

Abstract
Chronic hepatitis B virus (HBV) infection is a major health concern worldwide, frequently leading to liver cirrhosis, liver failure and hepatocellular carcinoma. Evidence exists that high viral antigen load may play a role in chronicity. Production of viral proteins is thought to depend on transcription of viral covalently closed circular DNA (cccDNA). In a human clinical trial with ARC-520, a RNA interference (RNAi)-based therapeutic targeting HBV transcripts, HBV S antigen (HBsAg) was strongly reduced in treatment-naïve patients positive for HBV e antigen (HBeAg) but was reduced significantly less in patients that were HBeAg negative or had received long-term therapy with nucleos(t)ide viral replication inhibitors (NUCs). The molecular basis for this unexpected differential response was investigated in chimpanzees chronically infected with HBV. Several independent lines of evidence demonstrated that HBsAg was expressed not only from the episomal cccDNA minichromosome, but also from transcripts arising from HBV DNA integrated into the host genome. The latter was the dominant source in HBeAg negative chimpanzees. Many of the integrants detected in chimpanzees lacked target sites for the siRNAs in ARC-520, explaining the reduced response in HBeAg negative chimpanzees and by extension in HBeAg negative patients. Our results uncover a heretofore under-recognized source of HBsAg that may represent a strategy adopted by HBV to maintain chronicity in the presence of host immune surveillance and could alter trial design and endpoint expectations of new therapies for chronic HBV.
INTRODUCTION
Globally an estimated 250–400 million people are chronically HBV infected and about one million die each year from HBV-related liver disease (1, 2). HBV is transmitted parenterally and infects hepatocytes. The immune system mounts an effective immune response that controls the virus in 95% of patients infected as adults. In contrast, infected neonates and young children usually become chronically HBV infected (CHB) following exposure. When untreated, 40% of men and 15% of women infected early in life later die of cirrhosis or hepatocellular carcinoma (HCC) or require liver transplantation (3).
The HBV virion contains a compact 3.2 kilobase (kb) genome that exists as a partially double-stranded, relaxed circular DNA (rcDNA) with a 7–9 base terminal redundancy that is converted into cccDNA within the nucleus of the hepatocyte, functioning as a minichromosome for HBV transcription (4). Host RNA polymerase II transcribes HBV genes on the cccDNA to produce 5 viral RNAs: transcripts that encode precore (serologically called HBeAg), the pregenomic RNA (pgRNA) that encodes the structural capsid protein (core) and polymerase and is reverse transcribed to produce rcDNA, the pre-S1 transcript that encodes the large S surface protein, another transcript that encodes pre-S2 and S surface proteins (middle and small S), and the X gene mRNA. The three surface proteins collectively comprise HBsAg. All HBV transcripts are encoded in overlapping reading frames, have a common 3′ end and utilize the same polyadenylation signal (PAS). HBsAg, in a lipid bilayer, forms an envelope around each core-encapsidated, partially double-stranded HBV genome.
Although cccDNA is considered the driver of HBV transcription and replication, HBV DNA may integrate into the host genome, which has been associated with the development of HCC, may disrupt host genes or change their transcription levels, and may result in oncogenic fusion proteins (5, 6). Integrated HBV genomes are found in non-HCC liver tissue of patients as well as in tumors (7). Levels of cccDNA are higher in HBeAg positive than HBeAg negative patients (8), whereas levels of integrated HBV are higher in HBeAg negative patients (7). The primary source of integrated HBV DNA is likely double-stranded linear HBV DNA (dslDNA), arising as an aberrant replication product due to mispriming of (+) strand synthesis (9). Although integrated HBV DNA is associated with HCC, its function in the maintenance of HBV infection is unknown.
HBeAg and HBsAg play important roles in chronic infection (10). Because HBeAg and core share common sequences, HBeAg is thought to induce T cell tolerance to both antigens and allow viral persistence (11). This tolerance predisposes the infant of an HBeAg positive mother to persistent replication following infection (12). In addition to enveloping the virion, HBsAg forms sub-viral particles at levels as much as 100,000-fold higher than the level of virions (13). High HBsAg levels are believed to contribute to T-cell exhaustion, resulting in limited or weak T-cell responses and even deletion of T-cells recognizing specific epitopes (14–16). For CHB patients the desired endpoint of treatment is seroclearance of HBsAg, referred to as a “functional cure”, resulting in improved long-term prognosis (17, 18). HBsAg loss is considered a hallmark of effective immune control of HBV (19).
Because high antigen load is believed to play a key role in maintaining chronicity, there is interest in directly reducing expression of viral antigens and regulatory proteins via RNAi (20). The RNAi therapeutic ARC-520 for treatment of chronic HBV infection comprises an equimolar mixture of liver-tropic cholesterol-conjugated siRNAs siHBV-74 and siHBV-77 plus an excipient that enables endosomal escape of the siRNAs into the cytoplasm where RNAi occurs (21). The mRNA target sites for siHBV-74 and siHBV-77 are 118 and 71 bases upstream, respectively, of the conventional HBV PAS (fig. S1). Because all cccDNA-derived HBV transcripts overlap at their 3′ end, the siRNAs in ARC-520 were designed to target all HBV mRNAs and reduce antigenemia, thus potentially enabling host immune responsiveness and functional cure.
Here we report results evaluating ARC-520 in a phase II clinical study in CHB patients and a complementary study in chimpanzees chronically infected with HBV. Our studies reveal a previously under-appreciated source of HBsAg, namely HBV DNA integrated into the host genome. This has implications for our understanding of HBV biology and host interactions, and for future development of drugs designed with curative intent for CHB patients.
RESULTS
Differential reduction of HBsAg associated with HBeAg status and prior exposure to NUCs in CHB patients treated with ARC-520
Heparc-2001 was a phase II clinical study to determine the safety and tolerability of ARC-520 and its effect on virologic parameters. Individual and mean patient demographics at study start are presented in tables S1 and S2, respectively. Initially, CHB patients receiving long-term entecavir (ETV) were enrolled into five 8-patient cohorts and randomized with a 6:2 (active:placebo) ratio. Patients in cohorts 1–4 were HBeAg negative, while patients in cohort 5 were HBeAg positive. ARC-520 was administered as a single intravenous infusion in cohorts 1–4, at ascending doses of 1, 2, 3 and 4 mg/kg. Patients in cohort 5 received 4 mg/kg. A sixth cohort of HBeAg positive patients received 2 doses of 2 mg/kg ARC-520 two weeks apart. Cohorts 1 to 6 had been on NUCs for 1.2 to 7.8 years prior to ARC-520 dosing. HBV serum DNA was below the lower limit of quantitation (<LLOQ) in these patients (table S2). Finally, a seventh, open-label cohort was added consisting of HBeAg negative and HBeAg positive patients that were NUC naïve. Daily ETV was initiated on the same day they received a single dose of 4 mg/kg ARC-520. A total of 58 CHB patients were successfully dosed in cohorts 1–7 of the study with 48 receiving drug and 10 placebo. There were few adverse events reported (table S3). No adverse trends were observed in vital signs, physical examinations or electrocardiograms. Laboratory values showed no clinically significant toxicity. Overall, ARC-520 was well tolerated when administered to CHB patients, as it was in normal volunteers (20).
We observed modest decreases in HBsAg levels in cohort 1–4 patients that correlated partially with dose (Fig. 1A). HBsAg levels decreased most sharply during the first 8 days after dosing in all cohorts, reaching a plateau before starting to relapse 6–8 weeks after dosing. At the highest dose level of 4 mg/kg (cohort 4), HBsAg was reduced by 0.3 log10 relative to baseline. This level of knockdown was substantially less than observed in preclinical studies using a mouse model of HBV infection (21). These mice were HBeAg positive; however, the HBeAg status of the patients did not appear to be a factor as cohort 5 (HBeAg positive) patients showed a similar muted response for HBsAg knockdown (Fig. 1B). Administration of two injections of 2 mg/kg two weeks apart in cohort 6, instead of a single 4 mg/kg dose, indicated that saturation of hepatocyte delivery of ARC-520 was an unlikely explanation for the low level of HBsAg knockdown and minimal dose-responsiveness (fig. S2).
Fig. 1. Serum HBsAg, HBcrAg and HBeAg reduction in human patients treated with a single dose of ARC-520.
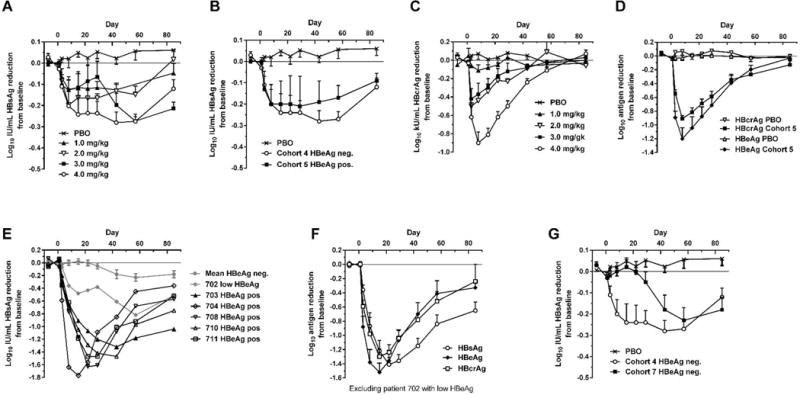
CHB patients were given a single intravenous dose of 1 to 4 mg/kg ARC-520 on a background of daily oral NUCs. HBsAg (A) or HBcrAg (C) reduction in CHB patients that were HBeAg negative, NUC-experienced and received single doses of 1 to 4 mg/kg (cohorts 1 to 4). (B) HBsAg reduction in CHB patients that were HBeAg negative and NUC-experienced (cohort 4) or HBeAg positive and NUC-experienced (cohort 5) that received a single dose of 4 mg/kg. (D) HBcrAg and HBeAg reduction in CHB patients that were HBeAg positive, NUC-experienced and received a single 4 mg/kg dose (cohort 5). (E) HBsAg reduction for individual CHB patients that were HBeAg positive in cohort 7. (F) HBsAg, HBeAg and HBcrAg reductions in CHB patients that were HBeAg positive, treatment naïve, and received a single 4 mg/kg dose (cohort 7). (G) HBsAg reduction in CHB patients that were HBeAg negative and treatment naïve (cohort 7) or NUC experienced (cohort 4). PBO, patients on NUC therapy given placebo injection; HBcrAg, HBV core-related antigen. Error bars show SEM.
The lack of high level HBsAg knockdown was unlikely due to inherent RNAi inefficiency or lack of ARC-520 efficacy in the CHB patients, as evidenced by measuring knockdown of other viral parameters. For example, measurement of hepatitis B core-related antigen (HBcrAg) in cohort 1–4 patients with quantifiable levels showed dose-dependent knockdown, reaching 0.9 log10 mean reduction in patients receiving 4 mg/kg (Fig. 1C). Similar results were observed in cohort 5 with mean HBcrAg reduction of 0.9 log10 and HBeAg reduction of 1.2 log10 at nadir (Fig. 1D).
Cohorts 1–6 comprised patients who had been on long-term NUC therapy. To understand whether prior NUC therapy affected HBsAg reduction following ARC-520 injection, an additional cohort consisting of NUC-naïve patients (cohort 7) was studied. Six of these patients were HBeAg positive and 6 HBeAg negative, but one of the positive patients had low HBeAg (<10 PEIU/mL) and was considered to be transitioning to HBeAg negative status. Cohort 7 patients received a single dose of 4 mg/kg ARC-520 and began daily ETV dosing on Day 1. HBV serum DNA in cohort 7 patients was higher at baseline in the HBeAg positive (8–9 log10 IU/mL, excluding the low HBeAg-positive patient) than HBeAg negative (3–5 log10) patients (table S1). HBV DNA levels decreased in HBeAg positive patients by 4.0 ± 0.6 log10 during the first three weeks of ARC-520 plus NUC treatment and became undetectable in most HBeAg negative patients (fig. S3).
Excluding the transitional patient, NUC-naive HBeAg positive patients in cohort 7 responded with deep HBsAg reduction: a 1.4 ± 0.1 log10 reduction in HBsAg (minimum of 1.3 log10 and a maximum of 1.8 log10 reduction) was observed, along with mean 1.5 ± 0.1 log10 reduction in HBeAg and 1.3 ± 0.1 log10 reduction in HBcrAg (Fig. 1, E and F). HBsAg in HBeAg negative cohort 7 patients decreased to a similar degree as in NUC-experienced HBeAg negative patients in cohort 4, but after a delay of approximately 3 weeks (Fig. 1G). We have no explanation for this delayed response, but it may be a secondary effect of knockdown of other viral components on HBsAg expression. The transitional HBeAg positive patient demonstrated HBsAg reductions intermediate to those observed in HBeAg positive and negative patients in cohort 7. Together, these results suggested that HBeAg negativity and previous long-term NUC therapy adversely impacted the ability of ARC-520 to reduce expression of HBsAg.
Differential HBsAg reduction in response to ARC-520 associated with HBeAg status in chronically infected chimpanzees
In addition to the Heparc-2001 clinical trial, we conducted a study of ARC-520 in chimpanzees chronically infected with HBV. Nine CHB chimpanzees were available for inclusion in this study: 5 males and 4 females, 5 initially HBeAg positive and 4 HBeAg negative (table S4). Safety and efficacy of repeat dosing of the chimpanzees with ARC-520 were monitored with regular blood collections for evaluation of safety parameters and HBV serum DNA, HBsAg, HBeAg, and anti-HBs and anti-HBe antibodies. Initial HBsAg levels varied by 4 orders of magnitude and HBV serum DNA levels by more than 6 orders of magnitude, providing an opportunity to evaluate the breadth of ARC-520 efficacy.
The chimpanzees were treated with daily oral NUCs (ETV for all, and for chimpanzee 4x0139 ETV + tenofovir) for 8–24 weeks to reduce viral replication prior to dosing with ARC-520 (Fig. 2A). While continuing NUC treatment, the chimpanzees were then given ARC-520 once every 4 weeks (Q4W) for a total of 6 to 11 injections. Monitoring of clinical chemistry and hematology parameters throughout the study showed treatment with ARC-520 and NUCs was well tolerated with no observed toxicity.
Fig. 2. Response to repeat dosing of chimpanzees with ARC-520.
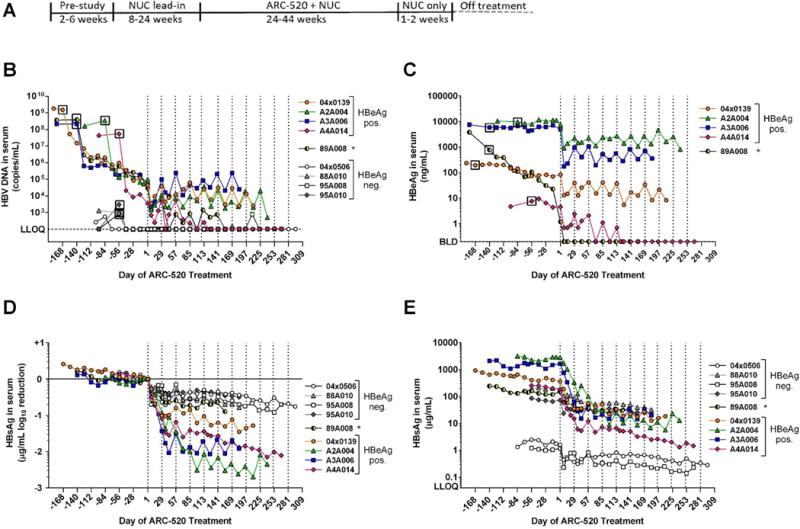
(A) Following a pre-study evaluation, nine chimpanzees began daily oral NUC dosing. After a variable NUC lead-in period to reduce viremia, NUC treatment continued concomitant with Q4W dosing of ARC-520 that began on Day 1. Dosing days are indicated by vertical dashed lines. Blood samples were collected periodically throughout the study and serum HBV DNA (B), HBeAg (C) and HBsAg (D, E) were measured. LLOQ, lower limit of quantitation. BLD, below the limit of detection. Asterisk, chimpanzee 89A008 transitioned from HBeAg positive to negative during the NUC lead-in period.
Mean serum HBV DNA levels in HBeAg positive chimpanzees decreased by 4.0 ± 0.2 log10 copies/mL during NUC lead-in (Fig. 2B). All HBeAg negative chimpanzees had serum DNA levels close to the lower limit of detection (LLOD) that became undetectable after two weeks of NUC treatment. Following NUC lead-in, the first ARC-520 injection reduced mean HBV serum DNA by an additional 1.3 ± 0.1 log10 in HBeAg positive chimpanzees (Fig. 2B). HBV DNA continued to be suppressed during ARC-520 treatment to levels near the nadir observed after the first dose. Two chimpanzees initially HBeAg positive, 89A008 and A4A014, seroconverted to HBeAg negative during the study and their serum HBV DNA become undetectable.
HBeAg levels changed negligibly during NUC lead-in, with the exception of HBeAg transitional chimpanzee 89A008 that seroconverted for HBeAg during NUC lead-in (Fig. 2C). In the 40 days between the health check and start of NUC lead-in, the HBeAg level in 89A008 decreased 5-fold and then continued to decline 3.5 log10 ng/mL during the 20-week NUC lead-in. In the other four HBeAg positive chimpanzees, mean HBeAg levels were reduced by 1.0 ± 0.1 log10 following the first ARC-520 injection and remained suppressed near these levels throughout ARC-520 dosing, though HBeAg levels partially rebounded from nadir after each injection (Fig. 2C). Chimpanzee A4A014 became positive for anti-HBe antibodies and then HBeAg became undetectable following the 5th ARC-520 injection.
At study initiation, HBsAg levels were an average 19-fold higher in the HBeAg positive than HBeAg negative chimpanzees (table S4). Two HBeAg negative chimpanzees (88A010 and 95A010) had levels of HBsAg similar to the two HBeAg positive chimpanzees that seroconverted for HBeAg during the study (A4A014 and 89A008), while the other two HBeAg negative chimpanzees had HBsAg levels that were two orders of magnitude lower (4x0506 and 95A008). HBsAg levels decreased minimally during NUC lead-in (Fig. 2, D and E). HBeAg positive animals responded to the first dose of ARC-520 (2–4 mg/kg) with HBsAg reductions of 0.9 to 1.4 log10 four weeks after injection, whereas the HBeAg negative chimpanzees responded less well, with 0.4 to 0.7 log10 reductions at nadir (Fig. 2D). The extent of HBsAg knockdown appeared not to correlate with starting levels of HBsAg, but rather with HBeAg status. (Fig. 2E). Unlike HBV DNA and HBeAg levels, HBsAg levels gradually decreased throughout the ARC-520 dosing period regardless of HBeAg status, with the exception of A3A006 (Fig. 2, D and E).
As observed in human patients that had been NUC-naïve prior to dosing with ARC-520, HBeAg positive chimpanzees responded with much deeper knockdown than HBeAg negative chimpanzees. Mean HBsAg values normalized to ARC-520 dosing Day 1 are shown in Fig. 3A. HBsAg knockdown following ARC-520 injection in the three chimpanzees that remained HBeAg positive throughout the study (4x0139, A2A004 and A3A006) was similar to that in A4A014, which became HBeAg negative, and there were no apparent differences in effect between 2 and 4 mg/kg ARC-520 (Fig. 2, D and E, and Fig. 3A).
Fig. 3. Serum HBsAg and liver HBV mRNA reduction in chimpanzees dosed with ARC-520.
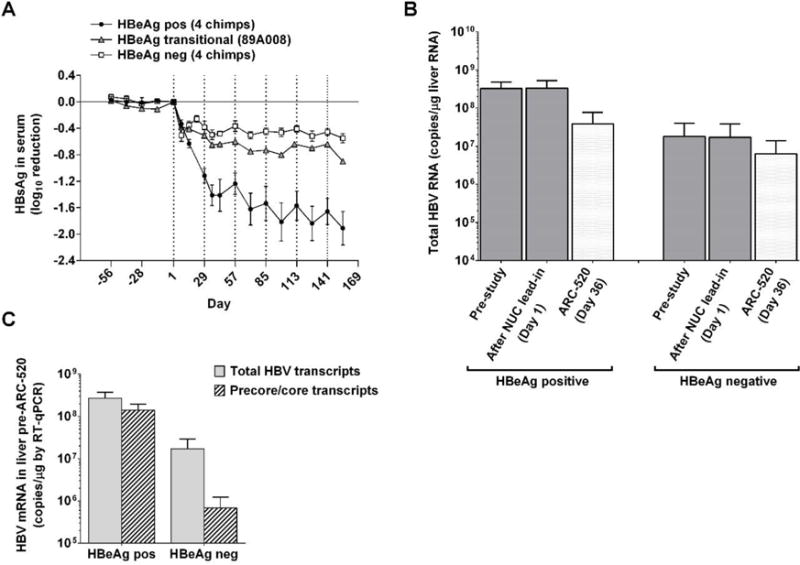
Following pre-study evaluation, nine chimpanzees began NUC dosing for a lead-in period of 8–24 weeks. Q4W dosing with ARC-520 began on Day 1. (A) HBsAg levels in serum. The mean log10 change in HBsAg is shown for the 8 weeks preceding the first dose of ARC-520 and during ARC-520 treatment of four HBeAg positive (●), four HBeAg negative (□), and in the HBeAg transitional chimpanzee (▲). (B, C) Liver mRNA from biopsies of HBeAg positive (A2A004, A3A006 and A4A014) and HBeAg negative (88A010, 95A008 and 95A010) chimpanzees was evaluated for HBV gene expression by RT-qPCR with probe sets in core to measure the levels of precore mRNA/pgRNA and in X to measure total HBV mRNA. (B) Mean total HBV mRNA levels were compared between HBeAg positive and negative chimpanzees pre-study, after the NUC lead-in (Day 1), and one week after the second ARC-520 injection (Day 36). (C) The numbers of total HBV transcripts and precore/pgRNA transcripts on Day 1 are compared for the HBeAg positive and negative chimpanzees. Error bars show SEM.
HBsAg reductions following ARC-520 treatment of HBeAg transitional chimpanzee 89A008 were intermediate between the HBeAg negative and positive chimpanzees (Fig. 2, D and E, and Fig. 3A). This is reminiscent of the HBsAg reduction in the low HBeAg expressing patient 702 from Heparc-2001 cohort 7 (Fig. 1E).
Abundance and characteristics of HBV DNA and RNA in the liver differ between HBeAg positive and negative chimpanzees
To understand the reason for the differential response to ARC-520 between HBeAg positive and negative chimpanzees, and by extension in HBeAg positive and negative humans, we characterized the molecular genetics of HBV found in these two chimpanzee serotypes. Liver biopsies were performed on eight of the chimpanzees at the pre-study health check, following NUC lead-in on Day 1, and at various time points following ARC-520 injections. These specimens were divided for isolation of total liver DNA, total liver RNA, and for histological evaluation of paraffin-embedded tissue. Chimpanzee 4x0506 was not biopsied due to low pre-study platelet counts.
Pre-study levels of total HBV DNA in the liver were measured using qPCR with a probe in the core region. The HBeAg positive chimpanzees had 5.9 – 7.8 log10 copies HBV/μg host DNA whereas the HBeAg negative chimpanzees had considerably less: 3.6 – 4.4 log10 copies/μg host DNA (fig. S4), consistent with the lower serum DNA levels in HBeAg negative chimpanzees. NUC treatment alone during lead-in reduced total HBV liver DNA in the HBeAg positive chimpanzees by an average 0.45 ± 0.06 log10 per month but did not reduce it in the HBeAg negative chimpanzees, suggesting that the majority of HBV DNA in the liver of the latter is not dependent on replication via viral polymerase. DNase evaluation of the liver further pointed to differences in the forms of liver HBV DNA in HBeAg positive compared to negative chimpanzees (supplementary materials).
Paired-end sequencing of fragmented liver DNA enriched for HBV-containing sequences revealed that HBeAg positive and negative chimpanzees had HBV DNA integrated into their genomes and the majority of integration sites were at direct repeat 1 (DR1) (fig. S5). Integrated HBV DNA in 4x0139 was previously characterized (22).
HBV transcripts in the liver were evaluated by three distinct approaches, each of which provides somewhat different but complementary information: quantitative reverse transcription PCR (RT-qPCR), paired-end next generation mRNA sequencing (mRNA-seq), and single molecule real-time (SMRT) sequencing (Iso-seq).
Total HBV transcripts in the liver specimens were quantified using a qPCR probe in the X gene (X probe) that hybridized to a region expected to be in all cccDNA-derived transcripts (precore, pgRNA, pre-S1, pre-S2/S and X). Transcript levels were compared between three HBeAg positive and three HBeAg negative chimpanzees (Fig. 3B). HBeAg positive chimpanzee 4x0139 was omitted due to low X probe efficiency in this animal and 89A008 was omitted because it was transitioning to HBeAg negative status. The mean number of total transcripts was 18-fold higher in HBeAg positive chimpanzees (7.8 – 8.7 log10 copies/μg RNA) than HBeAg negative chimpanzees (6.6 – 7.6 log10 copies/μg), similar to the difference in HBsAg expression between these groups. The difference in RNA reduction was similar to the difference in HBsAg reduction observed in these two groups of chimpanzees at this time point (Fig. 3A).
The precore plus pgRNA transcripts can be distinguished from the total HBV transcripts by use of a qPCR probe to a conserved sequence in the core region (Fig. 3C). Interestingly, the precore/pgRNA transcripts comprised only 3.5 ± 0.5% of the total HBV transcripts in HBeAg negative chimpanzees, whereas they comprised 52.6 ± 0.5% of the total in HBeAg positive chimpanzees.
Unlike PCR, which requires knowledge of target site sequences and is less efficient on variants with mismatches to primer or probe, mRNA-seq analysis accommodates variation (23). Using liver biopsy samples collected after NUC lead-in, approximately 40 million sequencing reads with a length of 50 bases were generated from each total liver RNA sample. Histograms were generated by aligning the reads from each chimpanzee to its consensus HBV DNA sequence obtained from liver DNA sequencing (Fig. 4). A steep decrease in the number of reads just downstream of the major HBV PAS (sequence TATAAA) in HBeAg positive chimpanzees suggests that the majority of HBV transcripts terminate near this polyadenylation signal. In the HBeAg negative chimpanzees, few reads extended beyond DR1 and thus they lacked the major HBV PAS that is located 81 bases downstream of DR1. There was no indication of resistance to ARC-520 developing after multiple doses (table S5 and fig. S6). These mRNA-seq results are consistent with HBsAg transcripts in HBeAg negative chimpanzees being produced from integrated dslDNA.
Fig. 4. Histogram of liver HBV mRNA paired-end sequencing reads in HBeAg positive and negative chimpanzees.
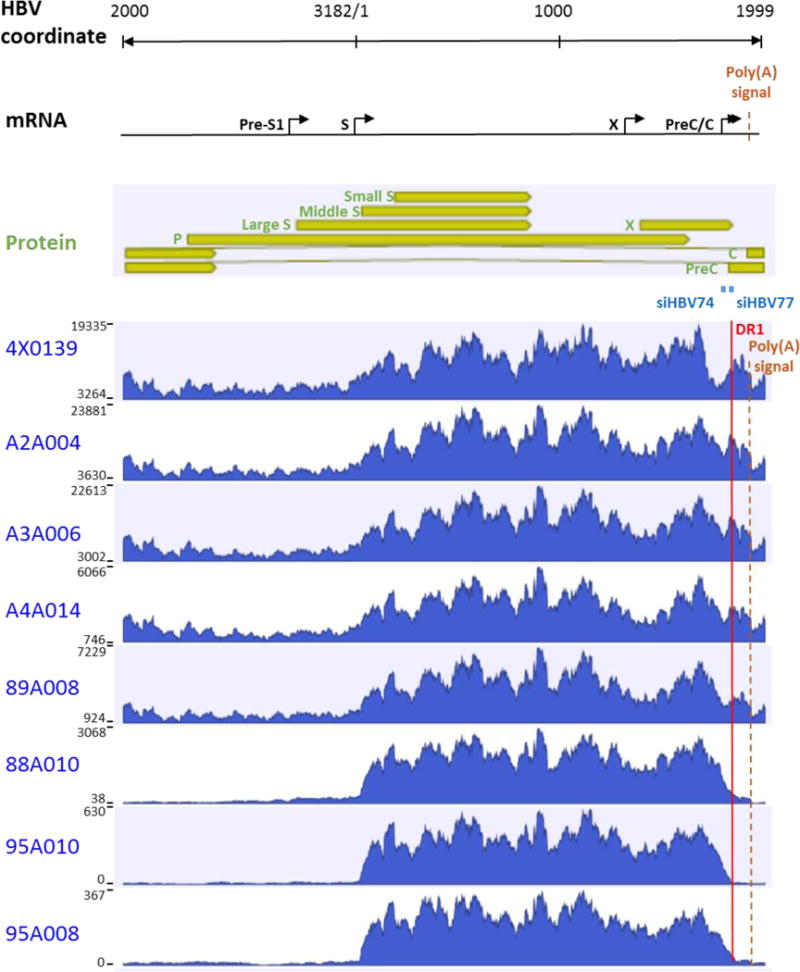
The HBV mRNA and HBV protein open reading frames are positioned relative to the coordinates of the HBV genome. The mRNA sequencing read histograms are shown for HBeAg positive chimpanzees 4x0139, A2A004, A3A006 and A4A014; for HBeAg transitional chimpanzee 89A008; and for HBeAg negative chimpanzees 88A010, 95A010 and 95A008. Locations of the DR1 sequence (red line), HBV PAS (brown dashed line) and binding sites for the siRNAs in ARC-520 (siHBV-74 and siHBV-77) are indicated.
We performed Iso-seq to allow sequencing of the entire full-length cDNA, using total liver RNA from one HBeAg positive (A2A004) and one HBeAg negative (88A010) chimpanzee. Alignment of the sequencing data from A2A004 to the HBV genome revealed that the vast majority of S transcripts had the expected 5′ and 3′ ends, originating from the expected promoters and terminating at the HBV PAS, respectively (Fig. 5A). In contrast, the 5′ ends of S transcripts from animal 88A010 mapped to the S promoters but in most cases the 3′ ends failed to align to HBV beginning in the DR2-DR1 region upstream of the HBV PAS (Fig. 5B). These were hybrid transcripts containing HBV sequence fused to non-HBV sequences, which were overwhelmingly of chimpanzee origin. While 90.5% of the HBV transcripts in A2A004 were non-fusion, only 22.7% were non-fusion from HBeAg negative 88A010 (Table 1). Consistent with our DNA-seq and mRNA-seq results, the Iso-seq data also demonstrate that HBV-chimpanzee integration points are located primarily near DR1 (see Supplementary information for elaboration of these results).
Fig. 5. Mapping of HBV S transcripts from an HBeAg positive and an HBeAg negative chimpanzee.
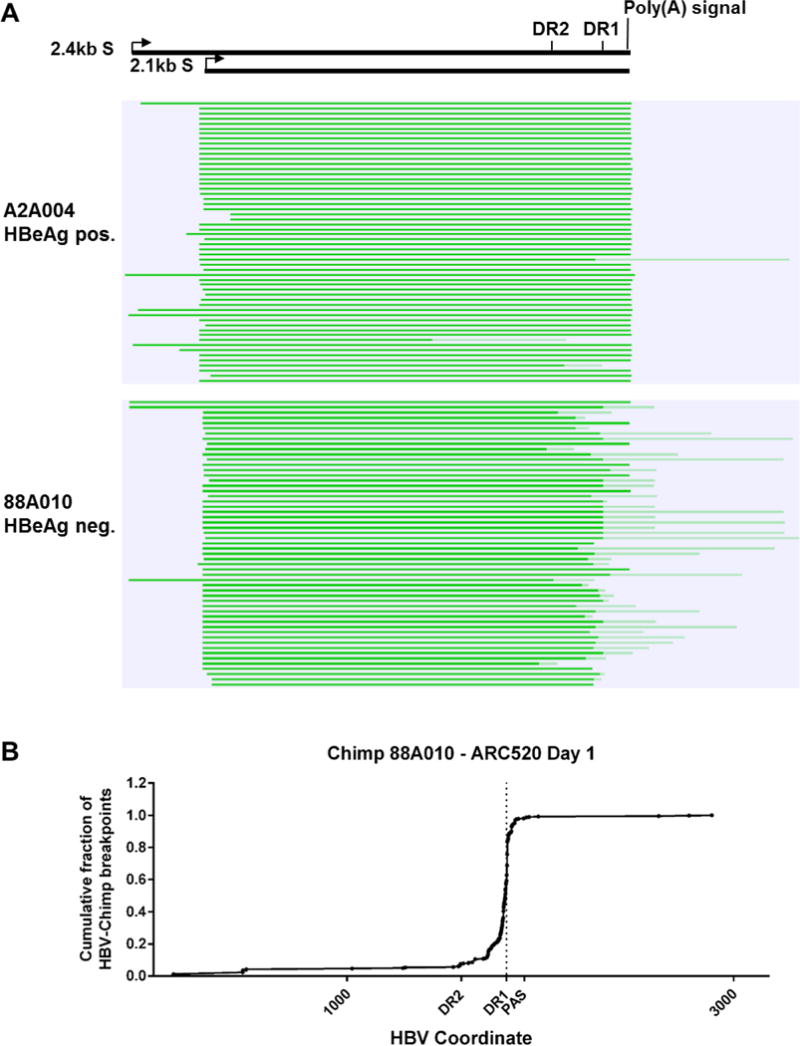
Total RNA isolated from the liver biopsies after the NUC lead-in and before ARC-520 dosing on Day 1 from HBeAg positive chimpanzee A2A004 and HBeAg negative chimpanzee 88A010 was reverse transcribed, size-selected and sequenced with single molecule real-time (SMRT) sequencing. (A) Full-length nonconcatemer reads (see methods) were aligned to each chimpanzee’s consensus HBV DNA sequence with CLC Genomics Workbench. Precore/pgRNA transcripts are not shown in this view. These were detected in A2A004 but were not in 88A010 at this sequencing depth. Green lines represent HBV-containing transcripts. Dark green represents sequences aligning to HBV and light green represents those not aligning to HBV. The HBV coordinates are shown with elements DR2, DR1 and the HBV polyadenylation signal. (B) Cumulative fraction of HBV-chimpanzee breakpoints were plotted against the HBV coordinate.
Table 1. Characterization of HBV transcripts and polyadenylation signal sequences in HBeAg negative and positive chimpanzees.
Total liver RNA collected after the NUC lead-in and prior to ARC-520 dosing (Day 1) from HBeAg negative chimpanzee 88A010 and HBeAg positive chimpanzee A2A004 was sequenced with the SMRT approach. All mRNA sequences that could be at least partially aligned with the HBV genome were analyzed to determine the number that contained entirely HBV sequence, HBV-chimpanzee fusion sequence, HBV-HBV fusion sequence, or that contained HBV fused to an unknown sequence.
| Characterization of HBV transcripts | ||||
|---|---|---|---|---|
| HBeAg negative 88A010 | HBeAg positive A2A004 | |||
| HBV transcripts | Number of transcripts | Percentage | Number of transcripts | Percentage |
| HBV non-fusion | 128 | 22.7% | 2466 | 90.5% |
| HBV-Chimpanzee | 375* | 66.4% | 35** | 1.3% |
| HBV-HBV | 57 | 10.1% | 218 † | 8.0% |
| HBV-Other | 5 | 0.9% | 7 | 0.3% |
| Totals | 565 | 100% | 2726 | 100% |
| Analysis of polyadenylation signal sequences in non-fusion HBV transcripts | ||||
| HBeAg negative 88A010 | HBeAg positive A2A004 | |||
| PAS sequence | Number of transcripts | Percentage | Number of transcripts | Percentage |
| TATAAA | 80 | 62.5% | 2396 | 97.2% |
| CATAAA | 42 | 32.8% | 7†† | 0.3% |
| Other | 1 | 0.8% | 18 | 0.7% |
| None detected | 5 | 3.9% | 45 | 1.8% |
| total | 128 | 100% | 2466 | 100% |
13 reads with reverse orientation were excluded from further analysis;
2 reads with reverse orientation were excluded from further analysis and 12 transcripts appear to have been misidentified as fusion transcripts (supplementary analysis);
166 transcripts contain gaps in HBV sequence consistent with splicing (36);
2 of 7 could be mutants or sequencing errors (TATAAA expected based on the HBV coordinate)
HBV-chimpanzee fusion transcripts in HBeAg negative 88A010 utilized cryptic chimpanzee polyadenylation signals identified as the sequence AATAAA or with a single base change (table S6). These results demonstrate that fusion transcripts can utilize chimpanzee sequences near their integration sites and do not require a bona fide HBV PAS.
Deep reduction of HBsAg in HBeAg negative chimpanzees treated with siRNA targeting outside the DR1-DR2 region
We tested the hypothesis that the reason for less efficient ARC-520 mediated HBsAg reduction in HBeAg negative chimpanzees was due to loss of ARC-520 siRNA target sites in the mRNA encoding their HBsAg. Two HBeAg negative chimpanzees were treated with siRNA siHBV-75 that targets a site upstream of the DR1-DR-2 region. The other two chimpanzees continued to receive ARC-520. After the 10th dose of ARC-520, chimpanzees 4x0506 and 95A008 both had 0.7 log10 reduction of HBsAg relative to Day 1. After seven doses of ARC-520, chimpanzees 88A010 and 95A010 had 0.8 and 0.5 log10 HBsAg reduction, respectively. These two chimpanzees were then dosed three times (Q4W) with 4 mg/kg siHBV-75. The HBsAg was reduced 1.4 and 1.7 log10; 1.9 and 2.4 log10; and finally 2.3 and 3.0 log10 relative to Day 1 following each of the three successive siHBV-75 doses in 88A010 and 95A010, respectively (Fig. 6). These reductions demonstrated that HBsAg could be just as effectively reduced by RNAi in the HBeAg negative chimpanzees as in the HBeAg positive chimpanzees, as long as the target site for the siRNA was present in the mRNA. HBeAg negative 88A010 and transitional 89A008 both had populations of hepatocytes in liver biopsies that appeared to be clonal and stained strongly for accumulated HBsAg, highly suggestive of integrated HBV (fig. S7). This HBsAg staining was greatly reduced following treatment with siHBV-75.
Fig. 6. Treatment of HBeAg negative chimpanzees with siRNA targeted outside the DR1-DR2 region.
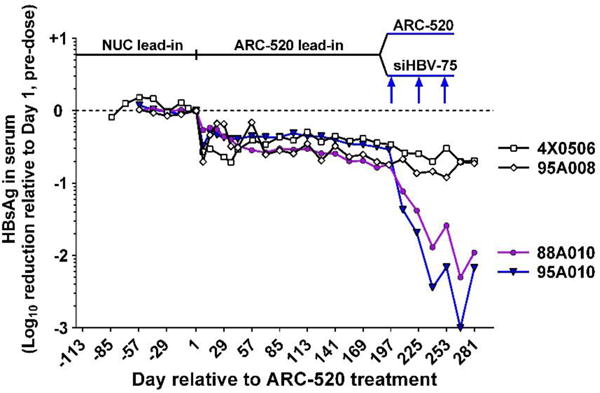
Following a NUC lead-in, all 4 HBeAg negative chimpanzees were given seven Q4W doses of ARC-520. Chimpanzees 4x0506 and 95A008 were then given an additional three doses of ARC-520 (4 mg/kg) while 95A010 and 88A010 were given three doses of 4 mg/kg siHBV-75 plus 4 mg/kg ARC-EX1 delivery reagent.
DISCUSSION
The differential response to ARC-520 reflected in the lower magnitude of HBsAg knockdown in HBeAg negative or NUC experienced HBeAg positive CHB patients in comparison to NUC-naïve HBeAg positive CHB patients could not have been predicted based on current models, which assume cccDNA is the only significant source of HBV transcripts. Instead, our data point to an additional, previously under-recognized source of HBsAg, namely integrated HBV DNA. Evidence for this comes from a variety of observations as well as direct experimentation. In humans, we show that the minimal HBsAg knockdown by ARC-520 in poor responders is not due to an inherent inefficiency of the RNAi machinery, as knockdown of HBcrAg in these patients was similar in magnitude to HBsAg knockdown in NUC-naïve HBeAg positive patients. HBcrAg transcripts can only be produced from HBV DNA in which the precore/core promoters are upstream and adjacent to the coding region. This is the spatial arrangement found in cccDNA, but not in integrated HBV DNA, hinting that HBcrAg and HBsAg may come from different HBV DNA sources. HBeAg data in NUC-treated and NUC-naïve HBeAg positive patients was consistent with this conclusion. Next, the availability of chronically infected chimpanzees, which also showed a differential pattern of response in HBeAg negative versus positive serotypes, allowed us to perform a series of investigations to more directly examine the molecular basis for this phenomenon.
First, we showed that total liver HBV DNA was present at a lower level in HBeAg negative compared to HBeAg positive chimpanzees, and the level of HBV DNA was largely unchanged by NUCs. This suggests that the majority of the liver HBV DNA in these HBeAg negative chimpanzees was not dependent on active HBV replication but rather represented HBV DNA integrated into the host chromosome. Second, we demonstrated that the proportion of precore/pgRNA transcripts, expected to arise from cccDNA, to the total amount of HBV transcripts was minimal in HBeAg negative chimpanzees, suggesting that most transcripts were produced from non-cccDNA such as integrated HBV DNA in the chimpanzees with this serotype. These data were confirmed using mRNA-seq, which additionally revealed that HBV sequences terminated prior to the HBV PAS in the vast majority of transcripts in HBeAg negative animals. This points to transcription from integrated HBV DNA, which has 3′ sequence deletions that would include deletion of the HBV PAS, and is consistent with our mapping of chimpanzee/HBV breakpoints in integrated HBV DNA. Third, sequencing of full-length HBV transcripts from liver of an HBeAg negative chimpanzee revealed that most contained HBV sequences fused to host chimpanzee sequence, and that these fusion points fell between DR1-DR2, which would be expected if they were expressed from integrated HBV DNA. Deletion of HBV sequences in this region, which include the target sequences for the RNAi triggers in ARC-520, would explain the relative lack of knockdown of HBsAg observed. Finally, treatment with an RNAi trigger that targeted a sequence upstream of these deletions resulted in levels of HBsAg knockdown in HBeAg negative chimpanzees that were comparable to those in HBeAg positive chimpanzees. We interpret the data in toto to indicate that integrated HBV DNA is a substantial source of HBsAg in HBeAg negative chimpanzees, HBeAg negative patients, and NUC-experienced HBeAg positive patients with low cccDNA.
Despite the well-documented occurrence of HBV DNA integration, only minimal, if any, HBsAg was thought to be produced from integrated HBV DNA. However, integrated HBV DNA has been detected in HBsAg positive individuals with no detectable HBV serum DNA (24). Others previously described in histological liver samples from CHB patients that the hepatocytes that stained for HBsAg were distinct from those that stained for core (25), and HBsAg containing hepatocytes were often in clusters consistent with clonal populations (26). We also observed prominent HBsAg staining suggestive of clonal hepatocyte populations in an HBeAg negative chimpanzee and the HBeAg transitional chimpanzee, but not in the HBeAg positive chimpanzees. These data could be explained by significant HBsAg expression from integrated HBV DNA without actively transcribed cccDNA in the same hepatocytes.
Integrated HBV DNA as a source of HBsAg may also help explain why levels of HBsAg and serum HBV DNA correlate in untreated HBeAg positive chronically HBV infected individuals, particularly those with high HBV serum DNA, but correlate less well or not at all in HBeAg negative patients and in those on NUC therapy (27–30).
Our finding that integrated HBV DNA can serve as a source of HBsAg has implications for design of next-generation antivirals to treat chronic hepatitis B. For example, that HBsAg appears to be produced from integrated HBV DNA rather than solely from cccDNA implies that eliminating cccDNA will likely not eliminate HBsAg. Considering that a single virion has the potential to re-establish infection, immune control that prevents re-infection and allows clearance of the virus remains the desired endpoint. Loss of HBsAg is the more relevant biomarker of immune control. This is likely the case whether the HBsAg is produced from cccDNA or from integrated HBV DNA. In a virus as evolutionarily successful as HBV, the abundant production of HBsAg may serve a critical function in the maintenance of CHB.
The increased understanding of HBsAg dynamics during chronic infection gained by the current investigation will lead to enhanced opportunities to reduce HBsAg levels and potentially allow the host immune response to eliminate residual infected cells. Direct RNAi-mediated reduction of HBsAg in the liver has the potential to improve the prognosis for infected individuals, but should take into account both cccDNA and integrated HBV DNA.
MATERIALS AND METHODS
RNAi triggers and ARC-EX1
ARC-520 comprises API-520, the Active Pharmaceutical Ingredient consisting of two equimolar cholesterol-conjugated RNAi triggers named “siHBV-74” and “siHBV-77” in a liquid solution, plus the novel delivery excipient ARC-EX1 containing hepatocyte-targeted N-acetylgalactosamine-conjugated melittin-like peptide in a powdered form (21). RNAi trigger siHBV-75 was previously described (21).
Study Designs
Clinical trial Heparc-2001
The study aimed to determine the safety, tolerability, and pharmacological effect of ARC-520 in CHB patients with or without preceding chronic NUC treatment. Initially, patients receiving chronic ETV were enrolled sequentially into five cohorts, 8 subjects per cohort, and randomized with a 6:2 (active:placebo) ratio (table S1). Each subject was assigned to either active (ARC-520) or placebo (0.9% normal saline) treatment using a block randomization algorithm. After completion of cohort 4 and while cohort 5 was recruiting, the protocol was amended to add cohort 6 and subsequently cohort 7 in open label fashion without placebo groups. Patient selection and additional study details are in supplementary materials.
Blood samples were collected pre-dose and at specified times after dosing to determine safety, pharmacokinetic (PK, cohorts 1–5), pharmacodynamic (PD) (cytokines and complement) and HBV serology parameters. Patients were pre-treated with antihistamine (chlorpheniramine, 8 mg p.o.) 2 hours prior to treatment administration and continued (cohorts 1–6) or started (cohort 7) daily oral ETV on Day 1 (0.5 mg/day p.o.), continuing throughout the study.
ARC-520, supplied by Arrowhead Pharmaceuticals, Inc. (Pasadena, California, USA) was administered as a single intravenous infusion at ascending doses of 1, 2, 3 and 4 mg/kg administered intravenously by clinical staff at the infusion rate of 10 mg/min.
The study was performed in accordance with the 2008 Declaration of Helsinki and good clinical practice guidelines and was approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (Queen Mary Hospital, Hong Kong) and the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee (Prince of Wales Hospital, Hong Kong). All subjects gave written informed consent before screening.
Chimpanzee study
This study aimed to determine the safety, tolerability, and pharmacological effect of ARC-520 in nine available HBsAg-positive CHB chimpanzees (Pan troglodytes) as shown in table S4. Chimpanzees were treated with daily oral NUC for a lead-in period and then in conjunction with ARC-520 dosing (table S4). All chimpanzees were given oral ETV, 0.5 mg daily for lamivudine-naïve chimpanzees, and 1.0 mg daily for chimpanzees with prior exposure to lamivudine. Beginning on study day 108 and continuing through the ARC-520 dosing period, chimpanzee 4x0139 was dosed in addition to ETV with daily tenofovir (300 mg pediatric formulation) to further reduce viral load in this high replicating chimpanzee (31). Following NUC lead-in, chimpanzees were dosed with ARC-520 Q4W for 6 to 11 doses. 5–10 minutes prior to ARC-520 dosing at 4 mg/kg, chimpanzees were given a 50 mg intravenous bolus of diphenhydramine as a precautionary measure to avoid infusion reaction. Dosing with siHBV-75 + ARC-EX1 was performed identically to dosing with ARC-520. Blood samples were collected pre-dose and at specified times after dosing to determine clinical chemistry, hematology and HBV serology parameters (supplementary materials). Animal experimentation was conducted according to the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committees of the Southwest National Primate Research Center at the Texas Biomedical Research Institute and the New Iberia Research Center at the University of Louisiana at Lafayette.
Statistical Analyses
Data are means ± SEM. Statistical analyses were performed using Microsoft Excel 2013 and GraphPad Prism software version 6.0.
Supplementary Material
Fig. S1. HBV sequence elements near DR1 and DR2.
Fig. S2: Serum HBsAg, HBeAg and HBcrAg reduction in human patients treated with two doses of ARC-520 two weeks apart.
Fig. S3. Reduction of HBV serum DNA in chronically HBV infected patients treated with a single dose of ARC-520 on a background of NUCs.
Fig. S4. Quantitation of liver HBV DNA in chimpanzees.
Fig. S5. Mapping of HBV DNA integration sites.
Fig. S6. Massive paired-end sequencing of chimpanzee liver mRNA and alignment to HBV reference genome.
Fig S7. Immunohistochemical staining of HBsAg in livers of HBeAg positive and HBeAg negative chimpanzees.
Table S1. Individual CHB patient demographics at screening in clinical trial Heparc-2001
Table S2. Mean CHB patient demographics at screening in clinical trial Heparc-2001
Table S3: Treatment emergent adverse events in cohorts 1–7 of the Heparc-2001 study
Table S4. Characteristics of chronically HBV-infected chimpanzees and treatment regimens.
Table S5. mRNA-seq data obtained from liver biopsies of chimpanzees chronically infected with HBV.
Table S6. Polyadenylation signals in HBV-chimpanzee fusion transcripts of HBeAg negative chimpanzee 88A010.
ONE SENTENCE SUMMARY.
The hepatitis B virus surface antigen, a key protein in maintaining chronic infection, is expressed from viral DNA integrated into the host chromosome.
Acknowledgments
The authors acknowledge Christopher Anzalone for conceiving of the chimpanzee study. We thank Kevin Liu, Ringo Wu, Grace Wong, Vincent Wong and Diamond Martin for assistance with clinical trial conduct; Jason Goetzmann, Kathleen Brasky and Dana Hasselschwert for chimpanzee trial conduct; Bernadette Guerra, Helen Lee, Lena Notvall-Elkey and Courtney Johnson for chimpanzee sample analysis; Julia Hegge and Jason Klein for supervision of chimpanzee dosing; Margaret Littlejohn for chimpanzee HBV sequence analysis; the University of Wisconsin Biotechnology Center DNA Sequencing Facility for targeted DNA sequencing service, the Great Lakes Genomics Center of the University of Wisconsin at Milwaukee for IsoSeq library preparation and sequencing, Amit Sinha and the Basepair LLC for customized sequence data analysis; Kathy Jackson, Renae Walsh and Ryan Peterson for additional data analysis.
Funding
The Southwest National Primate Research Center resources are supported by NIH grant P51-OD011133 from the Office of Research Infrastructure Programs/Office of the Director.
Footnotes
Author contributions
C.I.W. and D.L.L. designed and oversaw the chimpanzee study, analyzed data and wrote the initial manuscript; M.F.Y. and H.Y.C. performed the clinical trial, analyzed clinical data and reviewed manuscript; R.G.G., S.A.L., C.L.L., C.F. and J.Y.N.L. oversaw the clinical trial, analyzed clinical and chimpanzee data and reviewed manuscript; B.D.G., J.H. and T.S. initially designed the clinical trial, analyzed clinical and chimpanzee data and reviewed manuscript; Z.X. performed chimpanzee deep sequencing analysis and assisted in drafting manuscript, R.E.L. oversaw the chimpanzee study, analyzed data and reviewed manuscript; D.C. conducted and oversaw chimpanzee sample assays; S.B.K. analyzed data and reviewed manuscript.
Competing interests
C.I.W., D.L.L, Z.X., B.D.G., T.S., J.H., and S.B.K. are or were employees of Arrowhead Pharmaceuticals. R.G.G., S.A.L., C.L.L., C.F. and J.Y.N.L. receive compensation from Arrowhead. M.F.Y., H.Y.C., R.E.L. and D. C. performed work under contract with Arrowhead.
SUPPLEMENTARY MATERIALS
Supplemental Materials and Methods
References in supplementary material (5, 7, 9, 21, 32–36)
REFERENCES AND NOTES
- 1.G.B.D. Mortality. C. Causes of Death, Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. doi: 10.1016/S0140-6736(14)61682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuen MF, Ahn SH, Chen DS, Chen PJ, Dusheiko GM, Hou JL, Maddrey WC, Mizokami M, Seto WK, Zoulim F, Lai CL. Chronic Hepatitis B Virus Infection: Disease Revisit and Management Recommendations. J Clin Gastroenterol. 2016;50:286–294. doi: 10.1097/MCG.0000000000000478. [DOI] [PubMed] [Google Scholar]
- 3.Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2:1129–1133. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- 4.Bock CT, Schwinn S, Locarnini S, Fyfe J, Manns MP, Trautwein C, Zentgraf H. Structural organization of the hepatitis B virus minichromosome. J Mol Biol. 2001;307:183–196. doi: 10.1006/jmbi.2000.4481. [DOI] [PubMed] [Google Scholar]
- 5.Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, Diao J, Yeung S, Jubb A, Ye W, Wu TD, Kapadia SB, de Sauvage FJ, Gentleman RC, Stern HM, Seshagiri S, Pant KP, Modrusan Z, Ballinger DG, Zhang Z. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012;22:593–601. doi: 10.1101/gr.133926.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schluter V, Meyer M, Hofschneider PH, Koshy R, Caselmann WH. Integrated hepatitis B virus X and 3′ truncated preS/S sequences derived from human hepatomas encode functionally active transactivators. Oncogene. 1994;9:3335–3344. [PubMed] [Google Scholar]
- 7.Fowler MJ, Greenfield C, Chu CM, Karayiannis P, Dunk A, Lok AS, Lai CL, Yeoh EK, Monjardino JP, Wankya BM, et al. Integration of HBV-DNA may not be a prerequisite for the maintenance of the state of malignant transformation. An analysis of 110 liver biopsies. J Hepatol. 1986;2:218–229. doi: 10.1016/s0168-8278(86)80080-0. [DOI] [PubMed] [Google Scholar]
- 8.Wong DK, Yuen MF, Yuan H, Sum SS, Hui CK, Hall J, Lai CL. Quantitation of covalently closed circular hepatitis B virus DNA in chronic hepatitis B patients. Hepatology. 2004;40:727–737. doi: 10.1002/hep.20353. [DOI] [PubMed] [Google Scholar]
- 9.Staprans S, Loeb DD, Ganem D. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J Virol. 1991;65:1255–1262. doi: 10.1128/jvi.65.3.1255-1262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kakimi K, Isogawa M, Chung J, Sette A, Chisari FV. Immunogenicity and tolerogenicity of hepatitis B virus structural and nonstructural proteins: implications for immunotherapy of persistent viral infections. J Virol. 2002;76:8609–8620. doi: 10.1128/JVI.76.17.8609-8620.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen MT, Billaud JN, Sallberg M, Guidotti LG, Chisari FV, Jones J, Hughes J, Milich DR. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci U S A. 2004;101:14913–14918. doi: 10.1073/pnas.0406282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milich DR, Jones JE, Hughes JL, Price J, Raney AK, McLachlan A. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc Natl Acad Sci U S A. 1990;87:6599–6603. doi: 10.1073/pnas.87.17.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seeger C, Zoulim F, Mason WS. In: Fields Virology. Fields BN, Knipe DM, Howley PM, editors. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia: 2007. chap. 76. [Google Scholar]
- 14.Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, Laccabue D, Zerbini A, Cavalli A, Missale G, Bertoletti A, Ferrari C. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–4225. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol. 2005;79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut. 2012;61:1754–1764. doi: 10.1136/gutjnl-2011-301073. [DOI] [PubMed] [Google Scholar]
- 17.Kim GA, Lim YS, An J, Lee D, Shim JH, Kim KM, Lee HC, Chung YH, Lee YS, Suh DJ. HBsAg seroclearance after nucleoside analogue therapy in patients with chronic hepatitis B: clinical outcomes and durability. Gut. 2014;63:1325–1332. doi: 10.1136/gutjnl-2013-305517. [DOI] [PubMed] [Google Scholar]
- 18.Yuen MF, Wong DK, Fung J, Ip P, But D, Hung I, Lau K, Yuen JC, Lai CL. HBsAg Seroclearance in chronic hepatitis B in Asian patients: replicative level and risk of hepatocellular carcinoma. Gastroenterology. 2008;135:1192–1199. doi: 10.1053/j.gastro.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Chan HL, Wong VW, Wong GL, Tse CH, Chan HY, Sung JJ. A longitudinal study on the natural history of serum hepatitis B surface antigen changes in chronic hepatitis B. Hepatology. 2010;52:1232–1241. doi: 10.1002/hep.23803. [DOI] [PubMed] [Google Scholar]
- 20.Schluep T, Lickliter J, Hamilton J, Lewis DL, Lai CL, Lau JY, Locarnini SA, Gish RG, Given BD. Safety, Tolerability, and Pharmacokinetics of ARC-520 Injection, an RNA Interference-Based Therapeutic for the Treatment of Chronic Hepatitis B Virus Infection, in Healthy Volunteers. Clin Pharmacol Drug Dev. 2016 doi: 10.1002/cpdd.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, Hegge JO, Klein JJ, Wakefield DH, Oropeza CE, Deckert J, Roehl I, Jahn-Hofmann K, Hadwiger P, Vornlocher HP, McLachlan A, Lewis DL. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21:973–985. doi: 10.1038/mt.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mason WS, Low HC, Xu C, Aldrich CE, Scougall CA, Grosse A, Clouston A, Chavez D, Litwin S, Peri S, Jilbert AR, Lanford RE. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J Virol. 2009;83:8396–8408. doi: 10.1128/JVI.00700-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hadziyannis SJ, Lieberman HM, Karvountzis GG, Shafritz DA. Analysis of liver disease, nuclear HBcAg, viral replication, and hepatitis B virus DNA in liver and serum of HBeAg Vs. anti-HBe positive carriers of hepatitis B virus. Hepatology. 1983;3:656–662. doi: 10.1002/hep.1840030505. [DOI] [PubMed] [Google Scholar]
- 25.Naoumov NV, Portmann BC, Tedder RS, Ferns B, Eddleston AL, Alexander GJ, Williams R. Detection of hepatitis B virus antigens in liver tissue. A relation to viral replication and histology in chronic hepatitis B infection. Gastroenterology. 1990;99:1248–1253. doi: 10.1016/0016-5085(90)90811-e. [DOI] [PubMed] [Google Scholar]
- 26.Su IJ, Lai MY, Hsu HC, Chen DS, Yang PM, Chuang SM, Sung JL. Diverse virological, histopathological and prognostic implications of seroconversion from hepatitis B e antigen to anti-HBe in chronic hepatitis B virus infection. J Hepatol. 1986;3:182–189. doi: 10.1016/s0168-8278(86)80024-1. [DOI] [PubMed] [Google Scholar]
- 27.Chan HL, Wong VW, Tse AM, Tse CH, Chim AM, Chan HY, Wong GL, Sung JJ. Serum hepatitis B surface antigen quantitation can reflect hepatitis B virus in the liver and predict treatment response. Clin Gastroenterol Hepatol. 2007;5:1462–1468. doi: 10.1016/j.cgh.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Thompson AJ, Nguyen T, Iser D, Ayres A, Jackson K, Littlejohn M, Slavin J, Bowden S, Gane EJ, Abbott W, Lau GK, Lewin SR, Visvanathan K, Desmond PV, Locarnini SA. Serum hepatitis B surface antigen and hepatitis B e antigen titers: disease phase influences correlation with viral load and intrahepatic hepatitis B virus markers. Hepatology. 2010;51:1933–1944. doi: 10.1002/hep.23571. [DOI] [PubMed] [Google Scholar]
- 29.Karra VK, Chowdhury SJ, Ruttala R, Polipalli SK, Kar P. Clinical Significance of Quantitative HBsAg Titres and its Correlation With HBV DNA Levels in the Natural History of Hepatitis B Virus Infection. J Clin Exp Hepatol. 2016;6:209–215. doi: 10.1016/j.jceh.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai CL, Wong D, Ip P, Kopaniszen M, Seto WK, Fung J, Huang FY, Lee B, Cullaro G, Chong CK, Wu R, Cheng C, Yuen J, Ngai V, Yuen MF. Reduction of covalently closed circular DNA with long-term nucleos(t)ide analogue treatment in chronic hepatitis B. J Hepatol. 2016 doi: 10.1016/j.jhep.2016.08.022. [DOI] [PubMed] [Google Scholar]
- 31.Petersen J, Ratziu V, Buti M, Janssen HL, Brown A, Lampertico P, Schollmeyer J, Zoulim F, Wedemeyer H, Sterneck M, Berg T, Sarrazin C, Lutgehetmann M, Buggisch P. Entecavir plus tenofovir combination as rescue therapy in pre-treated chronic hepatitis B patients: an international multicenter cohort study. J Hepatol. 2012;56:520–526. doi: 10.1016/j.jhep.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 32.Tu T, Mason WS, Clouston AD, Shackel NA, McCaughan GW, Yeh MM, Schiff ER, Ruszkiewicz AR, Chen JW, Harley HA, Stroeher UH, Jilbert AR. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J Viral Hepat. 2015;22:737–753. doi: 10.1111/jvh.12380. [DOI] [PubMed] [Google Scholar]
- 33.Hilger C, Velhagen I, Zentgraf H, Schroder CH. Diversity of hepatitis B virus X gene-related transcripts in hepatocellular carcinoma: a novel polyadenylation site on viral DNA. J Virol. 1991;65:4284–4291. doi: 10.1128/jvi.65.8.4284-4291.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Bommel F, Bartens A, Mysickova A, Hofmann J, Kruger DH, Berg T, Edelmann A. Serum hepatitis B virus RNA levels as an early predictor of hepatitis B envelope antigen seroconversion during treatment with polymerase inhibitors. Hepatology. 2015;61:66–76. doi: 10.1002/hep.27381. [DOI] [PubMed] [Google Scholar]
- 35.Su Q, Wang SF, Chang TE, Breitkreutz R, Hennig H, Takegoshi K, Edler L, Schroder CH. Circulating hepatitis B virus nucleic acids in chronic infection : representation of differently polyadenylated viral transcripts during progression to nonreplicative stages. Clin Cancer Res. 2001;7:2005–2015. [PubMed] [Google Scholar]
- 36.Sommer G, Heise T. Posttranscriptional control of HBV gene expression. Front Biosci. 2008;13:5533–5547. doi: 10.2741/3097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. HBV sequence elements near DR1 and DR2.
Fig. S2: Serum HBsAg, HBeAg and HBcrAg reduction in human patients treated with two doses of ARC-520 two weeks apart.
Fig. S3. Reduction of HBV serum DNA in chronically HBV infected patients treated with a single dose of ARC-520 on a background of NUCs.
Fig. S4. Quantitation of liver HBV DNA in chimpanzees.
Fig. S5. Mapping of HBV DNA integration sites.
Fig. S6. Massive paired-end sequencing of chimpanzee liver mRNA and alignment to HBV reference genome.
Fig S7. Immunohistochemical staining of HBsAg in livers of HBeAg positive and HBeAg negative chimpanzees.
Table S1. Individual CHB patient demographics at screening in clinical trial Heparc-2001
Table S2. Mean CHB patient demographics at screening in clinical trial Heparc-2001
Table S3: Treatment emergent adverse events in cohorts 1–7 of the Heparc-2001 study
Table S4. Characteristics of chronically HBV-infected chimpanzees and treatment regimens.
Table S5. mRNA-seq data obtained from liver biopsies of chimpanzees chronically infected with HBV.
Table S6. Polyadenylation signals in HBV-chimpanzee fusion transcripts of HBeAg negative chimpanzee 88A010.