as single agents, we combined these epigenetic drugs to determine whether we could further improve the pro-angiogenic functions of ECFCs. These experiments show that a combined treatment consisting of 72 hr incubation with the EZH2 inhibitor GSK-343 at 5 μM and 2 hr incubation with the HDAC inhibitor panobinostat at 10 nM is significantly more efficient at increasing the migration kinetics of ECFCs (Figure 1C) and capillary network formation (Figure 1D) than either drug alone. Furthermore, the combined treatment is specific to H3K27 di- and tri-methylation and to H3- and H4-acetylation without affecting other histone marks (Figure 1A) and without altering the levels of the enzymes responsible for H3K27 methylation/demethylation and H3/H4 acetylation/deacetylation (Figure 1B). We also verified that the drug combination does not induce apoptosis as measured by annexin V staining (Figure 1E). Rather, we found that treatment with GSK-343 and panobinostat increases the resistance of ECFCs to serum starvation-induced apoptosis (Figure 1F). Finally, we noted a slight decrease in ECFC proliferation upon drug treatment (Figure 1G). Thus, a combined treatment using an EZH2 inhibitor followed by an HDAC inhibitor is an efficient strategy to enhance the pro-angiogenic properties of ECFCs and increase their resistance to apoptosis.
Combined Treatment of ECFCs with Epigenetic Drugs Activates Multiple Pro-angiogenic Pathways Simultaneously
To better understand the mechanism through which the combined treatment of GSK-343 and panobinostat increases the pro-angiogenic properties of ECFCs, we measured changes in gene expression by RNA sequencing (RNA-seq) immediately upon drug treatment (i.e., 72 hr incubation with the EZH2 inhibitor GSK-343 at 5 μM followed by 2 hr incubation with the HDAC inhibitor panobinostat at 10 nM). This experiment, performed using two independently generated ECFC clones from two distinct cord blood donors, identified 553 upregulated genes and 329 downregulated genes (p < 0.05) (Figure 2A). RNA-seq results were confirmed by qRT-PCR for all genes tested (Figures 3A, S4, S5A, and data not shown). Gene ontology (GO) analysis indicates that upregulated genes are enriched in categories related to blood-vessel development, cell motion, cell adhesion, and response to wounding (Figures 2B and S3A), which confirms that the epigenetic drug combination increases the pro-angiogenic properties of ECFCs. In contrast, downregulated genes are enriched in categories related to cell division and proliferation, consistent with the slight decrease in cell proliferation observed upon drug treatment (Figure 1G). See Tables S1 and S2 for a complete list of changing genes and GO categories. Most interestingly, a pathway analysis identified NOTCH and WNT (wingless-type mouse mammary tumor virus integration site) transduction pathways as being significantly enriched for activated genes (Figure S3B). Furthermore, we noticed that three additional pro-angiogenic pathways, CXCR4 (for C-X-C chemokine receptor type 4), VEGFR (for vascular endothelial growth factor receptor), and SHH (for sonic hedgehog), are also activated upon drug treatment (Figure 2C). All five transduction pathways activated by the epigenetic drug combination have previously demonstrated pro-angiogenic roles, with VEGFR being a master regulator of all aspects of angiogenesis and vasculogenesis (including cell survival, proliferation, migration, differentiation, and morphogenesis), CXCR4 promoting endothelial progenitor cell migration, and homing, while NOTCH, WNT, and SHH pathways play critical roles in regulating vessel formation, branching, and maturation (Carmeliet and Jain, 2011, Chavakis et al., 2008, Dejana, 2010, Herbert and Stainier, 2011, Le Bras et al., 2010, Park et al., 2013). Interestingly, for each signaling pathway, upregulated genes belong to ligands, receptors, and downstream target genes (Figure 2C), which indicates global activation upon drug treatment. For instance, ligands (DKK3, WNT5A, WNT9A, WNT4, and WNT11), receptors (FZD2, FZD1, FZD5, and FZD7), and target genes (TCF7L1, JAG1, MML1, and EGR1) of the WNT pathway are activated (Figure 2C and see Figures S3D and S4 for selected examples). Furthermore, negative regulators of WNT and VEGFR pathways (e.g., DKK1 as well as SEMA3A) are downregulated (Figure 2C). Noticeably, parsing the genes in categories according to their expression levels reveals that the drug combination activates low-to-moderately expressed genes (e.g., CXCL12, FZD7, DLL1, SHH, CXCR4, DLL4), whereas genes that are already expressed at high levels (e.g., the transcription elongation factor gene ELP3 or the endothelial nitric oxide synthase [eNOS] gene NOS3; Chan et al., 2004) are not significantly affected (Figure S3C). Finally, we confirmed activation of the identified pro-angiogenic pathways at the protein level by western blot where we detected increased levels of VEGFR1, VEGFR2, intracellular NOTCH, and CXCR4, as well as increased phosphorylation of VEGFR2, AKT, and ERK1/2 proteins (Figure 2D). Taken together, these results indicate that in ECFCs, pro-angiogenic pathways are maintained in a repressed/poised state. Most importantly, we found that five major pro-angiogenic transduction pathways can be activated in ECFCs through the combined action of the epigenetic drugs GSK-343 and panobinostat. Thus, multiple pro-angiogenic signaling pathways can be activated simultaneously in endothelial progenitors through ex vivo drug treatment.
Figure 2.

Pharmacologic Treatment of ECFCs with a Combination of EZH2 and HDAC Inhibitors Activates Multiple Pro-angiogenic Pathways Simultaneously
(A) Global changes in gene expression upon drug treatment versus vehicle treatment analyzed by RNA-seq. Scatterplot shows upregulated (red) and downregulated (green) genes (n = 3).
(B) Representative Gene Ontology (GO) Biological Process categories significantly enriched for up- and downregulated genes in ECFCs treated with GSK-343 and panobinostat.
(C) Heatmaps of up- and downregulated genes that belong to the indicated signaling pathways as identified by RNA-seq analysis and validated by qRT-PCR (∗). Data are expressed in log2(FPKM) (n = 3). The feedback arrow indicates a gene belonging to both receptor and target gene categories.
(D) Validation of cell-signaling pathway activation upon drug treatment at the protein level. Left: western blot analysis of ECFC extracts shows an increase in global protein levels of VEGFR1 (FLT1), VEGFR2 (KDR), cleaved intracellular domain of NOTCH1 (NOTCH1-NICD), and CXCR4 upon drug treatment. Middle: western blot analysis of VEGFR2 immunoprecipitates (IP) shows an increase in phospho-VEGFR2 upon drug treatment. Right: western blot analysis of ECFC extracts shows an increase in phospho-AKT and phospho-ERK1/2 upon drug treatment. Tubulin is used as a loading control. Molecular masses are indicated in kDa.
See also Figures S3 and S4 as well as Tables S1 and S2.
Figure 3.

Pharmacologic Treatment of ECFCs with a Combination of EZH2 and HDAC Inhibitors Increases Gene Expression through H3K27me3 Demethylation and Histone H3 Acetylation
(A) Transcript levels of indicated genes were measured by qRT-PCR following treatment of ECFCs with vehicle, GSK-343, panobinostat, or GSK-343-panobinostat combination, as indicated. qRT-PCR values are expressed as mean percentages of vehicle-treated cells ±SEM with B2M serving as internal control. (n ≥ 3). ∗∗∗p < 0.001; ∗p < 0.05; ns, non-significant.
(B) RNA polymerase II (pol II) binding to indicated gene promoters was assessed by ChIP-qPCR.
(C–E) Enrichment of H3K27me3 (C), H3K4me3 (D), and H3 acetyl (E) histone marks to indicated gene promoters was assessed by ChIP-qPCR.
(B–E) ChIPs were performed using indicated antibodies (Ab) or IgG as a negative control (Mock). qPCR values are expressed as mean fractions of input ±SD.
See also Figure S5.
Activation of Pro-angiogenic Pathways by Epigenetic Drugs Occurs through Resolving Bivalently Marked Genes
To better understand the mechanism of action of panobinostat and GSK-343, we further dissected the effect of these drugs (alone and in combination) on genes representative of various pathways. Interestingly, even though all genes tested are significantly increased upon treatment with the drug combination (validating our RNA-seq results), some genes respond to both drugs in a seemingly additive fashion (e.g., FZD7, KDR, DLL4, HEY2) while others appear to be more responsive to the HDAC inhibitor (e.g., DLL1, SHH) or to the EZH2 inhibitor (e.g., CXCR4, CXCL12, ANGPT2) with ELP3 serving as a control not altered by drug treatment (Figures 3A and S4). These differential effects on gene expression are reflected by differential RNA polymerase II (pol II) binding on gene promoters (Figure 3B). Then, we examined the effects of the drugs (alone and in combination) on histone post-translational modifications (PTMs). Consistent with their highly specific effects on global histone PTMs levels (Figures 1A, S1B, and S2C), we found that both drugs modify their respective targets without affecting other histone PTMs. Specifically, the EZH2 inhibitor GSK-343 decreases the repressive histone mark H3K27me3 on all three representative genes (Figure 3C) with no effect on H3K4me3 (Figure 3D) or H3-Ac (Figure 3E). Similarly, the HDAC inhibitor panobinostat increases histone H3 acetylation on all genes tested (Figure 3E) with no effect on H3K27me3 (Figure 3C) or H3K4me3 (Figure 3D). Taken together, these results indicate that the combination of GSK-343 and panobinostat increases gene expression in ECFCs through a mechanism that entails removal of the repressive histone mark H3K27me3 and increase in the active histone mark H3-Ac.
Interestingly, these experiments also revealed several properties of the genes that respond to the drug combination, including (1) the presence of both active (H3K4me3 and H3-Ac) and repressive (H3K27me3) histone marks on their promoter (Figures 3C, 3D, and 3E); (2) low, but detectable pol II binding on their promoter (Figure 3B); and (3) a low level of expression (Figure S3C). These properties characterize a particular sort of genes named “bivalent” that are maintained in a state of equilibrium between full repression and full activation (Benyoucef and Brand, 2015, Voigt et al., 2013). Even though bivalent genes are found in differentiated cells, they are more prevalent in stem/progenitor cells (e.g., Cui et al., 2009, Karmodiya et al., 2012, Mikkelsen et al., 2007), and they are poised to respond quickly (positively or negatively) to signaling cues from the environment (Benyoucef and Brand, 2015, Voigt et al., 2013). Bivalent genes differ both from highly expressed genes (e.g., ELP3 and NOS3) that are devoid of the repressive mark H3K27me3 (Figures 3 and S5) and from completely repressed genes (e.g., the non-endothelial genes PAX7 and MYOD) that are devoid of the active marks H3K4me3 and H3-Ac and do not display pol II binding (Figure S5).
To better understand the mechanism leading to H3K27me3 removal and H3-Ac increase upon drug treatment, we tested for binding of the epigenetic enzymes responsible for adding/removing these histone modifications. Interestingly, we found that bivalent gene promoters are co-bound by enzymes with antagonistic activities: (1) the H3K27 methyltransferase EZH2 and the H3K27 demethylase UTX, as well as (2) the histone acetyltransferase P300 and the histone deacetylase HDAC1 (Figure 4). Furthermore, these enzymes remain tethered to the genes upon drug treatment (data not shown). In contrast, EZH2 is not bound to the promoter of the highly expressed ELP3 gene (Figure 4A), consistent with the absence of H3K27me3 at this site (Figure 3C). Taken together, these results suggest that in non-treated ECFCs, EZH2 and UTX compete for methylation/demethylation of H3K27 at bivalent gene promoters, resulting in an enrichment of the repressive histone mark H3K27me3 (Figure 3C) that is sufficient to maintain low levels of gene expression (i.e., poised status) (Figures 3A, 4E, S3C, S3D, and S4). When EZH2 is inhibited (upon GSK-343 treatment), the equilibrium is displaced in favor of UTX, which rapidly removes the repressive mark H3K27me3 (Figure 3C), leading to increased gene transcription (Figures 3A, 4E, S3C, and S3D). A similar mechanism appears to be in place for histone acetylation/deacetylation where P300 and HDAC1 are co-bound to bivalent gene promoters (Figure 4) and compete for histone modification, resulting in increased acetylation upon panobinostat-mediated inhibition of HDAC activity (Figure 3E). Thus, ECFCs maintain specific genes in a repressed/poised state through the recruitment of antagonistic “activating” and “repressive” chromatin-modifying enzymes that establish a bivalent gene promoter status characterized by the presence of both active (H3K4me3 and H3-Ac) and repressive (H3K27me3) histone marks (Figure 4E). Importantly, this poised/bivalent status can be resolved through pharmacologic inhibition of the repressive enzymes EZH2 and HDAC leading to full gene activation.
Figure 4.

Co-binding of Histone H3K27 Methyltransferase/Demethylase and Histone Acetyltransferase/Deacetylase to Gene Promoters
(A–D) Binding of the H3K27 methyltransferase EZH2 (A), the H3K27 demethylase UTX (B), the histone deacetylase HDAC1 (C), and the histone acetyltransferase p300 (D) to indicated gene promoters was measured by ChIP-qPCR. ChIPs were performed using indicated antibodies (Ab) or IgG as a negative control (Mock). qPCR values are expressed as mean fractions of input ±SD.
(E) Model representing co-binding of antagonistic enzymes on gene promoters such that transcription is maintained at low levels (poised gene). Combined inhibition of the repressive enzymes EZH2 and HDAC1 displaces the equilibrium leading to transcriptional activation.
Gene Activation Mediated by HDAC and EZH2 Inhibitors Is a Reversible Process
Co-binding of antagonistic chromatin-modifying enzymes on bivalent gene promoters (Figure 4) predicts that gene activation through pharmacologic inhibition of HDACs and EZH2 will be reversible upon drug removal. To test this hypothesis, ECFCs were treated with a combination of GSK-343 and panobinostat. Then, treated ECFCs were harvested, washed, and re-plated in the absence of the epigenetic drug cocktail. Measuring gene expression and histone PTMs at several time points after re-plating, we observed both increased H3K27me3 and decreased H3-Ac at promoters of pro-angiogenic genes upon drug removal (Figures 5B and 5C). Furthermore, after 24 hr, the pro-angiogenic genes had reverted back to their native transcriptionally repressed status (Figure 5A). Taken together, these results confirm the transient nature of panobinostat-/GSK-343-mediated epigenetic modifications at pro-angiogenic genes in ECFCs.
Figure 5.

Gene Activation upon Treatment with a Combination of HDAC and EZH2 Inhibitors Is Reversible
(A) Transcript levels of indicated genes were measured by qRT-PCR at the indicated times after removal of the GSK-343/panobinostat drug combination. qRT-PCR values are expressed as mean percentages of vehicle-treated cells ±SEM with B2M serving as internal control. (n = 3).
(B and C) Enrichment of H3K27me3 (B) and H3 acetyl (C) histone marks is measured by ChIP-qPCR at the indicated times after removal of the GSK-343/panobinostat drug combination. qPCR values are expressed as mean fractions of input ±SD.
∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, non-significant.
Pharmacologic Inhibition of HDAC and EZH2 Increases ECFC-Mediated Vasculogenesis and Improves the Therapeutic Potential of Transplanted ECFCs in a Hindlimb Ischemia Mouse Model
The results described above revealed that several major pro-angiogenic transduction pathways are activated in ECFCs through pharmacologic inhibition of EZH2 and HDACs. To determine whether these effects translate into increased vasculogenesis in vivo, ECFCs were pretreated with GSK-343 and panobinostat prior to embedding into collagen matrices that were subcutaneously implanted into immunodeficient mice (Prasain et al., 2012). After 3 weeks, implants were harvested and analyzed for blood-vessel formation using a human-specific CD31 antibody as previously described (Prasain et al., 2012). This experiment revealed the formation of ECFC-derived micro-capillaries (Figure 6A) where the presence of surrounding murine smooth muscle cells suggests capillary maturation (Figure S6). Furthermore, the number of these human, ECFC-derived capillaries is significantly increased in implants from drug-treated ECFCs (Figure 6A, right panel), confirming the efficacy of combined GSK-343 and panobinostat treatment in improving ECFC-derived vasculogenesis in vivo.
Figure 6.

Pharmacologic Treatment with a Combination of EZH2 and HDAC Inhibitors Accelerates Vasculogenesis and Blood-Flow Recovery by Transplanted ECFCs in a Mouse Model of Hindlimb Ischemia
(A) H&E staining (left) and anti-human (h) CD31 staining (right) identify ECFCs that have formed micro-capillaries in collagen gels after 3 weeks of implantation in mice (scale bar, 200 μm). Numbers of fully closed ECFC-derived hCD31+ capillaries are indicated per measured explanted area as mean values ± SEM (n = 6 mice per group).
(B) Laser Doppler perfusion imaging was used to measure blood flow pre-operatively (pre-op), immediately after induction of hindlimb ischemia (day 0) and at the indicated time points after injection of ECFCs pretreated with vehicle or with the drug combination. Left: representative laser Doppler images (Isch., Ischemic leg; NI, non-ischemic leg). Right: perfusion ratios (Isch./NI) are indicated as mean values ± SEM (ECFCs: days 0–14, n = 7 mice; days 21–28, n = 3 mice. ECFCs treated: days 0–14, n = 8 mice; days 21–28, n = 4 mice).
(C) EZH2 and HDAC inhibitors upregulate multiple transduction pathways in ECFCs.
Schematic representation of cell-signaling molecules and processes that are upregulated (red) or downregulated (green) upon treatment of ECFCs with the GSK-343 and panobinostat combination.
∗∗p < 0.01; ∗p < 0.05. See also Figure S6.
Finally, we sought to determine the extent to which ex vivo treatment with epigenetic drugs enhances the vascular repair function of ECFCs. To address this question, we used a murine model of hindlimb ischemia (Limbourg et al., 2009, Palii et al., 2014), where ischemia is induced by ligation of the left femoral artery, followed by intramuscular injection of treated or non-treated ECFCs as cell therapy. This experiment revealed that ECFCs pretreated ex vivo with GSK-343 and panobinostat are more rapid at restoring blood flow in the ischemic leg compared with their non-treated counterparts (Figure 6B). Specifically, by measuring blood flow using Doppler analysis at several time points after cell therapy, we found that the kinetics of blood-flow recovery in the ischemic leg is significantly accelerated when GSK-343-panobinostat-treated ECFCs are used for transplantation (Figure 6B). This improvement is therapeutically relevant given that the speed of blood-flow recovery is one of the most important factors in saving organ function after acute ischemia (Callum and Bradbury, 2000, Creager et al., 2012). Furthermore, the improved perfusion persists for at least 4 weeks post transplantation in mice injected with either drug-treated or non-treated ECFCs (Figure 6B). Thus, pre-activation of ECFCs with a cocktail of epigenetic drugs (i.e., EZH2 and HDAC inhibitors) administered ex vivo is a promising strategy to increase the therapeutic potential of these cells and boost their vascular repair function in vivo through increased activity of multiple pro-angiogenic signaling pathways (Figure 6C).
Discussion
ECFCs are human endothelial progenitor cells with unique capabilities to form vascular structures in several animal models of ischemia (Palii et al., 2014, Prasain et al., 2014). However, these cells have not been fully characterized and additional strategies are needed to improve their therapeutic potential. Here, we show that ECFCs maintain several key pro-angiogenic pathways (i.e., NOTCH, WNT, CXCR4, VEGFR, and SHH) in a repressed/poised state through the establishment of bivalent gene promoters that are characterized by the simultaneous enrichment of active and repressive histone PTMs. This equilibrium is maintained by epigenetic enzymes with competing activities (i.e., EZH2/UTX and P300/HDAC1) that are co-bound to gene promoters, resulting in low expression levels. In vivo, ECFCs presumably require external signals from the environment to fully activate pro-angiogenic pathways in response to vascular injury. Interestingly, our results indicate that this activation process can be initiated ex vivo prior to transplantation through pharmacologic approaches with specific EZH2 and HDAC inhibitors. Importantly, this ex vivo “boosting” procedure that leads to the activation of multiple pro-angiogenic pathways in ECFCs is translated in vivo by enhanced vasculogenesis, angiogenesis, and accelerated vascular repair in a hindlimb ischemia model. Furthermore, the reversible nature of epigenetic modifications ensures that the injected cells remain responsive to signals from the environment, optimizing vascular repair to the host's specific needs and limiting potential adverse effects such as blood-vessel overgrowth.
In this study, we have focused on histone modifications enriched on promoters of specific genes that belong to pro-angiogenic pathways. Future studies, using genome-wide approaches, are necessary to identify the full extent of bivalent promoters in ECFCs. Furthermore, bivalent marks have also been identified on distal regulatory elements (Mikkelsen et al., 2007), suggesting that our epigenetic drug cocktail may also affect the epigenetic state of enhancers. A detailed understanding of enhancer/promoter communications in ECFCs, as determined by chromosome conformation capture (3C) approaches (Davies et al., 2017), will be required as a pre-requisite to appreciate the full extent of epigenetic changes occurring upon drug treatment and their consequences on gene expression and vascular repair.
Experimental Procedures
Cell Isolation and Culture
All procedures were approved by the Ottawa Hospital Research Ethics Board (2007804-01H). Human ECFCs were derived from umbilical cord blood obtained after informed consent from the Canadian Cord Blood for Research Program (2014.007) and cultured as previously described (Palii et al., 2014, Yoder et al., 2007). Briefly, mononuclear cells were isolated by Ficoll density gradient centrifugation and seeded onto CellBIND sux-well plates (Corning) at a density of 107 mononuclear cells/well in EGM-2 BulletKit medium (Lonza) supplemented with 10% fetal bovine serum. Individual ECFC colonies were re-plated separately and analyzed by flow cytometry (CD31+/CD34+/CD45−) after each passage. All experiments were performed with at least three biologically independent ECFC clones between passage 3 and 6.
Capillary-like Network Formation Assay
ECFCs, pretreated with epigenetic drugs or vehicle as indicated, were seeded into 96-well plates previously coated with 50 μL of phenol-red-free, growth factor reduced Matrigel (Corning) at a density of 5,000 cells/well in duplicates. After 24 hr, cells were stained with 5 μg/mL calcein (BD Bioscience) for 5 min at 37°C and photographed using an Axiovert S100 microscope (Zeiss) and AxioVision 4.6 software at 2.5× magnification. Total tube length was quantified using AngioQuant software as described (Fraineau et al., 2012).
Cell Migration Assay by Gap Closure
ECFC migration was measured by gap closure assay using the Radius 96-well cell migration assay (Cell Biolabs) according to the manufacturer’s instructions. Briefly, 2 × 104 ECFCs, pretreated with epigenetic drugs or vehicle, were seeded into a 96-well plate containing a hydrogel plug and allowed to adhere onto the plate. The plug was removed providing a cell-free area, and cell migration into the gap was monitored by taking pictures every 3 hr using an Axio Observer.A1 microscope (Zeiss) and AxioVision 4.6 software at 10× magnification. Once confluent, cells were labeled with crystal violet (Cell Biolabs). The cell-free area was measured using MATLAB software, which provides a simple graphical user interface through which a user can select an image to calculate the area automatically or manually. Automatic segmentation begins with gray-level thresholding of the input image, then SD filtering on the thresholded image, followed by a series of morphological operations to detect the cell-free area and calculate the area size in pixels.
Serum Starvation and Apoptosis Assay by Annexin V Staining
ECFCs were plated into 6-well plates containing EBM-2 medium without addition of EGM-2 SingleQuots (Lonza) and incubated overnight (16 hr) prior to harvesting and suspension in annexin V binding buffer (BD Pharmingen). Apoptosis was then assessed using the Annexin V PE Apoptosis Detection Kit I (BD Pharmingen) according to the manufacturer's instructions.
ECFC Transplant in NOD/SCID Mice
Three-dimensional cellularized collagen matrices were prepared as previously described using the Standardized Oligomer Polymerization Kit (GeniPhys, Indiana, USA) (Prasain et al., 2014). Briefly, type I collagen gels (1.5 mg/mL) were prepared on ice, mixed immediately with ECFCs (4 × 106 cells/mL) that had been treated (or not) with the epigenetic drug cocktail as indicated, and left to polymerize for 30 min at 37°C in a CO2 incubator prior to the addition of 500 μL of culture medium in the absence of GSK-343 and panobinostat. After 18 hr incubation, cellularized gels were implanted into the flanks of 6- to 12-week-old NOD/SCID mice, as described (Prasain et al., 2012). Surgical procedures were conducted under anesthesia and a constant supply of oxygen. Three weeks after implantation, gels were recovered from animals that had been humanely euthanized per approved IACUC protocol.
Histology was performed using H&E stain and immunohistochemistry using an anti-human CD31 antibody. Briefly, gel explants were formalin fixed, processed, and embedded in paraffin, followed by serial sectioning (4 μm) using a Leica microtome. Paraffin-embedded tissue sections were deparaffinized in xylene and rehydrated through a 100%–70% ethanol gradient. Heat-induced epitope retrieval was performed at 110°C, 12 min with EDTA buffer (pH 9.0), and 3% H2O2 was used to block endogenous peroxidases. Sections were blocked with Background Sniper reagent (BioCare Medical, CA, USA) and incubated with mouse anti-hCD31 Ab (DAKO, clone JC70A) for 1 hr at room temperature (1:2,000 dilution). The MACH4+ DAB detection system was used according to the manufacturer’s instructions (BioCare Medical, CA, USA). ECFC-derived hCD31+ blood vessels were imaged using an Aperio ScanScope XT (Leica Microsystems).
Mouse Model of Hindlimb Ischemia
ECFC-induced vascular repair was assessed using a hindlimb ischemia mouse model as described (Kuraitis et al., 2011, Palii et al., 2014). All procedures were approved by the University of Ottawa Animal Care Committee and performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. Ischemia was induced in the left hindlimb of 8-week-old athymic nude CD1 female mice by ligation of the proximal end of the femoral artery. ECFCs were injected immediately after skin closure. Specifically, 5 × 105 ECFCs, pretreated with the drug combination or vehicle as indicated, were re-suspended in 50 μL of PBS and delivered by two equivolumetric injections into the adductor muscle, downstream of the unilateral left hindlimb ligation site, using an insulin syringe. Blood perfusion was monitored pre-operatively, immediately after surgery, and at the indicated days post operation by laser Doppler perfusion imaging (moorLDI2; Moor Instruments). Results are expressed as the ratio of ischemic to non-ischemic hindlimb perfusion. For non-treated ECFCs, seven mice were monitored up to 14 days, three of which were maintained for up to 4 weeks to evaluate the persistence of blood perfusion. For drug-treated ECFCs, eight mice were monitored up to 14 days, four of which were maintained for up to 4 weeks to evaluate the persistence of blood perfusion.
RNA Extraction, Gene Expression Profiling by RNA Sequencing, and Bioinformatics Analysis
For RNA-seq, we used two independent ECFC clones (clone A and clone B) at passage 3 originating from two distinct cord blood samples, as well as clone A at passage 4 (i.e., clone A′). Clones A, A′, and B were treated with epigenetic drugs or vehicle independently of each other, and therefore represent three biological replicates. For each clone, total RNA was extracted from 106 ECFCs using the RNeasy Mini Kit (QIAGEN), including a genomic DNA digestion step with DNase I (QIAGEN), and analyzed by high-throughput sequencing as previously described (Benyoucef et al., 2016).
Chromatin Immunoprecipitation
Analysis of histone modifications was performed by native chromatin immunoprecipitation (ChIP) as described (Brand et al., 2008). Cofactor binding was assessed by crosslink ChIP as described (Palii et al., 2014). ChIPed DNA was purified by phenol-chloroform extraction, precipitated with ethanol, and specific DNA sequences were amplified with PerfeC TaSYBRGreen SuperMix (Quanta Biosciences) using a Rotor-Gene Q real-time PCR system (QIAGEN). ChIPed DNA quantity was calculated compared with a genomic DNA standard curve using Rotor-Gene Q Series Software 1.7 (QIAGEN) with R2 > 0.96.
Statistical Analyses
Data obtained from at least three independent experiments each in duplicate or triplicate were expressed either as mean values or percentages of control values ± SD or SEM. When indicated, statistical significance between data groups was determined by non-parametric Kruskal-Wallis test followed by Dunn’s post hoc test, or parametric one- or two-way ANOVA followed by Dunnett or Bonferroni post hoc test using GraphPad Prism 5 software. Use of the non-parametric Kruskal-Wallis test or ANOVA was determined depending on their distribution assessed by the Shapiro-Wilk normality test. Data are considered to be significantly different at p < 0.05. The p values are depicted with asterisks in the figures as follows: ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, non-significant; ND, non-determined.
Author Contributions
S.F. designed and performed most in vitro experiments, prepared cells for ischemia experiments, prepared figures, and contributed to the writing of the manuscript. C.G.P. designed and performed some in vitro experiments, prepared cells for ischemia experiments, and participated in collagen transplant experiments. B.M. designed and performed ischemia experiments under supervision of E.J.S. M.R. designed and performed some immunohistochemistry experiments under supervision of M.A.R. W.C.S. and N.P. designed and performed transplant of collagen matrices in NOD/SCID mice under supervision of M.C.Y. A.C. performed bioinformatics analyses. E.V. performed some in vitro experiments. K.R. participated in the initial drug screen. S.N. designed a program for analysis of cell migration under supervision of T.J.P. D.S.A. supervised the project. M.B. designed, coordinated, and supervised the project and contributed to the writing of the manuscript. All authors participated in data analyses and figure preparation and provided feedback on the manuscript.
Acknowledgments
We thank Y. Le, D. Yevstafiev, and C.-P. Chaturvedi (OHRI) for experimental help; S. Lavictoire, I.A.J. Lorimer, A.M. Gagnon, A. Sorisky, and W.L. Stanford (OHRI) for providing antibodies; and F.J. Dilworth (OHRI) for critically reading the manuscript. RNA sequencing was performed at the Institut de Recherches Cliniques de Montréal (Canada). This project was funded with grants from the Canadian Institutes of Health Research (CIHR) (MOP-82813 to M.B.); the Department of Medicine, University of Ottawa (to D.S.A.); and the Heart and Stroke Foundation of Canada (GIA-000225 to E.J.S.). S.F. is supported by a fellowship from the Cushing Fund.
Published: October 12, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2017.09.009.
Accession Numbers
RNA-seq data have been deposited in the public GEO database (GEO: GSE80819).
Supplemental Information
References
- Alphonse R.S., Vadivel A., Fung M., Shelley W.C., Critser P.J., Ionescu L., O'Reilly M., Ohls R.K., McConaghy S., Eaton F. Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation. 2014;129:2144–2157. doi: 10.1161/CIRCULATIONAHA.114.009124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu-Vieyra C.V., Berenson J.R. The potential of panobinostat as a treatment option in patients with relapsed and refractory multiple myeloma. Ther. Adv. Hematol. 2014;5:197–210. doi: 10.1177/2040620714552614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au P., Daheron L.M., Duda D.G., Cohen K.S., Tyrrell J.A., Lanning R.M., Fukumura D., Scadden D.T., Jain R.K. Differential in vivo potential of endothelial progenitor cells from human umbilical cord blood and adult peripheral blood to form functional long-lasting vessels. Blood. 2008;111:1302–1305. doi: 10.1182/blood-2007-06-094318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyoucef A., Brand M. Epigenetic gene regulation and stem cell function. In: Huang S., Litt M.D., Blakey C.A., editors. Epigenetic Gene Expression and Regulation. Elsevier; 2015. pp. 149–181. [Google Scholar]
- Benyoucef A., Palii C.G., Wang C., Porter C.J., Chu A., Dai F., Tremblay V., Rakopoulos P., Singh K., Huang S. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev. 2016;30:508–521. doi: 10.1101/gad.276790.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvard C., Gafsou B., Dizier B., Galy-Fauroux I., Lokajczyk A., Boisson-Vidal C., Fischer A.M., Helley D. alpha6-integrin subunit plays a major role in the proangiogenic properties of endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010;30:1569–1575. doi: 10.1161/ATVBAHA.110.209163. [DOI] [PubMed] [Google Scholar]
- Brand M., Rampalli S., Chaturvedi C.P., Dilworth F.J. Analysis of epigenetic modifications of chromatin at specific gene loci by native chromatin immunoprecipitation of nucleosomes isolated using hydroxyapatite chromatography. Nat. Protoc. 2008;3:398–409. doi: 10.1038/nprot.2008.8. [DOI] [PubMed] [Google Scholar]
- Callum K., Bradbury A. ABC of arterial and venous disease: acute limb ischaemia. BMJ. 2000;320:764–767. doi: 10.1136/bmj.320.7237.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P., Jain R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y., Fish J.E., D'Abreo C., Lin S., Robb G.B., Teichert A.M., Karantzoulis-Fegaras F., Keightley A., Steer B.M., Marsden P.A. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J. Biol. Chem. 2004;279:35087–35100. doi: 10.1074/jbc.M405063200. [DOI] [PubMed] [Google Scholar]
- Chavakis E., Urbich C., Dimmeler S. Homing and engraftment of progenitor cells: a prerequisite for cell therapy. J. Mol. Cell. Cardiol. 2008;45:514–522. doi: 10.1016/j.yjmcc.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Cooke J.P., Losordo D.W. Modulating the vascular response to limb ischemia: angiogenic and cell therapies. Circ. Res. 2015;116:1561–1578. doi: 10.1161/CIRCRESAHA.115.303565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creager M.A., Kaufman J.A., Conte M.S. Clinical practice. Acute limb ischemia. N. Engl. J. Med. 2012;366:2198–2206. doi: 10.1056/NEJMcp1006054. [DOI] [PubMed] [Google Scholar]
- Cui K., Zang C., Roh T.Y., Schones D.E., Childs R.W., Peng W., Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J.O., Oudelaar A.M., Higgs D.R., Hughes J.R. How best to identify chromosomal interactions: a comparison of approaches. Nat. Methods. 2017;14:125–134. doi: 10.1038/nmeth.4146. [DOI] [PubMed] [Google Scholar]
- Dejana E. The role of wnt signaling in physiological and pathological angiogenesis. Circ. Res. 2010;107:943–952. doi: 10.1161/CIRCRESAHA.110.223750. [DOI] [PubMed] [Google Scholar]
- Di Croce L., Helin K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013;20:1147–1155. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M., Clarke C., Marks P.A. Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- Fadini G.P., Losordo D., Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ. Res. 2012;110:624–637. doi: 10.1161/CIRCRESAHA.111.243386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraineau S., Monvoisin A., Clarhaut J., Talbot J., Simonneau C., Kanthou C., Kanse S.M., Philippe M., Benzakour O. The vitamin K-dependent anticoagulant factor, protein S, inhibits multiple VEGF-A-induced angiogenesis events in a Mer- and SHP2-dependent manner. Blood. 2012;120:5073–5083. doi: 10.1182/blood-2012-05-429183. [DOI] [PubMed] [Google Scholar]
- Fraineau S., Palii C.G., Allan D.S., Brand M. Epigenetic regulation of endothelial-cell-mediated vascular repair. FEBS J. 2015;282:1605–1629. doi: 10.1111/febs.13183. [DOI] [PubMed] [Google Scholar]
- Helin K., Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–488. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- Herbert S.P., Stainier D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011;12:551–564. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram D.A., Mead L.E., Tanaka H., Meade V., Fenoglio A., Mortell K., Pollok K., Ferkowicz M.J., Gilley D., Yoder M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104:2752–2760. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- Karmodiya K., Krebs A.R., Oulad-Abdelghani M., Kimura H., Tora L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics. 2012;13:424. doi: 10.1186/1471-2164-13-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Huang L., Critser P.J., Yang Z., Chan R.J., Wang L., Carlesso N., Voytik-Harbin S.L., Bernstein I.D., Yoder M.C. Notch ligand Delta-like 1 promotes in vivo vasculogenesis in human cord blood-derived endothelial colony forming cells. Cytotherapy. 2015;17:579–592. doi: 10.1016/j.jcyt.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraitis D., Hou C., Zhang Y., Vulesevic B., Sofrenovic T., McKee D., Sharif Z., Ruel M., Suuronen E.J. Ex vivo generation of a highly potent population of circulating angiogenic cells using a collagen matrix. J. Mol. Cell. Cardiol. 2011;51:187–197. doi: 10.1016/j.yjmcc.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Kwon S.M., Eguchi M., Wada M., Iwami Y., Hozumi K., Iwaguro H., Masuda H., Kawamoto A., Asahara T. Specific Jagged-1 signal from bone marrow microenvironment is required for endothelial progenitor cell development for neovascularization. Circulation. 2008;118:157–165. doi: 10.1161/CIRCULATIONAHA.107.754978. [DOI] [PubMed] [Google Scholar]
- Le Bras A., Vijayaraj P., Oettgen P. Molecular mechanisms of endothelial differentiation. Vasc. Med. 2010;15:321–331. doi: 10.1177/1358863X10371685. [DOI] [PubMed] [Google Scholar]
- Limbourg A., Korff T., Napp L.C., Schaper W., Drexler H., Limbourg F.P. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat. Protoc. 2009;4:1737–1746. doi: 10.1038/nprot.2009.185. [DOI] [PubMed] [Google Scholar]
- Lin R.Z., Moreno-Luna R., Li D., Jaminet S.C., Greene A.K., Melero-Martin J.M. Human endothelial colony-forming cells serve as trophic mediators for mesenchymal stem cell engraftment via paracrine signaling. Proc. Natl. Acad. Sci. USA. 2014;111:10137–10142. doi: 10.1073/pnas.1405388111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Weisdorf D.J., Solovey A., Hebbel R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Invest. 2000;105:71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R.J., O'Neill C.L., Sweeney M., Guduric-Fuchs J., Gardiner T.A., Simpson D.A., Stitt A.W. Molecular analysis of endothelial progenitor cell (EPC) subtypes reveals two distinct cell populations with different identities. BMC Med. Genomics. 2010;3:18. doi: 10.1186/1755-8794-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero-Martin J.M., Khan Z.A., Picard A., Wu X., Paruchuri S., Bischoff J. In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood. 2007;109:4761–4768. doi: 10.1182/blood-2006-12-062471. [DOI] [PubMed] [Google Scholar]
- Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda T.B., Cortez C.C., Yoo C.B., Liang G., Abe M., Kelly T.K., Marquez V.E., Jones P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K., Vlachojannis G.J., Koyanagi M., Boeckel J.N., Urbich C., Farcas R., Bonig H., Marquez V.E., Zeiher A.M., Dimmeler S. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ. Res. 2011;109:1219–1229. doi: 10.1161/CIRCRESAHA.111.247304. [DOI] [PubMed] [Google Scholar]
- Palii C.G., Vulesevic B., Fraineau S., Pranckeviciene E., Griffith A.J., Chu A., Faralli H., Li Y., McNeill B., Sun J. Trichostatin a enhances vascular repair by injected human endothelial progenitors through increasing the expression of TAL1-dependent genes. Cell Stem Cell. 2014;14:644–657. doi: 10.1016/j.stem.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Park C., Kim T.M., Malik A.B. Transcriptional regulation of endothelial cell and vascular development. Circ. Res. 2013;112:1380–1400. doi: 10.1161/CIRCRESAHA.113.301078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K.M., Gerecht S. Harnessing developmental processes for vascular engineering and regeneration. Development. 2014;141:2760–2769. doi: 10.1242/dev.102194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasain N., Lee M.R., Vemula S., Meador J.L., Yoshimoto M., Ferkowicz M.J., Fett A., Gupta M., Rapp B.M., Saadatzadeh M.R. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nat. Biotechnol. 2014;32:1151–1157. doi: 10.1038/nbt.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasain N., Meador J.L., Yoder M.C. Phenotypic and functional characterization of endothelial colony forming cells derived from human umbilical cord blood. J. Vis. Exp. 2012 doi: 10.3791/3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinisch A., Hofmann N.A., Obenauf A.C., Kashofer K., Rohde E., Schallmoser K., Flicker K., Lanzer G., Linkesch W., Speicher M.R. Humanized large-scale expanded endothelial colony-forming cells function in vitro and in vivo. Blood. 2009;113:6716–6725. doi: 10.1182/blood-2008-09-181362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saif J., Schwarz T.M., Chau D.Y., Henstock J., Sami P., Leicht S.F., Hermann P.C., Alcala S., Mulero F., Shakesheff K.M. Combination of injectable multiple growth factor-releasing scaffolds and cell therapy as an advanced modality to enhance tissue neovascularization. Arterioscler. Thromb. Vasc. Biol. 2010;30:1897–1904. doi: 10.1161/ATVBAHA.110.207928. [DOI] [PubMed] [Google Scholar]
- Schwarz T.M., Leicht S.F., Radic T., Rodriguez-Arabaolaza I., Hermann P.C., Berger F., Saif J., Bocker W., Ellwart J.W., Aicher A. Vascular incorporation of endothelial colony-forming cells is essential for functional recovery of murine ischemic tissue following cell therapy. Arterioscler. Thromb. Vasc. Biol. 2012;32:e13–21. doi: 10.1161/ATVBAHA.111.239822. [DOI] [PubMed] [Google Scholar]
- Shantsila E., Watson T., Lip G.Y. Endothelial progenitor cells in cardiovascular disorders. J. Am. Coll. Cardiol. 2007;49:741–752. doi: 10.1016/j.jacc.2006.09.050. [DOI] [PubMed] [Google Scholar]
- Tan J., Yang X., Zhuang L., Jiang X., Chen W., Lee P.L., Karuturi R.K., Tan P.B., Liu E.T., Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S.K., Tian X., LaFrance L.V., Duquenne C., Suarez D.P., Newlander K.A., Romeril S.P., Burgess J.L., Grant S.W., Brackley J.A. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 2012;3:1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P., Tee W.W., Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder M.C., Mead L.E., Prater D., Krier T.R., Mroueh K.N., Li F., Krasich R., Temm C.J., Prchal J.T., Ingram D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.