

SUMMARY
Correlative evidence suggests that polyploidization of heart muscle, which occurs naturally in post-natal mammals, creates a barrier to heart regeneration. Here, we move beyond a correlation by demonstrating that experimental polyploidization of zebrafish cardiomyocytes is sufficient to suppress their proliferative potential during regeneration. Initially, we determined that zebrafish myocardium becomes susceptible to polyploidization upon transient cytokinesis inhibition mediated by dominant negative Ect2. Using a transgenic strategy, we generated adult animals containing mosaic hearts composed of differentially labeled diploid and polyploid-enriched cardiomyocyte populations. Diploid cardiomyocytes outcompeted their polyploid neighbors in producing regenerated heart muscle. Moreover, hearts composed of equivalent proportions of diploid and polyploid cardiomyocytes failed to regenerate altogether, demonstrating that a critical percentage of diploid cardiomyocytes is required to achieve heart regeneration. Our data identify cardiomyocyte polyploidization as a barrier to heart regeneration and suggest that mobilizing rare diploid cardiomyocytes in the human heart will improve its regenerative capacity.
eTOC Blurb
It remains unclear why certain non-mammalian species efficiently regenerate their hearts, while mammals fail in this endeavor. Gonzalez-Rosa et al. demonstrate that simply increasing the DNA content of the highly regenerative zebrafish myocardium, to more closely resemble that in mammals, is sufficient to dampen cardiomyocyte proliferative capacity and organ regeneration.
INTRODUCTION
Cells containing two sets of homologous chromosomes are diploid, while those with greater than two complete sets, either enclosed within a single nucleus or separated into multiple nuclei, are termed polyploid (Davoli and de Lange, 2011). Polyploid cells arise naturally during the maturation of certain lineages when diploid cells fuse or complete DNA replication without mitosis and/or cytokinesis (Orr-Weaver, 2015). Although polyploidization correlates with terminal differentiation in some lineages, cell cycle exit is not a universal feature of polyploid cells. For instance, polyploid hepatocytes contribute robustly to regenerated liver segments through proliferation with or without ploidy reversal (Duncan et al., 2010). Therefore, the consequences of polyploidization are context specific.
Adult mammalian hearts lack any appreciable capacity to regenerate. By contrast, injured hearts of neonatal mice (Porrello et al., 2011) and some non-mammalian vertebrates, including salamanders (Becker et al., 1974; Nakamura et al., 2016; Oberpriller and Oberpriller, 1974) and zebrafish (González-Rosa and Mercader, 2012; Poss et al., 2002), mount an impressive regenerative response driven by myocardial proliferation (Foglia and Poss, 2016). Despite their clinical importance, the variables that promote or restrict myocardial regeneration remain obscure. One often-noted difference between non-regenerative and highly regenerative hearts is the DNA content of their cardiomyocytes (Bersell et al., 2009; Kikuchi, 2014; Vivien et al., 2016). Specifically, non-regenerative hearts contain a majority of polyploid cardiomyocytes (Brodsky et al., 1994) that form early in life when their diploid predecessors complete one round of DNA replication without mitosis and/or cytokinesis (Alkass et al., 2015; Brodsky et al., 1980; Li et al., 1996; Soonpaa et al., 1996). By contrast, highly regenerative hearts contain a majority of cardiomyocytes that are diploid (Oberpriller et al., 1988), or assumed to be diploid based on nucleation studies (Wills et al., 2008). Although these observations reveal an inverse correlation between ploidy and regeneration in the myocardium, a causal relationship has yet to be explored.
In rodents, the transition from diploid (1 × 2c) to polyploid myocardium occurs within the first post-natal week and results in myocardium composed of >90% polyploid cardiomyocytes that are largely binucleated (2 × 2c) (Li et al., 1996; Soonpaa et al., 1996). In humans, myocardial polyploidization occurs within the first two decades, which causes a similarly high percentage of cardiomyocytes to become polyploid (Mollova et al., 2013; Sandritter and Adler, 1976). However, the adult human heart contains 30% binucleated (2x2c) and ~60% mononucleated cardiomyocytes with DNA contents ranging from 4c to 16c (1x4c, 1x8c, 1x16c). Therefore, polyploid myocardium is a common feature of the non-regenerative hearts of adult mammals despite species-specific differences in DNA content and nuclear number.
In neonatal mice, the timing of myocardial polyploidization coincides with the disappearance of cardiac regenerative potential (Porrello et al., 2011), which has led to speculation of causality (Vivien et al., 2016). However, the post-natal maturation of the heart is accompanied by numerous transitions within cardiomyocytes, resident non-cardiomyocyte populations, extra-cardiac cells, and the extra-cellular matrix (Vivien et al., 2016). Despite the notion that one or more of these transitions are responsible for suppressing regenerative capacity, the causative factors remain largely unidentified.
In an effort to move beyond a correlation, we set out to design an experimental strategy for testing the hypothesis that myocardial polyploidization in zebrafish is sufficient to dampen heart regeneration. Important features of this system would include: 1) a mechanism for creating polyploid cardiomyocytes through inducible cytokinesis inhibition during hyperplastic growth of the heart, 2) a counter mechanism for re-enabling cytokinesis during adulthood both prior to cardiac injury and during regeneration, and 3) indelible reporter labeling of induced polyploid cardiomyocyte populations to compare their regenerative capabilities side by side with unlabeled diploid cardiomyocytes in vivo.
Our studies indicate that induced myocardial polyploidization is sufficient to dampen cardiomyocyte proliferation in a highly regenerative setting. Moreover, they uncover a requirement for a critical percentage (>50%) of diploid cardiomyocytes to achieve heart regeneration. Ultimately, these conclusions provide rationale for identifying therapeutic approaches to amplify the rare population of diploid cardiomyocytes in the human heart as a means to induce natural heart regeneration in disease states.
RESULTS
Zebrafish cardiomyocytes are mononucleated, diploid, and upregulate Ect2 during heart regeneration
Zebrafish cardiomyocytes were assumed to be diploid based on their predominantly mononuclear nature (Wills et al., 2008). However, because mononuclear cells can be either diploid or polyploid, we set out to make direct measurements of cardiomyocyte ploidy in adult animals. To that end, we developed an imaging and analysis platform to survey cardiomyocyte nucleation and DNA content in single-cell dissociations of adult ventricles. Cardiomyocytes containing a single 2c nucleus (1 × 2c) were categorized as diploid (Figure 1A), while those containing two ≥2c nuclei (2 × ≥2c) or a single ≥4c nucleus (1 × ≥4c) were classified as polyploid. To unequivocally identify cardiomyocytes and distinguish multinucleated cells from mononucleated aggregates, we analyzed dissociations from Tg(cmlc2:nucGFP), Tg(cmlc2:CAAXmKate) zebrafish in which cardiomyocyte nuclei and plasma membranes were labeled with GFP and mKate, respectively (Figures 1B–1D and Figure S1A–S1H). This approach virtually eliminated the possibility of falsely counting cell aggregates as polyploid cardiomyocytes, which is a drawback of flow cytometric analysis. Nucleation analysis alone revealed that ~99% of cardiomyocytes in homeostatic hearts are mononucleated (Figure 1E), consistent with a previous report (Wills et al., 2008). DAPI-fluorescence intensity was used to quantify the DNA contents of cardiomyocyte nuclei relative to a reference population of non-myocardial diploid cells (Figure S1I–S1Y). When plotted on a histogram, the distribution of cardiomyocyte ploidy, taking into account both nuclear number and DNA content (Figures 1F–1H), clustered into a single major peak that aligned with the diploid peak (Figure 1I). Altogether, this analysis revealed that ~99% of zebrafish cardiomyocytes are diploid (1x2c), whereas ~1% are polyploid with one (1x4c) or two (2x2c) nuclei (Figure 1K). Thus, in contrast to the non-regenerative polyploid hearts of adult mammals, the highly regenerative zebrafish myocardium is diploid, which further bolsters the inverse correlation observed between myocardial ploidy and regenerative capacity.
Figure 1. Zebrafish cardiomyocytes are mononucleated, diploid, and upregulate Ect2 during heart regeneration.

(A) Schematic depicting cardiomyocyte nucleation and ploidy. See also Figure S1. (B) Cardiomyocytes (white arrows) from dissociated Tg(cmlc2:nucGFP) hearts. (C,D) Mononucleated and binucleated Tg(cmlc2:nucGFP) cardiomyocytes. (E) Average percentages of mononucleated (MonoN) and binucleated (BiN) cardiomyocytes in uninjured (UI), 7 dpr and 60 dpr ventricles (mean±s.d., n=9309, 9269, 9578 total cells respectively from 4 biological replicates per group; 2 pooled ventricles per replicate; n.s., not significant by one-way ANOVA test). (F,H) DAPI-stained mononucleated diploid, binucleated tetraploid, and mononucleated tetraploid cardiomyocytes isolated from Tg(cmlc2:nucGFP) hearts. Insets show DAPI signal. (I) Distributions of non-cardiomyocyte (gray) and cardiomyocyte (green) DNA content in homeostatic ventricles (n=779 and 552 cells, respectively, from 3 biological replicates; 1 ventricle per replicate). (J) Distributions of cardiomyocyte DNA content in 7 dpr and 60 dpr ventricles (blue and orange bars, respectively; overlap appears in brown). Insets are magnifications to show low frequency events (black arrows, tetraploid cardiomyocytes). (K) Quantification of indicated cardiomyocyte populations from UI, 7 dpr and 60 dpr ventricles (mean±s.d., n=3250, 2699 and 4306 total cells from 7, 3 and 4 biological replicates per group, respectively; 3 ventricles per replicate; ****P<0.0001; **P<0.01 by one-way ANOVA followed by Tukey’s multiple comparisons test). (L) qPCR analysis showing relative expression of three genes involved in cytokinesis in UI, 7 dpr, and 60 dpr ventricles (mean±s.d, n=3 technical replicates, 10 biological replicates, 1 ventricle per replicate, ****P<0.0001; **P<0.01; *P<0.05 by one-way ANOVA followed by Tukey’s multiple comparisons test). See also Figure S2. (M,N) Single confocal sections of ect2 RNAScope (arrowheads) in UI and 7 dpr hearts. Boxed regions are shown at higher magnifications. n=4 hearts per group with 3 sections per heart. Asterisk indicates wound edge. Scale bars: 50 μm. CM, cardiomyocytes.
In adult mammals, a small number of cardiomyocytes surrounding infarcted myocardium re-enter the cell cycle, undergo DNA replication, but largely fail to complete cytokinesis, which increases ploidy further (Ebert and Pfitzer, 1977; Senyo et al., 2013). To determine if cardiac injury induces myocardial polyploidization in zebrafish, we surveyed cardiomyocyte nucleation and ploidy both during (7 days post-resection; dpr) and after (60 dpr) heart regeneration. While the percentage of binucleated cardiomyocytes remained constant at both time points (Figure 1E), the proportion of mononucleated 4c cells increased at 7 dpr (Figures 1J–1K), which we attribute to diploid cardiomyocytes cycling through G2 (Poss et al., 2002). At 60 dpr, the percentage of mononucleated 4c cells remained elevated (Figure 1K), despite lowered cardiomyocyte proliferation at this stage (Poss et al., 2002). These data suggest that a small proportion of bona fide polyploid cardiomyocytes are created during regeneration. Nonetheless, they constitute only ~1% of the total population, demonstrating that successful cytokinesis, rather than polyploidization, is the predominant outcome of zebrafish cardiomyocytes entering the cell cycle during regeneration.
We set out to test the hypothesis that doubling the DNA content of zebrafish cardiomyocytes would be sufficient to suppress their proliferative potential following injury. To that end, we sought an experimental tool to achieve transient inhibition of cardiomyocyte cytokinesis during cardiac growth, which occurs primarily by hyperplasia in zebrafish (Wills et al., 2008). We searched the literature to identify dominant-negative proteins that were previously demonstrated to promote cytokinesis failure. We learned that dominant-negative forms of Racgap1, Kif23, or Ect2 are potent inhibitors of cytokinesis in cultured mammalian cells (Hirose et al., 2001; Liu et al., 2004; Tatsumoto et al., 1999). Racgap1 and Kif23 perform essential functions in central spindle formation. At the spindle, Ect2 catalyzes the conversion of RhoA-GDP to RhoA-GTP, which is required for contractile ring assembly and cytokinesis initiation (Green et al., 2012). Previous studies, using gene knockdown and knockout approaches, have demonstrated that Ect2 is essential for cytokinesis both in cultured cells and in several model organisms including zebrafish (Green et al., 2012; Hoijman et al., 2015).
To determine whether racgap1, kif23, and ect2 are induced in proliferating cardiomyocytes, we performed qPCR on uninjured and regenerating hearts. We learned that all three genes are significantly upregulated at 7 dpr, with ect2 showing the strongest induction (Figure 1L), which persuaded us to evaluate its localization during regeneration. Using RNAScope in situ hybridization, we documented ect2 expression in cardiomyocytes along the wound edge (Figure 1M–1N) where proliferation is the highest (Sallin et al., 2015). These data are consistent with the notion that cardiomyocyte cytokinesis relies on Ect2 function in zebrafish.
Interestingly, by mining a publically available RNA-sequencing dataset (O’Meara et al., 2015), we discovered that Ect2 expression declines in mouse neonatal cardiomyocytes (Figure S2) as they become polyploid (Alkass et al., 2015; Soonpaa et al., 1996) and the regenerative window closes (Porrello et al., 2011). This observation suggests that Ect2 downregulation might be a natural mechanism to induce cardiomyocyte polyploidization in mammals.
Ect2 is required for zebrafish cardiomyocyte cytokinesis and for maintaining their diploid state
To learn if Ect2 is required for cardiomyocyte cytokinesis in zebrafish, we performed nucleation and ploidy analyses on cardiomyocytes from ect2 null-mutant embryos (Amsterdam et al., 2004) carrying the cmlc2:nucDsRed-Express transgene (Takeuchi et al., 2011), which labels cardiomyocyte nuclei with red fluorescence. Using brightfield microscopy, we identified ect2 mutant embryos by their pronounced body curvatures, small heads and necrotic brains (Figure S3). To measure cardiomyocyte ploidy in these animals, we first determined nuclear number for individual cardiomyocytes in confocal Z-stacks using nuclear red fluorescence and a plasma membrane marker. We then quantified the ploidy of each nucleus based on DAPI fluorescence intensity. Compared to control siblings, mutant hearts at 30 hours post fertilization (hpf) contained a 2-fold increase in polyploid cardiomyocytes (Figures S4A–S4C). This difference grew to 9-fold at 72 hpf (Figures 2A, 2B and 2D), which is soon after hyperplastic growth of the heart commences (Choi et al., 2013). These data demonstrate that proliferating zebrafish cardiomyocytes become polyploid in the absence of Ect2 activity.
Figure 2. Loss of Ect2 function causes polyploidization of zebrafish cardiomyocytes.

(A–C) Confocal projections of 72 hpf embryonic hearts from control (CTRL), ect2−/−, and Tg(cmlc2:GdnEct2) animals carrying the Tg(cmlc2:nucDsRed) transgene. Single confocal planes of boxed regions are shown at higher magnification with Alcama immunostaining to highlight plasma membranes. White and yellow arrows indicate diploid and polyploid cardiomyocytes, respectively. See also Figures S3 and S4A–S4F. (D) Quantification of indicated cardiomyocyte populations in the indicated cohorts at 72 hpf (mean±s.d, n=4, 4 and 4 embryos for ect2+/+, ect2−/−, non-Tg, and Tg(cmlc2:dnEct2), respectively. ****P<0.0001; ***P<0.001; **P<0.01; *P<0.05 by one-way ANOVA followed by Tukey’s multiple comparisons test. (E) 45 dpf (e19.5 mm SSL) Tg(cmlc2:GdnEct2) and non-Tg sibling animals with arrowheads highlighting scale bristling (green), blood pooling (white), and pericardial edema (purple) in Tg(cmlc2:dnEct2) zebrafish. 68/77 Tg(cmlc2:GdnEct2) and 0/88 non-Tg siblings developed these phenotypes. (F) Representative Kaplan-Meier plot for Tg(cmlc2:GdnEct2) and non-Tg cluchmates from one of three independent experiments. 77 total Tg(cmlc2:GdnEct2) animals and 88 total siblings were followed; ****P<0.0001, Log-rank test. (G,H) Confocal projections of DAPI-stained whole hearts from 30 dpf non-Tg or Tg(cmlc2:GdnEct2) animals carrying the Tg(cmlc2:nucDsRed) reporter. 19/25 transgenic animals showed this phenotype with 6/25 showing an intermediate phenotype. (I,J) Ventricular sections from 30 dpf non-Tg or Tg(cmlc2:GdnEct2) animals immunostained to detect Tropomyosin and counterstained with DAPI. Boxed regions show DAPI staining at higher magnifications. (K,L) Nuclear DNA content distributions of non-cardiomyocyte and cardiomyocyte populations from indicated cohorts. Representative data are shown from one of four replicates, one heart per replicate. See also Figure S4G–S4I. AT, atrium; BA, bulbus arteriosus; V, ventricle; Scale bars: 25 μm (A–C, G–H), 10 mm (E), 100 μm (I,J).
Next, we tested whether a dominant negative Ect2 protein (dnEct2) would inhibit cytokinesis and induce polyploidization in cardiomyocytes during cardiac growth. Previous reports demonstrated that overexpression of a C-terminally truncated mouse ECT2 protein, which lacks the GEF catalytic domain but retains RhoA-GDP binding activity, behaves as a dominant negative protein (dnECT2) by interfering with the catalytic conversion of RhoA-GDP to RhoA-GTP by wild-type ECT2 (Oceguera-Yanez, 2005; Sakata et al., 2000; Tatsumoto et al., 1999). We isolated and analyzed transgenic animals [Tg(cmlc2:GFP-P2A-dnEct2), abbreviated cmlc2:GdnEct2] continuously expressing GFP and an analogously-truncated zebrafish Ect2 protein (dnEct2) in all cardiomyocytes from embryonic stages. At 72 hpf, transgenic embryos displayed a 6-fold increase in polyploid cardiomyocytes (Figures 2C and 2D), a phenotype similar to that observed in ect2 mutants. To rule out perturbations in cell cycle entry caused by dnEct2 expression, we analyzed cardiomyocyte DNA synthesis and found no significant differences between cmlc2:GdnEct2 and sibling embryos (Figures S4D–S4F). During larval stages, cmlc2:GdnEct2 zebrafish were morphologically indistinguishable from siblings until 25 days post fertilization (dpf) (12.5 mm standardized standard length, SSL, Parichy et al., 2009) when their growth slowed, signs of heart failure emerged (Figure 2E), and survival plummeted (Figure 2F). An analysis of transgenic hearts at 30 dpf (14.5 mm SSL) revealed that ventricles of relatively normal size were composed of drastically fewer cardiomyocyte nuclei, 75% of which were polyploid with DNA contents ranging from 4c–16c (Figures 2G–2L). Cardiomyocyte diameter measurements revealed significant cellular hypertrophy (Figure S4G–S4I). These data demonstrate that continuous expression of dnEct2 expression in zebrafish cardiomyocytes impairs cytokinesis, induces polyploidization, and causes cells to grow by hypertrophy instead of hyperplasia. While tolerated initially, hypertrophic growth eventually becomes pathologic and heart failure ensures. We created these animals simply as a proof of concept that dnEct2 expression inhibits cardiomyocyte cytokinesis in zebrafish. They were not used for regeneration studies because ongoing experimental inhibition of cardiomyocyte cytokinesis after injury would mask any inherent proliferative potential polyploid cardiomyocytes might possess.
Design of a new transgenic system to create polyploid cardiomyocytes and track their contributions to regeneration
Therefore, we designed a double transgenic system, with two variations, to create adult zebrafish whose hearts contained cardiomyocyte populations that were: 1) significantly enriched in polyploid cells created by transient dnEct2 expression during cardiac growth and 2) permanently labeled with GFP. The first variation caused only a subset of the myocardium to become GFP+ and polyploid enriched, which permitted a side-by-side comparison of the regenerative capacities of diploid and polyploid cardiomyocyte populations in the same hearts. The second caused the entire myocardium to become GFP+ and polyploid enriched, which significantly boosted the overall polyploid proportion and allowed us to evaluate the consequences for cardiac regenerative fitness.
For the first variation, we generated the cmlc2:(iF-S)lox-mG transgene (Figure 3A), which contains a cardiomyocyte-specific promoter upstream of a “floxed” cassette for co-expression of mOrange fluorescent protein and 4-HT-inducible Flipase. This cassette also contains a polyadenylation signal to prevent downstream transcription of a second cassette for Cre-dependent expression of membrane GFP (mG or GFP) in cardiomyocytes. Next, we generated the hsp:(P*-S)FRT-Cre-dnEct2 transgene (Figure 3B), which contains the zebrafish heat shock promoter upstream of a “blank” expression cassette flanked by FRT recombination sites. This cassette also contains a polyadenylation signal to prevent downstream transcription of a second expression cassette for Flipase-dependent heat shock-mediated expression of Cre recombinase and dnEct2.
Figure 3. Experimental strategy to generate mosaic hearts containing permanently labeled polyploid cardiomyocytes through transient cytokinesis inhibition.

(A–C) Transgenes and experimental strategy used to create adult zebrafish with mosaic hearts composed of diploid (GFP−) and polyploid-enriched (GFP+) cardiomyocyte populations. Detailed description of the experimental strategy is provided in Figure S5. (D) External appearance of a double-transgenic adult zebrafish with a mosaic heart comprised of GFP− diploid and GFP+ polyploid-enriched cardiomyocytes. (E) Adjacent sections from an adult mosaic heart immunostained for Tropomyosin and GFP (left) or stained with AFOG (right). Boxed area shows apex region at higher magnification. (F) Quantification of the percentage of GFP+ myocardium relative to the total ventricular myocardium from mosaic adult hearts (n=19). (G) DNA content of GFP− (mO+) and GFP+ cardiomyocytes from mosaic hearts. (H) Quantification of indicated cardiomyocyte populations from (F) (mean±s.d., n=620 and 520 total cells from 4 and 3 biological replicates per group, respectively; 3 ventricles per replicate; ****P<0.0001; ***P<0.001 by one-way ANOVA followed by Tukey’s multiple comparisons test). (I) Dissociated cardiomyocytes from a mosaic heart immunostained for Tropomyosin and GFP. Boxed regions show DAPI staining. Shown are diploid (top), mononucleated tetraploid (middle) and binucleated tetraploid (bottom) cardiomyocytes. (J) Quantification of cardiomyocyte size from the indicated classes from ventricular dissociations (n=822, 84 and 97 cardiomyocytes; ****P<0.0001; Kruskal-Wallis test followed by Dunn’s multiple comparisons test). Scale bars: 5 mm (D) 100 μm (E), 25 μm (I). CM, cardiomyocyte; ER, early recombination; HS, heat-shocked.
To create adult animals whose hearts were composed of GFP− diploid and GFP+ polyploid enriched cardiomyocyte populations, we treated 24 hpf double transgenic embryos with 4-HT to induce Flipase-mediated excision of (P*-S) from the hsp:(P*-S)FRT-Cre-dnEct2 transgene in a random subset of cardiomyocytes (Figures 3C and S5). Thereafter, this myocardial subset carried the recombined hsp:Cre-dnEct2 transgene, which conferred heat shock dependent co-expression of Cre recombinase and dnEct2. It also became permanently labeled with GFP following a single heat shock at 48 hpf, which induced Cre-mediated excision of (iF-S)lox from cmlc2:(iF-S)lox-mG to create cmlc2:mG. Animals were grown for 3 weeks (up to ~10 mm SSL) at normal temperature to allow equivalent expansion of the GFP− diploid and GFP+ polyploid-susceptible cardiomyocyte populations through hyperplasia. Thereafter, animals were heat-shocked daily for 3 months to activate dnEct2 expression, inhibit cardiomyocyte cytokinesis, and induce polyploidization specifically within the GFP+ subset. Next, we injured adult hearts by ventricular apex amputation and analyzed regeneration in the absence of heat shocking and experimental cytokinesis inhibition (Figure 3C).
Before injury, these animals and their hearts were grossly indistinguishable from controls (Figures 3D and 3E). Prominent subsets of GFP+ myocardium were present, in both the trabecular and compact layers (Figure 3E), indicating that transient expression of dnEct2 during cardiac growth did not undermine cardiomyocyte viability. On average, approximately half of the myocardium was GFP+ in the mosaic hearts (50.28 ± 13.27 %, mean ± SD; Figure 3F, n=19).
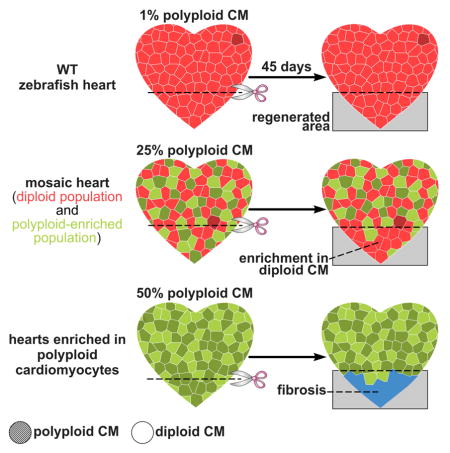
Next, we assessed the efficiency of our experimental strategy for inducing cardiomyocyte polyploidization within the GFP+ subset of cardiomyocytes. We learned that approximately half of the GFP+ cardiomyocytes were polyploid with 4c DNA content, which represents an ~50-fold enrichment over the native population (Figures 3G and 3H). The majority of the polyploid cardiomyocyte population was mononucleated (Figure 3H), similar to that in human hearts (Mollova et al., 2013; Sandritter and Adler, 1976). Given that ~50% of the myocardium was GFP+ (Figure 3F), and that ~50% of the GFP+ cardiomyocytes became polyploid (Figure 3G–3H), ~25% of the myocardium in mosaic hearts was polyploid. Importantly, GFP+ cardiomyocytes contained sarcomeres that were indistinguishable from neighboring cells (Figures 3I), ruling out a defect in cardiomyocyte differentiation. Polyploid cardiomyocytes in mosaic hearts were two times larger on average than their diploid counterparts (Figure 3I and 3J).
The second variation of our transgenic system conferred polyploid susceptibility to the entire myocardium during cardiac growth. To that end, we replaced inducible Flipase with constitutive Flipase to generate cmlc2:(F-S)lox-mG (Figure 4A). After heat shocking double transgenic embryos once during embryogenesis (Figure 4B), the entire myocardium turned GFP+ (data not shown), indicative of homogeneous excision of (P*-S) from hsp:(P*-S)FRT-Cre-dnEct2 to create hsp:Cre-dnEct2. After three weeks of growth without heat shock treatments followed by three months of daily heat shocking (Figure 4B), these animals and their GFP+ hearts were grossly indistinguishable from sibling animals not exposed to heat shocking during growth (Figures 4C and 4D). No evidence of DNA damage or apoptosis resulting from dnEct2 expression was detectable (Figures S6A–S6C and S6E–S6F). Control hearts were ~99% diploid (Figures 4E and 4F), indicating that polyploidization does not occur in the absence of heat shocking from transgene leakiness. Similar to the GFP+ population in partially enriched hearts (Figure 3F), approximately half of the homogenous GFP+ myocardium in heat-shocked animals was polyploid with 4c DNA content, with similar proportions containing one (1 × 4c) or two (2 × 2c) nuclei (Figures 4E and 4F).
Figure 4. Experimental strategy to generate hearts highly enriched in permanently labeled polyploid cardiomyocytes through transient cytokinesis inhibition.

(A,B) Transgenes and experimental strategy employed to maximize the percentage of cardiomyocytes susceptible to polyploidization. (C) External appearance of double-transgenic adult zebrafish, subjected to early recombination during embryogenesis, grown in the absence (top) or presence (bottom) of polyploid-inducing heat-shock treatments. (D) Adjacent sections from a fully recombined adult heart immunostained for Tropomyosin and GFP (left) or stained with AFOG (right). Boxed area shows apex region at higher magnification. (E) DNA content of GFP+ cardiomyocytes from the indicated cohorts. (F) Quantification of indicated cardiomyocyte populations from l (mean±s.d., n=564 and 406 total cardiomyocytes from 4 and 4 pooled ventricles, respectively, ***P<0.001; **P<0.01, by one-way ANOVA followed by Tukey’s multiple comparisons test). See also Figure S6. Scale bars: 5 mm (C) 100 μm (D). CM, cardiomyocyte; ER, early recombination; HS, heat-shocked.
Altogether, our two experimental strategies generated healthy adult zebrafish whose hearts contained a GFP+ cardiomyocyte population with approximately equivalent numbers of diploid and polyploid cardiomyocytes. In one strain, subsets of the myocardium were GFP+ and polyploid enriched (Figures 3A–3H). In the other strain, the entire myocardium was GFP+ and polyploid enriched (Figures 4A–4F). Importantly, we did not uncover evidence that cardiomyocyte polyploidization grossly undermines cellular (Figures 3E, 3I, 4D, S6A–S6C, and S6E–S6F) or animal (Figures 3D and 4C) health when analyzed prior to cardiac injury. Moreover, inducing dnEct2 expression transiently for one week in homeostatic adult diploid myocardium did not elicit signs of toxicity including DNA damage (Figure S6D) or cell death (Figure S6G), suggesting that dnEct2 targets cytokinesis very specifically without impinging on multiple cellular processes. In this experiment, cardiomyocyte ploidy was not altered (Figure S6H–S6K) because cellular proliferation is minimal during homeostasis (Wills et al., 2008).
We studied regeneration in both strains following ventricular amputation. Importantly, animals were never heat shocked during the regenerative window. In other words, dnEct2 expression and experimental cytokinesis inhibition were non-existent, which allowed us to monitor the inherent proliferative capacities of polyploid cardiomyocytes in an otherwise highly permissive environment in vivo.
Cardiomyocyte polyploidization is sufficient to impair regenerative proliferation
To evaluate the proliferative potential of polyploid cardiomyocytes relative to their diploid neighbors, we amputated ventricular apices of hearts containing subsets of GFP+ polyploid enriched cardiomyocytes (Figures 3C and 3E). After 45 days, we quantified the percentages of GFP+ polyploid enriched muscle in regenerated and surrounding myocardium. For each heart, we calculated a “GFP+ Cell Contribution Index” (GCCI), which represents the log2-fold change in the percentage of GFP+ myocardium that occurred during proliferative expansion of wound-edge cardiomyocytes. A negative GCCI would reflect an impaired relative contribution made by GFP+ cells. To establish a baseline, we generated adult animals whose hearts contained GFP+ “neutral” diploid cardiomyocytes by recombining cmlc2:(iF-S)lox-mG in a subset of embryonic cardiomyocytes with the cmlc2:CreERT2 transgene (Kikuchi et al., 2010). These hearts regenerated normally (Figure 5A) and GFP+ diploid cells contributed robustly to regenerated muscle (Figure 5E). Next, we confirmed that FlpOERT2 was not leaky and that three months of daily heat shocking during growth did not impair regeneration in animals carrying unrecombined transgenes (Figure 5B). Lastly, we documented that a second population of “neutral” cardiomyocytes, those carrying the hsp:Cre-dnEct2 transgene but never exposed to heat shocking, contributed normally to regenerated muscle (Figures 5C and 5E).
Figure 5. Myocardial polyploidization creates a barrier to cardiomyocyte proliferation and heart regeneration.

(A–D) Adjacent sections from 45 dpr hearts from the indicated cohorts, immunostained for Tropomyosin and GFP (left) or stained with AFOG (right). n = 14 (12), 10 (9), 9 (9), 19 (18) hearts (number of hearts that showed complete regeneration indicated within the brackets), respectively. (E) Relative change in percentage of GFP+ cardiomyocytes in the regenerate compared to the surrounding region in hearts from the indicated cohorts (n=12, 9 and 19, respectively). ***P<0.001; **P<0.01; Kruskal-Wallis test followed by Dunn’s multiple comparisons test. (F) Quantification of the scar area of hearts from (A–D) normalized to the ventricular area (n= 14, 10, 19 hearts; solid black line indicates the mean); n.s., not significant by Kruskal-Wallis test. (G) BrdU pulse-chase strategy used to evaluate cardiomyocyte proliferation during regeneration in experiments from G–H. (H) Section from a 14 dpr mosaic heart composed of diploid (GFP−) and polyploid-enriched (GFP+) cardiomyocyte populations, immunostained for Tropomyosin, GFP, Mef2 and BrdU. BrdU and Mef2/GFP signals of boxed region are shown at higher magnifications. Blue arrowheads indicate BrdU+ cardiomyocyte nuclei. (I) Cardiomyocyte BrdU labeling indices of GFP− and GFP+ populations in injury sites in experiments from F–G. Box-and-whisker plot. n = 7 hearts. **P < 0.01, Mann–Whitney test. See also Figure S6. CM, cardiomyocyte; ER, early recombination; HS, heat-shocked. Scale bars: 50 μm.
When we injured hearts containing subsets of GFP+ polyploid-enriched myocardium, regeneration proceeded normally as evidenced by a lack of accumulated scar tissue (Figure 5F). In the regenerated area, we observed a striking reduction in GFP+ cardiomyocytes (Figure 5D), which caused the GCCI to become significantly negative (Figures 5E). Importantly, BrdU incorporation analysis revealed that proliferation within the GFP+ polyploid enriched cardiomyocyte population was ~2-fold lower than that in the GFP− diploid fraction (Figures 5G–5I). Therefore, diploid cardiomyocytes outcompeted their polyploid neighbors in producing regenerated muscle through proliferative expansion, indicating that a single cytokinesis failure and resulting polyploidization during cardiac growth are sufficient to significantly reduce cardiomyocyte proliferation during regeneration.
Myocardial polyploidization creates a barrier to heart regeneration
During the course of analyzing regeneration of mosaic hearts containing a GFP+ polyploid-enriched cardiomyocyte population, we observed scar formation instead of myocardial regeneration in a small number of hearts (Figure 6A, n=2). In these hearts, ~80% of the myocardium surrounding the injury area was GFP+. Therefore, the polyploid percentage was ~40% (~80% GFP+ myocardium × ~50% polyploid cells in enriched population= ~40%). This observation suggested that the ploidy composition of wound edge myocardium might determine regenerative competence.
Figure 6. Increasing the proportion of polyploid cardiomyocytes impairs heart regeneration.

(A–C) Adjacent sections from 45 dpr hearts from the indicated cohorts, immunostained for Tropomyosin and GFP (left) or stained with AFOG (right). n = 2 (0), 5 (5), 9 (0) hearts (number of hearts that showed complete regeneration indicated within the brackets), respectively. (D) Quantification of the scar area of hearts from (B,C) normalized to the ventricular area (n= 5, 9 hearts; solid black line indicates the mean); ***P<0.001, two-tailed unpaired t-test. (E–G) Sections from 14 dpr hearts of the indicated cohorts, immunostained for Mef2 and BrdU, as described in Figure 5G–5H. White arrows indicate BrdU+ cardiomyocyte nuclei. (H) Cardiomyocyte BrdU labeling index in injury sites in experiments from (E–G). Box-and-whisker plot. n = 11, 10, 7 ventricles, respectively. ****P<0.0001; **P<0.01 by one-way ANOVA followed by Tukey’s multiple comparisons test). See also Figures S7 and S8. CM, cardiomyocyte; ER, early recombination; HS, heat-shocked. Scale bars: 50 μm.
To pursue this observation further, we injured hearts with homogenous GFP expression and 45% polyploid myocardium to determine if reducing the proportion of proliferation-competent diploid cardiomyocytes from 99% to 55% would undermine regenerative potential. First, we documented normal regeneration in animals grown at normal temperature to rule out the possibility that injury alone would activate the hsp:Cre-dnEct2 transgene (Figure 6B). Next, we learned that hearts composed of ~50% (45.04 ± 0.76 %, mean ± SD; Figure 4E) polyploid cardiomyocytes were incapable of completely regenerating and consistently scarred (Figure 6C). We found that the scar size in the polyploid-enriched hearts was 12-times bigger than that in control animals (Figure 6D). This was accompanied by a 61% reduction in BrdU incorporation between 7 dpr and 14 dpr (Figures 6E–6F and 6H). Importantly, heat shocking equivalent animals exclusively during adulthood, when the myocardium is minimally proliferative (Wills et al., 2008), did not undermine subsequent cardiomyocyte BrdU incorporation between 7 dpr and 14 dpr (Figures 6G and 6H), demonstrating that failed regeneration depends on dnEct2-mediated polyploidization that occurs specifically during cardiac growth (Figures 4F and S6J). Taken together, these data demonstrate that the ploidy composition of the myocardium is a critical determinant of regenerative capacity.
Importantly, the myocardium in these hearts naturally upregulated endogenous Ect2 following injury (Figure S7), which suggests that re-expression of Ect2 in polyploid cardiomyocytes is not sufficient to revert cardiomyocyte ploidy and that the proliferative barrier created by polyploidization is largely irreversible. This conclusion was further supported by the observation that overexpression of Ect2 in the highly polyploid myocardium of mice did not boost ventricular myocardial proliferation after experimental myocardial infarction (Figure S8).
DISCUSSION
Since the discovery of heart regeneration in zebrafish 15 years ago, substantial efforts have been expended to identify the signaling pathways required to initiate and achieve myocardial regeneration in this model organism (González-Rosa et al., 2017). However, it remains unclear whether the absence of cardiac regenerative potential in other vertebrate species, including adult mammals, can be ascribed to failed activation of pro-regenerative pathways, to cell-intrinsic properties that might prevent cardiomyocytes from participating in regeneration, or to a combination of both factors. To date, investigators have highlighted unique attributes of highly regenerative species as factors that might be permissive for cardiac regeneration. These include low blood pressure, hypoxic cardiac environment, low cardiac metabolic requirements, unique immune responses (Lai et al., 2017), and immature cardiomyocyte phenotypes including less organized sarcomeres and diploid DNA content (Vivien et al., 2016). Implicit in these arguments is that differing characteristics in non-regenerative species represent barriers to cardiac regeneration. However, these simple correlations do not establish causality. In most cases, experimental evidence has not been provided to demonstrate that modifying these properties in a highly regenerative species is sufficient to dampen cardiac regeneration.
We took advantage of the highly regenerative zebrafish heart to study the potentially negative effect of polyploidization on myocardial regenerative potential. In this regard, we attempted to mimic the natural process of cardiomyocyte polyploidization that occurs from failed cytokinesis during post-natal life in mammals and correlates temporally with the loss of regenerative potential (Porrello et al., 2011). As designed, we attempted to isolate polyploidization as an initiating event in regenerative decline without directly modifying other properties of the zebrafish heart, which might contribute to the loss of regenerative potential in post-natal mice including immune system maturation (Lai et al., 2017), cardiac fibroblast expansion (Banerjee et al., 2007), elevated production of reactive oxygen species (Puente et al., 2014), and extracellular matrix alterations (Bassat et al., 2017).
Despite the notion that cardiomyocyte polyploidization might be synonymous with cell-cycle exit, several counterexamples have been described in nature where polyploid cells re-enter the cell cycle and divide robustly. This is the case for mammalian hepatocytes during liver growth and regeneration (Duncan et al., 2010) and for rectal papillar cells in Drosophila (Fox et al., 2010). Moreover, polyploidization is required for regeneration of certain structures, such as the abdominal epithelium during Drosophila wound healing (Losick et al., 2013).
Recently, the percentage of mononuclear diploid cardiomyocytes in adult hearts of inbred mouse strains was reported to be variable and positively correlated with functional recovery after myocardial infarction (Patterson et al., 2017). Through genome wide association and genetic loss of function analyses, the authors identified TNNI3 interacting kinase (Tnni3k) activity as a positive regulator of myocardial polyploidization. Accordingly, continuous overexpression of Tnni3k in zebrafish cardiomyocytes elevated the percentage of mononuclear polyploid cardiomyocytes in adult animals from 0 to 7–20%, which correlated with compromised myocardial regeneration after amputation. While these data are consistent with a connection between myocardial polyploidization and reduced regenerative capacity, the potentially negative influence of a modest percentage of polyploid cardiomyocytes on heart regeneration in these animals cannot be disentangled from that of ongoing Tnni3k expression in spared cardiomyocytes (Vagnozzi et al., 2013). Lastly, this study was not designed to make direct comparisons between the regenerative capacities of diploid and polyploid cardiomyocytes as neither population carried a genetic label.
By establishing that the ploidy composition of zebrafish myocardium is diploid, we provided additional evidence to support the already-strong correlation between diploid myocardium and high regenerative capacity. To move beyond a correlation, we designed transgenic tools to transiently inhibit cytokinesis and induce cardiomyocyte polyploidization in zebrafish. To our knowledge, our experimental approach is unique in its ability to: 1) induce polyploidization through expression of dnEct2 exclusively in cardiomyocytes, without affecting other cell types; 2) enable lineage-tracking of polyploid enriched populations of cardiomyocytes during regeneration; and 3) discontinue dnEct2 expression during regeneration, which allowed us to assess the inherent proliferative capabilities of polyploid cardiomyocytes during heart regeneration. Importantly, our transgenic strategy allowed us to assess whether polyploidization resulting from cytokinesis failure, rather than dnEct2 expression itself, creates a barrier to regeneration. This strategy is therefore superior to alternatives where a factor is expressed under a heat-shock promoter, which allows for temporal control but lacks tissue specificity, or where a factor is expressed from a myocardial specific promoter, which remains active following injury.
While the most obvious consequence of cytokinesis failure is the increase in DNA content, it is important to emphasize that a number of downstream changes are triggered in polyploid cells. These changes include increases in cell size, centrosome amplification, or metabolic alterations (Schoenfelder and Fox, 2015). Importantly, some of these changes are precisely some of the cell-intrinsic properties that might explain the differences between regenerative and non-regenerative hearts (Vivien et al., 2016). Nonetheless, identifying the molecular and cellular events downstream of polyploidization that impinge on myocardial regeneration will require further investigation.
By exploiting the ability of our transgenic system to generate mosaic hearts composed of GFP− diploid and GFP+ polyploid enriched populations, we learned that decreasing the percentage of diploid cells in the GFP+ fraction, through polyploidization, caused quantitatively similar reductions in proliferation and contributions to regenerated muscle. Because these hearts were mosaic for the GFP+ population, the total percentage of polyploid myocardium was always less than 50% and regeneration was grossly unaffected. Remarkably, when we analyzed the regenerative potential of hearts containing >99% GFP+ myocardium, ~50% of which was polyploid, we observed a profound reduction in BrdU incorporation at 7 days post injury and scarring at later stages (Figure 7). Thus, while hearts composed of ~25% polyploid cardiomyocytes were able to regenerate, increasing the proportion to ~50% drastically reduced the regenerative ability of the zebrafish heart, suggesting that polyploid cardiomyocytes create a barrier to heart regeneration at a threshold within this range.
Figure 7. Model of cardiomyocyte polyploidization as a barrier to heart regeneration.

(A) Hearts composed almost exclusively of diploid cardiomyocytes (1x2c), such as those in the adult zebrafish and mouse neonate, regenerate efficiently after amputation through myocardial proliferation. (B) In mosaic hearts composed of diploid (GFP−) and polyploid-enriched (GFP+) cardiomyocyte populations, diploid cardiomyocytes proliferate actively to replace injured muscle, with minor contributions from the polyploid-enriched population. (C) Minimizing the proportion of diploid cardiomyocytes in the zebrafish heart, a situation similar to that in adult mammals, including humans, results in reduced cardiomyocyte proliferation and persistent scarring. Dashed line, plane of amputation; dark area, amputated tissue; blue boxes, regenerated myocardium; red and green cells, diploid and polyploid-enriched cardiomyocyte populations.
Further studies are required to understand why the substantial fraction (~50%) of diploid cardiomyocytes in these hearts did not support regeneration. One possibility is that diploid cardiomyocytes are endowed with a limited number cell divisions following injury. In agreement with this hypothesis, multicolor clonal analysis of the myocardium during zebrafish heart regeneration revealed that clones in the regenerated myocardium are composed of 2–4 cells, suggesting that cardiomyocytes along the wound edge divide 1–2 times (Gupta et al., 2013). As a result, the percentage of diploid cells might become rate limiting when a large enough fraction of cardiomyocytes become proliferation-compromised by polyploidization. Any given maximal number of divisions might be determined by telomere erosion upon division. This hypothesis is consistent with the observation that telomerase reexpression and telomere expansion upon injury is required for heart regeneration in zebrafish (Bednarek et al., 2015). An alternative hypothesis is based on the relative locations of polyploid and diploid cardiomyocyte populations in the mosaic hearts. If the GFP+ border zone myocardium is preferentially enriched in polyploid cells, then diploid cardiomyocytes might not be able to actively invade the injured area and contribute to regeneration. Lastly, it remains possible that regenerative capacity is regulated by non-autonomous signals released by diploid or polyploid cardiomyocytes that are pro- or anti-regenerative, respectively. If true, then increasing the polyploid fraction would tip the balance towards lowered regenerative potential.
In summary, we transiently inhibited cytokinesis to induce polyploidization of zebrafish cardiomyocytes to mimic one aspect of the post-natal maturation of mammalian hearts. Although several factors have been hypothesized to impair heart regeneration in mammals (Vivien et al., 2016), our data provide compelling evidence that cytokinesis failure and polyploidization are the initiating events in the loss of regenerative potential. Myocardial polyploidization appears to have evolved at the expense of regenerative ability, perhaps as a mechanism to enhance organ growth or physiology. However, the specific advantages of cardiomyocyte polyploidization remain unknown. Lastly, our data provide rationale to better characterize rare diploid cardiomyocytes in mammalian hearts and devise therapies to stimulate their deployment for boosting natural heart regeneration in humans.
STAR METHODS
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-DsRed | Clontech | Cat#632496; RRID:AB_10013483 |
| Mouse monoclonal anti-Dm-Grasp | Developmental Studies Hybridoma Bank | Clone: Zn-8; RRID:AB_531904 |
| Mouse monoclonal anti-BrdU | ROCHE / Sigma-Aldrich | Cat#BMC9318; RRID:AB_2313622 |
| Mouse monoclonal anti-GFP | Santa Cruz Biotechnology | Cat#sc-9996; RRID:AB_627695 |
| Chicken anti-GFP | Aves Labs | Cat#GFP-1010; RRID:AB_2307313 |
| Rat monoclonal anti-BrdU | Abcam | Cat#ab6326 RRID:AB_305426 |
| Mouse monoclonal anti-tropomyosin | Developmental Studies Hybridoma Bank | Cat#CH1; RRID:AB_2205770 |
| Rabbit polyclonal anti-Mef2 | Santa Cruz Biotechnology | Cat# sc-313; RRID:AB_631920 |
| Rabbit polyclonal anti-gH2A.X phosphor-Ser139 | GeneTex | Cat# GTX50293; RRID:AB_11176141 |
| Rabbit polyclonal anti-tRFP (mKate) | Evrogen | Cat# AB233 RRID:AB_2571743 |
| Rabbit polyclonal anti-Ect2 | Santa Cruz Biotechnology | Cat# sc-1005 RRID:AB_2246263 |
| Rabbit polyclonal anti-beta-galactosidase | Molecular Probes | Cat# A-11132; RRID:AB_221539 |
| Mouse monoclonal anti-BrdU (for mouse experiments) | Sigma-Aldrich | Cat# 11585860001 (clone BMG6H8); RRID:AB_514485 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11034; RRID:AB_2576217 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21428; RRID:AB_2535849 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21244; RRID:AB_2535812 |
| Goat anti-Chicken IgY (H+L) Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11039; RRID:AB_2534096 |
| Goat anti-Mouse IgG1 Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21121; RRID:AB_2535764 |
| Goat anti-Mouse IgG1 Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-21124; RRID:AB_2535766 |
| Goat anti-Mouse IgG1 Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21240; RRID:AB_2535809 |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21131; RRID:AB_2535771 |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21137; RRID:AB_2535776 |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 633 | Thermo Fisher Scientific | Cat# A-21136; RRID:AB_2535775 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 10% neutral buffered formalin | Sigma-Aldrich | Cat#HT501128 |
| 2,3-butanedione monoxime | Sigma-Aldrich | Cat#B0753 |
| Accumax | EMD Millipore | Cat#SCR006 |
| 5-Bromo-2′-deoxyuridine (BrdU), zebrafish experiments | EMD Millipore | Cat#19-160 |
| BrdU (for mice experiments) | Roche | Cat#280879 |
| IGEPAL CA-630 | Sigma-Aldrich | Cat#I8896 |
| RQ1 RNase Free-DNAse | Promega | Cat#M6101 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Trizol | Invitrogen | Cat#15596-026 |
| Trypsin / EDTA | Gibco | Cat#25200-056 |
| Critical Commercial Assays | ||
| Direct-zol RNA MicroPrep kit | Zymo Research | Cat#R2062 |
| Power SYBER Green PCR Master Mix | Thermo Fisher Scientific | Cat#4367659 |
| RNAScope 2.5 HD Assay - RED | ACD | Cat#322350 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | Cat#18080051 |
| Click-iT™ Plus TUNEL Assay for In Situ Apoptosis Detection, Alexa Fluor™ 647 dye | Thermo Fisher Scientific | Cat#C10619 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: ect2 null: ect2hi3820aTg | Amsterdam et al., 2004 | ZFIN: ZDB-GENO-040831-2 |
| Zebrafish: cmlc2:nucDsRed: Tg(cmlc2:nlsDsRed-Express)hsc4 | Takeuchi et al., 2011 | ZFIN: ZDB-GENO-110222-4 |
| Zebrafish: cmlc2:mKate-CAAX: Tg(cmlc2:mKate-CAAX)sd11 | Lin et al., 2012 | ZFIN: ZDB-GENO-120320-1 |
| Zebrafish: cmlc2:CreERT2: Tg(−5.1 cmlc2:CreERT2, cryaa:DsRed)pd10 | Kikuchi et al., 2010 | ZFIN ID: ZDB-GENO-120923-4 |
| Zebrafish: ubi:Zebrabow: Tg(ubb:Zebrabow-M)a131 | Pan et al., 2013 | ZFIN ID: ZDB-GENO-150205-4 |
| Zebrafish: cmlc2:nucGFP: Tg(cmlc2:nls-EGFP)fb18 | This paper | N/A |
| Zebrafish: cmlc2:GdnEct2: Tg(cmlc2:EGFP-P2A-dnEct2)fb19 | This paper | N/A |
| Zebrafish: cmlc2:(iF-S)lox-mG : Tg(cmlc2:loxP-nls-mOrange-P2A-FlpOERT2-STOP-loxP-memGFP)fb20 | This paper | N/A |
| Zebrafish: cmlc2:(F-S)lox-mG : Tg(cmlc2:loxP-nls-mOrange-P2A-FlpO-STOP-loxP-memGFP)fb21 | This paper | N/A |
| Zebrafish: hsp:(P*-S)FRT-Cre-dnEct2: Tg(hsp70l:FRT-nucPhiYFP*-STOP-FRT-nlsCre-P2A-dnEct2; cryaa:ZsYellow)fb22 | This paper | N/A |
| Mouse: MHC-nLAC | Soonpaa et al., 1994 | N/A |
| Mouse: MHC-ECT2 | This paper | N/A |
| Oligonucleotides | ||
| Primers for cloning dnEct2: For: 5′-ATGGCTGACAGCAGCATACT-3′ Rev: 5′-ACACAGCTTTCTTCATGGCAGG-3′ |
This paper | N/A |
| qPCR primers, rps11 For: 5′-GATGGCGGACACTCAGAAC -3′ Rev: 5′-CCAATCCAACGTTTCTGTGA -3′ |
González-Rosa et al., 2014 | N/A |
| qPCR primers, ect2 For: 5′-GCAGCTCCACACTGGCTACT -3′ Rev: 5′-GGTGTGGTACTTTCTCTCAAAGG-3′ |
This paper | N/A |
| qPCR primers, ect2 (splicing) For: 5′-CGCCTAAACAAAAACACTGATTG -3′ Rev: 5′-GCCAGTGAGAAAGCAGTGAA -3′ |
This paper | N/A |
| qPCR primers, kif23 For: 5′-CAGGGCATGGTCTCTGACA -3′ Rev: 5′-GCGTTTACCCTCCTCTCACA -3′ |
This paper | N/A |
| qPCR primers, racgap1 For: 5′-GCCTTCCTCATCATCCATCT -3′ For: 5′-TGTGGGTCCAAATACACGAG -3′ |
This paper | N/A |
| Recombinant DNA | ||
| MDR1734-202791320 (Danio rerio ect2) | GE Dharmacon | Cat# MDR1734-202791320 |
| MMM4769-202769099 (Mus musculus ect2) | GE Dharmacon | Cat# MMM4769-202769099 |
| p5E-hsp70l | Kwan et al., 2007 | N/A |
| pAAV-pgk-Cre | Gift from Patrick Aebischer | Addgene Cat#24593 |
| pBS-FlpOER | Lao et al., 2012 | N/A |
| pThy1-Brainbow3.2 | Cai et al., 2013 | Addgene Cat#45179 |
| Software and Algorithms | ||
| Fiji/ImageJ | NIH | RRID:SCR_002285 |
| GraphPad Prism 7 | GraphPad Software | N/A |
| NIS-Elements | Nikon Instruments | N/A |
| Other | ||
| 4′,6-diamidino-2-phenylindole dihydrochloride | Invitrogen | Cat#D1306 |
| FluorSave Reagent | Calbiochem | Cat#345789 |
| Hoechst 33342 | ThermoFisher Scientific | Cat#H3570 |
| Mouse-on-Mouse kit | Vector | Cat#BMK-2202 |
| Osmotic mini-pumps | Alzet | Cat#1002 |
| RNAscope probe Dr-ect2 | ACD | Cat#472701 |
| RNAscope probe Dr-ect2-O1 | ACD | Cat#504041 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Geoffrey C. Burns (gburns@cvrc.mgh.harvard.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Zebrafish
Zebrafish embryos, larvae, and adults were produced, grown, and maintained according to standard protocols approved by the Institutional Animal Care and Use Committees of Massachusetts General Hospital. Ethical approval was obtained from the Institutional Animal Care and Use Committees of Massachusetts General Hospital. For experiments with adult zebrafish, animals ranging in age from 3 to 18 months were used. Approximately equal sex ratios were used for experiments. Adult density was maintained at ~3–4 fish·l−1 for all experiments in Aquarius racks and fed three times daily. Water temperature was maintained at 28 °C except during heat shock treatments.
Published strains used in this study include: wild-type AB, ect2 mutants (ect2)hi3820aTg (Amsterdam et al., 2004), Tg(cmlc2:nlsDsRed-Express)hsc4 (Takeuchi et al., 2011), Tg(cmlc2:mKate-CAAX)sd11 (Lin et al., 2012), Tg(−5.1 cmlc2:CreERT2, cryaa:DsRed)pd10 (Kikuchi et al., 2010), Tg(ubb:Zebrabow-M)a131 (Pan et al., 2013). Details of the construction of the new lines generated in this study are described below. At least three independent founders of each line were isolated and tested to confirm the described expression patterns and phenotypes. Transgene sequences are available upon request.
Mice
Experimental mice were generated in an inbred DBA/2J background. Animals were housed under a 14-h light, 10-h dark cycle in the Laboratory Animal Research Center at the Indiana University Medical School. Experiments were initiated when mice reached 12 weeks of age. Animals were provided food and water ad libitum. All mice treatment, surgery, and euthanasia protocols utilized in this study were performed in accordance with National Institutes of Health Guidelines and were approved by the Institutional Animal Care and Use Committee (Study #10881). All surgeries were performed under isoflurane anesthesia, and all efforts were made to minimize suffering.
METHOD DETAILS
Ect2 mutant genotyping
The ect2hi3820aTg allele consists of a retroviral insertion in the first intron of ect2 that interrupts the expression of the gene (Figure S3). Heterozygous animals carrying the ect2hi3820aTg allele were identified by PCR using the primers 5′-CGCTTCTCGCTTCTGTTCG-3′ and 5′-GACATTGTGGAAGGAAGACACG-3′. Because the first primer anneals in the retroviral sequence, animals carrying the mutant allele produced a 471 bp amplicon.
Construction of cmlc2:nucEGFP
To generate the cmlc2:nucEGFP transgenic line, a construct containing the following DNA elements was assembled by Gibson cloning: (1) a ~0.9-kb cmlc2 promoter to drive specific expression in cardiomyocytes; (2) the coding sequence for a nuclear localized enhanced green fluorescent protein (nls-EGFP). The entire construct was flanked with Tol2 sites to facilitate transgenesis. In this line, all cardiomyocytes constitutively express nls-EGFP. The full name of this line is Tg(cmlc2:nls-EGFP)fb18.
Construction of cmlc2:GdnEct2
To generate the cmlc2:GdnEct2 transgenic line, a construct containing the following DNA elements was assembled by Gibson cloning: (1) a ~0.9-kb cmlc2 promoter to drive specific expression in cardiomyocytes; (2) a bicistronic cassette encoding EGFP and a truncated version of Danio rerio ect2 containing the first 337 amino acids (amplified from clone MDR1734-202791320, GE Dharmacon, using the primers 5′-ATGGCTGACAGCAGCATACT-3′ and 5′-ACACAGCTTTCTTCATGGCAGG-3′), separated by a P2A sequence (Kim et al., 2011). Expression of similar truncations have been described in mammals to strongly inhibit cytokinesis due to the lack of GEF activity (Oceguera-Yanez, 2005; Sakata et al., 2000; Tatsumoto et al., 1999). The entire construct was flanked with Tol2 sites to facilitate transgenesis. In this line, all cardiomyocytes constitutively express a dominant negative form of Ect2 and the fluorescent protein EGFP. The full name of this line is Tg(cmlc2:EGFP-P2A-dnEct2)fb19.
Construction of cmlc2:(iF-S)lox-mG zebrafish
To generate the cmlc2:(iF-S)lox-mG transgenic line, a construct containing the following DNA elements was assembled by Gibson cloning: (1) a ~0.9-kb cmlc2 promoter to drive specific expression in cardiomyocytes; (2) a floxed bicistronic nls-mOrange2-P2A-FlpOERT2-polyA cassette that was generated by subcloning mOrange2 from pThy1-Brainbow3.2 (Addgene #45179, (Cai et al., 2013)) and FlpOERT2 from pBS-FlpOER (kindly provided by Drs. Lao and Joyner (Lao et al., 2012)); and (3) a GFP containing a farnesylation tag to direct the fluorescent protein to the cell membrane (subcloned from pThy1-Brainbow3.2). The entire construct was flanked with Tol2 sites to facilitate transgenesis. In this line, all cardiomyocytes express the fluorescent protein mOrange2 directed to the nucleus and a tamoxifen-inducible Flipase. Upon Cre mediated recombination, the floxed cassette is eliminated and cardiomyocytes (and those cardiomyocytes that derive from them) are permanently labeled by the expression of a membrane-tagged GFP (Figure S5). The full name of this line is Tg(cmlc2:loxP-nls-mOrange-P2A-FlpOERT2-STOP-loxP-memGFP)fb20.
Construction of cmlc2:(F-S)lox-mG zebrafish
To generate the cmlc2:(F-S)lox-mG transgenic line, the cmlc2:(iF-S)lox-mG construct was modified by Gibson cloning as follows: (1) the bicistronic nls-mOrange2-P2A-FlpOERT2-polyA cassette was substituted with a nls-mOrange2-P2A-FlpO in which the recombinase lacks the ERT2 domain and is therefore constitutively active; and (2) a woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) – Xenopus β-globin polyadenylation signal cassette was added downstream of the FlpO to increase transcript stability. In this line, all cardiomyocytes express the fluorescent protein mOrange2 directed to the nucleus and a constitutively active Flipase. Upon Cre mediated recombination, the floxed cassette is eliminated and cardiomyocytes (and those cardiomyocytes that derive from them) are indelibly labeled by the expression of a membrane-tagged GFP. The full name of this line is Tg(cmlc2:loxP-nls-mOrange-P2A-FlpO-STOP-loxP-memGFP)fb21.
Construction of hsp:(P*-S)FRT-Cre-dnEct2 zebrafish
To generate the hs:(P*-S)FRT-Cre-dnEct2 transgenic line, a construct containing the following DNA elements was assembled by Gibson cloning: (1) a ~1.5-kb hsp70l promoter to drive expression in response to heat shock (Halloran et al., 2000), amplified from p5E-hsp70l (Kwan et al., 2007); (2) the coding sequence of a Y65A mutated version (non fluorescent) of the fluorescent protein PhiYFP amplified from pThy1-Brainbow3.2 (Cai et al., 2013) flanked by FRT sites; (3) a bicistronic nls-Cre-P2A-dnEct2 cassette generated by subcloning a codon optimized, nuclear directed Cre from pAAV-pgk-Cre (a gift from Patrick Aebischer, Addgene plasmid #24593) and the Danio rerio dnEct2 amplified from cmlc2:GdnEct2. A cryaa:ZsYellow transgenesis control was included in the opposite orientation to facilitate visualization of transgenic animals. The entire construct was flanked with Tol2 sites to facilitate transgenesis. In this line, cells express PhiYFP* only after heat shock. Upon Flipase mediated recombination, the FRT-flanked cassette is eliminated and cells exposed to heat shock then express nls-Cre and dnEct2 (Figure S5). The full name of this line is Tg(hsp70l:FRT-nucPhiYFP*-STOP-FRT-nlsCre-P2A-dnEct2; cryaa:ZsYellow)fb22.
Generation of MHC-ECT2 mice
The MHC-ECT2 transgene used the mouse alpha-cardiac myosin heavy chain (MHC) promoter and sequences encoding Mus musculus ECT2 (amplified from clone MMM4769-202769099, GE Dharmacon). The SV40 early region transcription terminator/poly-adenylation site (nucleotide residues #2586-2452) was inserted downstream from the ECT2 sequences. Transgene insert was purified and microinjected into inbred C3HeB/FeJ (Jackson Laboratories, Bar Harbor MA) zygotes as described (Soonpaa et al., 1994). The resulting pups were screened using diagnostic PCR amplification and transgenic lineages were established. Transgene expression was stratified initially via qPCR and then confirmed by anti-Ect2 immunofluorescence (Figure S8).
Zebrafish Cardiac Injuries
Regeneration experiments were conducted using adult zebrafish as described (Poss et al., 2002). Briefly, fishes were anesthetized in tricaine, placed with their ventral side up on a sponge and a small incision was created to expose the apex of the ventricle. Approximately 20% of the ventricle was amputated using iridectomy scissors. A Kimwipe was applied to the bleeding animal with gentle pressure for a few seconds, and the animals were retrieved to a fish tank. After surgery, animals were revived by gently directing water to their gills using a plastic Pasteur pipette.
Myocardial infarction and cardiomyocyte DNA synthesis assay in mouse
MHC-ECT2 line 1 mice were crossed with MHC-nLAC mice (Soonpaa et al., 1994), which express a nuclear cardiomyocyte-restricted β-galactosidase reporter to assist in cardiomyocyte nuclear identification in tissue sections. MHC-ECT2; MHC-nLAC double transgenic mice and MHC-nLAC single transgenic mice were intubated and ventilated with 2% isoflurane and supplemental oxygen. Via left thoracotomy, the left coronary artery was ligated at the inferior border of the left auricle as described previously (Murry et al., 2004). The mice were then implanted with osmotic mini-pumps (Alzet #1002; Cupertino, California) containing bromodeoxyuridine (16 mg/ml in phosphate buffered saline (PBS); Roche, #280879; Indianapolis, Indiana) as described (Soonpaa and Field, 1994). Tissues were harvested, fixed in cacodylic acid/ paraformaldehyde as described (Soonpaa and Field, 1997), and then cryoprotected in 30% sucrose in PBS, and sectioned at 10 microns using standard methodologies. After antigen retrieval (30 minute incubation in sodium citrate buffer at 100°C), samples were processed for β-galactosidase (Invitrogen Lifetechnologies #A-11132) and BrdU (Roche #11296736001; clone BMG 6H8 IgG1) immune reactivity using the Vector Mouse-on-Mouse kit (#BMK-2202). Signal was developed using Alexa 555-conjugated goat anti-rabbit and Alexa 488-conjugated goat anti-mouse secondary antibodies (Invitrogen Lifetechnologies #A21429 and #A11001, respectively). DNA was visualized with Hoechst 33342 (Invitrogen Lifetechnologies).
Tamoxifen and heat shock treatments
To induce Flipase-ERT2 mediated recombination, 24–30 hours post-fertilization (hpf) zebrafish larvae were exposed to 10 μM 4-hydroxy-tamoxifen (4-OHT, Sigma) for 12 hours. Treated embryos were washed in E3, raised to adulthood and used in the experiments described below. For mosaic experiments, embryos with highly recombined hearts were selected for growing. Heat-shock induction was conducted by placing embryos into a 39 °C water bath for 1 hour. When required, ~2–3 weeks post fertilization animals were transferred to an automated heat shock rack that exposed them daily to 60 minute elevations in temperature from 28 °C to 39 °C, as described (Lee et al., 2005). Adult heat shock treatments were performed using the same system.
Cardiomyocyte isolation from zebrafish ventricles
To generate single cell suspensions, zebrafish hearts were dissected as described previously (González-Rosa and Mercader, 2012) using ice-cold PBS supplemented with 0.3% bovine serum albumin (BSA, A3059, Sigma) and 20 mM glucose (G7528, Sigma) as dissection buffer. Hearts were collected in a 3 ml Petri Dish (153066, ThermoFisher Scientific) maintained on ice. After removing the atrium and the bulbous, ventricles were bisected to wash out any remaining blood. Tissues then were washed twice in dissection buffer and treated for 15 min in a solution of 0.2% trypsin, 0.8 mM EDTA (25200-056, Gibco) supplemented with 20 mM glucose and 10 mM 2,3-butanedione monoxime (B0753, Sigma). The first digestion was performed on ice and using gentle agitation. Next, the digestion solution was removed, the ventricles were washed three times using dissection buffer supplemented with 10 mM BDM and then digested for 45 min at room temperature in Accumax (SCR006, EMD Millipore) supplemented with 20 mM glucose and 10 mM BDM under mild agitation. Tissue fragments were dissociated by gentle pipetting and cell suspensions were immediately fixed in 10% neutral buffered formalin (HT501128, Sigma) for 1 hour at room temperature. Cells were pelleted by centrifugation at 400 × g for 5 minutes, resuspended in PBS, spread in Superfrost Plus slides (1255015, ThermoFisher Scientific) and air dried.
DNA quantification in cell spreads
DNA content was determined by quantifying the integrated nuclear density of cells stained with the DNA dye 4′,6-diamidino-2-phenylindole (DAPI), by adapting preexisting protocols (Ikenishi et al., 2012; Roukos et al., 2015; Tane et al., 2014). Briefly, cardiomyocyte spreads were rehydrated in PBS, permeabilized for 45 minutes in 0.1% IGEPAL CA-630 (I8896, Sigma), 3% BSA in PBS and stained for 30 min using 14.3 μM DAPI dihydrochloride (Invitrogen) in permeabilization solution. After washing in PBS, slides were mounted in FluorSave Reagent (345789, Calbiochem) and images were acquired using a Nikon 80i compound microscope (Nikon Instruments) with a 20x lens, a Retiga 2000R high/speed CCD camera (QImaging) and the NIS-Elements image acquisition and analysis system (Nikon Instruments).
For each field (592 μm × 444 μm), fluorescence was captured from adequate channels (e.g. red for mKate-CAAX and/or mCherry, green for nucGFP and blue for DAPI) and saved independently as 12-bit *.nd2 files. Individual channels were merged in ImageJ software (Schneider et al., 2012), exported as a composite *.tiff file and binary masks containing nuclear areas, based on the blue channel (DAPI), were generated manually in Adobe Photoshop (Adobe Systems Incorporated). Only intact cells were analyzed and isolated nuclei or damaged cells were excluded. Independent binary masks were generated according to the expression of fluorescent proteins to distinguish cell populations.
Masks were then processed using the ROI Manager tool of ImageJ and the following macro:
run(“RGB Color”);
run(“8-bit”);
setAutoThreshold(“Default”);
//setThreshold(0, 200);
setOption(“BlackBackground”, false);
run(“Convert to Mask”);
run(“Analyze Particles…”, “display exclude clear add”);
run(“Flatten”);
that allowed to recognize individual nuclei and to generate a copy of the binary mask with each nucleus receiving a number for further reference. Integrated density from each nucleus was then calculated using the Multimeasure tool in ImageJ in the original 12-bit *.nd2 DAPI image. Values were exported and collected in an Excel file.
Ploidy measurement in dissociated cardiomyocytes
Ploidy values were determined on a per-nucleus basis by using the following strategy (Figure S1): First, the average raw integrated density value from a reference cell population (i.e., uninjured cardiomyocytes expressing a distinctive fluorescent protein) was determined for every picture. Then, the integrated density value from each cell from the same picture was divided by the average value of the control population and multiplied by 2. Additionally, images were manually inspected to determine the presence of binucleated cells. To that end, binary masks were compared to images containing membrane labeling (i.e., mKate-CAAX) (Figure S1). In the case of binucleated cells, values from both nuclei were then added in Excel. Ploidy values were then transferred to GraphPad Prism 7 to generate frequency histograms using a bin size of 0.25c. As a consensus based on the reference population, we considered diploid “2c”, those cells with values between 1.25c and 2.75c.
Quantification of cardiomyocyte nucleation in adult zebrafish
Cardiomyocyte nucleation was scored manually using cell spreads counterstained with DAPI as described above. To efficiently distinguish bona fide binucleated cells from cell aggregates, cardiomyocytes were isolated from animals carrying a transgene that directed the expression of a fluorescent protein to the cell membrane (i.e., cmlc2:mKate-CAAX or cmlc2:mGFP) (Figure S1).
Quantification of cardiomyocyte nucleation and ploidy in zebrafish cardiomyocytes
Cardiomyocyte nucleation was analyzed in ect2 mutant, Tg(cmlc2:GdnEct2) and sibling embryos carrying the Tg(cmlc2:nlsDsRed-Express) transgene. Embryos were fixed at 30 and 72 hpf overnight in 4% paraformaldehyde (PFA) in PBS at 4°C. Fixed embryos were bleached and permeabilized as described (Jahangiri et al., 2016), and subjected to immunofluorescence staining with anti-DsRed (Clontech, 1:200) and anti-Alcama (Dm-Grasp, clone Zn-8, Developmental Studies Hybridoma Bank; 1:50) antibodies. Alexa conjugated secondary antibodies (Life Technologies, 1:500) were used to detect primary antibody signals. Nuclei were counterstained with DAPI. Stained embryos were mounted in low-melting agarose and imaged using a Nikon A1 confocal microscope equipped with a 40X water immersion lens. Hearts of stained embryos were imaged and analyzed using ImageJ software. To estimate cardiomyocyte nucleation, the number of DsRed+ nuclei per cardiomyocyte (whose borders were Almcam+) were quantified. Nuclear ploidy was calculated based on DAPI integrated density as described above and compared to non-cardiomyocyte populations present in the same optical sections. Cardiomyocyte ploidy combines both nucleation and nuclear ploidy analysis.
Analysis of cardiomyocyte proliferation in zebrafish embryos
Zebrafish embryos were incubated in 5 mg/ml BrdU, 1% DMSO in E3 medium from 48 hpf to 72 hpf at 28°C, rinsed three times i n E3 medium and fixed overnight in 4% PFA. Fixed embryos were rinsed in PBST, bleached in the dark for 20 min (using 0.8% KOH, 0.9% H2O2 and 1% Tween-20 in distilled water), permeabilized using 1% Triton-X100 in PBS for 2 h and equilibrated in DNase I buffer (40 mM Tris-HCl, pH 8.0, 10 mM MgSO4, 1 mM CaCl2) for 30 min at 37°C. Equili brated embryos were treated with DNase I (1:50 in equilibration buffer) for 2 h at 37°C, rinsed three times in PBSTw (PBS+0.1% Tween20) and subjected to immunofluorescent staining with anti-GFP (clone B-2, Santa Cruz Biotechnology; 1:200) and anti-BrdU (clone BMC9318, Roche; 1:100) antibodies. The ventricles of stained embryos were imaged and analyzed as described above. A ventricular proliferation index (number of BrdU+, GFP+ double positive cells divided by the total number of GFP+ cardiomyocytes) was manually calculated for each embryo using ImageJ and averaged.
Zebrafish histological analysis and imaging
Adult zebrafish were euthanized by immersion in 0.16% tricaine (Sigma) and hearts dissected as described (González-Rosa and Mercader, 2012). Samples were fixed overnight at 4°C in 4% PFA, included in paraf fin and sectioned following conventional histological procedures.
To detect ect2 transcripts, RNAscope (Advanced Cell Diagnostics) was performed on 7 μm paraffin sections following the manufacturer’s instructions. Advanced Cell Diagnostic designed an ect2 probe spanning nucleotides 859-1756. Following ect2 detection, sections were incubated overnight with anti-tropomyosin or GFP antibodies (see below) and nuclei were counterstained using DAPI. To confirm ect2 re-expression in polyploid-enriched hearts (Figure S7), a new probe spanning nucleotides 1920-2835 of ect2 NM_001003883.1 was generated to avoid cross-reaction with dnEct2 sequence.
Immunofluorescence and TUNEL in paraffin sections were performed as described (González-Rosa et al., 2011). Primary antibodies used were mouse anti-tropomyosin (clone CH1, Developmental Studies Hybridoma Bank; 1:50), chicken anti-GFP (AVES, 1:500), mouse anti-GFP (clone B-2, Santa Cruz Biotechnology; 1:200), rabbit anti-Mef2 (clone C-21, Santa Cruz Biotechnology; 1:100), rat anti-BrdU (ab6326, Abcam; 1:100), mouse anti-BrdU (clone BMC9318, Roche; 1:100), rabbit anti-γH2A.X phospho-Ser139 (GTX127342, GeneTex; 1:100), rabbit anti-mKate (AB233, Evrogen; 1:500) and rabbit anti-Ect2 (clone C-20, Santa Cruz Biotechnology; 1:100). Alexa conjugated secondary antibodies (Life Technologies, 1:500) were used to detect primary antibody signals. Nuclei were counterstained with DAPI and slides were mounted in FluorSafe. A Nikon A1 confocal microscope was used to image immunostained sections.
Acid fuchsin-orange G (AFOG) stain was used to detect fibrotic tissue. Muscle, fibrin/cell debris and collagen were stained brown-orange, red and blue, respectively.
BrdU pulse-chase experiments during zebrafish heart regeneration
For BrdU pulse-chase experiments (Figures 5G–5I and 6D–6G), adult zebrafish were used to perform apical ventricular resection. At 7 dpr, animals were injected intraperitoneally with 50 μl of 2.5 mg/ml, at 7 days post-resection (dpr), and hearts were extracted and processed for analysis at 14 dpr. To calculate BrdU cardiomyocyte labeling indices, ventricular sections were immunostained with anti-Mef2, anti-GFP, anti-Tropomyosin and anti-BrdU antibodies (see above). 2–3 ventricular sections containing the largest injury areas were imaged. Mef2+, GFP+Mef2+BrdU+ and GFP−Mef2+BrdU+ cells were counted manually using ImageJ software in defined regions (200 μm × 424.55 μm) that include the injury area and border zone. The percentages of GFP+Mef2+BrdU+/Mef2+ and GFP−Mef2+BrdU+/Mef2+ cells from individual sections were averaged to establish a BrdU labeling index for each animal.
Quantification of GFP+ contribution to regeneration
To quantify the percentage of regenerated myocardium occupied by cells expressing EGFP, histological sections containing regenerated muscle from each heart were systematically identified and analyzed. Every heart was sectioned in its entirety and pairs of consecutive sections were repeatedly distributed across the same five slides. As a result, five slides were created for every heart that contained equally spaced sections across the entire organ. For each heart, one slide was stained with AFOG and all sections were carefully examined under the microscope. Regenerated regions of muscle were identified in sections as localized thickenings of dense muscle containing evidence of residual fibrosis (blue) when compared to surrounding compact muscle. Next, a slide containing adjacent pairs of sections was immunostained with antibodies to recognize GFP and tropomyosin as described above. Guided by which section(s) contained regenerated muscle on the AFOG-stained slide, an adjacent section was identified on the immunostained slide. In that section, the location of regenerated muscle was identified as a localized thickening of dense muscle, which showed signs of myocardial disarray. This section was imaged by confocal microscopy as described elsewhere. In photoshop, three regions of 140 μm × 140 μm were defined. This region size was empirically determined to encompass the entire regenerated area in all cases. The middle region was centered over the regenerated muscle. Two adjacent, non-overlapping regions were aligned laterally on either side to define regions of spared muscle. The EGFP+ and tropomyosin+ areas were quantified in pixels for each region, and the percentage of EGFP versus tropomyosin was calculated. The average value of EGFP+/tropomyosin+ was calculated for the uninjured regions. To calculate the relative change in proportion of GFP between the regenerate and the surrounding area, the following expression was used: log2(%GFPregenerate/%GFPsurrounding).
Quantification of fibrotic tissue in regenerated hearts
To quantify the fibrotic area in regenerated hearts at 45 dpr, images of evenly-spaced AFOG-stained serial sections of the whole heart were captured using a Nikon 80i compound microscope (Nikon Instruments) with a 4x lens and a mounted Excelis AU600HDS HD Camera (Accu-Scope Inc. Commack, NY). On average, ~21 sections were analyzed per heart (min=13, max=30; 1427 total sections analyzed). Masks containing the entire ventricular area and the scar area were manually generated using Adobe Photoshop based on differential staining (uninjured/regenerated muscle=brown/orange; fibrotic area=(fibrin (red) + collagen (blue)). Selected areas were measured using ImageJ software. As described previously (Chablais et al., 2011; Schnabel et al., 2011), the fibrotic area was normalized to the total ventricular area to calculate the percentage of the scar size for each heart.
Quantitative real-time PCR
RNA from cardiac ventricles or zebrafish larvae was extracted using Trizol reagent (Life Technologies) and the Direct-zol RNA MicroPrep kit (Zymo Research). For adult expression experiments (Figure 1L), 1 ventricle was used per biological replicate and 10 biological replicates were analyzed per time point. For embryonic expression (Figure S3), 10-pooled control or mutant embryos were used per biological replicate. 6 biological replicates were analyzed. RNA was transcribed to cDNA using the SuperScript III First-Strand Synthesis System (Life Technologies). qRT-PCR was performed using Power SYBER Green PCR Master Mix (Applied Biosystems, Life Technologies). Expression of genes was normalized with the arithmetic mean of the expression level of the constitutive gene rps11 as described (González-Rosa et al., 2014). A complete list of the primers used in this study is provided in the Key Resources Table.
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample sizes were chosen based on previous publications and are indicated in each figure legend. No animal or sample was excluded from the analysis unless the animal died during the procedure. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment. All statistical values are displayed as mean ± standard deviation. Sample sizes, statistical test and P values are indicated in the figures or figure legends. Data distribution was determined before using parametric or non-parametric statistical test. Statistical significance was assigned at P < 0.05. All statistical tests were performed using Prism 7 software.
Supplementary Material
HIGHLIGHTS.
Cardiomyocytes in adult zebrafish are predominantly mononucleated and diploid.
Transient inhibition of Ect2 induces polyploidization of zebrafish cardiomyocytes.
Diploid cardiomyocytes outcompete polyploid cardiomyocytes during heart regeneration.
Hearts composed of >45% polyploid cardiomyocytes fail to regenerate.
Acknowledgments
We thank A. Roselló-Diez, H. Sánchez-Iranzo and A. Guarner for critical reading of the manuscript, E. Garcia for assistance with qPCR experiments, D. Yelon for cmlc2:mKate-CAAX zebrafish, K.D. Poss for cmlc2:CreERT2 zebrafish and Manu Beerens and Calum MacRae for access to the Cell Cycle Application Module within Metamorph Software. J.M.G.-R. was supported by an EMBO Long-Term Fellowship (ALTF 253-2014) and a Fund for Medical Discovery Award from the Executive Committee on Research at Massachusetts General Hospital. M.S. was supported by National Science Foundation Graduate Research Fellowship DGE1144152. This work was supported by NIH award R01 HL127067 to C.G.B. and C.E.B., R01 HL132927 to L.J.F. and by Hassenfeld and d’Arbeloff MGH Research Scholar Awards to C.E.B.
Footnotes
Author contributions
J.M.G.-R. designed, performed, and interpreted experiments. M.S. performed ex periments. D.F., M.H.S. and L.J.F. performed mice experiments. C.G.B. and C.E.B. directed the study, designed experiments, and interpreted data. J.M.G.-R., C.E.B., and C.G.B. conceived the study and wrote the manuscript. All authors commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could aFFect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkass K, Panula J, Westman M, Wu TD, Guerquin-Kern JL, Bergmann O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell. 2015;163:1026–1036. doi: 10.1016/j.cell.2015.10.035. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci USA. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. AJP: Heart and Circulatory Physiology. 2007;293:H1883–91. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Riabov Bassat D, Udi Y, Sarig R, Sagi I, Martin JF, Bursac N, Cohen S, Tzahor E. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179–184. doi: 10.1038/nature22978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RO, Chapin S, Sherry R. Regeneration of the ventricular myocardium in amphibians. Nature. 1974;248:145–147. doi: 10.1038/248145a0. [DOI] [PubMed] [Google Scholar]
- Bednarek D, González-Rosa JM, Guzmán-Martínez G, Gutiérrez-Gutiérrez Ó, Aguado T, Sánchez-Ferrer C, Marques IJ, Galardi-Castilla M, de Diego I, Gómez MJ, Cortés A, Zapata A, Jiménez-Borreguero LJ, Mercader N, Flores I. Telomerase Is Essential for Zebrafish Heart Regeneration. CellReports. 2015;12:1691–1703. doi: 10.1016/j.celrep.2015.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- Brodsky V, Sarkisov DS, Arefyeva AM, Panova NW, Gvasava IG. Polyploidy in cardiac myocytes of normal and hypertrophic human hearts; range of values. Virchows Arch. 1994;424:429–435. doi: 10.1007/BF00190566. [DOI] [PubMed] [Google Scholar]
- Brodsky WY, Arefyeva AM, Uryvaeva IV. Mitotic polyploidization of mouse heart myocytes during the first postnatal week. Cell Tissue Res. 1980;210:133–144. doi: 10.1007/BF00232149. [DOI] [PubMed] [Google Scholar]
- Cai D, Cohen KB, Luo T, Lichtman JW, Sanes JR. Improved tools for the Brainbow toolbox. Nat Methods. 2013;10:540–547. doi: 10.1038/nmeth.2450. [DOI] [PubMed] [Google Scholar]
- Chablais F, Veit J, Rainer G, JaŸwińska A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev Biol. 2011;11:1–21. doi: 10.1186/1471-213X-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WY, Gemberling M, Wang J, Holdway JE, Shen MC, Karlstrom RO, Poss KD. In vivo monitoring of cardiomyocyte proliferation to identify chemical modifiers of heart regeneration. Development. 2013;140:660–666. doi: 10.1242/dev.088526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585–610. doi: 10.1146/annurev-cellbio-092910-154234. [DOI] [PubMed] [Google Scholar]
- Duncan AW, Taylor MH, Hickey RD, Newell AEH, Lenzi ML, Olson SB, Finegold MJ, Grompe M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–710. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert L, Pfitzer P. Nuclear DNA of myocardial cells in the periphery of infarctions and scars. Virchows Arch, B, Cell Pathol. 1977;24:209–217. doi: 10.1007/BF02889280. [DOI] [PubMed] [Google Scholar]
- Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development. 2016;143:729–740. doi: 10.1242/dev.132910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DT, Gall JG, Spradling AC. Error-prone polyploid mitosis during normal Drosophila development. Genes Dev. 2010;24:2294–2302. doi: 10.1101/gad.1952710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rosa JM, Burns CE, Burns CG. Zebrafish heart regeneration: 15 years of discoveries. Regeneration (Oxf) 2017;4:105–123. doi: 10.1002/reg2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rosa JM, Guzmán-Martínez G, Marques IJ, Sánchez-Iranzo H, Jiménez-Borreguero LJ, Mercader N. Use of echocardiography reveals reestablishment of ventricular pumping efficiency and partial ventricular wall motion recovery upon ventricular cryoinjury in the zebrafish. PLoS ONE. 2014;9:e115604. doi: 10.1371/journal.pone.0115604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rosa JM, Martín V, Peralta M, Torres M, Mercader N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development. 2011;138:1663–1674. doi: 10.1242/dev.060897. [DOI] [PubMed] [Google Scholar]
- González-Rosa JM, Mercader N. Cryoinjury as a myocardial infarction model for the study of cardiac regeneration in the zebrafish. Nat Protoc. 2012;7:782–788. doi: 10.1038/nprot.2012.025. [DOI] [PubMed] [Google Scholar]
- Green RA, Paluch E, Oegema K. Cytokinesis in animal cells. Annu Rev Cell Dev Biol. 2012;28:29–58. doi: 10.1146/annurev-cellbio-101011-155718. [DOI] [PubMed] [Google Scholar]
- Gupta V, Gemberling M, Karra R, Rosenfeld GE, Evans T, Poss KD. An Injury-Responsive Gata4 Program Shapes the Zebrafish Cardiac Ventricle. Current Biology. 2013;23:1221–1227. doi: 10.1016/j.cub.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halloran MC, Sato-Maeda M, Warren JT, Su F, Lele Z, Krone PH, Kuwada JY, Shoji W. Laser-induced gene expression in specific cells of transgenic zebrafish. Development. 2000;127:1953–1960. doi: 10.1242/dev.127.9.1953. [DOI] [PubMed] [Google Scholar]
- Hirose K, Kawashima T, Iwamoto I, Nosaka T, Kitamura T. MgcRacGAP is involved in cytokinesis through associating with mitotic spindle and midbody. J Biol Chem. 2001;276:5821–5828. doi: 10.1074/jbc.M007252200. [DOI] [PubMed] [Google Scholar]
- Hoijman E, Rubbini D, Colombelli J, Alsina B. Mitotic cell rounding and epithelial thinning regulate lumen growth and shape. Nat Commun. 2015;6:7355. doi: 10.1038/ncomms8355. [DOI] [PubMed] [Google Scholar]
- Ikenishi A, Okayama H, Iwamoto N, Yoshitome S, Tane S, Nakamura K, Obayashi T, Hayashi T, Takeuchi T. Cell cycle regulation in mouse heart during embryonic and postnatal stages. Dev Growth Differ. 2012;54:731–738. doi: 10.1111/j.1440-169X.2012.01373.x. [DOI] [PubMed] [Google Scholar]
- Jahangiri L, Sharpe M, Novikov N, González-Rosa JM, Borikova A, Nevis K, Paffett-Lugassy N, Zhao L, Adams M, Guner-Ataman B, Burns CE, Burns CG. The AP-1 transcription factor component Fosl2 potentiates the rate of myocardial differentiation from the zebrafish second heart field. Development. 2016;143:113–122. doi: 10.1242/dev.126136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K. Advances in understanding the mechanism of zebrafish heart regeneration. Stem Cell Res. 2014 doi: 10.1016/j.scr.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DYR, Poss KD. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464:601–605. doi: 10.1038/nature08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, Choi SY. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE. 2011;6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Lai SL, Marín-Juez R, Moura PL, Kuenne C, Lai JKH, Tsedeke AT, Guenther S, Looso M, Stainier DY. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. Elife. 2017;6:e25605. doi: 10.7554/eLife.25605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao Z, Raju GP, Bai CB, Joyner AL. MASTR: a technique for mosaic mutant analysis with spatial and temporal control of recombination using conditional floxed alleles in mice. Cell Rep. 2012;2:386–396. doi: 10.1016/j.celrep.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Grill S, Sanchez A, Murphy-Ryan M, Poss KD. Fgf signaling instructs position-dependent growth rate during zebrafish fin regeneration. Development. 2005;132:5173–5183. doi: 10.1242/dev.02101. [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28:1737–1746. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- Lin YF, Swinburne I, Yelon D. Multiple influences of blood flow on cardiomyocyte hypertrophy in the embryonic zebrafish heart. Dev Biol. 2012;362:242–253. doi: 10.1016/j.ydbio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhou T, Kuriyama R, Erikson RL. Molecular interactions of Polo-like-kinase 1 with the mitotic kinesin-like protein CHO1/MKLP-1. J Cell Sci. 2004;117:3233–3246. doi: 10.1242/jcs.01173. [DOI] [PubMed] [Google Scholar]
- Losick VP, Fox DT, Spradling AC. Polyploidization and Cell Fusion Contribute to Wound Healing in the Adult Drosophila Epithelium. Curr Biol. 2013;23:2224–2232. doi: 10.1016/j.cub.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S, Kühn B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci USA. 2013;110:1446–1451. doi: 10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KBS, Virag JI, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004;428:664–668. doi: 10.1038/nature02446. [DOI] [PubMed] [Google Scholar]
- Nakamura R, Koshiba-Takeuchi K, Tsuchiya M, Kojima M, Miyazawa A, Ito K, Ogawa H, Takeuchi JK. Expression analysis of Baf60c during heart regeneration in axolotls and neonatal mice. Dev Growth Differ. 2016;58:367–382. doi: 10.1111/dgd.12281. [DOI] [PubMed] [Google Scholar]
- O’Meara CC, Wamstad JA, Gladstone RA, Fomovsky GM, Butty VL, Shrikumar A, Gannon JB, Boyer LA, Lee RT. Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration. Circulation Research. 2015;116:804–815. doi: 10.1161/CIRCRESAHA.116.304269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool. 1974;187:249–253. doi: 10.1002/jez.1401870208. [DOI] [PubMed] [Google Scholar]
- Oberpriller JO, Oberpriller JC, Arefyeva AM, Mitashov VI, Carlson BM. Nuclear characteristics of cardiac myocytes following the proliferative response to mincing of the myocardium in the adult newt, Notophthalmus viridescens. Cell Tissue Res. 1988;253:619–624. doi: 10.1007/BF00219752. [DOI] [PubMed] [Google Scholar]
- Oceguera-Yanez F. Ect2 and MgcRacGAP regulate the activation and function of Cdc42 in mitosis. J Cell Biol. 2005;168:221–232. doi: 10.1083/jcb.200408085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL. When bigger is better: the role of polyploidy in organogenesis. Trends Genet. 2015;31:307–315. doi: 10.1016/j.tig.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YA, Freundlich T, Weissman TA, Schoppik D, Wang XC, Zimmerman S, Ciruna B, Sanes JR, Lichtman JW, Schier AF. Zebrabow: multispectral cell labeling for cell tracing and lineage analysis in zebrafish. Development. 2013;140:2835–2846. doi: 10.1242/dev.094631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parichy DM, Elizondo MR, Mills MG, Gordon TN, Engeszer RE. Normal table of postembryonic zebrafish development: staging by externally visible anatomy of the living fish. Dev Dyn. 2009;238:2975–3015. doi: 10.1002/dvdy.22113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, Parikh S, Denholtz M, Huang Y, Yamaguchi Y, Shen H, Allayee H, Crump JG, Force TI, Lien CL, Makita T, Lusis AJ, Kumar SR, Sucov HM. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet. 2017;271:H2183. doi: 10.1038/ng.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roukos V, Pegoraro G, Voss TC, Misteli T. Cell cycle staging of individual cells by fluorescence microscopy. Nat Protoc. 2015;10:334–348. doi: 10.1038/nprot.2015.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata H, Rubin JS, Taylor WG, Miki T. A Rho-specific exchange factor Ect2 is induced from S to M phases in regenerating mouse liver. Hepatology. 2000;32:193–199. doi: 10.1053/jhep.2000.8271. [DOI] [PubMed] [Google Scholar]
- Sallin P, de Preux Charles AS, Duruz V, Pfefferli C, JaŸwińska A. A dual epimorphic and compensatory mode of heart regeneration in zebrafish. Dev Biol. 2015;399:27–40. doi: 10.1016/j.ydbio.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Sandritter W, Adler CP. Polyploidization of heart muscle nuclei as a prerequisite for heart growth and numerical hyperplasia in heart hypertrophy. Recent Adv Stud Cardiac Struct Metab. 1976;12:115–127. [PubMed] [Google Scholar]
- Schnabel K, Wu CC, Kurth T, Weidinger G. Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PLoS ONE. 2011;6:e18503. doi: 10.1371/journal.pone.0018503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder KP, Fox DT. The expanding implications of polyploidy. J Cell Biol. 2015;209:485–491. doi: 10.1083/jcb.201502016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–6. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis during hypertrophy in adult mice. Am J Physiol. 1994;266:H1439–45. doi: 10.1152/ajpheart.1994.266.4.H1439. [DOI] [PubMed] [Google Scholar]
- Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–9. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- Soonpaa MH, Koh GY, Klug MG, Field LJ. Formation of nascent intercalated disks between grafted fetal cardiomyocytes and host myocardium. Science. 1994;264:98–101. doi: 10.1126/science.8140423. [DOI] [PubMed] [Google Scholar]
- Takeuchi JK, Lou X, Alexander JM, Sugizaki H, Delgado-Olguín P, Holloway AK, Mori AD, Wylie JN, Munson C, Zhu Y, Zhou YQ, Yeh RF, Henkelman RM, Harvey RP, Metzger D, Chambon P, Stainier DYR, Pollard KS, Scott IC, Bruneau BG. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat Commun. 2011;2:187. doi: 10.1038/ncomms1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tane S, Kubota M, Okayama H, Ikenishi A, Yoshitome S, Iwamoto N, Satoh Y, Kusakabe A, Ogawa S, Kanai A, Molkentin JD, Nakamura K, Ohbayashi T, Takeuchi T. Repression of cyclin D1 expression is necessary for the maintenance of cell cycle exit in adult mammalian cardiomyocytes. J Biol Chem. 2014;289:18033–18044. doi: 10.1074/jbc.M113.541953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol. 1999;147:921–928. doi: 10.1083/jcb.147.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnozzi RJ, Gatto GJ, Kallander LS, Hoffman NE, Mallilankaraman K, Ballard VLT, Lawhorn BG, Stoy P, Philp J, Graves AP, Naito Y, Lepore JJ, Gao E, Madesh M, Force T. Inhibition of the cardiomyocyte-specific kinase TNNI3K limits oxidative stress, injury, and adverse remodeling in the ischemic heart. Science Translational Medicine. 2013;5:207ra141–207ra141. doi: 10.1126/scitranslmed.3006479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivien CJ, Hudson JE, Porrello ER. Evolution, comparative biology and ontogeny of vertebrate heart regeneration. Nature Publishing Group. 2016;1:1–14. doi: 10.1038/npjregenmed.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills AA, Holdway JE, Major RJ, Poss KD. Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development. 2008;135:183–192. doi: 10.1242/dev.010363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.