

Abstract
G protein-coupled receptors (GPCRs) comprise a large and diverse class of signal-transducing receptors that undergo dynamic and isoform-specific membrane trafficking. GPCRs thus have an inherent potential to initiate or regulate signaling reactions from multiple membrane locations. The present review discusses emerging insight to the subcellular organization of GPCR function in mammalian cells, focusing on signaling transduced by heterotrimeric G proteins and β-arrestins. We summarize recent evidence indicating that GPCR-mediated activation of G proteins occurs not only from the plasma membrane but also from endosomes and Golgi membranes, and that β-arrestin-dependent signaling can be transduced from the plasma membrane by β-arrestin trafficking to clathrin-coated pits after dissociating from a ligand-activated GPCR.
Keywords: Signaling, GPCR, G protein, arrestin, endosome, Golgi
A spatial dimension of cellular GPCR signaling
GPCRs mediate cellular signaling by stimulus-triggered coupling to cytoplasmic transducer proteins. Previously, the subcellular location of ligand-dependent GPCR signaling was of limited interest because it was assumed that activation by ligand is restricted to the plasma membrane (PM). Recently, additional sites of ligand-induced activation have been recognized, and interest has grown in the functional consequences of localized activation. Progress in this area has been facilitated by improved understanding of GPCR trafficking mechanisms and by the development of new approaches for achieving direct detection and spatiotemporal resolution of signaling reactions in living cells (e.g., [1–4]).
The present review focuses on spatial aspects of GPCR signal initiation through interactions with heterotrimeric G proteins and arrestins in mammalian cells. We first briefly outline early concepts regarding the location of GPCR-mediated signal initiation. We then review accumulating recent evidence supporting the current concept that ligand-dependent GPCRs activate G proteins and arrestin transducers at both PM and internal membrane locations. Finally, we discuss the evolving functional understanding of spatially organized GPCR signaling.
GPCR-mediated activation of heterotrimeric G proteins
GPCRs initiate canonical (G protein-dependent) signaling by stimulus-dependent rearrangements of the seven-transmembrane helical bundle that are allosterically coupled to a reduced affinity of guanine nucleotide binding to the Ras-like α subunit of the heterotrimeric G protein complex. This promotes release of bound GDP from the inactive Gα subunit and its replacement with GTP present in excess in the cytoplasm. GTP binding, in turn, stabilizes an activating conformational change in the Gα subunit that, depending on the G protein, promotes rearrangement or dissociation of the βγ subcomplex [5–8]. Both Gα and Gβγ regulate downstream effectors, with activity terminated by hydrolysis of GTP to GDP on Gα that promotes reformation of the inactive complex [9]. Accordingly, GPCRs trigger G protein signaling catalytically through an intrinsic ligand-dependent guanine nucleotide exchange factor (GEF) activity [10].
Recent studies make clear that considerably more remains to be learned about the molecular dynamics of this fundamental activation mechanism and its operation in living cells. Single molecule measurements of Förster resonance energy transfer between beta-2 adrenergic receptors (β2ARs) and the Gs heterotrimeric G protein in vitro revealed evidence for multiple conformational states depending on the bound ligand and identified a transient nucleotide-bound β2AR–Gs species that is distinct from presently known structures [11]. A recent study of single molecule trajectories in the PM of living cells revealed hot spots at which both the β2AR and Gs are transiently immobilized and proposed to couple [12].
Role of arrestins in terminating G protein activation
Many GPCR responses transduced by G proteins adapt over time with prolonged or repeated ligand exposure, exhibiting the phenomenon of desensitization. A well established mechanism of rapid desensitization was first recognized through studies of inactivation of the light-activated GPCR, rhodopsin, in retinal photoreceptor cells. Visual arrestin was shown to bind specifically to rhodopsin after its light-induced conformational activation and phosphorylation by rhodopsin kinase (GRK1). This interaction was found to produce a high-affinity (Kd~50 nM) GPCR-arrestin complex that terminates signaling by physical ‘arrest’ of the nucleotide exchange factor activity of the GPCR [13], [14].
Visual arrestin was then found to also inhibit the GEF activity of the β2AR in vitro. Maximal inhibition required ligand-induced phosphorylation of the β2AR by an enzyme that is homologous to rhodopsin kinase and expressed outside of photoreceptor cells (beta-adrenergic receptor kinase or GRK2). However, a relatively high concentration of visual arrestin was required to achieve inhibition of β2AR activity [15] and an arrestin homologue with higher apparent affinity for adrenergic receptors, called beta-arrestin (β-arrestin), was subsequently identified [16,17]. This mechanism for inactivating ligand-dependent GEF activity using GRKs and arrestins has proven to be relevant to many GPCRs, with two major β-arrestin isoforms (β-arrestin-1 and -2, also called Arrestin 2 and 3) and multiple GRK isoforms identified that are broadly expressed in mammalian tissues [17].
Additional trafficking and signaling functions of β-arrestins
In addition to terminating GEF activity by binding to GPCRs, β-arrestins were shown to function as endocytic adapters by interacting with phosphoinositides, clathrin, and AP-2 [18–22]. Through these interactions, β-arrestins promote the ligand-dependent concentration of GPCRs into clathrin-coated pits (CCPs) that form in the PM and pinch off by a dynamin-dependent membrane scission mechanism to produce receptor-containing endocytic vesicles [23–25].
Still more diversity of β-arrestin function was recognized by the discovery that β-arrestins can bind to over 20 different protein kinases [26–29]. These interactions are thought to support ‘non-canonical’ (G protein-independent) GPCR signaling by molecular scaffolding. In particular, β-arrestins have been shown to promote signaling by mitogen-activated protein kinases (MAPKs), serine/threonine kinases including ERK1/2, p38 kinases, and the c-Jun N-terminal kinases that are activated through phosphorylation mediated by an upstream protein kinase [26].
β-arrestin signaling from endosomes
Early evidence suggesting that ligand-dependent GPCR signaling can also occur from an internal membrane location emerged from investigations of the signal transducer function of β-arrestin. A correlation was noted between endocytosis and β-arrestin-dependent ERK activation triggered by ligand-dependent activation of several GPCRs [30,31], and dominant-negative versions of dynamin and β-arrestin-1 that inhibit β2AR internalization were shown to also inhibit ligand-dependent activation of ERK [32]. More support for activation at endosomes came from the demonstration that c-SRC recruitment to β2AR through binding to β-arrestin can promote ERK activation, and that ERK activation by the β2AR can be blocked by over-expression of a mutant version of β-arrestin-1 (V53D) that binds c-SRC but does not promote β2AR internalization [29]. Protease-activated receptor 2 was shown to form a complex after activation that includes β-arrestin-1, Raf-1, and phosphorylated ERK [33]. Ligand-activated AT1ARs were found to form a complex including β-arrestin-2, Raf-1, MEK1, and ERK1/2 [34]. Moreover, ERK and β-arrestins were found to colocalize on endocytic vesicles together with several GPCRs [34,35]. Together, such findings led to the concept that endosomes act as signalosomes specific to β-arrestin [36].
Endosomal activation of β-arrestin signaling is also consistent with kinetic data. A G protein-dependent component of ERK activation mediated by AT1ARs was found to peak ~2 minutes after the application of agonist ligand, corresponding to a time at which most receptors are still present in the PM. A β-arrestin-dependent component of the response became evident after 10 – 30 minutes, in line with the time required for AT1ARs to internalize [37]. Together, these observations solidified the view that GPCR-mediated activation of G proteins is restricted to the PM and that signaling transduced by β-arrestin requires GPCR-β-arrestin complex formation and trafficking of the complex to endosomes [38].
G protein signaling from internal membrane compartments
Rhodopsin activates transducin primarily from internal membrane compartments in vertebrate photoreceptor cells [39] but intracellular G protein activation by ligand-dependent GPCRs was doubted for many years due to a number of assumptions, as discussed previously [40]. This view began to change with the work of several groups [41–47]. Briefly summarized, these studies provided (1) colocalization and co-fractionation data indicating that G proteins and adenylyl cyclase are present in internal membrane compartments containing ligand-internalized GPCRs and (2) inhibitor data demonstrating that acute endocytic blockade attenuates some G protein-dependent cellular responses.
The development of conformational biosensors, a class of genetically encoded fluorescent detectors of specific GPCR and G protein conformational states, allowed the subcellular location of β2AR and Gs activation to be resolved dynamically in living cells. Application of agonist ligand produced conformational activation of both proteins at two locations sequentially, first the PM and then endosomes [48–50]. Some GPCRs, such as the β2AR and D1 dopamine receptor, produce transient Gs activation at endosomes [41,48]. Other GPCRs, such as the PTH and V2 vasopressin receptors, produce more prolonged activation at endosomes and appear to do so by forming an alternate activation complex containing the G protein together with β-arrestin [51,52].
Conformational biosensors subsequently resolved the Golgi apparatus and trans-Golgi network as additional locations of ligand-dependent GPCR and Gs activation. Two cellular mechanisms for Golgi-associated signal activation have been proposed. Activation of beta-1 adrenergic receptors (β1ARs) and Gs in the Golgi apparatus uses a preexisting receptor pool rather than receptors delivered from the cell surface and requires ligands to access the Golgi-localized receptor pool separately. Catecholamines gain access the Golgi-localized β1AR pool by facilitated transmembrane transport involving the organic cation transporter 3 whereas more hydrophobic drugs appear to access the Golgi pool by passive diffusion [53]. Activation of Gs by TSH receptors in the trans-Golgi network utilizes receptors internalized from the PM together with ligand, similar to activation of Gs at endosomes by other GPCRs such as the β2AR [54].
β-arrestin signaling from the PM
As noted above, there is considerable evidence that β-arrestin-dependent signaling can be initiated from endosomes. The possibility that β-arrestin can signal also from the PM was first suggested by a study demonstrating that engineered chemical recruitment of β-arrestin to the PM is sufficient to activate ERK in the absence of activated receptors [55]. It remained unknown for some time if β-arrestin signaling from the PM is triggered by ligand-induced activation of a GPCR. Suggesting that this might be the case, several studies showed that endocytic blockade imposed by chemical inhibition of dynamin enhances rather than inhibits β-arrestin-dependent activation of ERK by some GPCRs [56–58].
Live cell fluorescence microscopy provided insight into this initially puzzling result by revealing that chemical inhibition of dynamin stalls CCPs in the PM while leaving β-arrestin associated with them. Further, it was found that blockade of endocytosis by RNAi-mediated depletion of clathrin heavy chain – a distinct manipulation that prevents CCP formation altogether rather than stalling them in the PM – inhibits rather than enhances the MAPK response [57]. An additional property of GPCRs that promote β-arrestin-dependent ERK signaling from the PM is that their activation can prolong the lifetime of arrestin-associated CCPs at the cell surface before dissociation of the coat structure that occurs shortly after endocytic vesicle formation. Interestingly, this natural prolongation of CCP lifetime – produced by receptor activation rather than by chemical inhibition of dynamin – was similarly correlated with enhancement of the β-arrestin-dependent MAPK response. Together these observations revealed a discrete cellular mechanism of β-arrestin-dependent activation of MAPK that is initiated at the PM and uses CCPs as dynamically regulated PM signaling stations.
A discrete cellular mechanism of β-arrestin activation
Additional insight to β-arrestin activation at the PM emerged from efforts to resolve another biological conundrum. The β1AR was long recognized to promote β-arrestin signaling in vivo [59] but this GPCR, in contrast to the β2AR, typically internalizes poorly via CCPs. Consistent with this, live cell imaging demonstrated that β1ARs do not detectably accumulate in CCPs after ligand-induced activation. Nevertheless, β1AR activation was found to robustly promote the accumulation of β-arrestin in CCPs apparently without an associated receptor. Further, β1ARs were able to trigger ligand-dependent trafficking of β-arrestin to CCPs even under conditions in which the activating GPCR was laterally immobilized to assure its inability to move into CCPs [57]. These observations indicate that β-arrestin can operate as an autonomous transducer of GPCR activation, after dissociating from its upstream activating GPCR and trafficking to CCPs, and suggest that GPCRs can trigger β-arrestin signaling in a catalytic manner that does not require the GPCR and β-arrestin to remain stably bound.
In parallel with this emerging cell biological understanding, a confluence of recent structural and biophysical observations support additional flexibility of cellular β-arrestin function. Single particle electron microscopy has described two GPCR-arrestin complexes, a canonical complex in which both the GPCR tail and transmembrane core are engaged and another in which only the GPCR tail is engaged [60]. These discrete complexes may have functional relevance because mutations that selectively destabilize the GPCR transmembrane core interaction prevent functional desensitization of G protein signaling while maintaining receptor internalization and arrestin-dependent signaling [61–63]. Experiments using Förster resonance energy transfer have provided independent support for the hypothesis that β-arrestin can remain in an activated conformation after dissociation from a GPCR [64], and suggest that more than one conformational state of β-arrestin can be generated in living cells depending on the GPCR [65].
Concluding remarks and future directions
It is increasingly clear that ligand-dependent GPCRs can functionally couple to G protein and β-arrestin transducers at multiple subcellular membrane locations (summarized schematically in Figure 1). The PM, long recognized to be a major location of G protein activation, is also a site of β-arrestin-dependent signaling using CCPs. Endosomes, once thought to function only as a repository or sorting station for inactive or desensitized GPCRs, can also support G protein as well as β-arrestin -mediated signal initiation. In addition, some GPCRs mediate G protein activation at Golgi-associated membrane compartments. Together, these developments support a model in which GPCR signal initiation in mammalian cells is spatially and temporally organized across multiple membrane locations. This represents a significant change from the previous view and raises many new questions that remain to be investigated (see Outstanding Questions Box). Several of these are further elaborated below, placed in context of presently available data.
Figure 1.

Schematic summarizing subcellular locations of GPCR-mediated activation of arrestins and heterotrimeric G proteins supported by data discussed in the text. Top panel: Arrestin (β-arrestins -1 and -2) are thought to be subject to activation both in endosomes and the PM. Bottom panel: Gs activation by various GPCRs (see text for details) has been described in the PM, endosomes, Golgi apparatus and trans-Golgi network.
Outstanding questions box.
How is signaling from internal membranes regulated and terminated?
What are the functional consequences of localized G protein and β-arrestin activation?
How much diversity and specificity exists in localized GPCR signaling?
What is the spatiotemporal organization of cellular GPCR signaling in native tissues?
One important direction for future study is to determine how G protein and β-arrestin activities initiated from distinct membrane locations are terminated. As a general principle, cellular GPCR signaling is exquisitely regulated. Molecular mechanisms that terminate G protein signaling from the PM are now well understood, but much less is known about the control of G protein signal initiation from internal membranes. Endosome signaling may be terminated by acidification, receptor engagement of arrestins or arrestin-related proteins, or engagement of multi-protein regulatory complexes associated with the endosome membrane. In particular, an actin and sorting nexin 27 -linked version of the conserved retromer complex (ASRT) is interesting because it contains an arrestin-related protein (Vps26) and exerts receptor-specific control of the strength of endosomal G protein activation by determining the residence time of activated GPCRs in the endosome membrane [45,66–68]. GPCR - G protein activation at the Golgi apparatus is acutely inhibited by corticosterone through control of facilitated ligand transport activity [53], but clearly much more remains to be learned about termination of signaling from Golgi-associated membranes. Endosome signaling by β-arrestin depends on stability of the GPCR-β-arrestin complex and signaling from the PM is limited by the surface lifetime of arrestin-associated CCPs [57], but clearly much more remains to be learned also about the regulation and termination of arrestin-dependent signaling.
A second proposed future direction is to delineate the functional significance of location-based GPCR signal initiation. Endosome-initiated signals are delayed in onset and often more prolonged than signals initiated from the PM. Accordingly, endocytosis inherently affects the timing of cellular signal initiation as well as its location. In some cases location appears to be a primary determinant of downstream signaling selectivity. For example, location of β2AR-Gs activation in endosomes has been explicitly shown to be important for promoting cAMP-mediated transcriptional responses [69]. In other cases the primacy of one dimension over another is less clear, and it appears likely that both dimensions are salient. For example, TSH receptors drive a PKA-dependent phosphorylation of CREB and gene transcription in response to TSH that is delayed relative to PKA activation at the PM and occurs during, or upon, receptor relocalization from endosomes to the trans-Golgi network [54]. Endosomal activation of LH receptors in ovarian follicle cells sustains the cellular cAMP response elicited by the mid-cycle LH surge and also positions the site of cAMP generation near gap junctions; both effects are thought to be important for lifting meiotic arrest in the nucleus of the enclosed oocyte to drive oocyte maturation [70]. Clearly there is a great deal more to be learned about how spatial and temporal dimensions of GPCR-G protein signal initiation are interpreted functionally in living cells.
It is presently unclear if similar or different considerations apply to location-dependent signaling by β-arrestins. β-arrestin activation in endosomes is typically delayed in onset compared to activation in the PM, similar to what has been observed for endosomal G protein activation. However, β-arrestin-dependent activation of ERK from endosomes appears to restrict the activated kinase to the cytoplasm and prevent signaling to the nucleus [33,71,72]. This is opposite to endosomal Gs activation, which promotes nuclear responses. Thus, while endocytosis appears to affect the location and timing of β-arrestin activation in a similar manner as G protein activation, there may be significant differences in how location is interpreted to modify cellular function (Figure 2).
Figure 2.
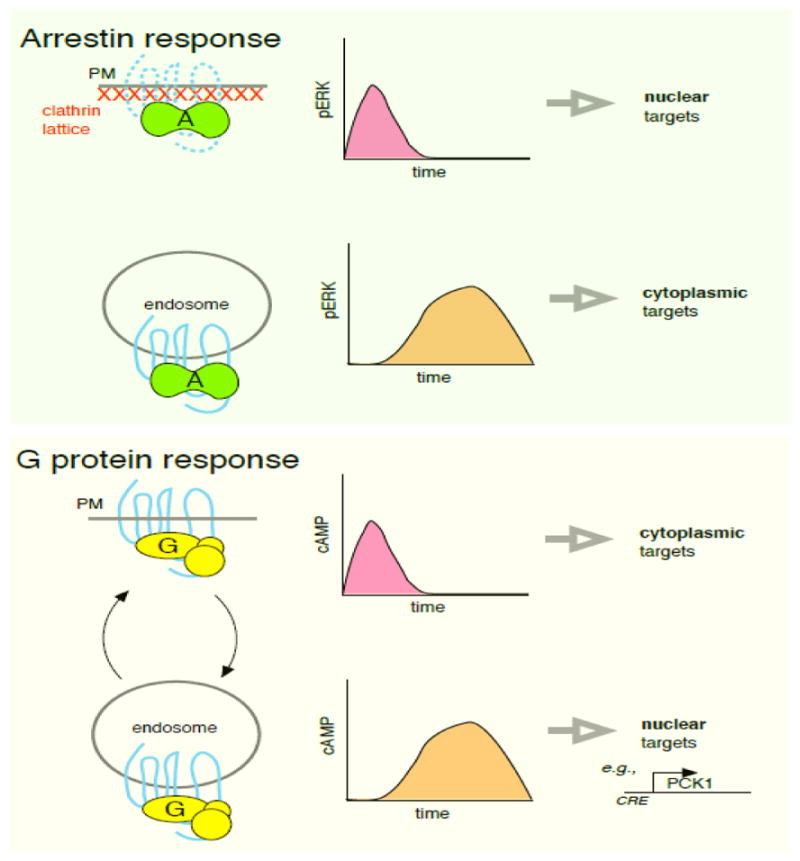
Schematic contrasting current view of functional impact of localized signaling transduced by β-arrestins and Gs. Top panel: GPCR-mediated activation of β-arrestins in the PM is thought to produce a rapid ERK response that can access nuclear targets. Activation in endosomes produces delayed ERK activation and sequesters ERK in the cytoplasm, thereby preventing access to nuclear targets. Bottom panel: GPCR-mediated activation of Gs in the PM produces a rapid and transient response that activates cAMP-dependent targets in the PM and cytoplasm. Activation in endosomes produces a delayed response that preferentially accesses nuclear targets (induction of the cAMP-responsive gene PCK1 is shown as an example. For GPCRs that are capable of efficient recycling, PM and endosome-initiated activation waves can occur repeatedly in the prolonged presence of agonist through multiple rounds of endocytosis and recycling (depicted as curved arrows).
A third future direction is to determine how broadly the presently recognized principles apply across the large GPCR family. Even closely related GPCR subtypes, such as the β1AR and β2AR, are already known to differ in membrane trafficking properties that impact signaling. This is likely only the beginning of diversity and specificity in the GPCR family. For example, LH receptors internalize and recycle in a generally similar manner as the β2AR but they recycle through a different population of endosomes that have been shown to change both the location and timing of downstream ERK activation [73]. β2AR-mediated activation of Gs in endosomes is terminated by receptor engagement with retromer [68], similar to termination of endosomal PTH receptor signaling [45], but retromer promotes rather than inhibits TSH receptor-mediated signaling transduced by Gs from the trans-Golgi network [54].
We also note that most of what is presently known about G protein activation from internal membranes is limited to activation of Gs. Other G protein isoforms have been observed at internal membranes but whether they undergo intracellular activation by GPCRs remains a largely open question. Much also remains to be learned about diversity of β-arrestin engagement by GPCRs and its functional significance, particularly with accumulating evidence that the subcellular location of β-arrestin-dependent signal initiation can differ even among closely related GPCRs such as the CB1 and CB2 cannabinoid receptors [74].
A fourth proposed direction is to elucidate the spatial organization of GPCR signaling in a native cellular environment and at endogenous protein expression levels. Our present understanding is derived largely from model cell systems in which at least one component of the cascade is overexpressed. Live cell imaging of fluorescently tagged proteins, although a powerful tool for direct examination of subcellular location, has oftentimes required overexpression of components due to technical limitations in gene transfer methodologies or detection sensitivity. Improved genetic tools and in situ fluorescent labeling methodologies are now being used to overcome this barrier. For example, a recent study using CRISPR-based gene editing and an improved version of split GFP was used to achieve real-time imaging of cellular proteins at endogenous levels in living cells, including several that function in GPCR signaling [75]. Laminated optical sheet microscopy was used to visualize the presence of endogenous TSH receptors in the Golgi region of primary mouse thyroid cells and activation of endogenous Gs at this location [54]. It was also previously shown that internal TSH receptor activation after endocytosis is evident in some cell types but not others [46]. Improved transgenic animal models, such as a cAMP biosensor-expressing mouse that has been used successfully to study endosomal signaling in thyroid cells as well as intact ovarian follicles [70], offer the potential to examine location-based signaling by GPCRs in a near-native tissue environment. A recent study that applied Talen and CRISPR -based gene editing to investigate β2AR-mediated ERK activation in an established cellular model system [76] indicates that such methods are also able to provide new mechanistic insight.
Trends box.
active signal initiation by GPCRs is not restricted to the plasma membrane
GPCR-mediated activation of Gs can occur in endosomes and the Golgi apparatus
β-arrestin-mediated activation of MAP kinase can occur from the plasma membrane
β-arrestin can mediate signaling after dissociating from its activating GPCR
clathrin-coated pits function as β-arrestin signaling stations in the plasma membrane
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Di Fiore PP, von Zastrow M. Endocytosis, signaling, and beyond. Cold Spring Harb Perspect Biol. 2014:6. doi: 10.1101/cshperspect.a016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irannejad R, von Zastrow M. GPCR signaling along the endocytic pathway. Curr Opin Cell Biol. 2014;27:109–116. doi: 10.1016/j.ceb.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irannejad R, et al. Effects of endocytosis on receptor-mediated signaling. Curr Opin Cell Biol. 2015;35:137–143. doi: 10.1016/j.ceb.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ni Q, et al. Live-cell imaging of cell signaling using genetically encoded fluorescent reporters. FEBS J. 2017 doi: 10.1111/febs.14134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasmussen SGF, et al. Structure of a nanobody-stabilized active state of the beta2 adrenoceptor. 2011 doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen SGF, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. 2011 doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenbaum DM, et al. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank M, et al. G Protein activation without subunit dissociation depends on a G{alpha}(i)-specific region. J Biol Chem. 2005;280:24584–24590. doi: 10.1074/jbc.M414630200. [DOI] [PubMed] [Google Scholar]
- 9.Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 10.Ross EM. G Protein-coupled receptors: Multi-turnover GDP/GTP exchange catalysis on heterotrimeric G proteins. Cell Logist. 2014;4:e29391. doi: 10.4161/cl.29391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregorio GG, et al. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature. 2017;547:68–73. doi: 10.1038/nature22354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sungkaworn T, et al. Single-molecule imaging reveals receptor-G protein interactions at cell surface hot spots. Nature. 2017;550:543–547. doi: 10.1038/nature24264. [DOI] [PubMed] [Google Scholar]
- 13.Wilden U, et al. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci U S A. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuckerman R, et al. Arrestin mediates ATP/ADP exchange and quench of cGMP phosphodiesterase activation. Invest Ophthalmol Vis Sci. 1985;26:45. [Google Scholar]
- 15.Benovic JL, et al. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proceedings of the National Academy of Sciences. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohse MJ, et al. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 17.Attramadal H, et al. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- 18.Shenoy SK, Lefkowitz RJ. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaidarov I, et al. Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J. 1999;18:871–881. doi: 10.1093/emboj/18.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laporte SA, et al. The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proceedings of the National Academy of Sciences. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laporte SA, et al. The interaction of β-arrestin with the AP-2 adaptor is required for the clustering of β2-adrenergic receptor into clathrin-coated pits. J Biol Chem. 2000;275:23120–23126. doi: 10.1074/jbc.M002581200. [DOI] [PubMed] [Google Scholar]
- 22.Goodman OB, Jr, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson SS, et al. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 24.Kirchhausen T. Clathrin. Annu Rev Biochem. 2000;69:699–727. doi: 10.1146/annurev.biochem.69.1.699. [DOI] [PubMed] [Google Scholar]
- 25.Goodman OB, et al. Role of Arrestins in G-Protein-Coupled Receptor Endocytosis. In: Goldstein DS, et al., editors. Advances in Pharmacology. Supplement C. Vol. 42. Academic Press; 1997. pp. 429–433. [DOI] [PubMed] [Google Scholar]
- 26.DeWire SM, et al. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 27.Gurevich EV, Gurevich VV. Arrestins: ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kook S, et al. Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J Biol Chem. 2013;288:37332–37342. doi: 10.1074/jbc.M113.510412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luttrell LM, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 30.Luttrell LM, et al. G protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat 1a fibroblasts. Shc phosphorylation and receptor endocytosis correlate with activation of Erk kinases. J Biol Chem. 1997;272:31648–31656. doi: 10.1074/jbc.272.50.31648. [DOI] [PubMed] [Google Scholar]
- 31.Lin FT, et al. β-Arrestins Regulate Mitogenic Signaling and Clathrin-mediated Endocytosis of the Insulin-like Growth Factor I Receptor. J Biol Chem. 1998;273:31640–31643. doi: 10.1074/jbc.273.48.31640. [DOI] [PubMed] [Google Scholar]
- 32.Daaka Y, et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 33.DeFea KA, et al. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a β-arrestin-dependent scaffolding complex. Proceedings of the. 2000 doi: 10.1073/pnas.190276697. at < http://www.pnas.org/content/97/20/11086.short>. [DOI] [PMC free article] [PubMed]
- 34.Luttrell LM, et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci U S A. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tohgo A, et al. The Stability of the G Protein-coupled Receptor-β-Arrestin Interaction Determines the Mechanism and Functional Consequence of ERK Activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 36.Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J. 2003;375:503–515. doi: 10.1042/BJ20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahn S, et al. Differential Kinetic and Spatial Patterns of β-Arrestin and G Protein-mediated ERK Activation by the Angiotensin II Receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- 38.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 39.Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsvetanova NG, et al. G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J Biol Chem. 2015;290:6689–6696. doi: 10.1074/jbc.R114.617951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kotowski SJ, et al. Endocytosis promotes rapid dopaminergic signaling. Neuron. 2011;71:278–290. doi: 10.1016/j.neuron.2011.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullershausen F, et al. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol. 2009;5:428–434. doi: 10.1038/nchembio.173. [DOI] [PubMed] [Google Scholar]
- 43.Calebiro D, et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7:e1000172. doi: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrandon S, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742. doi: 10.1038/nchembio.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feinstein TN, et al. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol. 2011;7:278–284. doi: 10.1038/nchembio.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Werthmann RC, et al. Persistent cAMP signaling by internalized TSH receptors occurs in thyroid but not in HEK293 cells. FASEB J. 2012;26:2043–2048. doi: 10.1096/fj.11-195248. [DOI] [PubMed] [Google Scholar]
- 47.Jong YJI, et al. Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J Biol Chem. 2009;284:35827–35838. doi: 10.1074/jbc.M109.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Irannejad R, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495:534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rasmussen SGF, et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol. 2011;21:567–572. doi: 10.1016/j.sbi.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jean-Alphonse FG, et al. β2-adrenergic receptor control of endosomal PTH receptor signaling via Gβγ. Nat Chem Biol. 2017;13:259–261. doi: 10.1038/nchembio.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomsen ARB, et al. GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell. 2016;166:907–919. doi: 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Irannejad R, et al. Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol. 2017;13:799–806. doi: 10.1038/nchembio.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Godbole A, et al. Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat Commun. 2017;8:443. doi: 10.1038/s41467-017-00357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terrillon S, Bouvier M. Receptor activity-independent recruitment of betaarrestin2 reveals specific signalling modes. EMBO J. 2004;23:3950–3961. doi: 10.1038/sj.emboj.7600387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flores-Otero J, et al. Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat Commun. 2014;5:4589. doi: 10.1038/ncomms5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eichel K, et al. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat Cell Biol. 2016;18:303–310. doi: 10.1038/ncb3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinberg ZY, et al. Sequence-Specific Regulation of Endocytic Lifetimes Modulates Arrestin-Mediated Signaling at the μ Opioid Receptor. Mol Pharmacol. 2017;91:416–427. doi: 10.1124/mol.116.106633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel PA, et al. Physiologic and cardiac roles of β-arrestins. J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2008.11.015. at < http://www.sciencedirect.com/science/article/pii/S0022282808014089>. [DOI] [PubMed]
- 60.Shukla AK, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumari P, et al. Functional competence of a partially engaged GPCR-β-arrestin complex. Nat Commun. 2016;7:13416. doi: 10.1038/ncomms13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumari P, et al. Core engagement with β-arrestin is dispensable for agonist-induced vasopressin receptor endocytosis and ERK activation. Mol Biol Cell. 2017;28:1003–1010. doi: 10.1091/mbc.E16-12-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cahill TJ, et al. Distinct conformations of GPCR β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proceedings of the National Academy of Sciences. 2017;114:2562–2567. doi: 10.1073/pnas.1701529114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nuber S, et al. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature. 2016;531:661–664. doi: 10.1038/nature17198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee MH, et al. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature. 2016;531:665–668. doi: 10.1038/nature17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bowman SL, et al. Distinct G protein-coupled receptor recycling pathways allow spatial control of downstream G protein signaling. J Cell Biol. 2016;214:797–806. doi: 10.1083/jcb.201512068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tian X, et al. The α-Arrestin ARRDC3 Regulates the Endosomal Residence Time and Intracellular Signaling of the β2-Adrenergic Receptor. J Biol Chem. 2016;291:14510–14525. doi: 10.1074/jbc.M116.716589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varandas KC, et al. Retromer Endosome Exit Domains Serve Multiple Trafficking Destinations and Regulate Local G Protein Activation by GPCRs. Curr Biol. 2016;26:3129–3142. doi: 10.1016/j.cub.2016.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsvetanova NG, von Zastrow M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol. 2014;10:1061–1065. doi: 10.1038/nchembio.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lyga S, et al. Persistent cAMP Signaling by Internalized LH Receptors in Ovarian Follicles. Endocrinology. 2016;157:1613–1621. doi: 10.1210/en.2015-1945. [DOI] [PubMed] [Google Scholar]
- 71.DeFea KA, et al. β-Arrestin–Dependent Endocytosis of Proteinase-Activated Receptor 2 Is Required for Intracellular Targeting of Activated Erk1/2. J Cell Biol. 2000;148:1267–1282. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tohgo A, et al. β-Arrestin Scaffolding of the ERK Cascade Enhances Cytosolic ERK Activity but Inhibits ERK-mediated Transcription following Angiotensin AT1a Receptor Stimulation. J Biol Chem. 2002;277:9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- 73.Jean-Alphonse F, et al. Spatially restricted G protein-coupled receptor activity via divergent endocytic compartments. J Biol Chem. 2014;289:3960–3977. doi: 10.1074/jbc.M113.526350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nogueras-Ortiz C, et al. Retromer stops beta-arrestin 1 mediated signaling from internalized cannabinoid 2 receptors. Mol Biol Cell. 2017 doi: 10.1091/mbc.E17-03-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leonetti MD, et al. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci U S A. 2016;113:E3501–8. doi: 10.1073/pnas.1606731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O’Hayre M, et al. Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci Signal. 2017;10(484) doi: 10.1126/scisignal.aal3395. pii: eaal3395. [DOI] [PMC free article] [PubMed] [Google Scholar]