

Abstract
Dendritic cell migration to the T-cell-rich areas of the lymph node is essential for their ability to initiate the adaptive immune response. While it has been shown that the actin cytoskeleton is required for normal DC migration, the role of many of the individual cytoskeletal molecules is poorly understood. In this study, we investigated the contribution of the Arp2/3 complex binding protein, haematopoietic lineage cell-specific protein 1 (HS1), to DC migration and force generation. We quantified the random migration of HS1−/− DCs on 2D micro-contact printed surfaces and found that in the absence of HS1, DCs have greatly reduced motility and speed. This same reduction in motility was recapitulated when adding Arp2/3 complex inhibitor to WT DCs or using DCs deficient in WASP, an activator of Arp2/3 complex-dependent actin polymerization. We further investigated the importance of HS1 by measuring the traction forces of HS1−/− DCs on micropost array detectors (mPADs). In HS1 deficient DCs, there was a significant reduction in force generation (3.96 ± 0.40 nN/cell) compared to WT DCs (13.76 ± 0.84 nN/cell). Interestingly, the forces generated in DCs lacking WASP were only slightly reduced compared to WT DCs. Taken together, these findings show that HS1 and Arp2/3 complex-mediated actin polymerization are essential for the most efficient DC random migration and force generation.
Graphical abstract
Introduction
Dendritic cells (DCs) are potent antigen presenting cells (APCs) which possess the unique ability of linking the innate and adaptive immune responses (1,2). Immature DCs reside in a variety of tissues and continually sample their environment for antigens. Once DCs encounter a pathogen, they capture it and load antigenic fragments onto MHC molecules on their surface for presentation to other immune cells. Pathogen recognition also triggers DC maturation, which is characterized by a variety of phenotypic and functional changes (reviewed in (3,4)). Of particular interest to us is a dramatic switch from a slowly moving, tissue resident cell to a highly migratory cell (5). This increased level of migration allows DCs to exit peripheral tissues and travel via the lymphatic system to T cell rich regions of the lymph nodes, where they activate T cells and launch a specialized immune response (6).
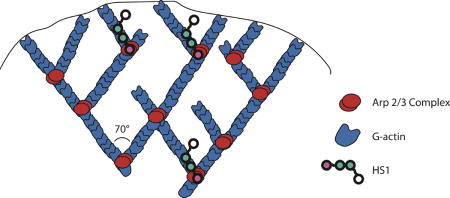
Cell migration depends on coordinated interactions among the different components of the actin cytoskeleton (7,8). At the front of a migrating cell, rapid actin polymerization forms protrusive structures that help to push the plasma membrane forward. One such structure found in DCs is the lamellipodium (Figure 2a) (9), which is formed by branched actin polymerization driven by the actin related protein (Arp) 2/3 complex (Figure 1b) (10,11). While the Arp2/3 complex alone is a weak actin nucleator, the rate of Arp2/3 mediated actin polymerization is increased in the presence of a class of proteins known as nucleation promoting factors (NPFs) (12–15). These proteins bind to both actin and the Arp2/3 complex and enhance Arp2/3 complex-dependent nucleation of branched actin filaments. One such NPF is hematopoietic lineage cell-specific protein 1 (HS1).
Figure 2.
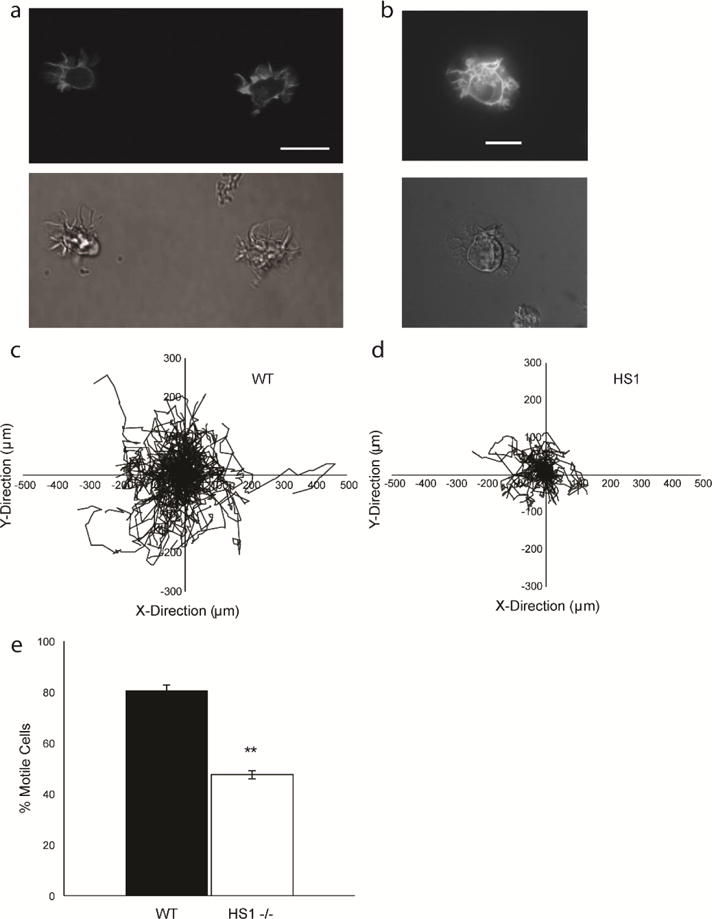
DCs contain actin rich structures at the leading edge and show defects in migration when HS1 is eliminated. (a) Sample images of GFP-Life-act DCs showing GFP-labeled actin filaments (top) and phase contrast images of the same cells (bottom). Scale bar equals 20 μm. Asterisk denotes broad lamellipodium. (b) Sample images of GFP-Life-act transduced HS1−/− DC showing GFP-labeled actin filaments (top) and bright field images of the same cell (bottom). Cell tracks for (c) WT and (d) HS1−/− DCs during a representative chemokinesis experiment. ≥ 110 cells were tracked per condition for a period of 2–3 hours. (c) Average percentage of motile cells. Figures represent average values ± SEM, for > 1000 DCs from at least three independent experiments per condition. Statistical significance calculated with single factor ANOVA and post hoc Tukey test. Indicates significant difference compared to WT DCs. *p<0.05, **p<0.01
Figure 1.
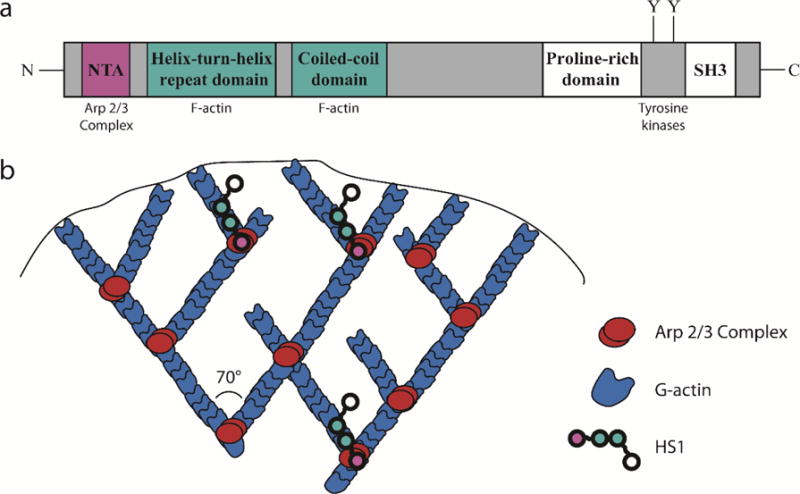
HS1 stabilizes branched actin filaments at the leading edge of DCs. (a) Structure of HS1. The NTA domain, shown in red, binds to the Arp 2/3 complex. The helix-turn-helix domain and coiled-coil domains, shown in blue, bind to F-actin. A proline rich domain, SH3 domain and important tyrosine residues are located at the C-terminus. (b) Cartoon representation of branched actin structure in lamellipodia. Red represents the Arp 2/3 Complex and blue represents actin. HS1 (chain of colored circles) is thought to stabilize actin branch points by simultaneously binding the Arp 2/3 Complex and neighboring F-actin.
HS1 (also known as HCLS1 and LckBP1) is a 75 kDa protein expressed exclusively in hematopoietic cells (16,17). HS1 is the only known Class II NPF in hematopoietic cells and is highly homologous with the widely studied protein cortactin (18). It contains an N-terminal acidic (NTA) region, which binds to the Arp2/3 complex, followed by a helix-turn-helix region and a coiled-coil domain, both of which bind to F-actin and are required for proper Arp2/3 complex activation and branched actin polymerization (Figure 1a) (14,19). HS1 has been shown to increase the rate of Arp2/3 complex mediated actin polymerization, prolong the half-life of existing branched actin filaments and regulate lamellipodial dynamics (14,19–21). It has also been shown that HS1 interacts with another NPF—Wiskott Aldrich Syndrome Protein (WASP) (22). Unlike HS1, WASP is a Class I NPF, which uses its VCA domain to bind to monomeric actin and the Arp2/3 complex (23). It is one of multiple Class I NPFs in DCs and may be redundant in some of its functions (23). Through its interactions with the Arp2/3 complex and WASP, HS1 is able to shape the dynamics of branched actin networks.
HS1 is expressed at high levels in mature dendritic cells (24), yet the exact role it plays in DC migration is not clear. Initial studies revealed that HS1 could be eliminated from dendritic cells without altering their morphology or viability (22,24). Nonetheless, there are a few differences in the structure and function of HS1−/− DCs. HS1−/− DCs adhere and spread normally, but they form disorganized and overly dynamic actin structures, such as podosomes and lamellipodia, and have defective antigen capture via receptor-mediated endocytosis (22,24). Previous work by Klos Dehring et al. highlighted the importance of HS1 in dendritic cell migration, in particular during chemotaxis (22). The authors found that HS1−/− DCs are able to migrate across transwells in response to CCL19 or CCL21 to the same degree as WT DCs, and have unaltered expression of chemokine receptors. However, when placed in a microfluidic gradient generator, HS1−/− DCs migrate significantly faster than WT DCs and are less able to persistently migrate in the direction of the chemokine gradient. These findings reveal that the long-term ability of HS1−/− DCs to reach a target location is not significantly altered, but that the manner in which DCs migrate to the target is affected. Building upon this study, we sought to determine if HS1 impacts other aspects of DC migration. Using motility assays and micropost array detectors (mPADs), we quantified the random migration and force generation of HS1−/− DCs, respectively. In addition, we used both Arp2/3 complex inhibitor and WASP−/− DCs to elucidate the molecular mechanisms through which HS1 acts. We found that HS1 is required for efficient migration and force generation, and we hypothesize that HS1 likely influences DC migration through its interaction with the Arp2/3 complex.
Materials and Methods
Reagents
Recombinant murine CCL19 was purchased from R&D Systems (Minneapolis, MN). Lipopolysaccharide (LPS; L4516), CK-666 (Lot: 043M4606V) DMSO, Pluronics F127, silane (Trichloro(1H,1H,2H,2H-perfluorooctyl)silane) and bovine fibronectin were obtained from Sigma-Aldrich (St. Louis, MO). Penicillin-Streptomycin (10,000 U/mL), RPMI 1640 (1x, with L-Glutamine and 25 mM HEPES) and PBS were obtained from Thermo Fisher Scientific (Hampton, NH). Poly(dimethylsiloxane) (Sylgard 184 Silicone Elastomer) was purchased from Dow Corning, Midland, MI. 200 proof ethanol was obtained from Decon Laboratories (King of Prussia, PA). Fetal bovine serum was obtained from Atlanta Biologicals (Flowery Branch, GA). Recombinant GM-CSF was produced from the B78Hi/GMCSF.1 cell line provided by T. Laufer (University of Pennsylvania, Philadelphia, PA).
Mice
C57BL/6J mice, WASP−/− mice bred on the C57BL/6J background (25) (both from Jackson Laboratories), HS1−/− mice bred on the C57BL/6J background (26) and GFP-Lifeact mice bred on the C57BL/6J background (26) were reared at Children’s Hospital of Philadelphia. All mice were housed under pathogen-free conditions in the Children’s Hospital of Philadelphia animal facility. All studies involving animals were reviewed and approved by the Children’s Hospital of Philadelphia Institutional Animal Care and Use Committee.
Culture of bone marrow derived DCs (BMDCs)
Primary dendritic cells were prepared from murine bone marrow, following the protocol of Sixt and Lämmermann (27). Bone marrow was flushed from the femurs and tibiae with sterile PBS. Cells were centrifuged at 1500 rpm for 10 min at 4°C and resuspended at 2.5 × 106 cells/mL in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin and streptomycin. 1 mL cell suspension was combined with 9 mL media and 100 μL GM-CSF in a 10 cm Petri dish. On day 3, 10 mL fresh media and 100 μL GM-CSF were added, and on day 6, 100 μL GM-CSF was added after half of the media was gently replaced. 200 ng/mL LPS was added between day 7 to 9 and DCs were matured for 24 hours. After maturation, DCs were centrifuged at 1500 rpm for 10 min at 4°C and resuspended at 100,000 cells/mL for random migration experiments or 50,000 cells/mL for force measurement experiments. Cells were maintained at 37°C and 5% CO2.
Surface Preparation for Random Migration Experiments
25 mm round glass coverslips (Thermo Fisher Scientific) were coated with a thin layer of degassed poly(dimethylsiloxane) (PDMS, 10:1 base to cure by weight) (using a Laurell spinner (4000 rpm, 1 minute). Coverslips were cured for at least three hours at 65°C. Circular holes were laser cut from the bottom of each well of a 6 well dish. The PDMS coated coverslips were affixed to the bottom of each well using 10:1 PDMS and the seal was cured overnight at 65°C. The coverslips were functionalized by microcontact printing following the protocol of Desai et al (28). Stamps for printing were prepared by casting degassed PDMS against a flat silicon wafer. The PDMS was allowed to cure in contact with the wafer overnight at 65°C. 1 cm2 square stamps were cut from the block of PDMS using a premade grid pattern as a guide. Stamps were sonicated in 200 proof ethanol for 5 minutes, rinsed twice in diH2O and gently dried with N2 gas. 200 μL of a 10 μg/mL solution of fibronectin was added to each stamp and incubated at room temperature for 2 hours. Stamps were again rinsed twice in diH2O and gently dried with N2 gas. The 6 well dish containing the PDMS coverslips was treated for 7 minutes with ultraviolet ozone (UVO Cleaner Model 342, Jelight, Irvine, CA) and PDMS stamps were used to transfer protein to the coverslip surface. The printed coverslips were blocked with 0.2 % Pluronic F127 for 1 hour at room temperature, rinsed twice with sterile PBS and stored at 4°C overnight.
The following day, 1 mL of mature DCs (100,000 cells/mL) were seeded on each printed coverslip. In inhibition studies, the desired concentration of CK-666 or equivalent volume of DMSO was added 1 hour prior to imaging and the cells were incubated at 37°C and 5% CO2. Motility was stimulated with 10 nM CCL19 immediately before moving the plate to the microscope stage.
Live Cell Imaging for Random Migration Experiments
DCs were imaged at 10× by phase microscopy on a Nikon Eclipse TE300 (Nikon, Melville, NY) with custom-built Labview (National Instruments, Austin, TX) software. A motorized stage was used to capture images at multiple positions. Time-lapse experiments were performed by acquiring images every two to three minutes for up to three hours. Cells were maintained at 37°C and 5% CO2 during imaging.
Random Motility Data Analysis
Cell migration was analyzed with the Manual Tracking plugin in ImageJ (http://rsbweb.nih.gov/ij). Cells were tracked over time by manually specifying their centroid positions in each frame. Cells that were apoptotic or completely stationary were excluded, as well as any cells in contact with other cells. The list of x- and y-coordinates obtained from ImageJ was further analyzed with a custom-written MATLAB (Mathworks, Natick, MA) script. The MATLAB script computed the MSD of the compiled list of cell tracks and used the Dunn equation to fit speed (S) and persistence (P) (29). The random motility coefficient was calculated from the speed and persistence as follows: , where n specifies the dimensionality (30).
mPAD Preparation
Micropost array detectors (mPADs) were prepared following the protocol of Yang et al. (31). Micropost arrays were prepared from a silicon master by replica molding. Negative molds were created by casting degassed PDMS (10:1 base to cure) against a silanized silicon master mold and curing for 12 minutes at 110°C. The negative molds were gently peeled away from the silicon master, plasma treated (SPI Supplies Plasma Prep II, West Chester, PA) for 7 seconds, and silanized overnight. PDMS (10:1 base to cure) was added to the negative molds and degassed for 30 minutes to remove any air bubbles. 25 mm round glass coverslips were cleaned with N2 gas and plasma treated for 90 seconds. The molds were placed, fresh PDMS side down, onto the cleaned coverslip and cured at 110°C for 20 hours. The mPADs were gently peeled away from the negative molds, immediately submerged in 200 proof ethanol and supercritical dried (SAMDRI-PVT-3D, Tousimis Corporation, Rockville, MD) in liquid CO2. Circular holes were laser cut from the bottom of one well dishes. mPAD coverslips were secured to the bottom of the one well dishes with 10:1 PDMS and the seal was cured overnight at 65°C.
10 μg/mL fibronectin was microcontact printed onto the post tips, following the same procedure as above with minor modifications. After stamping the mPADs, 1 mL of 200 proof ethanol was added and the stamp was gently flicked off the micropost array. The ethanol was immediately diluted to 60% with diH2O. The mPADs were rinsed three times with diH2O and were incubated with 1x Δ9-DiI (1,1′-dioleyl-3,3,3′,3′-tetramethylindocarbocyanine methanesulfonate; Invitrogen Carlsbad, CA) for one hour, at room temperature. To prevent photobleaching of the fluorescent label, the mPAD was covered and lights were dimmed, when possible, for the rest of the preparation process. The mPADs were gently rinsed three times with diH2O. The post tips were blocked with 0.2 % Pluronic F127 for 1 hour at room temperature, rinsed three times with sterile diH2O and stored at 4°C overnight. The following day 1 mL of mature DCs (50,000 cells/mL) was seeded on the mPAD. Motility was stimulated with 10 nM CCL19 immediately before moving the dish to the microscope stage, and cells were allowed to settle on the micropost array before imaging began.
Live Cell Imaging for Force Measurement Experiments
DCs were imaged at 40× by phase and fluorescence microscopy on a Nikon Eclipse TE300 with custom-built Labview software. A motorized stage was used to capture images at multiple positions. Time-lapse experiments were performed by acquiring images every minute for up to an hour. Cells were maintained at 37°C and 5% CO2 during imaging.
Traction Force Analysis
A custom MATLAB (Mathworks, Natick, MA) script was used to analyze a series of paired phase and fluorescent images. Phase images were used to manually delineate the area occupied by the cell of interest. The corresponding fluorescent image was then opened and the position of the posts in the area of interest was recorded. The script determines the ideal position of each post based on the theoretical post packing geometry. Deflection distance was calculated for the posts in the area of interest as the 2-dimensional distance between the ideal position the measured position. The deflections were then converted into vector forces using Hooke’s law ( ) and the known spring constant of the micropost array (k=1.92 nN/μm) for the given post height and diameter. These forces were then used to calculate the total scalar force per cell, which was averaged over the entire ensemble of cells for each type of cell (WT, HS1−/− and WASP−/−).
Statistical Analysis
We used a one way ANOVA along with a Tukey’s Post Hoc Test to determine statistical significance. Statistically significant differences at p < 0.05 were marked with an asterisk on plots (*) and statistically significant differences at p < 0.01 were marked with a double asterisk (**).
Results
HS1 is Required for Efficient DC Random Migration
It was previously shown that HS1 contributes to DC speed and persistence during chemotaxis (22), but its role in random migration was not tested. It has been revealed in a variety of immune cell types that HS1 can have different effects on different modes of migration (32–36). Therefore, we hypothesized that we would see defects in DC chemokinesis that were absent during DC chemotaxis. To assess this, we performed parallel experiments with WT and HS1−/− DCs undergoing chemokinesis. Briefly, we plated cells on PDMS surfaces that had been printed with bovine fibronectin and blocked with the Pluronic F127. We acquired time lapse images of each cell type in phase contrast and used the Manual Tracking Plugin in ImageJ to quantify cell position as a function of time.
In these low magnification, phase contrast experiments, the appearance of HS1−/− DCs was qualitatively indistinguishable from that of WT DCs (Supplementary Figure 1, Supplementary Video 1, and Supplementary Video 2). HS1−/− DCs were viable and still formed extensive membrane veils and protrusions. However, while HS1−/− DCs retained some ability to migrate, the extent of their migration was significantly reduced. Figure 2c–d shows the migration patterns of representative populations of WT and HS1−/− DCs. The start position of each cell was set at the origin to allow for visualization of the distance traveled by all cells in a given field of view. As expected, both WT and HS1−/− DCs migrated randomly, with no directional bias. While both populations of cells were able to migrate, we saw a large difference in the area explored during migration, with HS1−/− DCs restricted to a smaller region than WT DCs. From this initial observation, it appears that HS1 is an important component of the DC migratory machinery.
While some HS1−/− DCs were able to move, many HS1−/− DCs appeared to vacillate around a fixed point. We designated cells as “motile” if they traveled at least two cell diameters over the course of the experiment. Cells that traveled less than this were designated as “stationary.” During chemokinesis, 80.45 ± 2.22 % of WT DCs were “motile,” compared to only 47.61 ± 1.69% for HS1−/− DCs (Figure 2e). This shows that HS1−/− DCs have a significant defect in translocation, as compared to their WT counterparts.
We further quantified the migration of HS1−/− DCs by calculating the mean squared displacement (MSD) as a function of time for both WT and HS1−/− DCs (Figure 3a). When plotting MSD vs. time on a log-log scale, we would expect to see a slope of one for a population of cells moving randomly or diffusively. This is indeed what we observed for WT DCs. However, HS1−/− DCs failed to migrate diffusively over the entire course of the experiment, as shown by an initial linear rise in MSD followed by a plateau. It has been shown that HS1 is necessary for the stability of DC lamellipodia (22) and we have observed dynamic ruffling in our HS1 KO DCs (Supplementary Video 3). We hypothesize that the decrease in migration that we observed at long times could be related to the inability of these cells to form persistent actin structures.
Figure 3.
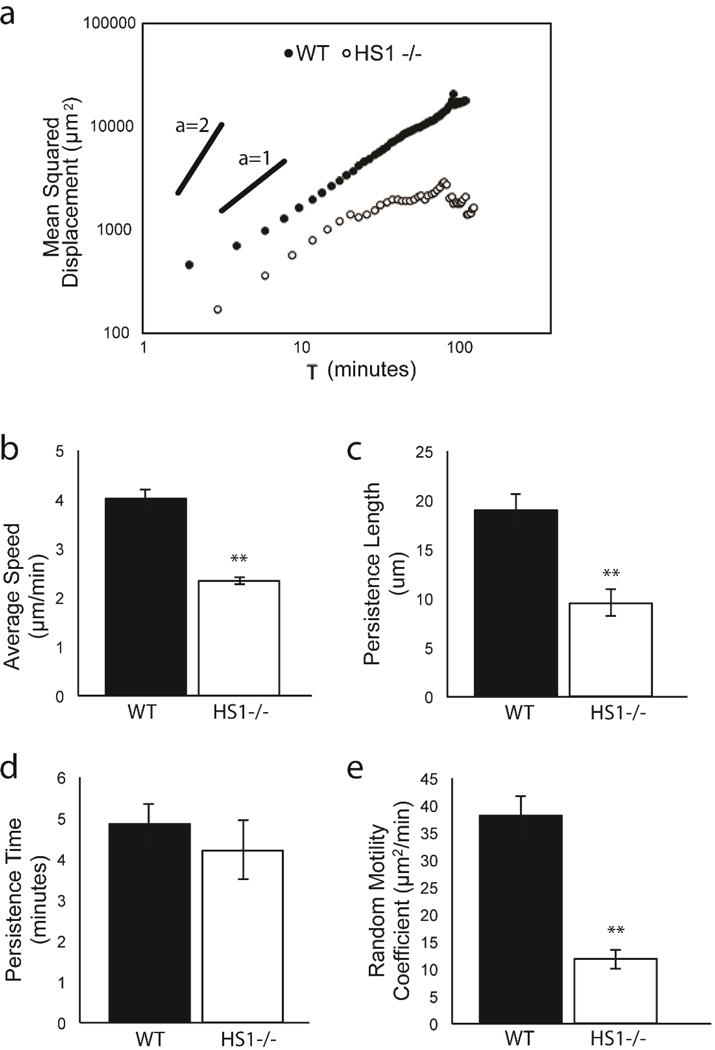
HS1−/− DCs exhibit multiple defects in migration during chemokinesis (a) MSD vs. time on log-log scale for WT and HS1−/− DCs. Quantification of DC random migration: (b) average speed, (c) persistence length, (d) persistence time, (e) random motility coefficient. Figures represent average values ± SEM, for > 1000 DCs from at least three independent experiments per condition. Statistical significance calculated with single factor ANOVA and post hoc Tukey test. Indicates significant difference compared to WT DCs. *p<0.05, **p<0.01
We next fit the curves for MSD vs. time to the Dunn equation (29). This allowed us to calculate a variety of useful motility metrics, such as average speed (S), persistence length (the distance traveled before a cell changed direction), persistence time (P, length of time a cell traveled before changing direction) and random motility coefficient (μ). WT DCs undergoing chemokinesis moved at speeds of 4.00 ± 0.18 μm/min (Figure 3b). HS1−/− DCs showed a significant reduction in migration speed, traveling at only 2.31 ± 0.08 μm/min. HS1−/− DCs also showed significant reductions in persistence length, but their persistence time was unaffected (Figure 3c–d). This suggests that HS1−/− DCs were able to turn at the same frequency as WT DCs, but their reduced speed led to shorter distances traveled between subsequent turns. These effects were extended to the random motility coefficient (Figure 3e), which is a quantitative measure of cell diffusivity and is related to speed and persistence as follows: . HS1−/− DCs had a much lower random motility coefficient (11.80 ± 1.75 μm2/min) than WT DCs (38.20 ± 3.41 μm2/min). This agreed with our MSD calculation and confirmed that HS1−/− DCs were less capable of diffusive migration. From these initial experiments, we conclude that HS1 expression is required for efficient DC chemokinesis.
HS1 and the Arp2/3 Complex Work Together to Coordinate DC Random Migration
HS1 binds to both the Arp2/3 complex and polymerized actin via its N terminus, and is thought to stabilize the branched actin network through these interactions (14,19). We hypothesize that the reduction (but not total elimination) of HS1−/− DC motility could be due to the failure of HS1−/− DCs to stabilize branched actin filaments. To determine how inhibition of the Arp2/3 complex compares with loss of HS1 function, we incubated WT DCs with CK-666 (Figure 4a). CK-666 is a small molecule inhibitor that binds to the Arp2/3 complex and locks it in an inactive conformation, thereby preventing actin binding and polymerization (37). We tested three different concentrations of CK-666 - 1 μM, 10 μM and 100 μM. WT DCs were added to PDMS surfaces microcontact printed with fibronectin and were incubated with CK-666 for one hour before imaging. As predicted, inhibiting Arp2/3 complex function decreased migration, and it did so in a dose dependent manner. At the highest concentration of CK-666 tested, migration was quantitatively indistinguishable from HS1−/− DC migration, as shown by reductions in average speed, persistence length and random motility coefficient (Figure 4b–d). Inhibition with CK-666 at any level did not affect the persistence time, suggesting that the Arp2/3 complex is not required for DCs to change direction at the proper frequency (data not shown). DMSO exposed cells were used as a control and showed no significant defects in persistence or random motility (Supplementary Figure 2). To further explore the importance of the Arp2/3 Complex on DC random migration, we inhibited HS1−/− DCs with 100 μM CK-666. Addition of Arp2/3 complex inhibitor to HS1 −/− DCs led to a reduction in average speed, but the persistence length and random motility coefficient were not significantly affected (Figure 5). Since HS1−/− DC migration is not greatly affected with addition of Arp2/3 complex inhibitor, this suggests that the defects observed in HS1−/− migration are partly due to interaction with the Arp2/3 complex.
Figure 4.
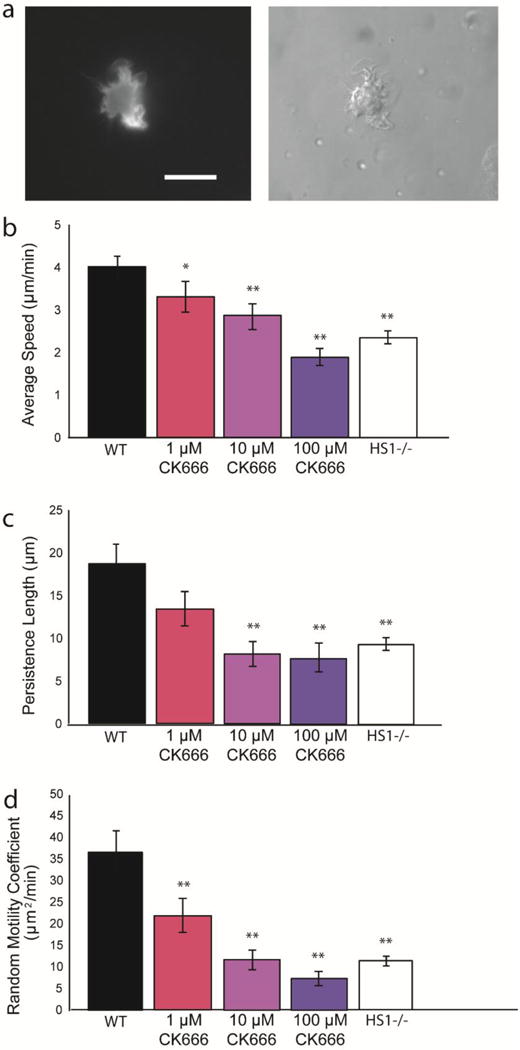
(a) Sample images of GFP-Life-act DC inhibited with CK-666, showing GFP-labeled actin filaments (left) and bright field images of the same cell (right). Scale bar equals 20 μm. Quantification of WT DC chemokinesis in the presence of CK-666, an Arp2/3 inhibitor (b) average speed, (c) persistence length and (d) random motility coefficient (values taken from Figure 3 and included for reference). Three different CK-666 concentrations were used: from left to right 1 μM, 10 μM and 100 μM. Figures represent average values ± SEM, for > 290 DCs from at least three independent experiments per condition. Statistical significance calculated with single factor ANOVA and post hoc Tukey test. Indicates significant difference compared to WT DCs. *p<0.05, **p<0.01
Figure 5.
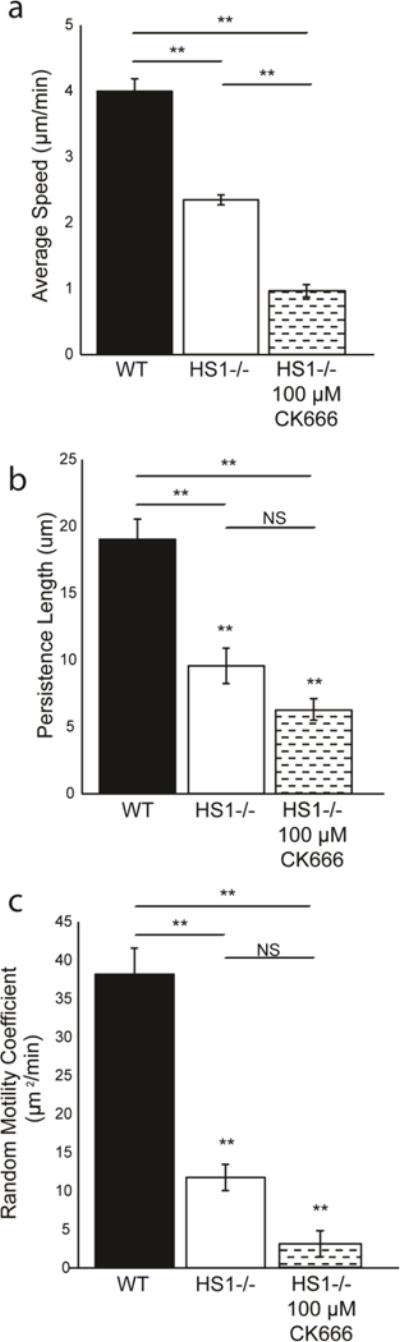
Quantification of HS1−/− DC chemokinesis in the presence and absence of CK-666 (a) average speed, (b) persistence length and (c) random motility coefficient (values taken from Figure 3 and included for reference). Figures represent average values ± SEM, for > 1000 DCs from at least three independent experiments per condition. Statistical significance calculated with single factor ANOVA and post hoc Tukey test. *p<0.05, **p<0.01, with lines showing conditions being compared.
Next we investigated the role of WASP (Wiskott-Aldrich Syndrome protein), another Arp 2/3 complex activating protein found in hematopoietic cells (38,39). Previous studies in DCs have shown that WASP is required for proper morphology and cytoskeletal organization (22,40,41), antigen processing (42), activation of the innate and adaptive immune systems (43–45), and migration (22,25,40,46). While various aspects of WASP−/− DC migration have been studied, to our knowledge no one has yet determined how WASP impacts the random migration of mature DCs. Since we hypothesize that defective HS1−/− DC chemokinesis is due to impaired Arp2/3 complex activity and branched actin instability, we expected to observe similar defects in WASP−/− DC chemokinesis. As we previously observed with HS1−/− DCs, WASP−/− DCs had significantly impaired migration, with reduced average speed, persistence length and random motility coefficient (Figure 6, previous HS1−/− data included as a reference). The persistence length and random motility coefficient were comparable to the values calculated for HS1−/− DCs. The average speed of WASP−/− DCs (3.07 ± 0.15 μm/min) was significantly lower than WT DCs (4.01 ± 0.18 μm/min), but was also significantly higher than HS1−/− DCs (2.35 ± 0.08 μm/min). As with HS1−/− DCs, WASP−/− DCs had no significant reduction in persistence time. Since HS1 and WASP have similar but non-redundant functions within the cell, it is not surprising that we observe similar but non-identical results after their knockout.
Figure 6.
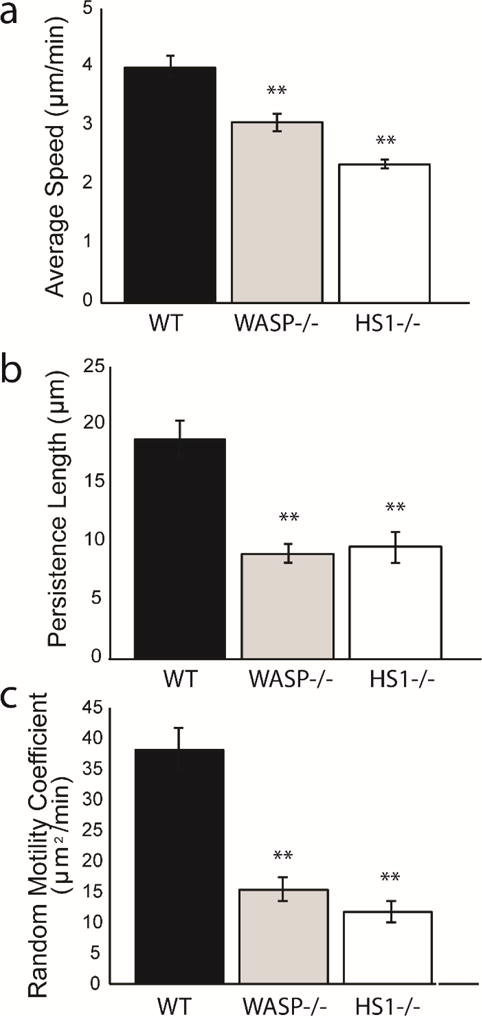
Comparison of chemokinesis of DCs lacking WASP and HS1 (a) average speed, (b) persistence length and (c) random motility coefficient. WASP−/− DCs are shown in gray. WT DCs are shown in blue and HS1−/− DCs are shown in red (values taken from Figure 3 and included for reference). Figures represent average values ± SEM, for > 1000 DCs from at least three independent experiments per condition. Statistical significance calculated with single factor ANOVA and post hoc Tukey test. Indicates significant difference compared to WT DCs. *p<0.05, **p<0.01
HS1 is Required for Maximal Dendritic Cell Force Generation
Previous work from our group has shown that inhibition of actin polymerization results in a drastic reduction in magnitude of DC traction forces (47). In that study, we used a nonspecific actin polymerization inhibitor, and it is unknown how individual actin-binding proteins are involved in DC force production. In the present study, we wanted to determine whether HS1 is involved in force generation. In the preceding sections, we have highlighted the impact of HS1 on the kinematics of DC migration. Since cell migration and force generation are intimately linked processes, we expected the impact of HS1 on random migration to correlate with a reduction in traction force strength (48). We also posited that a defect in HS1−/− DC force generation would be a direct result of its interaction with the Arp2/3 complex, and would therefore extend to other proteins interacting with the Arp2/3 complex, such as WASP.
To test this hypothesis, we used micropost array detectors (mPADs) to measure the forces exerted by WT, HS1−/− and WASP−/− DCs during chemokinesis. mPADs are arrays of elastic micropillars which are sensitive enough to measure the weak forces exerted by DCs and other amoeboid cell types (49). We used a 10:1 solution of PDMS to replica mold mPADs from silicon master molds. Before adding cells to the mPADs, we stamped the post tips with bovine fibronectin, and stained them with a lipophilic dye in order to visualize post deflection. The mPADs were also blocked with Pluronic F127 to ensure that the cells were interacting with the post tips and not with the sides of the posts or interpost regions. We added DCs to the mPADs at a concentration of 50,000 cells/mL along with 10 nM CCL19, and allowed the cells to settle on the posts before imaging. A series of fluorescence and phase images were captured in order to record post deflections and cell position over time. A representative cell imaged in phase and fluorescence is shown in Figure 7a and b. The arrow in Figure 7b points to the location of the cell, where post deflections were observed and quantified.
Figure 7.
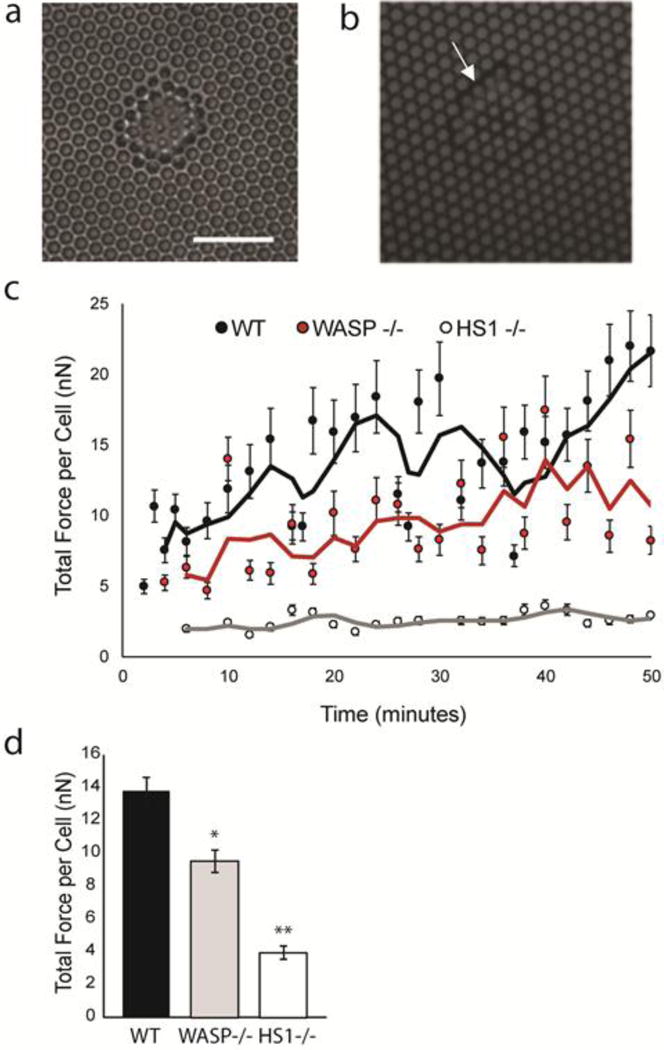
Calculation of DC traction forces using mPADs. (a) Phase contrast image of DC on mPAD surface. Scale bar equals 20 μm. Fluorescent and phase images correspond to same cell and same position. (b) Sample image of fluorescent micropost tips in area occupied by DC. Post diameter and height are 1.83 μm and 12.9 μm, respectively. (c). Average traction forces for ensemble of WT DCs, HS1−/− DCs and WASP−/− DCs. The forces were calculated from images such as those shown in (a) and (b). Displacement of mPADs was converted to traction force using Hooke’s law and known spring constant (1.92 nN/μm) for the mPAD array. The x-axis indicates time in minutes, with 0 corresponding to the start of imaging. Lines indicate 3 point moving averages for each condition. (d) Time and ensemble averages of DC traction forces, corresponding to values shown in (c). Figures represent average values ± SEM, for at least 28 DCs per condition. Statistical significance calculated with single factor ANOVA and post hoc Tukey test. Indicates significant difference compared to WT DCs. *p<0.05, **p<0.01
Individual traction forces were calculated using a custom MATLAB code that converted post deflections into vector forces using Hooke’s law, given the spring constant for the array (1.92 nN/μm). We then calculated the total force per cell, as the sum of all the scalar forces exerted on the posts by a given DC. Since there was considerable variation within each population of cells, we chose to compute an ensemble average as a function of time (Figure 7c). WT DCs produced traction forces on the order of 10 nN/cell, which agrees well with our previous measurements (47). We saw an initial rise in WT traction forces, followed by a plateau. We hypothesize that this initial rise in force is due to imaging shortly after adding the cells, before they had a chance to fully engage the post array. Both WASP−/− DCs and HS1−/− DCs are capable of interacting with the mPAD array, but their force generation appeared to be lower than what we observed in WT cells.
We next performed a time and ensemble average for all three populations of DCs (Figure 7d). Compared to WT DCs, WASP−/− and HS1−/− DCs produced significantly less force per cell. WT DCs produced an average force of 13.76 ± 0.84 nN/cell, whereas WASP−/− DCs produced an average force of 9.55 ± 0.69 nN/cell and HS1−/− DCs produced an average force of 3.96 ± 0.40 nN/cell. The force generation was most affected in HS1−/− DCs, whose forces were more than 3 times lower than WT forces. While WASP−/− DCs produced significantly less force than WT DCs, they produced significantly greater force than HS1−/− DCs. These results show that both WASP and HS1 are involved in the DC force transduction pathway, but that HS1 is more important for force generation than WASP.
Discussion
Due to the importance of DC migration and the involvement of the actin cytoskeleton in this process (7), we sought to identify whether the actin regulatory protein HS1is required for DC random migration and force generation. We used microcontact printed PDMS coverslips to assess random migration and mPADs to measure cellular traction forces. We found that HS1 contributed to random migration through its effect on speed but not persistence time. HS1−/− DCs migrate more slowly, and often fail to translocate over a 3 hour period, leading to a reduction in area explored during migration. One of the ways that HS1 interacts with actin is through activation of the Arp2/3 complex. This interaction appears to be important in DCs, as inhibition of the Arp2/3 complex in WT DCs or elimination of other Arp2/3 complex nucleating proteins led to similar reductions in migration. Migration and force generation are often intimately linked and we observed a concomitant reduction in force generation in HS1−/− DCs along with defects in migration. Interestingly, while migration seems to have a general dependence on the Arp2/3 complex, force generation is affected to a different degree by different Arp2/3 complex activators. Since DCs need to migrate quickly and efficiently to the lymph nodes to launch an adaptive immune response (6), it is possible that defects in the Arp2/3 complex, HS1 or other similar proteins could lead to immune dysregulation. This is indeed the case for mutated WASP, which leads to the X-linked autoimmune disease Wiskott-Aldrich syndrome (WAS) (38,39). WAS leads to complete immune system dysregulation (reviewed in (40)) and is characterized by thrombocytopenia, easy bruising, frequent and prolonged bleeding, eczema and recurrent infections (41). To date, abnormalities in HS1 have been associated with both chronic lymphocytic leukemia (50) and systemic lupus erythematosus, (51) and it is possible that HS1 will be linked to more diseases in the future.
Two parameters that describe DC random migration are cell speed and cell persistence. In DCs, we saw that HS1 does not affect the persistence time, or frequency of turning. It does, however, contribute a great deal to the speed of migration. HS1 is the hematopoietic homologue of the more widely studied NPF cortactin (18). Many of the migration results that we observe in HS1−/− DCs mirror what is seen in cells expressing reduced levels of cortactin (52). Cortactin, unlike WASP or other NPFs, has the unique ability to stabilize actin branch points and prevent disassembly (53,54). We hypothesize that this stabilizing effect is responsible for the reduction in speed we observe in HS1−/− DCs. WASP is still present in these cells, allowing Arp2/3 complex branch points to form, but without HS1 holding the branch point together, the structure disassembles too quickly for a cell to gain much speed.
We saw that HS1 and WASP play a role in DC random migration to different degrees. One of the first differences we observed was the extent of reduction in average speed for each type of cell. Both HS1 and WASP−/− DCs move more slowly, but WASP−/− DCs were significantly faster than HS1−/− DCs. These disparate results could be explained by differences between the two proteins. WASP is a Class I NPF, with a VCA domain that binds to monomeric actin and the Arp2/3 complex, while HS1 is a Class II NPF with an NTA domain that binds to polymerized F actin and the Arp2/3 complex (23). Class I NPFs like WASP are thought to initiate Arp2/3 complex-mediated actin polymerization by bringing monomeric G-actin in close proximity to the Arp2/3 complex, while Class II NPFs like HS1 are thought stabilize a pre-formed branch point by binding to both F-actin and the Arp2/3 complex. There are multiple Class I NPFs in hematopoietic cells, but HS1 is the only known Class II NPF. Perhaps the speed is not affected as much in WASP−/− DCs due to Class I NPF redundancy, whereas elimination of HS1 rids the cell of all stabilizing Class II NPFs. Another possibility could be related to differences in downstream binding partners of the two proteins.
We expected to see similar force reductions in WASP and HS1−/− DCs, since they are both known to play a role in adhesive structures, such as podosomes (22). Furthermore, the Arp2/3 complex, which is activated by both HS1 and WASP, has been shown to transiently associate with vinculin in focal complex like structures (48). Interestingly, we saw much smaller forces in HS1−/− DCs than in WASP−/− DCs. This difference could again be explained by the slight differences in function of the two proteins. Since HS1−/− DCs have unstable branched actin networks, it is possible that their actin networks disassemble before they are able to generate substantial force. It has also been shown that reducing levels of cortactin inhibits the assembly of adhesion structures (52), whereas loss of WASP−/− seemingly has no effect on integrin organization (5). Therefore, it is possible that adhesion and force generation are influenced by Class II NPFs (i.e. HS1) more strongly than they are by Class I NPFs (i.e. WASP). Another factor that likely contributes to these differences in force generation is NPF redundancy. As discussed in the previous paragraph, HS1 is the only Class II NPF in DCs, while WASP is one of multiple Class I NPFs in DCs. It is possible that other Class I NPFs compensate for WASP during force generation.
In this study, we used CK-666 to identify whether the migration effects seen in HS1−/− DCs were due to HS1’s ability to activate and stabilize Arp2/3 complex mediated actin polymerization. CK-666 is a potent inhibitor of the Arp2/3 complex, with an IC50 of 17 μM and 4 μM, for bovine and human Arp2/3 complex, respectively (55). We chose a range of concentrations surrounding these IC50 values (1 μM, 10 μM and 100 μM), since we expected the IC50 for murine Arp2/3 complex to lie somewhere in this range. Previous cytotoxicity studies have indicated that concentrations up to 200 μM CK-666 with incubation times similar to ours did not negatively impact cell viability (56). We saw a graded decrease in migration as more CK-666 was added to DCs, with the highest concentration (100 μM) approximating the behavior of HS1−/− DCs. We still saw low levels of migration, even at high levels of inhibitor, which is likely due to incomplete inhibition of cellular Arp2/3 complex. While the speed of HS1−/− DCs is further decreased upon Arp2/3 complex inhibition, the persistence length and random motility coefficient are unaffected. Removing HS1 from DCs greatly reduces Arp2/3 complex activity without completely eliminating its ability to polymerize actin. When inhibitor is added, the remaining Arp2/3 complex activity is further reduced.
In combination with previous studies, this work reveals that HS1 is involved in various aspects of DC migration. Klos Dehring et al. showed that during chemotaxis, DCs lacking HS1 form highly dynamic lamellipodia, move well through transwells and travel through microfluidic devices with lower directionality and higher speed (22). This description of overly dynamic, unstable lamellipodia agrees well with our observations that HS1−/− DCs form extensive membrane ruffles during chemokinesis and are often observed to quickly vacillate around fixed points. This suggests that new branched actin structures are actively being formed, but due to the loss of HS1, and the resultant instability of the actin network, they are often too inefficient to allow for DC translocation. During chemotaxis, HS1−/− DCs are less able to migrate persistently up a gradient than WT DCs (22). Since chemokine receptor expression is unaltered by HS1 removal, and endpoint studies of HS1−/− DCs show that they are able to properly respond to a gradient (transwell), this defect in directional persistence is likely due to instability and not chemokine responsiveness (22). It is possible that HS1−/− DCs, are unable to maintain protrusive actin structures at the front of the cell while traveling up a gradient. Dynamic lamellipodial protrusions at other areas of the cell body may lead to deviations in directionality. These deviations would then be corrected upon additional chemokine signaling, but the inherent actin instability would lead to more frequent missteps and lower observed persistence in HS1−/− DCs. In our chemokinesis experiments, we observed no significant differences in persistence time. While this may seem contradictory, DCs behave quite differently in chemokinesis and chemotaxis. In chemokinesis, DC motion is completely random, whereas in chemotaxis, DCs are guided in a specific direction. Klos Dehring et al. showed that DCs seemed to have a higher propensity for deviations from directed migration, and in essence moved more randomly than their WT counterparts (22). Therefore, it is quite possible that in a scenario where DCs are being encouraged to migrate randomly, HS1−/− DCs will do so as efficiently as WT DCs.
Another major difference between our study and the work of Klos Dehring et al. is the effect of HS1 on cell speed. Klos Dehring et al observe a significant increase in speed during chemotaxis (22), while we observed a significant decrease during chemokinesis. While it is clear that HS1−/− DCs migrate towards a chemokine gradient during chemotaxis, the spread of HS1−/− DC trajectories is not identical to the spread of WT DCs trajectories (22). The cell tracks for WT DCs traveling towards a gradient in the y-direction are symmetric about the y-axis (22). However, the cell tracks for HS1−/− DCs traveling towards a gradient in the y-direction are biased towards the positive x-direction (22). The directional bias seen in HS1−/− DCs could be due to an increased susceptibility to flow in the microfluidic gradient generator. Therefore, it is possible that the increase in HS1−/− DC speed during chemotaxis is due the cells being more easily pushed along by flow in the system. This is quite likely given the effect of HS1 on DC adhesive strength (defective podosomes in immature HS1−/− DCs (22) and significantly reduced traction forces in mature HS1−/− DCs). In the present study, we observed a significant decrease in speed, in an environment free of flow and directional cues. Therefore, it is possible that innate, random migratory speed is impaired in HS1−/− DCs when they are not presented with any additional stimuli, but that reductions in adhesion and subsequent susceptibility to flow could help DCs to compensate for defects in speed during chemotaxis. Additional experiments should be performed using chemotactic chambers to fully characterize HS1−/− DC chemotaxis and resolve the disparities observed in speed during chemokinesis and chemotaxis.
While we found the Arp2/3 complex to be important for DC random migration on 2D surfaces, it appears to be dispensable for other forms of migration. Recently Vargas et al. showed that Arp2/3 complex inhibition had no effect on DC chemotaxis in confinement (57). DCs in confined channels did not form leading edge branched actin networks but instead had extensive actin cables at the cell rear. This finding does not negate our findings, since many of the experimental variables were different between their study and ours. Rather, this suggests that DCs can employ multiple methods of migration based on the characteristics of their microenvironment.
Conclusions
The migration of dendritic cells is essential for launching a proper immune response. The biomolecular coordination within a migrating DC is complex. In this study, we explored the contributions of one of these molecules, HS1, and determined that it is important for both migration and force generation, likely through its interaction with the Arp2/3 complex. With such drastic reductions in speed and force generation in HS1−/− DCs, we would expect to see other aspects of DC function impaired. Future work on the role of HS1 in vivo could help to elucidate its importance for health and immune function.
Supplementary Material
Insight, Innovation, Integration.
Dendritic cell migration is required for initiation of the adaptive immune response. However, the molecular mechanisms underlying dendritic cell migration are not fully understood. We used a combination of genetically modified dendritic cells and an Arp2/3 complex inhibitor to characterize the role of HS1 during DC migration and we used micropost array detectors (mPADs) to characterize the ability of these cells to generate forces during migration. From this study, we conclude that DC random migration and force generation are intimately linked and both depend on HS1. This dependence likely stems from the influence of both HS1 and the Arp2/3 complex on branched actin polymerization.
Acknowledgments
This work was supported by NIH GM104287. ACB acknowledges support from the NSF GRFP under Grant No. DGE-1321851.
HS1−/− DCs transduced with GFP Life-act were kindly provided by Daniel Blumenthal.
References
- 1.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu Y, et al. Immunobiology of Dendritic Cells. Annu RevImmunology. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM. The Dendritic Cell System and Its Role in Immunogenicity. Annu RevImmunology. 1991;9:271–96. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 3.Watts C, Amigorena S. Antigen traffic pathways in dendritic cells. Traffic [Internet] 2000;1(4):312–7. doi: 10.1034/j.1600-0854.2000.010404.x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11208116. [DOI] [PubMed] [Google Scholar]
- 4.Reis e Sousa C, Sher A, Kaye P. The role of dendritic cells in the induction and regulation immunity to microbial infection. Curr Opin Immunol. 1999;11:392–9. doi: 10.1016/S0952-7915(99)80066-1. [DOI] [PubMed] [Google Scholar]
- 5.Burns S, Hardy SJ, Buddle J, Yong KL, Jones GE, Thrasher AJ. Maturation of DC Is Associated with Changes in Motile Characteristics and Adherence. Cell Motil Cytoskeleton. 2004;57:118–32. doi: 10.1002/cm.10163. October 2003. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez D, Vollmann EH, von Andrian UH. Mechanisms and consequences of dendritic cell migration. Immunity [Internet] 2008;29(3):325–42. doi: 10.1016/j.immuni.2008.08.006. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18799141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell [Internet] 2003;112(4):453–65. doi: 10.1016/s0092-8674(03)00120-x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12600310. [DOI] [PubMed] [Google Scholar]
- 8.Lauffenburger DA, Horwitz AF. Cell migration: A physically integrated molecular process. Cell. 1996;84(3):359–69. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 9.Abraham VC, Krishnamurthi V, Taylor DL, Lanni F. The actin-based nanomachine at the leading edge of migrating cells. Biophys J. 1999;77(3):1721–32. doi: 10.1016/S0006-3495(99)77018-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol [Internet] 2012;197(2):239–51. doi: 10.1083/jcb.201112113. Available from: http://www.jcb.org/cgi/doi/10.1083/jcb.201112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailly M, Ichetovkin I, Grant W, Zebda N, Machesky LM, Segall JE, et al. The F-actin side binding activity of the Arp2/3 complex is essential for actin nucleation and lamellipod extension. Curr Biol [Internet] 2001;11(8):620–5. doi: 10.1016/s0960-9822(01)00152-x. Available from: http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6VRT-430G2NG-Y&_user=501045&_coverDate=04/17/2001&_rdoc=1&_fmt=high&_orig=gateway&_origin=gateway&_sort=d&_docanchor=&view=c&_acct=C000022659&_version=1&_urlVersion=0&_userid=501045&md5=35f97e5ebec. [DOI] [PubMed] [Google Scholar]
- 12.Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A. 1998;95(11):6181–6. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarar D, To W, Abo A, Welch MD. The Wiskott–Aldrich syndrome protein directs actin-based motility by stimulating actin nucleation with the Arp2/3 complex. Curr Biol [Internet] 1999;9(10):555–S1. doi: 10.1016/s0960-9822(99)80243-7. Available from: http://www.sciencedirect.com/science/article/pii/S0960982299802437. [DOI] [PubMed] [Google Scholar]
- 14.Uruno T, Zhang P, Liu J, Hao J-J, Zhan X. Haematopoietic lineage cell-specific protein 1 (HS1) promotes actin-related protein (Arp) 2/3 complex-mediated actin polymerization. Biochem J [Internet] 2003;371(Pt 2):485–93. doi: 10.1042/BJ20021791. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1223309/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uruno T, Liu J, Zhang P, Fan Yx, Egile C, Li R, et al. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat Cell Biol. 2001;3(3):259–66. doi: 10.1038/35060051. [DOI] [PubMed] [Google Scholar]
- 16.Takemoto Y, Furuta M, Li XK, Strong-Sparks WJ, Hashimoto Y. LckBP1, a proline-rich protein expressed in haematopoietic lineage cells, directly associates with the SH3 domain of protein tyrosine kinase p56lck. EMBO J. 1995;14(14):3403–14. doi: 10.1002/j.1460-2075.1995.tb07346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitamura D, Kaneko H, Miyagoe Y, Ariyasu T, Watanabe T. Isolation and characterization of a novel human gene expressed specifically in the cells of hematopoietic lineage. Nucleic Acids Res [Internet] 1989;17(22):9367–79. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC335138/ [PMC free article] [PubMed] [Google Scholar]
- 18.Zhan X, Hu X, Hampton B, Burgess WH, Friesel R, Maciag T. Murine cortactin is phosphorylated in response to fibroblast growth factor-1 on tyrosine residues late in the G1 phase of the BALB/c 3T3 cell cycle. J Biol Chem. 1993;268(32):24427–31. [PubMed] [Google Scholar]
- 19.Hao JJ, Zhu J, Zhou K, Smith N, Zhan X. The coiled-coil domain is required for HS1 to bind to F-actin and activate Arp2/3 complex. J Biol Chem. 2005;280(45):37988–94. doi: 10.1074/jbc.M504552200. [DOI] [PubMed] [Google Scholar]
- 20.Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, Nolz JC, et al. HS1 Functions as an Essential Actin-Regulatory Adaptor Protein at the Immune Synapse. Immunity [Internet] 2006;24(6):741–52. doi: 10.1016/j.immuni.2006.03.022. Available from: http://www.sciencedirect.com/science/article/pii/S1074761306002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muzio M, Scielzo C, Frenquelli M, Bachi A, De Palma M, Alessio M, et al. HS1 complexes with cytoskeleton adapters in normal and malignant chronic lymphocytic leukemia B cells. Leukemia [Internet] 2007;21(9):2067–70. doi: 10.1038/sj.leu.2404744. Available from: http://dx.doi.org/10.1038/sj.leu.2404744. [DOI] [PubMed] [Google Scholar]
- 22.Dehring DAK, Clarke F, Ricart BG, Huang Y, Gomez TS, Williamson EK, et al. Hematopoietic Lineage Cell-Specific Protein 1 Functions in Concert with the Wiskott–Aldrich Syndrome Protein To Promote Podosome Array Organization and Chemotaxis in Dendritic Cells. J Immunol [Internet] 2011;186(8):4805–18. doi: 10.4049/jimmunol.1003102. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3467106/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol [Internet] 2006;7(10):713–26. doi: 10.1038/nrm2026. Available from: http://www.nature.com/doifinder/10.1038/nrm2026. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Biswas C, Dehring DAK, Sriram U, Williamson EK, Li S, et al. The actin regulatory protein HS1 is required for antigen uptake and presentation by dendritic cells. J Immunol [Internet] 2011;187(11):5952–63. doi: 10.4049/jimmunol.1100870. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3221870/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snapper SB, Meelu P, Nguyen D, Stockton BM, Bozza P, Alt FW, et al. WASP deficiency leads to global defects of directed leukocyte migration in vitro and in vivo. J Leukoc Biol. 2005 Jun;77:993–8. doi: 10.1189/jlb.0804444. [DOI] [PubMed] [Google Scholar]
- 26.Taniuchi I, Kitamura D, Maekawa Y, Fukuda T, Kishi H, Watanabe T. Antigen-receptor induced clonal expansion and deletion of lymphocytes are impaired in mice lacking HS1 protein, a substrate of the antigen-receptor-coupled tyrosine kinases. EMBO J [Internet] 1995;14(15):3664–78. doi: 10.1002/j.1460-2075.1995.tb00036.x. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC394441/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sixt M, Lämmermann T. Vitro Analysis of Chemotactic Leukocyte Migration in 3D Environments. In: Wells CM, Parsons M, editors. Cell Migration [Internet] Humana Press; 2011. pp. 149–65. Available from: http://dx.doi.org/10.1007/978–1–61779-207-6_11. [DOI] [PubMed] [Google Scholar]
- 28.Desai RA, Khan MK, Gopal SB, Chen CS. Subcellular spatial segregation of integrin subtypes by patterned multicomponent surfaces. Integr Biol (Camb) 2011;3(5):560–7. doi: 10.1039/c0ib00129e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunn GA. Characterising a kinesis response: time averaged measures of cell speed and directional persistence. [Internet] Agents and actions. 1983:14–33. doi: 10.1007/978-3-0348-9352-7_1. Supplements. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6573115. [DOI] [PubMed]
- 30.Henry SJ, Crocker JC, Hammer DA. Motile Human Neutrophils Sense Ligand Density Over Their Entire Contact Area. Ann Biomed Eng [Internet] 2015 doi: 10.1007/s10439-015-1408-2. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26219404. [DOI] [PMC free article] [PubMed]
- 31.Yang MT, Fu J, Wang Y-K, Desai RA, Chen CS. Nat Protoc [Internet] 2. Vol. 6. Nature Publishing Group; 2011. Assaying stem cell mechanobiology on microfabricated elastomeric substrates with geometrically modulated rigidity; pp. 187–213. Available from: http://www.nature.com/doifinder/10.1038/nprot.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scielzo C, Bertilaccio MTS, Simonetti G, Dagklis A, ten Hacken E, Fazi C, et al. HS1 has a central role in the trafficking and homing of leukemic B cells HS1 has a central role in the trafficking and homing of leukemic B cells. Blood. 2010;116(18):3537–46. doi: 10.1182/blood-2009-12-258814. [DOI] [PubMed] [Google Scholar]
- 33.Cavnar PJ, Mogen K, Berthier E, Beebe DJ, Huttenlocher A. J Biol Chem [Internet] 30. Vol. 287. 9650 Rockville Pike, Bethesda, MD 20814, U.S.A.: American Society for Biochemistry and Molecular Biology; 2012. The Actin Regulatory Protein HS1 Interacts with Arp2/3 and Mediates Efficient Neutrophil Chemotaxis; pp. 25466–77. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3408136/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butler B, Kastendieck DH, Cooper JA. Nat Immunol [Internet] 8. Vol. 9. Nature Publishing Group; 2008. Differently phosphorylated forms of the cortactin homolog HS1 mediate distinct functions in natural killer cells; pp. 887–97. Available from: http://dx.doi.org/10.1038/ni.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lettau M, Kabelitz D, Janssen O. SDF1α-induced interaction of the adapter proteins Nck and HS1 facilitates actin polymerization and migration in T cells. Eur J Immunol [Internet] 2015;45(2):551–61. doi: 10.1002/eji.201444473. Available from: http://doi.wiley.com/10.1002/eji.201444473. [DOI] [PubMed] [Google Scholar]
- 36.Mukherjee S, Kim J, Mooren OL, Shahan ST, Cohan M, Cooper JA. Role of Cortactin Homolog HS1 in Transendothelial Migration of Natural Killer Cells. PLoS One [Internet] 2015;10(2):e0118153. doi: 10.1371/journal.pone.0118153. Available from: http://dx.plos.org/10.1371/journal.pone.0118153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hetrick B, Han MS, Helgeson LA, Nolen BJ. Chem Biol [Internet] 5. Vol. 20. Elsevier Ltd; 2013. Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change; pp. 701–12. Available from: http://www.sciencedirect.com/science/article/pii/S1074552113001245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Higgs HN, Blanchoin L, Pollard TD. Influence of the C terminus of Wiskott-Aldrich syndrome protein (WASp) and the Arp2/3 complex on actin polymerization. Biochemistry [Internet] 1999;38(46):15212–22. doi: 10.1021/bi991843+. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10563804. [DOI] [PubMed] [Google Scholar]
- 39.Blanchoin L, Amann K, Higgs H, Marchand J, Kaiser D, Pollard T. Direct observation of dendritic actin complex and WASP/Scar proteins. Nature. 2000;171(1994):1007–11. doi: 10.1038/35010008. [DOI] [PubMed] [Google Scholar]
- 40.Binks M, Jones GE, Brickell PM, Kinnon C, Katz DR, Thrasher a J. Intrinsic dendritic cell abnormalities in Wiskott-Aldrich syndrome. Eur J Immunol [Internet] 1998;28(10):3259–67. doi: 10.1002/(SICI)1521-4141(199810)28:10<3259::AID-IMMU3259>3.0.CO;2-B. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9808195. [DOI] [PubMed] [Google Scholar]
- 41.Burns S, Thrasher AJ, Blundell MP, Machesky LM, Jones GE. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood. 2001;98(4):1142–9. doi: 10.1182/blood.v98.4.1142. [DOI] [PubMed] [Google Scholar]
- 42.Westerberg L, Wallin RPA, Greicius G, Ljunggren H-G, Severinson E. Efficient antigen presentation of soluble, but not particulate, antigen in the absence of Wiskott-Aldrich syndrome protein. Immunology [Internet] 2003;109(3):384–91. doi: 10.1046/j.1365-2567.2003.01668.x. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1782978&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borg C, Abdelali J, Laderach D, Maruyama K, Wakasugi H, Charrier S, et al. NK Cell Activation by Dendritic Cells (DC) Require The Formation of a Synapse leading to IL-12 Polarization in DC. Blood [Internet] 2004;104(10):3267–76. doi: 10.1182/blood-2004-01-0380. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15242871. [DOI] [PubMed] [Google Scholar]
- 44.Bouma G, Burns S, Thrasher AJ. Impaired T-cell priming in vivo resulting from dysfunction of WASp-deficient dendritic cells. Blood. 2007;110(13):4278–84. doi: 10.1182/blood-2007-06-096875. [DOI] [PubMed] [Google Scholar]
- 45.Pulecio J, Tagliani E, Scholer A, Prete F, Fetler L, Burrone OR, et al. Expression of Wiskott-Aldrich syndrome protein in dendritic cells regulates synapse formation and activation of naive CD8+ T cells. J Immunol. 2008;181(2):1135–42. doi: 10.4049/jimmunol.181.2.1135. [DOI] [PubMed] [Google Scholar]
- 46.de Noronha S, Hardy S, Sinclair J, Blundell MP, Strid J, Schulz O, et al. Impaired dendritic-cell homing in vivo in the absence of Wiskott-Aldrich syndrome protein. Blood [Internet] 2005;105(4):1590–7. doi: 10.1182/blood-2004-06-2332. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15494425. [DOI] [PubMed] [Google Scholar]
- 47.Ricart BG, Yang MT, Hunter CA, Chen CS, Hammer DA. Biophys J [Internet] 11. Vol. 101. Biophysical Society; 2011. Measuring traction forces of motile dendritic cells on micropost arrays; pp. 2620–8. Available from: http://dx.doi.org/10.1016/j.bpj.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeMali KA, Barlow CA, Burridge K. Recruitment of the Arp2/3 complex to vinculin: Coupling membrane protrusion to matrix adhesion. J Cell Biol. 2002;159(5):881–91. doi: 10.1083/jcb.200206043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci U S A [Internet] 2003;100(4):1484–9. doi: 10.1073/pnas.0235407100. Available from: http://www.pnas.org/content/100/4/1484.full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frezzato F, Gattazzo C, Martini V, Trimarco V, Teramo A, Carraro S, et al. HS1, a lyn kinase substrate, is abnormally expressed in B-chronic lymphocytic leukemia and correlates with response to fludarabine-based regimen. PLoS One. 2012;7(6):1–11. doi: 10.1371/journal.pone.0039902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sawabe T, Horiuchi T, Koga R, Tsukamoto H, Kojima T, Harashima S, et al. Aberrant HS1 molecule in a patient with systemic lupus erythematosus. Genes Immun. 2003;4(2):122–31. doi: 10.1038/sj.gene.6363932. [DOI] [PubMed] [Google Scholar]
- 52.Bryce NS, Clark ES, Leysath JL, Currie JD, Webb DJ, Weaver AM. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr Biol. 2005;15(14):1276–85. doi: 10.1016/j.cub.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 53.Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, et al. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol [Internet] 2001;11(5):370–4. doi: 10.1016/s0960-9822(01)00098-7. Available from: http://www.sciencedirect.com/science/article/pii/S0960982201000987. [DOI] [PubMed] [Google Scholar]
- 54.Egile C, Rouiller I, Xu XP, Volkmann N, Li R, Hanein D. Mechanism of filament nucleation and branch stability revealed by the structure of the Arp2/3 complex at actin branch junctions. PLoS Biol. 2005;3(11):1902–9. doi: 10.1371/journal.pbio.0030383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, et al. Nature [Internet] 7258. Vol. 460. Nature Publishing Group; 2009. Characterization of two classes of small molecule inhibitors of Arp2/3 complex; pp. 1031–4. Available from: http://dx.doi.org/10.1038/nature08231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ilatovskaya DV, Chubinskiy-Nadezhdin V, Pavlov TS, Shuyskiy LS, Tomilin V, Palygin O, et al. Arp2/3 complex inhibitors adversely affect actin cytoskeleton remodeling i the cultured murine kidney collecting duct M-1 cells. Cell Tissue Res. 2013;354(3):783–92. doi: 10.1007/s00441-013-1710-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vargas P, Maiuri P, Bretou M, Saez PJ, Pierobon P, Maurin M, et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat cell Biol. 18(1):43–53. doi: 10.1038/ncb3284. 15AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.