

Abstract
The mammalian liver is one of the most regenerative tissues in the body, capable of fully recovering mass and function after a variety of injuries. This factor alone makes the liver unusual among mammalian tissues, but even more atypical is the widely held notion that the method of repair depends on the manner of injury. Specifically, the liver is believed to regenerate via replication of existing cells under certain conditions and via differentiation from specialized cells—so-called facultative stem cells—under others. Nevertheless, despite the liver’s dramatic and unique regenerative response, the cellular and molecular features of liver homeostasis and regeneration are only now starting to come into relief. This review provides an overview of normal liver function and development and focuses on the evidence for and against various models of liver homeostasis and regeneration.
Keywords: liver regeneration, homeostasis, injury, cellular reprogramming
INTRODUCTION
Because vertebrate animals can live for decades, they must be able to maintain and repair tissues across long time intervals. With few exceptions, vertebrate organs undergo a regular process of renewal whereby aged cells are replaced by new cells over the life of the animal—the phenomenon of tissue homeostasis. Upon injury, some tissues have an additional capacity to increase rates of cellular turnover, leading to the accelerated formation of new cells that are needed to repair underlying damage through regeneration.
During evolution, vertebrates adopted a range of cellular mechanisms for maintaining normal homeostasis and for achieving tissue recovery following injury. These mechanisms vary across species and cell types, but they fall into three general categories: (a) replication of existing cells, (b) expansion and differentiation of stem/progenitor cells, and (c) transdifferentiation (i.e., cellular reprogramming) or dedifferentiation of cells from one cell type to another. Replication is straightforward: Bone, kidney, cartilage, and other tissues use simple replication as the major mechanism of repair. Indeed, tissues in which replicative potential is limited—e.g., the central nervous system and heart—also have a limited ability to regenerate. Stem cells are distinctive cells that have the ability to self-renew (give rise to more stem cells) and to differentiate into other cell types. Both of these properties become progressively restricted with development, resulting in a stem/progenitor hierarchy based on the extent of differentiation potency (1). The skin, intestine, and blood are the archetypal examples of tissues that continually generate new cells from stem cells, but there remains significant uncertainty concerning which other tissues use stem cells (2). Finally, transdifferentiation and dedifferentiation involve changes in cellular identity upon injury. These processes are most readily observed in amphibians and fish, in which terminally differentiated cells acquire new identities (3, 4), but emerging data suggest that some mammalian tissues may exhibit similar cellular plasticity (5–7).
The mammalian liver has several features that make it an interesting case study for regeneration. Under homeostatic conditions, the liver has a low rate of cellular turnover, with cells persisting for weeks to months without dividing. Following injury, by contrast, the liver can produce vast numbers of new cells. The liver is most famous for its ability to regenerate following partial resection of the organ, rapidly recovering its preinjury size through a combination of cellular hypertrophy and cell division. By contrast, liver injuries that occur in the absence of a surgical intervention are thought to mobilize a different kind of regenerative response—one dependent upon stem/progenitor cells. Specifically, exposure to these more physiological injuries—toxins, viral infection, and immune attack—results in the production of vast numbers of small epithelial cells with presumed progenitor activity. Below, we review the basic functions of the mammalian liver, describe how it develops during embryogenesis and early postnatal life to accomplish those functions, and consider the current state of thinking regarding the mechanisms that promote regeneration in this essential organ.
LIVER FUNCTION AND STRUCTURE
General
The liver participates in multiple functions related to digestion, detoxification, fluid and electrolyte balance, and hemostasis. These activities can be roughly divided into (a) synthetic function, (b) bile secretion, and (c) detoxification. The essential nature of these functions become apparent in patients with liver cirrhosis (scarring) or liver failure, in whom inadequate bile drainage, faulty detoxification, or defects in synthesis of plasma proteins result in the clinical stigmata of liver disease. These functions of the liver are carried out by its two parenchymal epithelial cell types: hepatocytes and biliary epithelial cells (BECs, also known as cholangiocytes).
Hepatocytes
Hepatocytes constitute approximately 60% of the liver by cell number and approximately 80% by mass. They are cellular factories, equipped with abundant mitochondria and endoplasmic reticulum to produce large quantities of albumin, clotting factors, and other serum proteins. Among the products of hepatocytes are bile acids, the amphipathic products of cholesterol metabolism. In addition to these synthetic activities, hepatocytes play a major role in detoxification. Molecules absorbed from the intestine are carried by blood vessels that feed the portal vein, a pattern of circulation that causes almost all potentially toxic compounds to pass through the liver (so-called first-pass metabolism). Hepatocytes contain a vast arsenal of detoxifying enzymes (collectively termed the P450 enzymes) that recognize and modify a wide variety of chemicals, allowing for their elimination in bile or urine. In addition to processing toxins and drugs, hepatocytes have other metabolic functions, including glutamine synthesis, urea formation, and gluconeogenesis.
Biliary epithelial cells
The other major cells involved in liver functions are BECs. These small cuboidal cells line bile ducts, the conduits for the passage of bile from the hepatocyte to the intestine. Small bile ducts embedded deep within the liver are termed intrahepatic bile ducts (IHBDs), whereas the larger bile ducts that exit the liver and fuse with the intestine are termed extrahepatic bile ducts (EHBDs). BECs are less metabolically active than hepatocytes but have some metabolic functions, including bicarbonate synthesis.
Nonparenchymal cells
Like all epithelial tissues, the liver contains a number of nonepithelial cell types that perform various other functions. These other cells are collectively referred to as the nonparenchymal cells (NPCs) of the liver to distinguish them from the parenchymal cell populations (i.e., hepatocytes and BECs). The most important of the NPCs are the fibroblasts, whose extracellular matrix gives the liver its substance, thereby maintaining normal liver architecture (see below). There are two types of matrix-producing cells in the liver: stellate cells and portal fibroblasts. Both cell types are thought to contribute to liver fibrosis and cirrhosis following chronic injury, although murine lineage-tracing studies suggest that stellate cells are the major contributor (8). In addition to their fibrogenic effects, both cell types have an array of additional functions (9). Importantly, these cells, as well as other NPCs—endothelial cells and macrophages (Kupffer cells)—are not derived from hepatocytes or BECs and arise instead from nonendodermal origins (10, 11).
Liver Architecture
All of the liver’s functional activity occurs within microscopic functional units termed lobules (Figure 1a). Importantly, the lobular arrangement—with proper placement of hepatocytes, BECs, and NPCs, including capillary endothelial cells—permits the liver to conduct its diverse synthetic, metabolic, and detoxifying activities.
Figure 1.
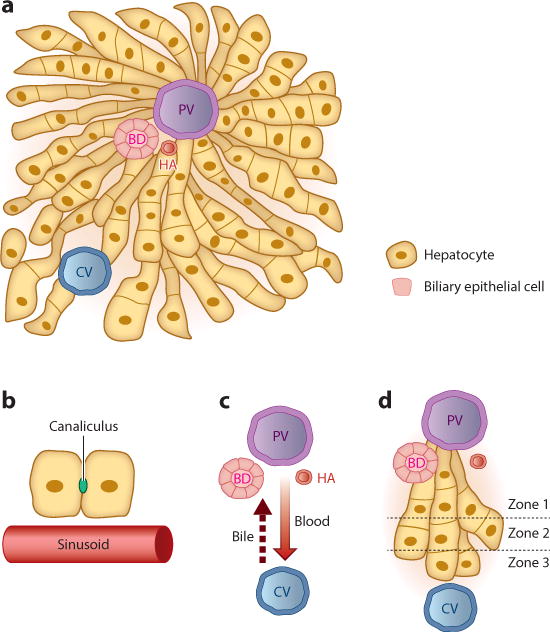
The liver lobule. (a) The liver is composed largely of two endoderm-derived cell types—hepatocytes and biliary epithelial cells—that are organized into functional units known as lobules. Blood enters the lobule through branches of the PV and HA and exits through branches of the CV to be returned to the systemic circulation. Bile makes its way to the BD, which in turn drains into the intestine to aid in digestion. (b) Blood passes through the lobule in specialized vascular channels (sinusoids). Bile is handled separately and is secreted into thin channels (canaliculi) between hepatocytes that carry the bile through the lobule and into the BD. (c) There is countercurrent flow of blood and bile: Blood flows in a portal-to-central direction, whereas bile flows in a central-to-portal direction. (d) The lobule is divided into zones, with hepatocytes closest to the PV composing zone 1 and those closest to the CV composing zone 3. Hepatocytes in different zones have different sets of functions. Abbreviations: BD, bile duct; CV, central vein; HA, hepatic artery; PV, portal vein.
Lobules
The liver is unique among mammalian organs in that it is fed independently by two blood supplies. As noted above, the portal vein and its branches are the major circulatory source, delivering roughly two-thirds of the liver’s blood supply. The remaining third comes from branches of the hepatic artery, which in turn is a branch of the celiac artery. Venous blood and arterial blood from these two sources mix together as they percolate through a series of liver capillaries known as the sinusoids, until they come in contact with branches of the central vein, the vascular egress system of the liver.
In parallel, hepatocytes synthesize and transport bile acids into the canalicular space—a tiny channel formed by indented apical membranes on two adjacent hepatocytes—where they combine with cholesterol and bilirubin to form bile (Figure 1b). Bile passes from the canaliculi into the bile ducts before draining into the small intestine, where it aids in the emulsification of dietary fats. Bile acids are reabsorbed by the distal small intestine and returned to the liver, a circuit that constitutes the enterohepatic recycling system. In contrast to blood, which moves in a portal-to-centrilobular direction, bile moves in a centrilobular-to-portal direction, resulting in a countercurrent pattern of flow (Figure 1c).
Zonation and ploidy
Although hepatocytes located in different parts of the lobule have a similar appearance, hepatocytes located near the portal tracts carry out functions that are often different from those of more centrally located hepatocytes. On the basis of these distinctions, the lobule can be conceptually divided into three regions, or zones: zone 1, which contains hepatocytes close to the portal tracts; zone 3, which contains hepatocytes close to the central veins; and zone 2, which contains hepatocytes in between the portal tracts and central veins (Figure 1d). Importantly, many of the functional differences that distinguish hepatocytes in these three zones concern metabolism. For example, hepatocytes in the periportal region (zone 1) are engaged principally in gluconeogenesis, whereas those in the pericentral region (zone 3) conduct glycolysis. Likewise, nitrogen and fatty acid metabolism are also zonally regulated, as zone 1 hepatocytes are engaged in urea production and β-oxidation of fatty acids, whereas zone 3 hepatocytes use glutamine synthetase to remove nitrogen and engage in lipogenesis. Zonation is under the control of Wnt signaling (12). Whether disrupting the liver’s normal zonal organization has major physiological consequences remains unclear.
Another interesting feature of the liver is the high frequency of hepatocyte polyploidy, the presence of more than two genomes per cell. Polyploidy is the result of defective cytokinesis, leading to tetraploid or octoploid cells that can be either mononucleated or binucleated. The process of polyploidization is regulated and under developmental control, with more than half of all hepatocytes becoming polyploid by 4–5 weeks of age (13). Activation of insulin-Akt signaling at the time of weaning seems to be critical for this emergence of polyploidy (14). Although the precise function of polyploidy remains to be determined, it has been proposed that polyploidy (along with resulting aneuploidy) may allow subsets of hepatocytes to adapt to chronic injury (15).
LIVER DEVELOPMENT
“It is important to consider tissue responses to injury in the context of development, since the mechanisms that ensure…” Considering tissue responses to injury in the context of development is important; the mechanisms that ensure proper tissue differentiation, morphogenesis, and growth in the embryo are often reused during regeneration. The liver has long been a favored organ for studying developmental mechanisms, both because of its size (which simplifies biochemical studies) and because of the availability of distinctive differentiation markers. As a result, much is known about the signals leading to proper liver formation. A comprehensive treatment of the subject is beyond the scope of this review (the reader is referred to several excellent papers on the topic, including References 16 and 17). Here we outline some of the key morphological and molecular events that occur during normal liver development.
Specification
Like other parts of the gastrointestinal tract, the liver is embryonically derived from the endoderm germ layer. In the French biologist Nicole Le Douarin performed a series of pioneering studies that laid out the nature of signaling that leads to the formation of the liver. Using the presence of hepatic glycogen granules as a readout for cells that had been assigned to become liver, Le Douarin microdissected small pieces of chick endoderm and mesoderm and cultured each alone or together. Only when the endoderm was cultured together with mesoderm from the future heart did glycogen granules appear (18). This result indicated that signals from adjacent cells induce endoderm cells to commit to a liver fate. Moreover, these signals act before any overt manifestations of liver differentiation are present. This developmental step is known as specification, and subsequent studies revealed the identity of the signals involved in liver specification to include members of the fibroblast growth factor (FGF) and transforming growth factor β (TGFβ) families of soluble factors (19). These signals provide cues to the newly formed cells, tutoring them to become liver cells as opposed to any of the other fates available to endoderm cells (e.g., pancreatic or intestinal cells). Among the critical steps in liver specification is the induction of two FoxA transcription factors—FoxA1 and FoxA2—within the hepatic domain (20). Most, if not all, of the early steps in liver specification can now be recapitulated in vitro by adding soluble factors, in the correct combination and sequence, to embryonic stem cells (ESCs) (21).
Differentiation and Morphogenesis
Following specification, committed liver cells leave the endoderm and stream into the adjacent mesoderm, forming a primordial liver bud. Although this bud lacks any recognizable tissue architecture, the cells within it—hepatoblasts—represent a proliferative precursor compartment. Importantly, hepatoblasts are bona fide liver progenitor cells, as they have the capacity to differentiate into either hepatocytes or BECs. During the fetal period, the liver becomes the major source of hematopoiesis, and nests of differentiating blood cells are apparent throughout midgestation. Also during this period, branches of the portal vein expand into the liver parenchyma, bringing with them a collection of fibroblast-like cells known as the portal vein mesenchyme. These vascular branches establish the location of future portal tracts, as bile ducts and hepatic arteries form secondarily following the pattern laid down by the portal veins.
Formation of the IHBDs, like liver specification, is the result of signaling between two distinct cell types. The signals for bile duct development again emerge from a mesoderm derivative—the portal vein mesenchyme—and act upon an endoderm derivative (hepatoblasts). Since the late 1990s, it has been known that cues involving the Notch signaling pathway are pertinent to bile duct development, as patients with a bile duct paucity condition known as Alagille syndrome harbor mutations in the Notch pathway ligand Jagged1 (22, 23). Subsequent work has delineated the relevant signaling molecules and cellular relay in the differentiation of BECs: a two-step process in which Jagged1 expressed on portal tract mesenchymal cells confers a biliary differentiation signal (24). Because Jagged1, like all Notch ligands, is membrane associated, only those hepatoblasts immediately adjacent to the portal vein mesenchyme receive a Notch signal. Hence, a ring of Notch2-expressing hepatoblasts surrounding the portal veins known as the ductal plate receive this biliary signal (25, 26).
During IHBD development, differentiation and morphogenesis are coordinated. Following formation of the ductal plate, lumens appear at discrete locations to give rise to primitive ductal structures lined asymmetrically by BECs on one side and hepatoblasts on the other (27, 28). Several signals, including the Notch, TGFβ, Wnt, and Hippo pathways, ensure normal IHBD development (29–32). With time, primitive ductal structures resolve into mature bile ducts that are lined symmetrically by BECs. Importantly, the EHBDs have distinct origins, arising from a patch of Sox17-expressing endoderm cells in the ventral pancreatic region (33). This developmental arrangement seems crucial for ensuring that ductal systems of the liver and pancreas merge proximally to their connection with the intestine.
Maturation and Growth
As we see from the above discussion, hepatocytes are responsible for a number of distinct functions. Although embryonic hepatoblasts contain many of the transcription factors that define adult hepatocytes, they require further maturation to carry out their specialized functions. Among the critical factors involved in hepatocyte differentiation is the nuclear factor HNF4α, as HNF4α-deficient embryos initiate liver development but fail to activate transcription of liver-specific genes (34). Further investigation has revealed the existence of a self-reinforcing network of liver-enriched transcription factors that includes HNF4α, HNF6, HNF1α, FoxA2, and others (35). Notably, although embryonic inactivation of HNF4α results in reduced hepatic gene expression, inactivation in the adult has a minimal effect (36). This result indicates that the transcriptional network that defines mature hepatocytes becomes stable and less dependent on any single transcription factor over time. MicroRNAs also play a role in hepatocyte differentiation and maturation, as exemplified by the likely participation of miR-122 in a positive feedback loop regulating HNF6 expression (37).
An increasingly powerful tool for studying hepatocyte maturation and biology is the use of ESCs and iPSCs (induced pluripotent cells). These two types of pluripotent cells—derived from blastocyst-stage embryos and from the introduction of defined factors into terminally differentiated cells, respectively—can be coaxed to pass through the various stages of hepatocyte differentiation, ultimately yielding cells with some features of mature hepatocytes (38). This approach has been useful for modeling human liver diseases in vitro, resulting in the identification of additional requirements for hepatocyte maturation (e.g., cellular aggregation and cAMP signaling) (39). Moreover, such methods permit the identification of small molecules that can induce stem cell–derived hepatocytes—termed iHep cells—to undergo maturation in vitro (40), an approach that may have therapeutic implications for augmenting or modifying hepatocyte function in the future.
CELLULAR HOMEOSTASIS IN THE NORMAL LIVER
In contrast to tissues with a high degree of cellular turnover—such as the skin, intestine, and blood—the liver has a low rate of turnover. At any given time, less than 1–2% of hepatocytes are cycling, with the remainder resting in a quiescent (G0) state (41). Nevertheless, hepatocytes retain a dramatic ability to reenter the cell cycle upon injury, although this replicative ability decreases with age (42). Tissues maintain cellular homeostasis primarily by two mechanisms: replication of existing cells and differentiation from stem/progenitor cells. In the liver, the mechanisms underlying normal homeostasis, and the extent to which stem/progenitor cells versus simple replication governs the process, have fascinated investigators for decades, with strong proponents for and against both models of homeostasis.
One model for liver homeostasis, posed more than three decades ago, is the so-called streaming liver hypothesis (43), which posits that hepatocytes near the portal tracts have an enhanced replicative potential relative to all other hepatocytes. According to the hypothesis, the progeny of these cells stream in a portal-to-central fashion so that over time the entire lobule comes to be derived from this periportal population. In 2011, Furuyama and colleagues (44) used a tamoxifen-inducible lineage-tracing system to provide dramatic evidence for the streaming liver hypothesis. The authors reported that, over a period of weeks to months, lineage-traced cells expanded across the lobule (in a portal-to-central direction), in some cases occupying the vast majority of the liver. Taken at face value, these data suggested that the bulk of the liver parenchyma is derived from the much smaller biliary compartment.
However, most studies have not supported the streaming liver model. For example, β-galactosidase-expressing hepatocytes transplanted into the liver remain in the same location for more than a year (45), and radioactive tracing experiments show no evidence for hepatocyte movement (46). In one recent study, more than 99% of hepatocytes were labeled and assessed for a decrease in the labeling index, as would be expected if new hepatocytes were robustly generated from BECs (47). No such decrease was observed, consistent with the notion that new hepatocytes come from preexisting hepatocytes. Likewise, lineage-tracing studies in which BECs were labeled using independent methods have also failed to find evidence that BECs give rise to hepatocytes under homeostatic conditions (48, 49). Thus, at present there is limited evidence for liver streaming, and it appears that under homeostatic conditions that the liver is maintained by simple replication of existing cells.
TYPES OF LIVER DAMAGE
In contrast to the relative tranquility of the normal liver, the damaged liver abounds with activity. Parenchymal cell death provokes a number of adaptive cellular changes, including infiltration of inflammatory cells, activation of hepatic stellate cells and/or portal fibroblasts, and vascular alterations. It is worth briefly considering some of the insults to which the human liver is routinely subjected.
Patterns of Injury
There are many different causes of liver damage. These can be grouped into various categories depending on the timing of the insult and the types of cells most severely affected. As the nature of injury often dictates patient outcome, these patterns of injury have important clinical implications.
Acute versus chronic
Acute liver injury is a common clinical problem. In the most severe cases, acute injury results in overwhelming hepatocyte loss, a clinical condition known as acute liver failure. More frequently, however, acute injuries are self-limiting (assuming that the underlying pathophysiological process resolves) and thus generally do not result in long-term damage. By contrast, chronic liver injuries occur over years, leading to liver dysfunction through the development of severe scarring, or cirrhosis. Although patients can live with chronic liver disease for long periods of time, progressive damage that results in loss of more than ∼70–80% of hepatocyte function can also result in liver failure. A common consequence of chronic liver injury from all causes is the development of hepatocellular carcinoma.
Hepatitis versus cholestasis
Liver injuries can also be categorized on the basis of the pattern of damage: injuries that result principally in hepatocyte death (hepatitis) and those that result principally in defective drainage of bile (cholestasis). Hepatocyte injury, or hepatitis, is diagnosed clinically by the presence of the hepatocyte proteins AST and ALT in the bloodstream, whereas cholestasis is diagnosed by the presence of bilirubin, a heme breakdown product that is normally cleared in bile.
Agents of Injury
Given the vast number of insults to which the liver is routinely exposed, the tissue has developed a number of contingencies for dealing with such injuries. Here, we list the most common categories of liver injury, only some of which are routinely modeled in rodent studies.
Toxin-induced injury
Substances absorbed from the intestine are taken up by the intestinal (splanchnic) vasculature and are delivered to the liver via the portal vein. Thus, foreign substances undergo so-called first-pass metabolism before they reach other organs in the body. Some hepatotoxins, including alcohol and many commonly used drugs, directly cause damage to hepatocytes. However, other toxins become toxic only as a result of enzymatic modification by the liver’s detoxification machinery—the P450 enzymes. For example, acetaminophen (Tylenol) has no toxic properties on its own but becomes noxious upon conversion to the mitochondrial toxin NAPQI (N-acetyl-p-benzoquinone imine) through the activity of the P450 enzyme. Toxins can cause injury to hepatocytes, BECs, or both. P450 enzymes are most abundant in zone 3 hepatocytes, accounting for the higher rate of drug toxicity in that portion of the lobule. Toxins can cause either acute or chronic damage, with the most common offenders being alcohol, acetaminophen, antibiotics, and nonsteroidal anti-inflammatory drugs.
Viral hepatitis
Viral agents ranging from the common (e.g., Epstein-Barr virus, HIV) to the obscure (e.g., echovirus, measles virus) can infect the liver as part of a systemic infection, typically causing short-term damage with few long-term consequences. By contrast, the so-called hepatitis viruses—hepatitis A, B, C, and E viruses—have a more specific tropism for the liver. Infection with hepatitis A or E viruses typically results in an acute, self-limited illness, whereas hepatitis B and C viruses tend to establish chronic infection, leading to a protracted inflammatory state. Like chronic toxin injury, chronic hepatitis B or C infection can lead to cirrhosis, hepatocellular carcinoma, and liver failure.
Biliary obstruction
Conditions that result in bile duct damage or obstruction lead to the backup of bile, or cholestasis. Gallstones are responsible for most cases of acute bile duct obstruction in the Western world. However, many other conditions cause cholestasis, including immunological diseases that target BECs (e.g., primary biliary cirrhosis and primary sclerosing cholangitis), chronic obstruction due to tumor or parasitic infection, and congenital malformation of the biliary system. Because bile acids can have toxic effects in high concentrations, hepatocyte damage is a secondary consequence of chronic biliary obstruction.
Metabolic, vascular, and immune-mediated injury
A variety of other disorders—including metabolic disturbances, vascular abnormalities, and immune attack—can lead to liver injury. As the liver is one of the major sites of fatty acid synthesis, excess fat accumulation in the liver (steatosis) commonly occurs in the setting of obesity. With additional factors, steatosis can result in hepatocyte death (steatohepatitis), a major source of liver injury in the Western world. Liver damage can also be caused by vascular conditions, particularly clots (e.g., vascular thrombosis), hyperimmunity (e.g., autoimmune hepatitis), and immune incompatibility (e.g., graft-versus-host disease).
THE CELLULAR RESPONSE TO LIVER DAMAGE
As noted above, the liver has a vigorous injury response. This phenomenon has important clinical implications, as most patients who suffer acute liver injury fully recover function. This exquisite ability to recover raises several important questions. (a) How does the liver know that it has been damaged—what signals stimulate the tissue to initiate a regenerative response? (b) Which cells drive the regenerative response—does regeneration involve stem/progenitor cells or differentiated cells? (c) What terminates the regenerative response—what causes cessation of regeneration upon recovery of mass or function? (d) What accounts for failures in regeneration—why are patients with chronic liver damage no longer able to recover function?
Rodent Models of Injury
These questions of cellular and molecular mechanism are difficult to address in humans. Hence, most of our mechanistic understanding of liver regeneration comes from animal studies assessing the effects of acute or chronic damage to hepatocytes and/or biliary cells. A major advantage of these animal models has been the ability to exploit genetic tools—including gene knockout and lineage-tracing technology—to delineate how the liver responds to injury. One important point is that the commonly used injury models represent only a fraction of the types of injuries to which the human liver is normally subjected. Thus, the liver’s response to some of the most common forms of injury—particularly viral hepatitis—has not been well studied in animal models. Moreover, attempts to create robust models of cirrhosis have largely fallen short; for the most part, chronic toxin exposure leads to fibrosis but does not lead to the severe decline in liver function seen in human cirrhotic patients.
Surgical injury
The first experimental system to explore liver injury and regeneration was surgical removal of a portion of the liver, so-called partial hepatectomy (PHx) (50). In rodents, removing approximately two-thirds of the organ (three of the five rodent lobes) is technically straightforward. Following this procedure, the remaining liver tissue expands rapidly so that liver mass is fully recovered within a week of surgery. This response is stereotyped, consisting of waves of growth and proliferation of hepatocytes followed by proliferation of other cell types in the liver (BECs, endothelial cells, and stellate cells) (51, 52). Following 30% PHx, by contrast, liver re-generation occurs largely through cellular growth rather than through cellular proliferation (51). Another widely used surgical model is bile duct ligation (BDL). In this procedure, the common bile duct is tied off with a surgical suture, resulting in a backup of bile, hepatocellular injury, BEC proliferation, and stellate cell activation (53). BDL—with or without concomitant toxin treatment—reproducibly causes liver fibrosis in mice (54).
Toxin-mediated injury
In the wild, the liver is not normally exposed to a surgeon’s scalpel; thus, PHx is a nonphysiological model of liver injury (modeling obstruction via BDL is more physiological). In nature, the liver is routinely exposed to an array of natural compounds with hepatotoxic activity. Two of the first toxins to be used experimentally were the carcinogens 2-acetylaminofluorene (2-AAF) and ethionine (55, 56). Subsequently, a host of hepatotoxins, including carbon tetrachloride (CCl4) (57), 5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) (58), and choline-deficient diet supplemented with ethionine (CDE) (59), were employed in rodent models. Although these toxins result in different patterns of injury, it was recognized early on that a common feature following ingestion of most dietary toxins is the emergence of small, biliary-type cells of uncertain origins and differentiation potentials (60).
Replication and Growth of Existing Cells
Following Higgins & Anderson’s (50) description of PHx in 1931, removing a portion of the liver became the preeminent model for studying liver regeneration. As a result, hepatocyte proliferation—the dominant mechanism for liver regrowth following surgical resection—has been the focus of most liver regeneration studies (52). The prevailing view has been that hepatocyte replication is the liver’s preferred method for regeneration. The high efficiency of hepatocyte replication in response to a variety of injuries was recently documented by Malato and colleagues (47), who used genetic labeling to show that, following a variety of injuries, most hepatocytes come from preexisting hepatocytes.
Surprisingly, and in contrast to the case of the robust proliferative capacity of hepatocytes in vivo, growing hepatocytes in vitro is quite difficult; upon plating, primary hepatocytes rapidly lose their functional properties (40). This discrepancy between the behavior of cells in vivo and ex vivo provides an important cautionary note: Inferring cellular performance in the tissue on the basis of studies in culture is not always possible. Similarly, cellular differentiation states do not remain stable in vitro, and thus studies examining lineage relationships using tissue culture techniques may not reflect the situation in vivo.
Contributions from Putative Liver Stem/Progenitor Cells
Given the liver’s dramatic regenerative capacity, it did not take long for investigators to imagine that the liver might use stem cells for regeneration. However, the liver’s normally low rate of proliferation, and the finding that most hepatocytes synthesize DNA following PHx, prompted a need for an alternative to the standard stem cell model. This alternative came in the form of the facultative stem cell hypothesis, a model in which stem cells are not used during homeostasis but are called into service only upon injury (61).
In the mid-1950s, Farber (55) and Popper et al. (56) described the emergence of small oval-shaped cells in response to hepatotoxins, a tissue response known as the ductal reaction. Such cells arise in many or most models of hepatocarcinogenesis or toxin-induced injury (62, 63) and have been given various names, including ductular hepatocytes, intermediate hepatobiliary cells, and atypical ductal cell proliferation, to reflect the fact that they often have mixed features of hepatocytes and BECs (58, 64–68). Oval cells are characterized by an oval shape, small size, scant cytoplasm, and small lumens (69–71). On the basis of these observations, oval cells gradually came to be viewed as the presumptive facultative stem cells of the liver (72, 73).
Oval cells are thought to emerge from cells that reside in a specialized portion of the lobule known as the canal of Hering. Schematically, liver lobules are commonly portrayed as polygonal or hexagonal structures, with the portal tracts at the periphery and the central veins at the center (Figure 2a). At higher resolution, the canal of Hering can be observed as the transitional zone where hepatocyte-lined canaliculi drain into BEC-lined ducts. The canal of Hering is a tubular unit lined by hepatocytes on one side and by BECs on the other, and this strategic position has led many investigators to conclude that oval cells arise from BECs that exist in this location (Figure 2a) (74).
Figure 2.
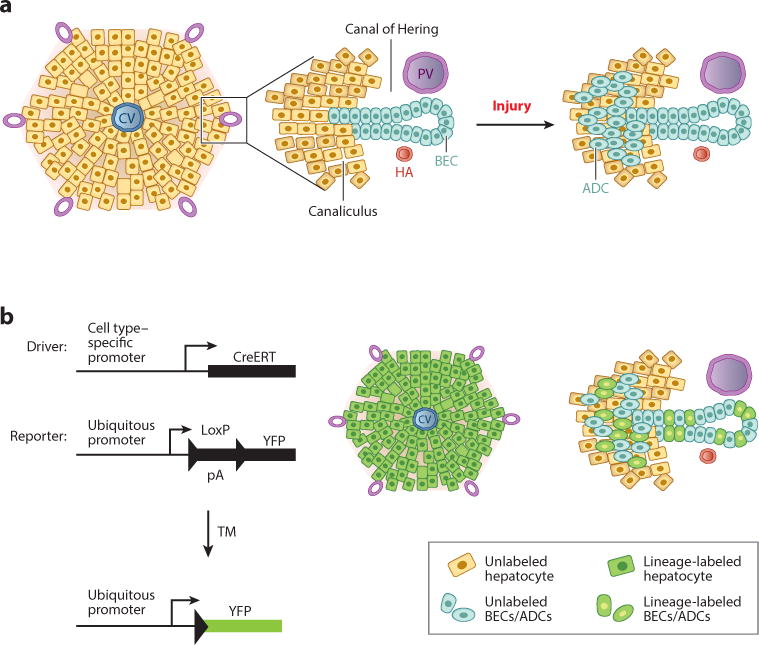
Use of lineage tracing to study cellular dynamics in liver regeneration. (a) (Left) Schematic view of a liver lobule, with a CV in the center and the portal tracts at the periphery. (Middle) An enlargement of a portal tract reveals the canal of Hering: a transitional zone between the canaliculi and the bile ducts that is asymmetrically lined by hepatocytes and BECs. (Right) Upon injury, ADCs (oval cells) emerge near the portal tracts. (b) In vivo Cre-based lineage tracing requires the use of a Cre-expressing genetic driver, which provides a means of marking cells in a specific manner, and a reporter, which allows for the detection of cells that have been marked as a result of Cre activity. In one iteration, a TM-inducible Cre (CreERT) becomes active only in the presence of the synthetic estrogen analog TM, allowing for labeling of cells (green) that have active CreERT transcription from the cell type–specific promoter employed. On the right are examples of the types of labeling that could be envisioned with a driver that permits labeling of hepatocytes with high efficiency (key, top) or with one that permits labeling of ADCs and BECs with medium efficiency (key, bottom). Abbreviations: ADC, atypical ductal cell; BEC, biliary epithelial cell; CV, central vein; HA, hepatic artery; pA, RNA polyadenylation signal; PV, portal vein; TM, tamoxifen; YFP, yellow fluorescent protein.
Three assays, each with its own advantages and disadvantages, can be used to document stem cell activity: clonogenic (in vitro) growth, cellular transplantation, and lineage tracing (1). For example, clonogenic growth (the ability to derive clones from single cells in culture) is technically straightforward and can provide information about self-renewal and the capacity for differentiation, the two defining features of stem cells. Indeed, much of the evidence that oval cells act as bona fide liver progenitor cells comes from in vitro studies (reviewed in References 72, 75, and 76). Importantly, however, in vitro assays may not accurately reflect the in vivo situation, and there are many reasons for cautiously interpreting such studies. Cellular transplantation experiments have also supported the facultative stem cell hypothesis, as transplanted oval cells appear competent to differentiate into hepatocytes (77). However, transplantation assays can also have pitfalls, including difficulties obtaining pure populations of cells for transplant and the possible confounding effects of cellular fusion, which can give a false impression regarding cellular potential (78). For these reasons, I will focus on the lineage-tracing methods that have been used to study putative liver stem/progenitor cells.
One of the first applications of lineage tracing to study liver regeneration in vivo came from Tatematsu et al. (79), who examined cellular dynamics in rats subjected to the combination of PHx and 2-AAF treatment. By giving the injured rats 3H-thymidine, which labels dividing cells and their progeny, the authors labeled most oval cells but few hepatocytes (a consequence of the antiproliferative effects of 2-AAF). Over time, very few hepatocytes became labeled with 3H-thymidine, leading to the hypothesis that oval cells and hepatocytes do not have a precursor-product relationship. By contrast, a similar study by Evarts et al. (80) came to the opposite conclusion: 3H-thymidine first appeared in oval cells and subsequently in hepatocytes. Importantly, the PHx/2-AAF injury paradigm—which is considered by some to be the gold standard for oval cell biology (because of the block in hepatocyte replication)—cannot be studied in mice because mouse cells lack the N-sulfotransferase that renders 2-AAF active (81).
Over the past several years, there has been a burst of activity in the use of lineage tracing with the application of Cre-Lox methodology (Figure 2b). When used carefully, Cre-Lox technology is a powerful technique for determining the origin of new cells. In particular, a variant of Cre recombinase fused to the estrogen receptor (CreER™) permits cell labeling in a temporally controlled manner (Figure 2b) (82). Specifically, when a cell type–specific promoter is used to mark a specific population of cells, one can rigorously trace the progeny of such cells to determine lineage.
Several groups have used Cre-based lineage tracing to study the contribution of progenitor cells to hepatocyte neogenesis. The first and most dramatic was a report by Furuyama and colleagues (44), who used Sox9 as a biliary-specific marker for lineage tracing. In this study, biliary cells were labeled with Sox9-CreER™ under homeostatic or injury conditions. Remarkably, the authors observed that labeled Sox9+ cells differentiated into hepatocytes under homeostatic conditions, with more than 80–90% of hepatocytes becoming labeled over prolonged periods (months). Thus, taken at face value, this paper offered strong support for the notion that virtually all hepatocytes are repopulated over time from a small population of Sox9-expressing biliary cells.
Subsequent studies have resulted in more tempered claims of stem/progenitor cell activity. As noted above, Malato et al. (47) marked a large percentage of hepatocytes and looked for evidence of newly born unlabeled hepatocytes, which might be evidence that new hepatocytes were being generated from nonhepatocytes. Although no decrease was observed under homeostatic conditions, injury with CCl4 (but no other injury conditions) resulted in a 1–2% decrease, consistent with a small contribution of progenitors to hepatocyte neogenesis in this setting (47). Similarly, Español-Suñer et al. (48) used the biliary marker osteopontin (OPN) to label oval cells during various injury paradigms and found that with CDE diet (but not with CCl4 or DDC treatment) a small percentage (1–2%) of hepatocytes originate from OPN+ cells during injury or following recovery. At even further odds with Furuyama et al. (44), a recent study using an orthogonal approach to label Krt19+ BECs, hepatocytes, and rapidly cycling cells found no evidence that hepatocytes arise from nonhepatocytes under homeostatic or injury conditions (83).
In addition to studies utilizing the known BEC markers Sox9, OPN, and Krt19, there have been attempts to find markers that would specifically identify stem/progenitor cells. One such putative marker is Foxl1, which is undetectable in normal liver but is strongly induced following BDL or DDC treatment (84). A fraction of Foxl1+ cells can be expanded in culture and differentiated into cells with features of either hepatocytes or BECs (85). Similarly, the Wnt target gene Lgr5 is expressed by liver cells selectively upon injury. Originally identified as a marker of rapidly dividing intestinal stem cells (86), Lgr5 has been reported to be a marker of other stem/progenitor cells in the gastrointestinal tract (87), and Lgr5+ cells isolated from the liver can grow and expand in culture and give rise to cells with features of BECs or hepatocytes (88). Similar clonogenic and differentiation properties have also been observed for cells expressing the cell surface markers MIC1-1C3 (89), EpCAM (90), or CD133 (91, 92). Notably, Lgr5+ cells expanded clonally in vitro can give rise to hepatocytes upon transplantation into a conditioned host in vivo (88).
Thus, some studies report that stem/progenitors play a minor (or even major) role in liver homeostasis or regeneration, and other studies find negligible evidence. What accounts for these discrepancies? One explanation is technical and is related to the fact that Cre-Lox-based labeling relies on tissue-specific promoters. The interpretation of all Cre-Lox experiments is thus subject to promoter specificity. Moreover, Cre-based lineage-tracing experiments are extraordinarily sensitive (because only a single Cre molecule needs to act for a cell and all of its progeny to be irreversibly marked). Thus, if any of the aforementioned reagents (Sox9-CreER, OPN-CreER, Krt19-CreER, and Lgr5-CreER) label even a small number of hepatocytes in addition to BECs, one could be left with the impression that new hepatocytes are derived from BECs when they are merely derived from previously labeled hepatocytes. The critical nature of the specificity of labeling is exemplified by the fact that Tarlow et al. (93), using a Sox9-CreER different from the one employed by Furuyama et al. (44), found that almost no hepatocytes were derived from Sox9+ cells in the setting of injury with CCl4, DDC, or CDE diet.
Although oval cells appear to have a limited (if any) capacity to differentiate into hepatocytes with these classical methods of injury, their activity may be different under other conditions. As discussed above, the PHx/2-AAF model does not efficiently block hepatocyte proliferation in mice, and biliary cells might be able to undergo conversion to hepatocytes if such a state were achieved. This notion is supported by the finding that, following near-total loss of hepatocytes in zebrafish, new hepatocytes are derived mostly from BECs rather than from preexisting hepatocytes (94, 95). A similar paradigm has been observed in pancreatic islets, where insulin-producing β cells form through the replication of preexisting β cells under most circumstances (96) but can be derived from glucagon-producing α cells following extreme β cell loss (5).
Transdifferentiation or Reprogramming
As noted above, cells with features of both “...into another. Although the presumption has been that cells transition from a biliary state to hepatocyte state, an alternative explanation, of course, is that...” hepatocytes and BECs are readily observed after injury, a sign that one cell type is turning into another; the presumption is that cells transition from a biliary state to a hepatocyte state. An alternative explanation, of course, is that cells change identity in the opposite direction, undergoing conversion from hepatocytes into BECs. Michalopoulos et al. (97) were the first to provide in vivo support for this relationship through a series of rat experiments in which marked hepatocytes were transferred into recipients treated with the biliary toxin methylene diamiline. Upon transplantation, the marker was present in BECs, suggesting that the hepatocytes had undergone biliary transdifferentiation (97). Transdifferentiation of hepatocytes into BECs has also been documented in unmanipulated mouse livers using Cre-Lox-based lineage tracing (6). In response to a variety of injuries (including toxins and BDL), hepatocytes activate a biliary program characterized by the early BEC markers OPN and Sox9; with time, such cells become polarized, coalesce around central lumens, and express the terminal BEC marker Krt19 (6, 98, 99).
MOLECULAR MECHANISMS OF LIVER REGENERATION
Mechanisms of Hepatocyte Replication
Liver regrowth following PHx has been the major liver regeneration paradigm. As such, it has been the subject of hundreds, if not thousands, of studies, making it a topic well beyond the scope of this review. Nevertheless, a few points are worth mentioning. First, it is well established that recovery of hepatocyte mass following PHx is achieved through the growth and proliferation of the remaining hepatocytes (51), a process mediated by the core cell cycle machinery, particularly cyclin D1 and cyclin E (70). Remarkably, hepatocytes seem to have an almost unlimited replicative capacity, as serial transplantation studies estimate that hepatocytes can undergo at least 69 cell doublings (corresponding to a 7 × 1020–fold expansion) (100). As noted above, hepatocytes are frequently polyploid. As a consequence, dividing hepatocytes form error-prone, multipolar mitotic spindles, resulting in aneuploidy of a large fraction of both murine and human hepatocytes under normal conditions (101, 102).
A second important feature of liver regeneration concerns the mechanism of liver size control. Despite decades of study, the signals that permit the liver to know that it is too small—resulting in the activation of the cell cycle regulators noted above—remain unclear. Likewise, it is unknown how the liver terminates these programs of growth and proliferation after it has fully recovered its mass. One attractive model is that specific growth factors, particularly epidermal growth factor (EGF) and hepatocyte growth factor (HGF), that are trapped in the extracellular matrix of the liver become mobilized upon PHx, leading to mitotic signaling (52). This model is supported by the finding that genetic ablation of the receptor for either EGF or HGF results in impaired regeneration following PHx (103, 104). A number of accessory or priming pathways with a role in regeneration following PHx, including pathways involving tumor necrosis factor (105), interleukin-6 (106), and bile acids (107), have also been reported. There appears to be considerable redundancy in the signals that drive liver regrowth, as even the most successful attempts to block the process delay but do not abrogate regeneration. Thus, it will be particularly interesting to see whether double mutants lacking both EGF signaling and HGF signaling can regenerate liver mass, as these signaling pathways were previously shown to be redundant in other settings (108).
Third, it is unclear whether all hepatocytes have an equivalent replicative capacity or whether a certain subset of hepatocytes has an enhanced capacity to divide. The above discussion shows that there is significant functional heterogeneity among hepatocytes, depending on their zonal position within the lobule, and it is intriguing to think that this heterogeneity extends to liver-repopulating activity, as has been suggested (51). This provocative possibility is further supported by the observation that Wnt-responsive zone 3 hepatocytes exhibit a greater capacity for cell division than does the bulk population (B. Wang & R. Nusse, personal communication). Such enhanced replicative potential may operate during both normal liver homeostasis and the regenerative response.
Mechanisms of Ductal Expansion and Reprogramming
As discussed above, hepatocyte-to-BEC reprogramming frequently accompanies toxin-mediated biliary injury. Although the molecular mechanisms underlying this transition in cellular identity remain poorly understood, some general principles have emerged. The first comes from the finding that Notch signaling—which controls the hepatoblast cell fate decision to become a hepatocyte or a BEC—is also involved in the conversion of adult hepatocytes into BECs. Misexpression of the intracellular domain of Notch1, which confers a constitutive Notch signal, leads to the conversion of hepatocytes into BECs (6, 98). Conversely, deletion of either of the essential Notch signaling mediators RBP-Jκ and Hes1 blunts reprogramming (6, 98). Interestingly, reprogramming occurs in a gradient fashion within the lobule, with periportal hepatocytes having a high propensity to become BECs and pericentral hepatocytes exhibiting complete resistance (6). This finding suggests that zone 3 cells adjacent to the central vein either lack a necessary reprogramming signal or are exposed to an inhibitor of reprogramming. In vitro studies have suggested that HGF and EGF play a role in biliary reprogramming (109), and thus these factors may influence Notch responsiveness.
The Hippo pathway, an evolutionarily conserved signaling module involved in tissue growth control, also appears to mediate biliary reprogramming. Hippo signaling regulates proliferation and survival in a wide variety of tissues, including the liver, through the transcriptional coactivator YAP (110–112), with liver cancer a highly penetrant consequence of dysregulated YAP activity (113). The Hippo pathway also plays an important role in normal bile duct development, as liver-specific deletion of YAP results in bile duct paucity (30). Recent work has shown that hepatocyte-specific misexpression of YAP leads to hepatocyte-to-BEC reprogramming through a Notch-dependent mechanism (114). Thus, two pathways that depend on direct cell-cell contact, the Notch and Hippo pathways, seem to be important regulators of plasticity, whereby hepatocytes can be converted into BECs.
Finally, what upstream signals drive the biliary response to injury? Toxins induce many changes in the liver, including leukocyte infiltration, qualitative changes in the vasculature, myofibroblast activation, and disruption of the extracellular matrix; thus, many candidate mediators (presumably Notch and Hippo ligands among them) may influence ductal expansion and hepatocyte-to-BEC reprogramming. Fgf7—produced by Thy1+ mesenchymal cells—appears to be one critical mediator, as Fgf7-deficient mice had a completely blunted ductal reaction following injury (115).
SUMMARY AND IMPLICATIONS FOR THERAPY
The liver has a well-organized lobular structure that subserves its many functions in metabolism, synthesis of critical biomolecules, and detoxification, making it indispensable for life. No artificial devices come close to replacing the function of the liver, and none are on the horizon. The field has focused for many years on liver progenitor cells: cells not utilized during normal liver homeostasis but that are called into action when necessary to repair damage. Cells with clonal growth properties can be readily expanded in culture from injured livers, and these cells may find some clinical application in the future. However, such progenitors seem to have a limited role in vivo under most injury conditions.
What lessons can we take from understanding how the liver regenerates so robustly? A recurrent theme in the field of liver regeneration is the central role of the hepatocyte, which seems capable of replicating to an almost unlimited degree. Given this capacity for expansion, it is important to recognize that the failure of regeneration in the setting of cirrhosis—the most clinically significant cause of liver dysfunction or liver failure—probably has more to do with environment (e.g., fibrosis) than with problems intrinsic to the parenchymal cells of the liver. To the extent that hepatocytes and BECs retain normal function in cirrhosis, therefore, one must question the rationale for cell therapy: What is the utility of introducing healthy cells into an unhealthy environment? Moreover, cell therapy–based approaches will need to overcome the challenge of creating a normal structure in a diseased microenvironment. Thus, for now at least, it is hard to imagine using cell therapy for clinical applications beyond the treatment of inborn errors of metabolism, for which delivering a fraction of normally functioning hepatocytes can result in clinical benefit (116).
One interesting finding is the recent observation that the adult liver retains substantial plasticity into adulthood, with hepatocytes poised to take on a ductal phenotype under a variety of circumstances. This phenotypic may be a protective mechanism, whereby a loss of differentiation may allow hepatocytes to endure certain toxic conditions, or it may serve as fuel for the formation of a more extensive ductal system to more efficiently clear the toxin. In either case, these new findings should spur liver research to look for innovative alternatives to transplantation of cells into a diseased liver, including but not limited to making livers at ectopic sites (117).
WHICH CELLS GIVE RISE TO LIVER CANCER?
This so-called cell-of-origin question for cancer presupposes that only certain cells in normal tissues are capable of giving rise to a given type of cancer. The two types of cancer in the liver—hepatocellular carcinoma (HCC) and cholangiocarcinoma (CC)—have been presumed to arise from hepatocytes and BECs, respectively. However, other possibilities exist. For example, it has been proposed that cancers can arise from resident tissue stem/progenitor cells, with the notion that such cells already harbor the molecular machinery for self-renewal. Notwithstanding remaining questions about the existence of functional stem/progenitor cells in the mammalian liver, and the extraordinary self-renewal capacity of normal hepatocytes, a progenitor origin for liver cancer cannot be excluded. Nevertheless, the cell of origin of HCC remains unknown. More recently, mouse lineage-tracing experiments have suggested that CC can arise from hepatocytes. Studies using Cre-based technology to mark hepatocytes have shown that hepatocytes, and not BECs, are the source of experimental CCs induced by Notch and Akt overexpression (118) or chemical carcinogens (119). Notch activation alone leads to HCC (120), indicating that the specific mutational spectrum plays an important role in dictating tumor histology.
SUMMARY POINTS.
The liver has a three-dimensional structure that underlies its normal function.
During development, hepatocytes and BECs arise from bona fide progenitor cells: hepatoblasts.
Liver injury has many etiologies, including toxin-mediated damage, autoimmunity, and viral infection.
The liver is maintained during normal homeostasis through replication of existing cells.
Growth and replication of existing cells also account for recovery following most types of liver injury.
Hepatocytes may differ with respect to their replicative capacity, and nonhepatocytes may give rise to hepatocytes under particular injury conditions.
The adult liver retains significant cellular plasticity, with adult hepatocytes capable of transdifferentiating into BECs.
FUTURE ISSUES.
How does the liver achieve its intricate three-dimensional lobular architecture?
What are the defining epigenetic characteristics of hepatocytes and BECs that make them distinct?
How does the liver know what size to become, either normally or during regeneration?
Under which circumstances do liver stem/progenitor cells emerge and contribute to repair?
What accounts for failures in liver regeneration? Why don’t cirrhotic livers regenerate?
Acknowledgments
I am grateful to my colleagues at the University of Pennsylvania, particularly Ken Zaret, Klaus Kaestner, Mike Pack, and Becky Wells, for helpful discussions regarding liver biology. This review was supported in part by grant R01-DK083355 from the NIH/NIDDK and by a grant from the Pew Charitable Trusts.
Glossary
- Homeostasis
normal tissue turnover, whereby new cells are generated at a pace that equals the rate of cell death
- Regeneration
the reacquisition of cellular mass and function following tissue injury
- Stem cells
cells having the capacity to create more stem cells (self-renewal) and to differentiate into other cell types (differentiation)
- Progenitor cells
cells that have self-renewal and differentiation properties, but to a lesser extent than do stem cells
- Cellular reprogramming, or transdifferentiation
the process by which cells are interconverted between two terminally differentiated states
- Dedifferentiation
the process by which a terminally differentiated cell acquires less differentiated properties (e.g., conversion to a progenitor state)
- Parenchymal cells
hepatocytes and cholangiocytes (referred to as biliary epithelial cells), the functional cells of the liver
- Nonparenchymal cells (NPCs)
all other cellular components other than parenchymal cells; NPCs include fibroblasts (stellate cells and portal fibroblasts), macrophages (Kupffer cells), and endothelial cells
- Liver zonation
each liver lobule is divisible into different regions, or zones, with varying metabolic functions with respect to glucose or nitrogen metabolism
Footnotes
The Annual Review of Physiology is online at physiol.annualreviews.org
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Slack JM. Origin of stem cells in organogenesis. Science. 2008;322:1498–501. doi: 10.1126/science.1162782. [DOI] [PubMed] [Google Scholar]
- 2.Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell. 2000;100:157–68. doi: 10.1016/s0092-8674(00)81692-x. [DOI] [PubMed] [Google Scholar]
- 3.Simon A, Tanaka EM. Limb regeneration. Wiley Interdiscip Rev Dev Biol. 2013;2:291–300. doi: 10.1002/wdev.73. [DOI] [PubMed] [Google Scholar]
- 4.Tornini VA, Poss KD. Keeping at arm’s length during regeneration. Dev Cell. 2014;29:139–45. doi: 10.1016/j.devcel.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, et al. Conversion of adult pancreatic α-cells to β-cells after extreme β-cell loss. Nature. 2010;464:1149–54. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27:719–24. doi: 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–23. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wells RG. The portal fibroblast: not just a poor man’s stellate cell. Gastroenterology. 2014;147:41–47. doi: 10.1053/j.gastro.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scholten D, Osterreicher CH, Scholten A, Iwaisako K, Gu G, et al. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology. 2010;139:987–98. doi: 10.1053/j.gastro.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu AS, Diaz R, Hui JJ, Yanger K, Zong Y, et al. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology. 2011;53:1685–95. doi: 10.1002/hep.24206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Mowry LE, Nejak-Bowen KN, Okabe H, Diegel CR, et al. Beta-catenin signaling in murine liver zonation and regeneration: a Wnt-Wnt situation! Hepatology. 2014;60(3):964–76. doi: 10.1002/hep.27082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Celton-Morizur S, Desdouets C. Polyploidization of liver cells. Adv Exp Med Biol. 2010;676:123–35. doi: 10.1007/978-1-4419-6199-0_8. [DOI] [PubMed] [Google Scholar]
- 14.Celton-Morizur S, Merlen G, Couton D, Margall-Ducos G, Desdouets C. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J Clin Investig. 2009;119:1880–87. doi: 10.1172/JCI38677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duncan AW, Hanlon Newell AE, Bi W, Finegold MJ, Olson SB, et al. Aneuploidy as a mechanism for stress-induced liver adaptation. J Clin Investig. 2012;122:3307–15. doi: 10.1172/JCI64026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zorn AM. Liver development. StemBook, editor. The Stem Cell Research Community. 2008 doi: 10.3824/stembook.1.25.1. http://www.stembook.org/node/512. [DOI]
- 17.Si-Tayeb K, Lemaigre FP, Duncan SA. Organogenesis and development of the liver. Dev Cell. 2010;18:175–89. doi: 10.1016/j.devcel.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 18.Le Douarin N. Synthesis of glycogen in hepatocytes undergoing differentiation: role of homologous and heterologous mesenchyma. Dev Biol. 1968;17:101–14. doi: 10.1016/0012-1606(68)90092-4. [DOI] [PubMed] [Google Scholar]
- 19.Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322:1490–94. doi: 10.1126/science.1161431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–47. doi: 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- 21.Gouon-Evans V, Boussemart L, Gadue P, Nierhoff D, Koehler CI, et al. BMP-4 is required for hepatic specification of mouse embryonic stem cell–derived definitive endoderm. Nat Biotechnol. 2006;24:1402–11. doi: 10.1038/nbt1258. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Krantz ID, Deng Y, Genin A, Banta AB, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–51. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 23.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–42. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela-Arispe ML. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development. 2010;137:4061–72. doi: 10.1242/dev.052118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geisler F, Nagl F, Mazur PK, Lee M, Zimber-Strobl U, et al. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology. 2008;48:607–16. doi: 10.1002/hep.22381. [DOI] [PubMed] [Google Scholar]
- 26.Kodama Y, Hijikata M, Kageyama R, Shimotohno K, Chiba T. The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology. 2004;127:1775–86. doi: 10.1053/j.gastro.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 27.Antoniou A, Raynaud P, Cordi S, Zong Y, Tronche F, et al. Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology. 2009;136:2325–33. doi: 10.1053/j.gastro.2009.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, et al. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136:1727–39. doi: 10.1242/dev.029140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clotman F, Jacquemin P, Plumb-Rudewiez N, Pierreux CE, Van der Smissen P, et al. Control of liver cell fate decision by a gradient of TGFβ signaling modulated by Onecut transcription factors. Genes Dev. 2005;19:1849–54. doi: 10.1101/gad.340305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang N, Bai H, David KK, Dong J, Zheng Y, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan X, Yuan Y, Zeng G, Apte U, Thompson MD, et al. β-Catenin deletion in hepatoblasts disrupts hepatic morphogenesis and survival during mouse development. Hepatology. 2008;47:1667–79. doi: 10.1002/hep.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Decaens T, Godard C, de Reynies A, Rickman DS, Tronche F, et al. Stabilization of β-catenin affects mouse embryonic liver growth and hepatoblast fate. Hepatology. 2008;47:247–58. doi: 10.1002/hep.21952. [DOI] [PubMed] [Google Scholar]
- 33.Spence JR, Lange AW, Lin SC, Kaestner KH, Lowy AM, et al. Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell. 2009;17:62–74. doi: 10.1016/j.devcel.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, Ning G, Duncan SA. Mammalian hepatocyte differentiation requires the transcription factor HNF-4α. Genes Dev. 2000;14:464–74. [PMC free article] [PubMed] [Google Scholar]
- 35.Odom DT, Dowell RD, Jacobsen ES, Nekludova L, Rolfe PA, et al. Core transcriptional regulatory circuitry in human hepatocytes. Mol Syst Biol. 2006;2:2006.0017. doi: 10.1038/msb4100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kyrmizi I, Hatzis P, Katrakili N, Tronche F, Gonzalez FJ, Talianidis I. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev. 2006;20:2293–305. doi: 10.1101/gad.390906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laudadio I, Manfroid I, Achouri Y, Schmidt D, Wilson MD, et al. A feedback loop between the liver-enriched transcription factor network and miR-122 controls hepatocyte differentiation. Gastroenterology. 2012;142:119–29. doi: 10.1053/j.gastro.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Cai J, DeLaForest A, Fisher J, Urick A, Wagner T, et al. Protocol for directed differentiation of human pluripotent stem cells toward a hepatocyte fate. StemBook, editor. The Stem Cell Research Community. 2008 doi: 10.3824/stembook.1.52.1. http://www.stembook.org/node/721. [DOI] [PubMed]
- 39.Ogawa S, Surapisitchat J, Virtanen C, Ogawa M, Niapour M, et al. Three-dimensional culture and cAMP signaling promote the maturation of human pluripotent stem cell–derived hepatocytes. Development. 2013;140:3285–96. doi: 10.1242/dev.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shan J, Schwartz RE, Ross NT, Logan DJ, Thomas D, et al. Identification of small molecules for human hepatocyte expansion and iPS differentiation. Nat Chem Biol. 2013;9:514–20. doi: 10.1038/nchembio.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacDonald RA. “Lifespan” of liver cells. Autoradio-graphic study using tritiated thymidine in normal, cirrhotic, and partially hepatectomized rats. Arch Intern Med. 1961;107:335–43. doi: 10.1001/archinte.1961.03620030023003. [DOI] [PubMed] [Google Scholar]
- 42.Sawada N, Ishikawa T. Reduction of potential for replicative but not unscheduled DNA synthesis in hepatocytes isolated from aged as compared to young rats. Cancer Res. 1988;48:1618–22. [PubMed] [Google Scholar]
- 43.Zajicek G, Oren R, Weinreb M., Jr The streaming liver. Liver. 1985;5:293–300. doi: 10.1111/j.1600-0676.1985.tb00252.x. [DOI] [PubMed] [Google Scholar]
- 44.Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2011;43:34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- 45.Bralet MP, Branchereau S, Brechot C, Ferry N. Cell lineage study in the liver using retroviral mediated gene transfer. Evidence against the streaming of hepatocytes in normal liver. Am J Pathol. 1994;144:896–905. [PMC free article] [PubMed] [Google Scholar]
- 46.Magami Y, Azuma T, Inokuchi H, Kokuno S, Moriyasu F, et al. Cell proliferation and renewal of normal hepatocytes and bile duct cells in adult mouse liver. Liver. 2002;22:419–25. doi: 10.1034/j.1600-0676.2002.01702.x. [DOI] [PubMed] [Google Scholar]
- 47.Malato Y, Naqvi S, Schürmann N, Ng R, Wang B, et al. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J Clin Investig. 2011;121:1850–60. doi: 10.1172/JCI59261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Español-Suñer R, Carpentier R, Van Hul N, Legry V, Achouri Y, et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology. 2012;143:1564–75.e7. doi: 10.1053/j.gastro.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 49.Yanger K, Stanger BZ. Liver cell reprogramming: parallels with iPSC biology. Cell Cycle. 2014;13:1211–12. doi: 10.4161/cc.28381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Higgins GM, Anderson RM. Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol. 1931;12:186–202. [Google Scholar]
- 51.Miyaoka Y, Ebato K, Kato H, Arakawa S, Shimizu S, Miyajima A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol. 2012;22:1166–75. doi: 10.1016/j.cub.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 52.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Georgiev P, Jochum W, Heinrich S, Jang JH, Nocito A, et al. Characterization of time-related changes after experimental bile duct ligation. Br J Surg. 2008;95:646–56. doi: 10.1002/bjs.6050. [DOI] [PubMed] [Google Scholar]
- 54.Chang ML, Yeh CT, Chang PY, Chen JC. Comparison of murine cirrhosis models induced by hepatotoxin administration and common bile duct ligation. World J Gastroenterol. 2005;11:4167–72. doi: 10.3748/wjg.v11.i27.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farber E. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3′-methyl-4-dimethylaminoazobenzene. Cancer Res. 1956;16:142–48. [PubMed] [Google Scholar]
- 56.Popper H, Kent G, Stein R. Ductular cell reaction in the liver in hepatic injury. J Mt Sinai Hosp N Y. 1957;24:551–56. [PubMed] [Google Scholar]
- 57.Leduc EH, Wilson JW. Injury to liver cells in carbon tetrachloride poisoning; histochemical changes induced by carbon tetrachloride in mouse liver protected by sulfaguanidine. AMA Arch Pathol. 1958;65:147–57. [PubMed] [Google Scholar]
- 58.Preisegger KH, Factor VM, Fuchsbichler A, Stumptner C, Denk H, Thorgeirsson SS. Atypical ductular proliferation and its inhibition by transforming growth factor β1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Investig. 1999;79:103–9. [PubMed] [Google Scholar]
- 59.Akhurst B, Croager EJ, Farley-Roche CA, Ong JK, Dumble ML, et al. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology. 2001;34:519–22. doi: 10.1053/jhep.2001.26751. [DOI] [PubMed] [Google Scholar]
- 60.Wilson JW, Leduc EH. Role of cholangioles in restoration of the liver of the mouse after dietary injury. J Pathol Bacteriol. 1958;76:441–49. doi: 10.1002/path.1700760213. [DOI] [PubMed] [Google Scholar]
- 61.Zipori D. The nature of stem cells: state rather than entity. Nat Rev Genet. 2004;5:873–78. doi: 10.1038/nrg1475. [DOI] [PubMed] [Google Scholar]
- 62.Roskams TA, Libbrecht L, Desmet VJ. Progenitor cells in diseased human liver. Semin Liver Dis. 2003;23:385–96. doi: 10.1055/s-2004-815564. [DOI] [PubMed] [Google Scholar]
- 63.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–16. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 64.Grisham JW, Porta EA. Origin and fate of proliferated hepatic ductal cells in the rat: electron microscopic and autoradiographic studies. Exp Mol Pathol. 1964;86:242–61. doi: 10.1016/0014-4800(64)90057-7. [DOI] [PubMed] [Google Scholar]
- 65.Evarts RP, Nagy P, Nakatsukasa H, Marsden E, Thorgeirsson SS. In vivo differentiation of rat liver oval cells into hepatocytes. Cancer Res. 1989;49:1541–47. [PubMed] [Google Scholar]
- 66.Gerber MA, Thung SN, Shen S, Stromeyer FW, Ishak KG. Phenotypic characterization of hepatic proliferation. Antigenic expression by proliferating epithelial cells in fetal liver, massive hepatic necrosis, and nodular transformation of the liver. Am J Pathol. 1983;110:70–74. [PMC free article] [PubMed] [Google Scholar]
- 67.Factor VM, Radaeva SA, Thorgeirsson SS. Origin and fate of oval cells in dipin-induced hepatocarcinogenesis in the mouse. Am J Pathol. 1994;145:409–22. [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou H, Rogler LE, Teperman L, Morgan G, Rogler CE. Identification of hepatocytic and bile ductular cell lineages and candidate stem cells in bipolar ductular reactions in cirrhotic human liver. Hepatology. 2007;45:716–24. doi: 10.1002/hep.21557. [DOI] [PubMed] [Google Scholar]
- 69.Paku S, Schnur J, Nagy P, Thorgeirsson SS. Origin and structural evolution of the early proliferating oval cells in rat liver. Am J Pathol. 2001;158:1313–23. doi: 10.1016/S0002-9440(10)64082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fausto N, Campbell JS. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev. 2003;120:117–30. doi: 10.1016/s0925-4773(02)00338-6. [DOI] [PubMed] [Google Scholar]
- 71.Dorrell C, Grompe M. Liver repair by intra- and extrahepatic progenitors. Stem Cell Rev. 2005;1:61–64. doi: 10.1385/SCR:1:1:061. [DOI] [PubMed] [Google Scholar]
- 72.Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39:1477–87. doi: 10.1002/hep.20214. [DOI] [PubMed] [Google Scholar]
- 73.Yanger K, Stanger BZ. Facultative stem cells in liver and pancreas: fact and fancy. Dev Dyn. 2011;240:521–29. doi: 10.1002/dvdy.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saxena R, Theise N. Canals of Hering: recent insights and current knowledge. Semin Liver Dis. 2004;24:43–48. doi: 10.1055/s-2004-823100. [DOI] [PubMed] [Google Scholar]
- 75.Tanimizu N, Miyajima A. Molecular mechanism of liver development and regeneration. Int Rev Cytol. 2007;259:1–48. doi: 10.1016/S0074-7696(06)59001-1. [DOI] [PubMed] [Google Scholar]
- 76.Miyajima A, Tanaka M, Itoh T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell. 2014;14:561–74. doi: 10.1016/j.stem.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 77.Wang X, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Grompe M. The origin and liver repopulating capacity of murine oval cells. Proc Natl Acad Sci USA. 2003;100(Suppl. 1):11881–88. doi: 10.1073/pnas.1734199100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wagers AJ, Weissman IL. Plasticity of adult stem cells. Cell. 2004;116:639–48. doi: 10.1016/s0092-8674(04)00208-9. [DOI] [PubMed] [Google Scholar]
- 79.Tatematsu M, Ho RH, Kaku T, Ekem JK, Farber E. Studies on the proliferation and fate of oval cells in the liver of rats treated with 2-acetylaminofluorene and partial hepatectomy. Am J Pathol. 1984;114:418–30. [PMC free article] [PubMed] [Google Scholar]
- 80.Evarts RP, Nagy P, Marsden E, Thorgeirsson SS. A precursor-product relationship exists between oval cells and hepatocytes in rat liver. Carcinogenesis. 1987;8:1737–40. doi: 10.1093/carcin/8.11.1737. [DOI] [PubMed] [Google Scholar]
- 81.Michalopoulos GK. The liver is a peculiar organ when it comes to stem cells. Am J Pathol. 2014;184:1263–67. doi: 10.1016/j.ajpath.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–26. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 83.Yanger K, Knigin D, Zong Y, Maggs L, Gu G, et al. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell. 2014;15(3):340–49. doi: 10.1016/j.stem.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sackett S, Li Z, Hurtt R, Gao Y, Wells R, et al. Foxl1 is a marker of bipotential hepatic progenitor cells in mice. Hepatology. 2009;49:920–29. doi: 10.1002/hep.22705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shin S, Walton G, Aoki R, Brondell K, Schug J, et al. Foxl1-Cre-marked adult hepatic progenitors have clonogenic and bilineage differentiation potential. Genes Dev. 2011;25:1185–92. doi: 10.1101/gad.2027811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 87.Barker N, Tan S, Clevers H. Lgr proteins in epithelial stem cell biology. Development. 2013;140:2484–94. doi: 10.1242/dev.083113. [DOI] [PubMed] [Google Scholar]
- 88.Huch M, Dorrell C, Boj SF, van Es JH, Li VS, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–50. doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dorrell C, Erker L, Schug J, Kopp JL, Canaday PS, et al. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes Dev. 2011;25:1193–203. doi: 10.1101/gad.2029411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Okabe M, Tsukahara Y, Tanaka M, Suzuki K, Saito S, et al. Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development. 2009;136:1951–60. doi: 10.1242/dev.031369. [DOI] [PubMed] [Google Scholar]
- 91.Kamiya A, Kakinuma S, Yamazaki Y, Nakauchi H. Enrichment and clonal culture of progenitor cells during mouse postnatal liver development in mice. Gastroenterology. 2009;137:1114–26.e14. doi: 10.1053/j.gastro.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 92.Suzuki A, Sekiya S, Onishi M, Oshima N, Kiyonari H, et al. Flow cytometric isolation and clonal identification of self-renewing bipotent hepatic progenitor cells in adult mouse liver. Hepatology. 2008;48:1964–78. doi: 10.1002/hep.22558. [DOI] [PubMed] [Google Scholar]
- 93.Tarlow BD, Finegold MJ, Grompe M. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatology. 2014;60:278–89. doi: 10.1002/hep.27084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choi TY, Ninov N, Stainier DY, Shin D. Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish. Gastroenterology. 2014;146:776–88. doi: 10.1053/j.gastro.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.He J, Lu H, Zou Q, Luo L. Regeneration of liver after extreme hepatocyte loss occurs mainly via biliary transdifferentiation in zebrafish. Gastroenterology. 2014;146:789–800. doi: 10.1053/j.gastro.2013.11.045. [DOI] [PubMed] [Google Scholar]
- 96.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 97.Michalopoulos G, Barua L, Bowen W. Transdifferentiation of rat hepatocytes into biliary cells after bile duct ligation and toxic biliary injury. Hepatology. 2005;41:535–44. doi: 10.1002/hep.20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sekiya S, Suzuki A. Hepatocytes, rather than cholangiocytes, can be the major source of primitive ductules in the chronically injured mouse liver. Am J Pathol. 2014;184:1468–78. doi: 10.1016/j.ajpath.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 99.Tanimizu N, Nishikawa Y, Ichinohe N, Akiyama H, Mitaka T. Sry HMG box protein 9-positive (Sox9+) epithelial cell adhesion molecule-negative (EpCAM−) biphenotypic cells derived from hepatocytes are involved in mouse liver regeneration. J Biol Chem. 2014;289:7589–98. doi: 10.1074/jbc.M113.517243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Overturf K, Al-Dhalimy M, Ou CN, Finegold M, Grompe M. Serial transplantation reveals the stem-cell-like regenerative potential of adult mouse hepatocytes. Am J Pathol. 1997;151:1273–80. [PMC free article] [PubMed] [Google Scholar]
- 101.Duncan AW, Hanlon Newell AE, Smith L, Wilson EM, Olson SB, et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology. 2012;142:25–28. doi: 10.1053/j.gastro.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–10. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci USA. 2007;104:17081–86. doi: 10.1073/pnas.0704126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci USA. 2004;101:10608–13. doi: 10.1073/pnas.0403412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Webber EM, Bruix J, Pierce RH, Fausto N. Tumor necrosis factor primes hepatocytes for DNA replication in the rat. Hepatology. 1998;28:1226–34. doi: 10.1002/hep.510280509. [DOI] [PubMed] [Google Scholar]
- 106.Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–83. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 107.Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, et al. Nuclear receptor–dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233–36. doi: 10.1126/science.1121435. [DOI] [PubMed] [Google Scholar]
- 108.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 109.Limaye PB, Bowen WC, Orr AV, Luo J, Tseng GC, Michalopoulos GK. Mechanisms of hepatocyte growth factor–mediated and epidermal growth factor–mediated signaling in transdifferentiation of rat hepatocytes to biliary epithelium. Hepatology. 2008;47:1702–13. doi: 10.1002/hep.22221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13:877–83. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dong J, Feldmann G, Huang J, Wu S, Zhang N, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–33. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–60. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 113.Avruch J, Zhou D, Fitamant J, Bardeesy N. Mst1/2 signalling to Yap: gatekeeper for liver size and tumour development. Br J Cancer. 2011;104:24–32. doi: 10.1038/sj.bjc.6606011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–38. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Takase HM, Itoh T, Ino S, Wang T, Koji T, et al. FGF7 is a functional niche signal required for stimulation of adult liver progenitor cells that support liver regeneration. Genes Dev. 2013;27:169–81. doi: 10.1101/gad.204776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, et al. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. N Engl J Med. 1998;338:1422–26. doi: 10.1056/NEJM199805143382004. [DOI] [PubMed] [Google Scholar]
- 117.DeWard AD, Komori J, Lagasse E. Ectopic transplantation sites for cell-based therapy. Curr Opin Organ Transplant. 2014;19:169–74. doi: 10.1097/MOT.0000000000000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Investig. 2012;122:2911–15. doi: 10.1172/JCI63212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Investig. 2012;122:3914–18. doi: 10.1172/JCI63065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143:1660–69.e7. doi: 10.1053/j.gastro.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]