

Epithelial-to-mesenchymal transition (EMT) is a cellular program that operates in the context of embryogenesis, wound-healing and carcinoma pathogenesis to drive epithelial cells toward a mesenchymal state. During carcinoma progression, EMT enables the cells forming these tumours to acquire the traits of highly malignant cells, notably motility, invasiveness and an ability to disseminate to form distant metastases. Indeed, a number of published reports have associated EMT with a variety of malignant carcinoma cells. Recently, however, Zheng et al. (1) reported that in genetically engineered mouse models of pancreatic adenocarcinoma development, carcinoma cells could metastasize without activating EMT programs. Their conclusions, if sustained by the evidence presented, would prompt a major change in how we conceptualize malignant progression and metastasis of carcinoma cells, including the neoplastic cells in human carcinomas.
To assess the role of EMT in metastasis, Zheng et al. deleted Snail or Twist – two important EMT-associated transcription factors (EMT-TFs). Despite this deletion, neither conditional knockout (CKO) exhibited a decrease in the rate of metastasis of pancreatic carcinoma cells. The authors concluded that EMT is dispensable for metastasis based on their claim that deletion of either Snail or Twist significantly attenuates EMT. However, evidence that EMT programs were eliminated is lacking in their report, and thus this claim is not supported by their data.
It has been well-established that EMT is not a single, stereotypical program but instead has multiple manifestations, many of which endow epithelial cells – both normal and neoplastic – with a variety of traits normally expressed by mesenchymal cells (2, 3). Importantly, EMT is achieved through the action of multiple context-dependent signals and EMT-TFs; furthermore, cells activating an EMT program may lose their epithelial properties and acquire invasive properties by progressing only part way through an EMT program (“partial EMT”). These facts help to understand the EMT programs that likely persisted in the pancreatic carcinoma cells studied by Zheng et al.
To prove that EMT is indeed suppressed in the CKO tumours, Zheng et al. used a lineage traced mouse model of pancreatic cancer similar to the model previously reported by Rhim et al. (4), in which all carcinoma cells are unambiguously marked by the fluorescent protein YFP. They show that tumours in TwistcKO animals exhibited a significant reduction in the percentage of YFP+ cells that co-expressed the mesenchymal marker α-smooth muscle actin (αSMA) relative to tumours with an intact Twist locus. At face value, this would seem to be a good indicator that the EMT was indeed suppressed in the pancreatic carcinoma cells. However, unlike several other mesenchymal markers, αSMA expression is rarely induced upon activation of EMT in this mouse tumour model (Fig. 1).
Figure 1.
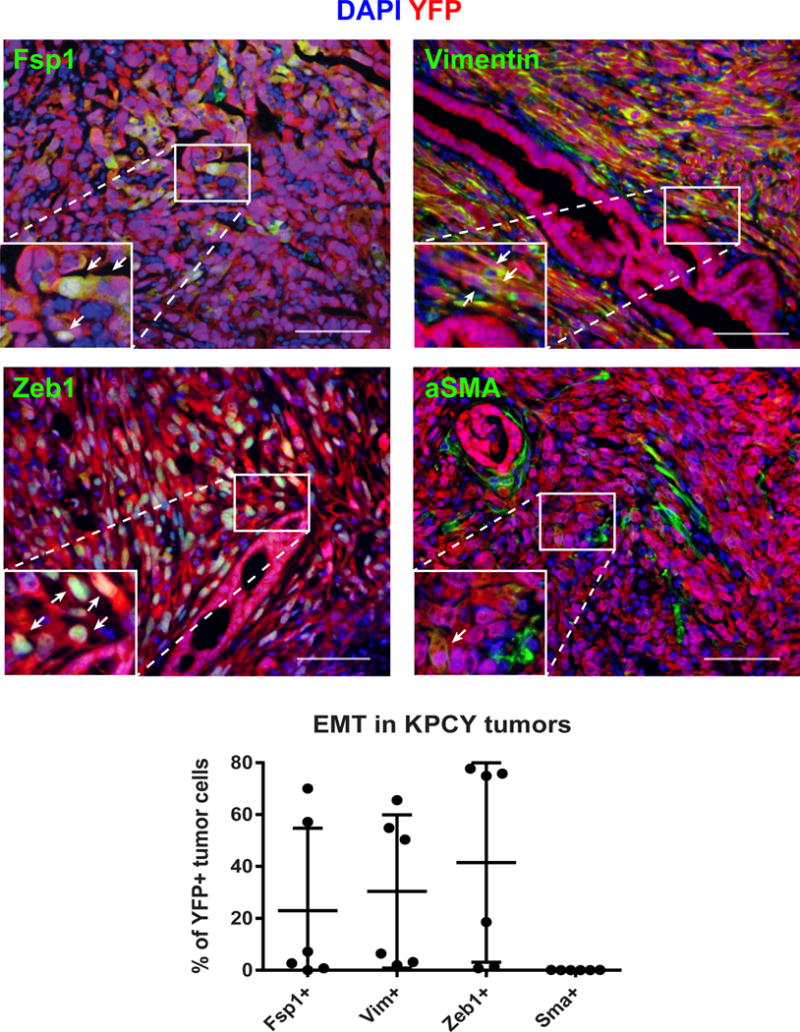
Six pancreatic tumours from KPCY mice (4) were stained for YFP (tumour cells, red) and the mesenchymal markers Fsp1, vimentin (Vim), Zeb1, or αSMA (green). The analysis included three poorly differentiated tumors (with a high degree of EMT) and three well differentiated tumors (with a low degree of EMT) to reflect the heterogeneity seen in the KPCY model; all images were taken from the poorly differentiated tumors. Although there was wide variation in the frequency of Fsp1, Vimentin, and Zeb1 staining in the tumour cells, virtually all of the αSMA staining was confined to the non-tumour (YFP-negative) stroma. This was true in both well-differentiated and poorly differentiated tumours (arrows indicate YFP+ tumour cells expressing the indicated marker; αSMA+YFP+ cells were extremely rare). These data were quantified in the graph below, demonstrating that αSMA expression is uncommon in the pancreatic carcinoma cells including those that have undergone an EMT. A minimum of 2,500 tumour cells were counted per mouse for each marker.
Although the authors also stained for other EMT markers, including Zeb1, Zeb2, Slug, and Sox4, these stainings were done without marking the tumour cells with a lineage-specific tracer. Consequently, one cannot discern whether any particular detected staining was present in a carcinoma cell or in a non-neoplastic stromal cell that was recruited to a pancreatic tumour; for this reason, the impact on EMT of the Snail and the Twist deletion remains undefined. Indeed, there is abundant evidence that the tumours described in Zheng et al. continued to exhibit EMT phenotypes despite loss of either Snail or Twist. For example, poorly differentiated regions of these tumours – histologically defined zones reflecting EMT – were undiminished upon Snail or Twist deletion. Moreover, the tumour cells of the primary tumours still expressed considerable levels of the other EMT-TFs, as shown for the Zeb1, Sox4, and Slug EMT-TFs following deletion of either Snail or Twist.
Likewise, in carcinoma cells isolated from both of these primary CKO tumours, quantitative PCR demonstrated that expression of the Zeb1 and Slug mRNAs was reduced by ~2 fold, a decrease that is plausibly insignificant functionally; importantly, expression of the mesenchymal markers vimentin and N-cadherin was only slightly down-regulated and the down-regulation was statistically insignificant.
Thus, while the results of Zheng et al. are interesting and may speak to redundancy within the transcriptional network that defines and orchestrates EMT in pancreatic carcinomas, the authors nevertheless failed to achieve what they set out to do, namely to completely suppress activation of EMT. Hence, the conclusion that EMT is not required for metastatic dissemination cannot be sustained, simply because the authors’ genetic manipulations failed to suppress expression of versions of the EMT program. Accordingly, we continue to embrace the notion that pancreatic carcinoma cells utilize an EMT program or programs during metastatic dissemination.
METHODS
Three well differentiated and three poorly differentiated tumours from LSL-Kras; p53fl/+; Pdxl-Cre; Rosa-YFP (KPCY) animals were embedded in paraffin following overnight fixation. Sections were stained with antibodies against GFP (Abcam ab6673) and either αSMA (Sigma F3777), Fsp1 (DAKO A5114), Vimentin (Cell Signaling D21H3), or Zeb1 (Santa Cruz H102) using standard techniques. Images were obtained with an Olympus IX71 fluorescent microscope and quantified using Fiji software. Staining for αSMA with Abcam ab5694 (as employed by Zheng et al.) gave similar results.
Acknowledgments
We thank Amine Sahmoud for assistance in preparing the figure.
Footnotes
Author Contributions
N.A. produced and analysed the experimental data; T.B., Y.K., M.A.N., R.A.W., and B.Z.S. conceived and wrote the manuscript.
Competing Financial Interests Declared none
References
- 1.Zheng X, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 3.Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342:1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- 4.Rhim AD, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]