

Abstract
Colibactin is a genotoxic hybrid nonribosomal peptide/polyketide secondary metabolite produced by various pathogenic and probiotic bacteria residing in the human gut. The presence of colibactin metabolites has been correlated to colorectal cancer formation in several studies. The specific function of many gene products in the colibactin gene cluster can be predicted. However, the role of ClbQ, a type II editing thioesterase, has not been established. The importance of ClbQ has been demonstrated by genetic deletions that abolish colibactin cytotoxic activity, and recent studies suggest an atypical role in releasing pathway intermediates from the assembly line. Here we report the 2.0 Å crystal structure and biochemical characterization of ClbQ. Our data reveal that ClbQ exhibits greater catalytic efficiency toward acyl-thioester substrates as compared to precolibactin intermediates and does not discriminate among carrier proteins. Cyclized pyridone-containing colibactins, which are off-pathway derivatives, are not viable substrates for ClbQ, while linear precursors are, supporting a role of ClbQ in facilitating the promiscuous off-loading of premature precolibactin metabolites and novel insights into colibactin biosynthesis.
Graphical Abstract
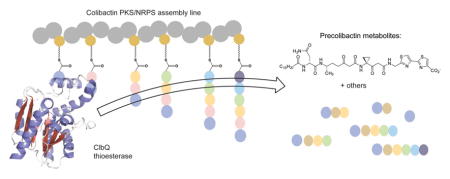
The intestinal microbiota has rapidly emerged as a key modulator of human health and disease.1–5 Microbial-produced natural products likely play a significant role in host/ microbe and microbe/microbe interactions. Colibactin is established as a genotoxic natural product produced by certain strains of gut microbiome-associated bacteria possessing the clb (pks) genomic island.6–8 Paradoxically, the clb cluster is found in the probiotic Nissle 1917, which is used in Europe for the treatment of gastrointestinal disorders.9,10 The gene cluster encodes a hybrid nonribosomal peptide synthetase (NRPS)/ polyketide synthase (PKS) pathway along with accessory proteins (Figure 1 and Figure S1).6,11,12 Modular NRPSs and PKSs are multidomain enzymes responsible for the generation of a wide range of structurally diverse natural products.13–15 Common to both systems, pathway intermediates are covalently linked to the synthases or synthetases via a thioester bond to a 4'-phosphopantethiene (Ppant) cofactor of the carrier protein domains (acyl carrier protein, ACP, or peptidyl carrier protein, PCP), facilitating the directed transport of the growing chain through the assembly line.
Figure 1.

Biosynthetic pathways to precolibactins. Compounds 1–13 (complete structures are shown in Figure S1) are proposed to be generated by the NRPS/PKS hybrid proteins ClbNBCHIJ. The extender unit 2-aminomalonyl (AM, highlighted in red) is produced by ClbDEF together with the first A domain of ClbH. The absence of ClbQ (in a ΔclbP ΔclbQ mutant of clb+ E. coli), allowed identification of 2-AM containing compounds 14–16, via pathway A. The presence of ClbQ (in a ΔclbP background) directs to pathway B, compounds 17–19. A, adenylation; ACP, acyl carrier protein; AT, acyl transferase; C condensation; Cy, cyclization; DH, dehydratase; E, epimerase; KS, keto synthase; Ox, oxidase; PCP, peptidyl carrier protein.
The tumor promoting activity of colibactin correlates with DNA damage induced by clb+ bacteria.6–8,16 The biosynthesis of colibactins follows a prodrug-type pathway where inactive precolibactins are processed by the periplasmic peptidase ClbP.17–20 ClbP cleaves precolibactins to release N-acyl-D-Asn along with active genotoxic colibactin products.17,19 Although the structures of mature and processed colibactin(s) have not been completely defined, various prodrug metabolites have been characterized from ΔclbP strains or ΔclbPQ strains of Escherichia coli, revealing a mixture of precolibactins and, consequently, new theoretical intermediates, with varying structural complexity (1–19, Figure 1, Figure S1).21–27 Insights gained from these isolation studies, along with gene deletion experiments,6,18,28 suggest that the varied biological activities (pathogenic and probiotic) reported for colibactin might result from a mixture of compounds produced through the hybrid NRPS-PKS pathway. Therefore, understanding the off-loading mechanism of such biosynthetic intermediates is essential to gain insights into the biosynthesis and biological activities of pathway metabolites. In NRPS/PKS pathways, the off-loading of biosynthetic products from the assembly line is commonly mediated by a thioesterase (TE) domain encoded in the pathway.29 However, the colibactin pathway unusually does not contain a terminal releasing domain as part of the assembly line. The candidate TE in the pks gene cluster is the standalone gene product ClbQ.
Sequence homology analysis suggests that ClbQ belongs to the type II family of editing TEs (TEIIs).23 TEIIs have a corrective (editing or proofreading) role, removing undesirable substrates and aberrantly loaded intermediates that would otherwise stall the NRPS/PKS biosynthetic pathway.30–33 In addition to an editing role, TEIIs have also been shown to participate in the selection of starter units incorporated into a pathway,34–36 release intermediates or final products,37,38 regulate the yield of products,39 and influence the overall performance of the synthases or synthetases.30,34 Two models have been reported to account for the activity of TEIIs, a high specificity model where TEIIs hydrolyze only aberrant units and a low specificity model where the hydrolases act on both “correct” and “incorrect” intermediates with the “correct” released at a slower rate.40 Deletion of TEIIs in NRPS/PKS biosynthetic pathways commonly result in a decrease in the yield of the final natural product.30,38,39,41 Although a large number of TEIIs have been identified, four TEIIs have been structurally characterized: RifR from the rifamycin hybrid NRPS/PKS pathway,36 RedJ from the prodiginine hybrid NRPS/PKS,39 SrfD from the surfactin NRPS,42 and the human TEII involved in fatty acid synthesis.43 Collectively these structures have revealed that unique structural conformations of TEIIs enable selective interactions with individual enzyme domains within the multidomain systems. Also, a flexible lid region in each structure controls the size and shape of the active site, regulating the enzyme-based selectivity for various substrates. Therefore, determining the substrate selectivity based on sequence analysis alone is not yet feasible. Additional structural information will more broadly aid in defining the selectivities and roles of TEs in a specific NRPS/PKS pathway.
Prior studies have demonstrated the importance of clbQ in the colibactin pathway. Deletion of clbQ in clb+ E. coli was shown to abolish the cytotoxic effects of clb+ E. coli.6,16,44 In addition, a recent report has suggested a unique function of ClbQ in off-loading pathway intermediates rather than removing aberrant intermediates.23 Characterization of a clbPQ double mutant allowed the identification of the late-stage precolibactin candidate 16, incorporating a 2-aminomalonyl unit (AM) (pathway A, Figure 1), whereas a clbP single mutant provided compound 19 (pathway B, Figure 1).23 In the same report, in vitro studies of ClbQ against N-acetylcysteamine (SNAC) precolibactin thioester derivatives revealed ClbQ off-loading the early stage clb pathway intermediates. Together, these studies suggest that ClbQ plays a key role in directing or controlling the flux of colibactin products and thus may affect the generation of bacterial molecules implicated in host genotoxicity. However, the structural and molecular basis of the unique TE activity of ClbQ remained unclear.
Here we report structural and biochemical characterization of ClbQ. Our data highlight unique structural features of ClbQ and provide insights into the substrate selectivity of ClbQ for several key precolibactin metabolites.
RESULTS AND DISCUSSION
X-ray Structure of ClbQ
ClbQ crystallized in the space group P21 with cell dimensions of a = 45.0 Å, b = 95.0 Å, c = 68.4 Å, β = 109.2°. The overall structure contains two independent monomers in the asymmetric unit (Figure 2A) and was isotropically refined to a resolution of 1.98 Å with a final a Rfree of 21% (Table S1). Native ClbQ crystals exhibited severe twinning (twin law = h, –k, –h – l) allowing an incomplete structure with electron density missing for amino acid residues 110–148 and 183–200, this including the active site aspartate, D186 (data not shown). Cocrystallization with synthetic precolibactin substrates produced several crystals with significantly less twinning, suggesting that ClbQ is more structured in the presence of ligands. Several data sets were collected (data not shown) to obtain protein–ligand complexes from crystals soaked or cocrystallized with precolibactin derivatives (9, 18, 19, 20, 26, 27). Data for precolibactin–ClbQ complexes were collected to high resolution, but no clear density for the bound ligand was apparent. However, the density for the disordered loops and missing regions from the native crystals was observed in the cocrystals, allowing a complete model (residues 2–240) of ClbQ, suggesting that transient ligand binding aids in crystal growth and formation.
Figure 2.

Structure of ClbQ. (A) The asymmetric unit with two monomers of ClbQ, βME is shown as orange sticks. (B) The active site of ClbQ. The catalytic triad, Ser78, Asp186, and His215, are shown as gold sticks, the oxyanion hole (Ser12 and Leu79) and surrounding active site residues are colored salmon. (C) Superimposition of the chain A and chain B monomers. The core regions of both domains (cyan) is constant, and the flexible lid domains are represented (chain A, green; chain B, orange). Side chains of the residues on the flexible lid domain are labeled in the expanded view and the movement and orientations between the two monomers are depicted with blue arrows. The catalytic triad is shown as pink sticks.
The ClbQ structure reveals, as expected, a canonical α/β hydrolase core domain (Ser2–Pro110 and Ile165–His240) and a cap domain (Asp111–Thr164) (Figure 2A). Five α helices of the core domain surround a six-stranded parallel β-sheet, and a flexible cap domain is composed of helices α'4–α'5, lid loop 1, and lid loop 2 inserted between β4 and β5 of the core. The active site of ClbQ shows a classic α/β hydrolase signature sequence Gly76-His77-Ser78-Xaa79-Gly80 of the “nucleophilic elbow” between strands β4 and the α3 helix. The catalytic triad residues of ClbQ, Ser78, Asp186, and His215 (Figure 2B), are on loop regions, following strands β3 (Ser), β5 (Asp), and β6 (His) of the α/β-hydrolase fold. The catalytic Ser78 in both monomers of ClbQ is a conformational outlier based on Ramachandran analysis, as commonly observed in the structures of α/β hydrolase fold enzymes.45 In addition, several significant changes in the residues adjacent to the catalytic site as well as hydrogen bond positioning of residues in the catalytic region were observed in the ClbQ structure. His77 of the signature motif forms a hydrogen bond with the backbone carbonyl of the catalytic His215, stabilizing its alignment within the triad (Figure 2B). The variable amino acid Xaa79 of the signature sequence is a methionine in most TEIIs. However, in ClbQ, it is a leucine. The backbone amides of Leu79 and Ser12 (Met95 and Ala29, respectively, in RifR)36 are reported to be essential in stabilizing the tetrahedral intermediate in the oxyanion hole, which is occupied by a water molecule in our structure. The catalytic histidine (His215 in ClbQ) in most TEIIs is present in the motif GXHF (where X= G, N, or D). In ClbQ, a significant change is observed in this signature motif containing AADHF. An additional alanine residue results in a longer loop region (indicated by purple arrow in the Figure 3A) presumably contributing to greater flexibility of the catalytic histidine in the active site.
Figure 3.

Conformational flexibility of ClbQ: (A) Alignment of ClbQ (chain A, cyan) with RedJ (salmon, PDB code 3QMV, chain B) and RifR (gray, PDB code 3FLB, chain A). The extended loop region in ClbQ formed by the motif PAADH is indicated (purple arrow). The catalytic triad is labeled. (B) Modeling of a Ppant arm (green) into ClbQ active site based on the EntF PCP (light pink)–TE (wheat) didomain structure (PDB code 3TEJ) with ClbQ (chain A, blue/cyan). (C) Surface diagram of the two monomers of ClbQ. The substrate binding site is indicated by a red circle, and Ppant entrance is indicated by red arrow. (D) Cartoon and surface representative of the active site entrance channel. The narrow tunnel-like entrance channel of chain A (circled and open) and chain B (closed, largely by His115 and Glu116).
We also observed clear electron density for a β-mercaptoethanol (βME) molecule forming a disulfide bond with Cys49 of each monomer. This βME originates from the protein purification process during the protein dialysis step. The βME molecule attached to Cys49 of chain B is located at the dimer interface and makes extensive contacts with chain A (Figure S2A), while the βME molecule attached to chain A points away from chain B (Figure S2B). As compared with other TEIIs, significant changes around the Cys49 region were observed in ClbQ. Additional differences between ClbQ and TEIIs include position 157. While this position is conserved as Ala or Ser in other TEIIs, it is an Asn in ClbQ with the side chain interacting with the backbone amide of Cys49, and in turn, the amide NH forms a hydrogen bond with the carbonyl of Leu50. This arrangement looks to stabilize the structural conformation of the loop region between β2 and α2 (Figure S2B). A cysteine at position 49 is not present in classic TEII enzymes, and structural changes around this region are unique to ClbQ.
A structural homology search using the DALI server46 reveals that ClbQ shares the highest structural similarity (Z-score of 29.9) with RedJ (PDB code 3QMV). The RMSD is 1.7 Å for 227 aligned αC atoms between the N-terminal domain of RedJ and ClbQ. ClbQ also shares high structural similarity with RifR (Z-score of 27.0, PDB code 3FLB, RMSD of 2.3 Å for 223 aligned αC atoms). Interestingly, ClbQ also shares significant structural homology to the recently published crystal structures of human TEII (Z score of 22.4, PDB code 4XJV) (Figure S3). ClbQ has equal similarities with RedJ and RifR in the core region; however, the flexible lid region of chain A in ClbQ is more similar to RedJ than RifR (Figure 3A).
Conformational Changes of ClbQ
The two monomers in the asymmetric unit are highly similar in structure (RMSDall3atoms = 1.5 Å), with observed conformational changes around the cap domain (RMSD111–164 = 2.9 Å) (Figures 2C, S4), suggesting flexibility of the lid region. Similar to other TEIIs, lid loop 1 is associated with the size and shape of the substrate chamber, whereas lid loop 2 contributes to the Ppant entrance tunnel. In chain A, lid loop 1 is positioned toward the active site, and lid loop 2 is in an open conformation with respect to the chain B structure, where lid loop 1 is positioned away from the active site and lid loop 2 is in a closed conformation (Figure 2C).
In order to model a structural basis for a ClbQ–carrier protein domain interaction, the structure of EntF PCP-TE (PDB code 3TEJ) was aligned to ClbQ providing a proposed protein/protein interaction surface along with a basis for the positioning of the Ppant arm into the TE active site (Figure 3B). We previously reported the structure of the PCP-TE didomain from the E. coli EntF NRPS with a conjugated Ppant based inhibitor providing a structural basis for TE/PCP interactions.47 As common with carrier protein domains, the PCP is a four-helical domain with the conserved Ser covalently linked to the Ppant arm. Structural alignment suggests the position of Ser78 of ClbQ is in a catalytically relevant form and provides insights for the observed structural differences between the two monomers. The alignment and interaction of the two ClbQ monomers is similar to the interaction of the two domains in the EntF PCP-TE structure. Contact residues of ClbQ suggest that chain B acts as a PCP mimic to chain A, leading to the movement of flexible lid loop 2 to an open conformation. The backbone amides of residues Tyr20 and Lys21 of the α1 helix of chain A interact with the βME molecule attached to the Cys49 of chain B, while the residues of α'5 and loop region between β2 and α2 of chain A make contacts with the residues of α2 and α3 of chain B (Figure S2A).
Movement of lid loop 1 affects the size and shape of the substrate chamber in both monomers. The side chains of residues Ala113 and Glu116 of lid loop 1 are pointed inward to the active site, forming a narrow and longer substrate chamber in chain A. In contrast, these residues are in an opposite configuration forming a wider, short substrate chamber in chain B (Figure 2C and Figure 3C). The Ppant entrance site and general access to the catalytic triad were affected by the movement of flexible lid loop 2 (Figure 3D). The side chains of residues Val139 and Asp140 are pointed to the active site covering the Ppant entrance in chain B, whereas these residues are pointed outward opening the Ppant entrance and forming a narrow tunnel-like entrance in chain A. This active site entrance tunnel is blocked by the side chains of residues His115 and Gly116 in chain B (Figure 3D).
Biochemical Characterization of ClbQ
Previously reported in vitro studies on the substrate specificity of ClbQ against precolibactin N-acetyl cysteamine (SNAC) thioester derivatives (Figure S1) revealed that the TE readily hydrolyzed the early stage intermediate derivatives 21 and 22 to generate the corresponding precolibactins 2 and 3, respectively.23 However, hydrolysis of the late-stage, cyclized precolibactin derivatives 23, 24, or 25 was not observed. This study did not include linear late-stage intermediates, suggesting one or a combination of the following possibilities. First, the cyclized compounds (pyridone-containing) 23, 24, and 25 are not the actual substrates for ClbQ, and cyclization is an artifact of the in vitro isolation procedures. Second, ClbQ discriminates alternate substrates based on chain length, as has been observed for other TEIIs. Third, the substrate specificity is a result of ClbQ’s selectivity toward various ACP/PCP domains in the pathway. To address these possibilities, we conducted studies using both wild-type ClbQ and a S78A active site mutant, in which the catalytic serine residue was substituted with alanine.
Substrate Specificity of ClbQ
To test the first possibility, we synthesized new compounds 26 and 27 (SNAC derivatives of 18 and 19) as well as compound 20 (SNAC derivative of 1), as a positive control, and assayed their thioester hydrolysis activities in the presence of ClbQ (Figure 4). Using a LCMS based assay, we observed hydrolysis of compound 20 by wild-type ClbQ, but we did not observe hydrolysis of 26 or 27 (30 min incubation). Given the poor solubility of late stage intermediates in the assay buffers, we repeated the assays for 18 h and observed slow hydrolysis of linear substrate 26 but no hydrolysis of the cyclized substrate 27 (Figure 4). To confirm that thioester hydrolysis of 26 is specifically catalyzed by ClbQ, the enzymatic reaction was compared with an active site ClbQ mutant and a buffer control. LCMS analysis data showed that the linear substrate 26 was weakly hydrolyzed by wild-type ClbQ, and the hydrolysis product 18 cyclized over time to compound 19. Spontaneous cyclodehydration of 18 to 19 was experimentally demonstrated in earlier studies.48 Partial hydrolysis of 26 at a decreased rate was observed in the samples incubated with the ClbQ S78A mutant (approximately 2-fold less compared to WT), suggesting that the substrate 26 may be capable of binding in the active site. In place of an active site serine residue, bound water can act as a nucleophile, allowing the hydrolysis of 26 albeit at a slower rate (Figure 4). Similar observations have been reported for other TEII active-site serine mutants.36 The ClbQ S78A mutant showed only minor or no hydrolysis of SNAC derivatives 20, 26, and 27 (Figure 4). Together with previous studies, these results support ClbQ as a low specificity TEII enzyme with a stronger preference for early stage intermediates. In addition, our observations of ClbQ hydrolysis of late-stage linear precolibactin intermediates support our hypothesis that late-stage linear precolibactin intermediates and not the stable cyclized precolibactin intermediates are indeed colibactin biosynthetic intermediates.
Figure 4.

Hydrolysis of precolibactin thioester SNAC derivatives with ClbQ. (A) LC-MS based assay showing hydrolysis of a linear precolibactin SNAC thioester (26) catalyzed by WT ClbQ and partial thioester hydrolysis by the S78A mutant. (B) Extracted ion chromatograms (EIC) EIC+ = 467.30 ± 0.01 (top) and 398.33 ± 0.01 (bottom), corresponding to 26 and the hydrolyzed and cyclized product precolibactin 19, respectively. (C) LC-MS extracted ion chromatogram (EIC) traces displaying the hydrolysis of compound 20 catalyzed by recombinant ClbQ and not by ClbQ S78A (middle). EIC+ = 444.28 ± 0.01 and 343.28 ± 0.01, corresponding to SNAC thioester 20 and the hydrolyzed product precolibactin 1, respectively. (D) LC-MS extracted ion chromatogram (EIC) traces displaying no hydrolysis of cyclized precolibactin 27. EIC+ = 449.28 ± 0.01 [M + H]+2, corresponding to SNAC thioester 27.
Additionally, we tested the ability of ClbQ to hydrolyze common acyl-SNACs of varying chain lengths (acetyl, lauroyl, myristoyl) in order to understand whether ClbQ performs a generic editing function (common to TEIIs) or is specific to early stage precolibactin metabolites. Using the previously reported DTNB-based spectrophotometric assay,39 the activity of ClbQ against acetyl-, lauroyl-, and myristoyl-SNACs, as well as precolibactin SNACs 20, 26, and 27, were compared. The assays revealed that ClbQ has thioesterase activity with model straight-chain acyl-SNACs, and the rates of hydrolysis were greater than that measured for precolibactin-SNACs. However, the slow hydrolysis rate of lauryl-SNAC and precolibactin-SNAC thioesters 20 and 26 compared to acetyl-SNAC indicate that these are relatively poorer substrates for ClbQ. The catalytic efficiencies (kcat/KM) of acetyl-, lauroyl-, myristoyl-, 20-, and 26-SNACs are 27.3, 7.3, 38.8, 5.2, and 1.6 M−1 s−1, respectively. Additionally, we did not observe hydrolysis of cyclized precolibactin SNAC derivative 27 as anticipated from our LC-MS data and previous reports, suggesting that these compounds are not the optimal substrates.
Selectivity toward Acyl ACPs
ClbQ hydrolysis of specific precolibactin SNAC derivatives posed the question as to whether the thioesterase is selective toward carrier protein domains in the colibactin pathway. ClbQ shares high structural homology to RedJ, a type II TE that was shown to have specificity for ACPs in the prodiginine pathway.39 Therefore, we investigated the possibility of ClbQ discrimination between ACP and PCP domains. There are a total of 12 carrier protein domains (5-ACPs/7-PCPs) in the colibactin assembly line; all of these domains are embedded in multimodule enzymes, except for the stand alone, ClbE (Figure 1, S1). Our initial strategy was to utilize the standalone ClbE carrier protein in acyl-ACP assays. However, our efforts to load various acyl substrates on ClbE were undetected in our assays (data not shown) with ClbE only accepting acetyl-CoA and not lauroyl-or myristoyl-CoA using the Ppant transferase (PPTase), Sfp. This is an interesting observation given that ClbE has a unique role in the colibactin pathway acting as an in trans carrier protein to the first ClbH adenylation domain, which is further processed to a 2-aminomalonyl unit (AM) by accessory enzymes ClbDEF.24,28 Accepting only short-chain acyl-CoA substrates but not longer ones suggest that ClbE unusually displays strict specificity toward small amino acids like L-Ser. Mass analysis (MALDI-TOF) did show that ClbQ was able to hydrolyze acetyl-ClbE consistent with its editing role (Figure 5A).
Figure 5.

Acyl-ACP hydrolysis by ClbQ. (A) MALDI (matrix-assisted laser desorption/ionization) analysis of acetyl-ClbE hydrolysis by ClbQ. Mass spectrum of acetyl-ClbE (top panel), reaction with WT ClbQ (middle panel), and the ClbQ S78A mutant (bottom). The reference m/z peak at 11783.7 is shown as vertical dotted line. (B) LC-MS analysis of hydrolysis of acetyl-FscF (top), lauroyl-FscF (middle), and myristoyl-FscF (bottom) incubated with ClbQ at 37 °C for 15 min. Acylation reactions of FscF are shown as solid lines, and the hydrolysis reactions of acyl-FscF catalyzed by ClbQ are represented as dotted lines in all three spectra.
Unfortunately, attempts to solubly express the colibactin gene cluster specific ACP/PCPs of ClbB, ClbK, and ClbO as discrete, standalone domains were unsuccessful under the conditions of our experiments (data not shown). Given the difficulty in soluble expression of individual in cis ACP/PCPs from the colibactin gene cluster, we chose an alternative approach. We tested the ability of ClbQ to recognize and hydrolyze acyl-CoA tethered to a model standalone carrier domain, FscF, a well behaved carrier protein from the fuscachelin NRPS biosynthetic pathway.49 We tested the hydrolysis of various acyl-FscFs by ClbQ and observed all conjugates were substrates (Figure 5B). This suggests that ClbQ may lack selectivity toward specific colibactin synthase or synthetase carrier protein domains, implying that the differences in the hydrolysis of precolibactin SNAC derivatives is due primarily to the substrate chain-length specificity of ClbQ. To further substantiate these results, we conducted a sequence homology analysis of the 12 carrier protein domains in the colibactin pathway to examine differences in the regions that have previously been reported47 to be essential for TE binding (Figure S5). Consistent with our assay results, no significant differences between the ACP/PCP domains in the TE binding region were apparent. However, consistent with its specialized role, ClbE showed the most differences as compared to the other carrier protein domains.
We further carried out secondary structure predictions of all 12 carrier protein domains using JPred4.50 In general, PCP/ ACP domains adopt the expected four-helix structure, in which residues in α3 and α4 helices interact with partner domains.51 Our prediction showed that all carrier protein domains in the colibactin pathway similarly possess the canonical four-helix structure, except ClbE, in which α3 is interestingly replaced with a β strand (Figure S5). We speculate that this atypical change in overall predicted structural topology likely plays a critical role in ClbE’s unique activity with AM production.
The colibactin pathway is a unique PKS/NRPS assembly line with novel, noncanonical biosynthetic elements. Although a number of precolibactin metabolites have been reported, all these structures have been identified from either clbP mutant strains or clbPQ double mutant strains23 to facilitate isolation and identification of precolibactins. Two studies have reported that deletion of clbQ in E. coli CCR20 or E. coli 11G5 strains abolished colibactin cytopathogenic effects including in vitro megalocytosis, DNA damage, cellular senescence, and tumor growth in infected human HCT116 cells.16,44 Both of these studies suggested that colibactin biosynthesis and bioactivity are strongly influenced by the unique TE ClbQ. Additionally, a recent report demonstrated the effect of ClbQ on the production yields of upstream and downstream precolibactin intermediates.23,27 In one study, the ΔclbPQ strain produced higher yields of late-stage intermediates and aided in the isolation of the advanced metabolite 16.23 Taken together these studies suggest ClbQ plays an important regulatory role in colibactin production and cytotoxic activity.
Our structural and biochemical data revealed that ClbQ is a unique TEII enzyme. The crystal structure shows a characteristic α/β hydrolase fold and flexible lid region consistent with reported TEII structures. However, ClbQ displayed some unique structural modifications. The conformational changes around the flexible lid domain of the two monomers provide structural insights for the broad substrate specificity of the enzyme. Consistent with our biochemical studies, the crystal structure revealed alternate size and shape of the substrate binding pocket allowing the enzyme to accommodate a range of substrates. Also, consistent with observed colibactin product formation, ClbQ could possibly interact with all 12 carrier proteins of the assembly line given the relaxed specificity with a foreign carrier protein domain, FscF.
ClbQ is an atypical TEII enzyme, utilizing a low specificity mechanism in which the enzyme releases both “correct” and “incorrect” intermediates in the NRPS/PKS assembly line. In contrast to an earlier report where the late-stage cyclic (pyridone) precolibactin intermediate SNAC compounds were not hydrolyzed by ClbQ,23 we observed slow hydrolysis of the late-stage linear intermediate SNAC thioester 26. This suggests that the cyclized precolibactin derivatives are not the actual substrates of ClbQ but are rather off-path cyclization products. Synthetic colibactin lactams are potent DNA alkylators, whereas the colibactin pyridones were inactive.52 Cyclized colibactins are likely produced by a facile double cyclodehydration route during the fermentation and isolation process.25,48,52
Our in vitro studies suggest that ClbQ can off-load the AM unit from the assembly line, which is consistent with the observed accumulation of AM-containing compound 16 in a ΔclbPΔclbQ pks mutant (Figures S6, S7).23 In addition, the putative substrate binding pocket of ClbQ reveals a general negatively charged surface (Figure S8) similar to the previously reported ClbP structure,18 providing additional evidence that fully mature (pre)colibactins are positively charged due to the use of AM substrates. Confirming the structural basis of ClbQ for multiprotein interactions is our current focus. As a slow acting TEII enzyme, ClbQ was shown to process late-stage linear intermediates in the colibactin NRPS/PKS pathway, suggesting that increased production of 16 is in part dependent on the lifetime of a bound AM unit.
Presented is the structure and function of the atypical type II thioesterase ClbQ from the colibactin pathway providing novel insights into the colibactin biosynthetic pathway. The complex roles host–pathogen interactions play in the gut microbiome and their relation to human health are emerging. Detailed understanding of clb metabolites along with the biosynthetic machinery provides a framework to understand colibactin-mediated carcinogenesis and probiotic activity and to rationally manipulate the system using small molecule probes.
METHODS
Unless otherwise noted, all chemicals and general reagents were purchased from Sigma-Aldrich or Fisher Scientific.
Expression Plasmids
The genes encoding the proteins ClbQ, ClbE, ClbH, ClbD, and ClbF were amplified by PCR from a bacterial artificial chromosome (BAC) harboring the pks genomic DNA6 using the primers listed in Supplementary Table 1. Amplified fragments were digested with appropriate restriction enzymes and were purified using agarose gel electrophoresis. The digests were ligated into respective linearized expression vectors (pET28a or pET30b) using T4 DNA ligase and transformed into competent E. coli TOP10 cells. All plasmids were verified by DNA sequencing. A ClbQ S78A mutant was generated using the QuikChange site-directed mutagenesis kit (Stratagene). The Sfp containing plasmid was obtained using a previously reported protocol.53
Protein Expression and Purification
Unless otherwise stated, all the proteins were expressed and purified using the following protocol. Expression vectors were transformed into E. coli BL21(DE3), and bacteria were grown in 2 L of LB medium containing 50 μg mL−1 kanamycin at 37 °C and 155 rpm to an optical density of 0.5, equilibrated to 18 °C, and induced with 0.2 mM IPTG, and expression continued at 18 °C for ~18 h. Cells were harvested, suspended in 25 mL of lysis buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl), and lysed with a microfluidizer, and the lysate was clarified by centrifugation at 14000g for 40 min. The supernatant was incubated with 1 mL of Ni-NTA resin (Qiagen) for 45 min at 4 °C and washed with one column volume of lysis buffer and then one column volume of wash buffer (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 25 mM imidazole). The tagged protein was eluted with elution buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 250 mM imidazole) and dialyzed overnight in the buffer containing 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM βME (β-mercaptoethanol), and 10% glycerol. The protein was further purified by gel filtration chromatography (HiLoad 16/60 SuperDex-200 column, AKTA FPLC System, GE Healthcare) with 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM βME. The fractions containing protein, as detected by SDS-PAGE analysis, were pooled, concentrated, and used for crystallization experiments. FscF was also expressed and purified as described above except that it was induced at 37 °C with 0.25 mM IPTG and allowed to grow at 37 °C for ~5 h before harvesting and purification. Sfp was expressed and purified following the reported protocol.54 Proteins ClbH, ClbD, ClbF, and ClbE (C-terminal His tag) were expressed and purified according to the reported protocol.24
Crystallization
Purified ClbQ was concentrated to 5 mg mL−1 and incubated with 2 mM precolibactin SNAC (27) for 2 h at 37 °C. Cocrystallization trials were conducted using the hanging drop vapor diffusion method with 2 μL drops containing equal volumes of protein stock and reservoir solution (30% PEG-4000 and 0.1 M Tris-HCl, pH 8.5, 0.2 M lithium sulfate monohydrate) at 20 °C. Crystals formed in ~12 h and were harvested in loops, cryoprotected with corresponding well solution supplemented with 20% glycerol, and frozen in liquid nitrogen for data collection.
Data Collection and Structure Determination
Diffraction data were collected on beamline 21-ID-G of the Life Sciences Collaborative Access Team (LS-CAT) facility at the Advanced Photon Source (APS), Argonne National Laboratory (Argonne, IL). Data were collected at 100 K with a wavelength of 0.9786 Å (1 Å = 0.1 nm), integrated, merged, and scaled using XDS55 to a resolution of 1.98 Å in space group P1211, with two protein molecules per asymmetric unit. A sequence homology search indicated that the RifR (30% identity) and RedJ (32%) TEs had the highest sequence identity.36,39 We tested these α/β hydrolase domains (PDB codes 3FLB and 3QMV), along with the human TEII (4XJV; 25% identity)43 as molecular replacement search models with PHASER.56 Initial refinement of the diffraction data (ClbQ crystallized with 27) using RifR as the initial search model provided a partial structure of ClbQ containing residues 2–110, 150–182, and 204–240. Missing residues were built manually, and the final atomic model containing residues 2–240 was completed by several rebuilding and refinement cycles using PHENIX.REFINE57 and COOT.58 A standard distance restraint between SβME and SCys49 was added (2.05 Å) during the refinement. Water molecules were placed in the structure based on manual inspection of the 2mFo–DFc and mFo–DFc electron density maps, and the refined coordinates have been deposited in the PDB (accession code 5UGZ). Statistics on data collection and atomic structure refinement are given in Supplementary Table 2. PyMOL (https://www.pymol.org) was used for structural illustrations.
Acylation of Carrier Protein Domains
Sfp from Bacillus subtilis was used for the conversion of apo-ACP to corresponding holo-ACP (acyl-ACPs) using a previously reported procedure.53 Briefly, each reaction mixture of 100 μL contained 50 μM apo-ACP, 150 μM acyl-CoA, 2 μM Sfp in Tris HCl buffer, pH 7.5, 0.5 mM TCEP, and 2.5 mM MgCl2 and was incubated for 2 h at 37 °C. Acyl-ACP formation was confirmed by MALDI and ESI-LC-MS. The reaction mixtures were then filtered using Microcon YM-3 filter (Millipore) to remove excess acyl-CoA substrate and exchange the buffer to 100 mM Tris HCl, pH 7.5, 20 mM NaCl and concentrate the Acyl-ACP product to 90 μL.
Acyl-ACP Hydrolysis
An LC-MS and MALDI-TOF based assay was used to determine the hydrolysis of Acyl-ACP catalyzed by ClbQ. In a standard assay reaction, 10 μL of the concentrated Acyl-ACP product was incubated with 5 μL of 1 μM ClbQ or ClbQ S78A mutant or no enzyme for 15 min at 37 °C. Reactions were quenched with 10 μL of 10% formic acid, and the loss of acyl-ACP and formation of holo-ACP were analyzed by LC-MS. Samples for LC-MS analysis were injected via auto sampler, and mass analysis was performed using an Agilent 6130 quadruple LC-MS with an Agilent Zorbax SB-C18 1.8 μm (2.1 mm × 50 mm) column operated at a flow rate of 0.2 mL/min. The running method was 0–2 min (H2O/0.1% formic acid), 2–22 min (0–100% acetonitrile (ACN)/0.1% formic acid). Retention times were as follows: Holo-FscF (FscF-Ppant), 13.74 min; Acetyl-FscF, 13.84 min; Lauroyl-FscF, 14.36 min; Myristoyl-FscF, 14.41 min.
ClbE-Acylation and acyl-ClbE hydrolysis catalyzed by ClbQ and its mutant was confirmed by MALDI-TOF MS-based assay using benchtop MicroFlex LT mass spectrometer (Bruker Daltonics) following the previously reported method.43
AM-ClbE Hydrolysis Assays
An in vitro reconstitution of AM-ClbE was generated using a previously reported protocol.24 Each 40 μL of the reaction mixture containing seryl-ClbE or AM-ClbE was incubated with 1 μM of ClbQ or ClbQ S78A mutant or no enzyme for 15 min at 37 °C. Reactions were quenched with 10 μL of 10% formic acid, and the loss of acyl-ACP and formation of holo-ACP were analyzed by LC-MS. Samples for LC-MS analysis were injected via auto sampler, and mass analysis was performed using an Agilent 6130 quadruple LC-MS with an Agilent Zorbax SB-C18 1.8 μm (2.1 mm × 50 mm) column operated at a flow rate of 0.2 mL/min. The running method was 0–2 min (H2O/0.1% formic acid), 2–50 min (0–100% acetonitrile (ACN)/0.1% formic acid). Deconvolution of protein raw mass spectra was performed using Agilent MassHunter software (version B.06.00). The masses detected correspond to [M + H]+ ions.
Precolibactin SNAC Hydrolysis Assays
An LC-MS based assay was used to determine the TE activity of ClbQ toward precolibactin SNAC thioesters using a previously reported method.23,38 Each assay was carried out in triplicate. In a standard 100 μL assay reaction containing 50 mM Tris HCl, pH 8.0, 80 μM substrate (in DMSO) was incubated with 4 μM ClbQ or ClbQ S78A mutant or no enzyme at 37 °C for 30 min and 18 h. Each reaction mixture was processed and analyzed according to a previously reported method.23
Acyl-SNAC Hydrolysis versus Precolibactin SNAC Hydrolysis
The DTNB (5,5'-dithiobis (2- nitrobenzoic acid)-based continuous spectrophotometric assay was used to examine substrate specificity of ClbQ with various acyl-SNACs and precolibactin SNACs using a previously reported protocol.39 In brief, each experiment (in triplicate) was carried out at 37 °C in a 96-well flat bottom microtiter plate. Each reaction mixture containing varying concentrations of acyl-SNACs was added to 100 μM DTNB in reaction buffer (100 mM Tris HCl, pH 7.4; 20 mM NaCl) and incubated with 100 nM ClbQ or ClbQ S78A or no enzyme. All data points were collected in triplicate, and the amount of free thiol released is measured at 412 nM. Data analysis and calculation of kcat and KM values were performed using GraFit 4.012 (Middlesex, UK).
Supplementary Material
Acknowledgments
Financial support from the National Science Foundation (Grant 1411991 to S.D.B) and the National Institutes of Health (Grant R01GM110506 to S.B.H. and Grant 1DP2-CA 186575 to J.M.C) is gratefully acknowledged. We are grateful to the staff of LS-CAT 21-ID-G beamline at Argonne National Laboratory, IL, U.S.A., for assistance with X-ray data collection and processing. We also thank W. H. Chen for assistance in the crystal structure refinement. We thank E. Trautman for synthesizing N-myristoyl-D-Asn-SNAC.
ABBREVIATIONS
- NRPS
nonribosomal peptide synthetase
- PKS
polyketide synthase
- Ppant
phosphopantetheine
- PPtase
Ppant transferase
- PCP
peptidyl carrier protein
- ACP
acyl carrier protein
- TE
thioesterase
- TEII
thioesterase type II
- IPTG
isopropyl β-D-thiogalactopyranoside
- βME
β-mercaptoethanol
- SNAC
N-acetyl cysteamine
- BAC
bacterial artificial chromosome
- AM
2-amino malonyl
- MALDI-TOF
matrix-assisted laser desorption/ ionization-time of flight
- ESI
electrospray ionization
- LCMS
liquid chromatography-mass spectrometry
- DTNB
5,5'-dithiobis(2-nitro-benzoic acid)
- EIC
extracted-ion chromatogram
- TCEP
tris(2-carboxyethyl)phosphine
Footnotes
The authors declare no competing financial interest.
Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession number 5UGZ.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.7b00479.
Primers used for cloning, crystallographic data of ClbQ, colibactin biosynthetic pathway with reported precolibactin structures, interactions of bME with Cys49 of each monomer of ClbQ, structure-based sequence alignment of ClbQ, sausage representation of B-factors of the ClbQ, sequence alignment of carrier domains in the colibactin biosynthetic pathway, proposed role of ClbQ in the colibactin biosynthetic pathway, hydrolysis of AM-ClbE with the ClbQ, electrostatic potential map of the active site of ClbQ, and synthesis and NMR data of thioester SNAC derivatives (PDF)
References
- 1.Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800–812. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garrett WS. Cancer and the microbiota. Science. 2015;348:80–86. doi: 10.1126/science.aaa4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geuking MB, Köller Y, Rupp S, McCoy KD. The interplay between the gut microbiota and the immune system. Gut Microbes. 2014;5:411–418. doi: 10.4161/gmic.29330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hooper LV, Littman DR, Macpherson AJ. Interactions Between the Microbiota and the Immune System. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 7.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède J-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010;107:11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olier M, Marcq I, Salvador-Cartier C, Secher T, Dobrindt U, Boury M, Bacquié V, Penary M, Gaultier E, Nougayrède JP, Fioramonti J, Oswald E. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes. 2012;3:501–509. doi: 10.4161/gmic.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scaldaferri F, Gerardi V, Mangiola F, Lopetuso LR, Pizzoferrato M, Petito V, Papa A, Stojanovic J, Poscia A, Cammarota G, Gasbarrini A. Role and mechanisms of action of Escherichia coli Nissle 1917 in the maintenance of remission in ulcerative colitis patients: an update. World J Gastroenterol. 2016;22:5505–5511. doi: 10.3748/wjg.v22.i24.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Putze J, Hennequin C, Nougayrède JP, Zhang W, Homburg S, Karch H, Bringer MA, Fayolle C, Carniel E, Rabsch W, Oelschlaeger TA, Oswald E, Forestier C, Hacker J, Dobrindt U. Genetic structure and distribution of the colibactin genomic island among members of the family. Enter-obacteriaceae Infect Immun. 2009;77:4696–4703. doi: 10.1128/IAI.00522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Homburg S, Oswald E, Hacker J, Dobrindt U. Expression analysis of the colibactin gene cluster coding for a novel polyketide in Escherichia coli. FEMS Microbiol Lett. 2007;275:255–62. doi: 10.1111/j.1574-6968.2007.00889.x. [DOI] [PubMed] [Google Scholar]
- 13.Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 14.Felnagle EA, Jackson EE, Chan YA, Podevels AM, Berti AD, McMahon MD, Thomas MG. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol Pharmaceutics. 2008;5:191–198. doi: 10.1021/mp700137g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marahiel MA, Stachelhaus T, Mootz HD. Modular Peptide Synthetases Involved in Nonribosomal Peptide Synthesis. Chem Rev. 1997;97:2651–2674. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 16.Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, Sauvanet P, Darcha C, Déchelotte P, Bonnet M, Pezet D, Wodrich H, Darfeuille-Michaud A, Bonnet R. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014;63:1932–1942. doi: 10.1136/gutjnl-2013-305257. [DOI] [PubMed] [Google Scholar]
- 17.Dubois D, Baron O, Cougnoux A, Delmas J, Pradel N, Boury M, Bouchon B, Bringer M-A, Nougayrède J-P, Oswald E, Bonnet R. ClbP is a prototype of a peptidase subgroup involved in biosynthesis of nonribosomal peptides. J Biol Chem. 2011;286:35562–35570. doi: 10.1074/jbc.M111.221960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cougnoux A, Gibold L, Robin F, Dubois D, Pradel N, Darfeuille-Michaud A, Dalmasso G, Delmas J, Bonnet R. Analysis of structure-function relationships in the colibactin-maturating enzyme ClbP. J Mol Biol. 2012;424:203–214. doi: 10.1016/j.jmb.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 19.Brotherton CA, Balskus EP. A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J Am Chem Soc. 2013;135:3359–3362. doi: 10.1021/ja312154m. [DOI] [PubMed] [Google Scholar]
- 20.Bian X, Fu J, Plaza A, Herrmann J, Pistorius D, Stewart AF, Zhang Y, Müller R. In Vivo Evidence for a Prodrug Activation Mechanism during Colibactin Maturation. ChemBioChem. 2013;14:1194–1197. doi: 10.1002/cbic.201300208. [DOI] [PubMed] [Google Scholar]
- 21.Balskus EP. Colibactin: understanding an elusive gut bacterial genotoxin. Nat Prod Rep. 2015;32:1534–1540. doi: 10.1039/c5np00091b. [DOI] [PubMed] [Google Scholar]
- 22.Vizcaino MI, Engel P, Trautman E, Crawford JM. Comparative metabolomics and structural characterizations illuminate colibactin pathway-dependent small molecules. J Am Chem Soc. 2014;136:9244–9247. doi: 10.1021/ja503450q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z-R, Li J, Gu J-P, Lai JYH, Duggan BM, Zhang W-P, Li Z-L, Li Y-X, Tong R-B, Xu Y, Lin D-H, Moore BS, Qian P-Y. Divergent biosynthesis yields a cytotoxic aminomalonate-containing precolibactin. Nat Chem Biol. 2016;12:773–775. doi: 10.1038/nchembio.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zha L, Wilson MR, Brotherton CA, Balskus EP. Characterization of Polyketide Synthase Machinery from the pks Island Facilitates Isolation of a Candidate Precolibactin. ACS Chem Biol. 2016;11:1287–1295. doi: 10.1021/acschembio.6b00014. [DOI] [PubMed] [Google Scholar]
- 25.Li ZR, Li Y, Lai JYH, Tang J, Wang B, Lu L, Zhu G, Wu X, Xu Y, Qian PY. Critical Intermediates Reveal New Biosynthetic Events in the Enigmatic Colibactin Pathway. ChemBioChem. 2015;16:1715–1719. doi: 10.1002/cbic.201500239. [DOI] [PubMed] [Google Scholar]
- 26.Zha L, Jiang Y, Henke MT, Wilson MR, Wang JX, Kelleher NL, Balskus EP. Colibactin assembly line enzymes use S-adenosylmethionine to build a cyclopropane ring. Nat Chem Biol. 2017 doi: 10.1038/nchembio.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trautman EP, Healy AR, Shine EE, Herzon SB, Crawford JM. Domain-Targeted Metabolomics Delineates the Heterocycle Assembly Steps of Colibactin Biosynthesis. J Am Chem Soc. 2017;139:4195–4201. doi: 10.1021/jacs.7b00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brachmann AO, Garcie C, Wu V, Martin P, Ueoka R, Oswald E, Piel J. Colibactin biosynthesis and biological activity depend on the rare aminomalonyl polyketide precursor. Chem Commun. 2015;51:13138–13141. doi: 10.1039/c5cc02718g. [DOI] [PubMed] [Google Scholar]
- 29.Horsman ME, Hari TPa, Boddy CN. Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: logic gate or a victim of fate? Nat Prod Rep. 2016;33:183–202. doi: 10.1039/c4np00148f. [DOI] [PubMed] [Google Scholar]
- 30.Kotowska M, Pawlik K. Roles of type II thioesterases and their application for secondary metabolite yield improvement. Appl Microbiol Biotechnol. 2014;98:7735–7746. doi: 10.1007/s00253-014-5952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwarzer D, Mootz HD, Linne U, Marahiel MA. Regeneration of misprimed nonribosomal peptide synthetases by type II thioesterases. Proc Natl Acad Sci U S A. 2002;99:14083– 14088. doi: 10.1073/pnas.212382199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Meng Q, You D, Li J, Chen S, Ding D, Zhou X, Zhou H, Bai L, Deng Z. Selective removal of aberrant extender units by a type II thioesterase for efficient FR-008/candicidin biosynthesis in Streptomyces sp. strain FR-008. Appl Environ Microbiol. 2008;74:7235–7242. doi: 10.1128/AEM.01012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh E, Kohli RM, Bruner SD, Walsh CT. Type II thioesterase restores activity of a NRPS module stalled with an aminoacyl-S-enzyme that cannot be elongated. ChemBioChem. 2004;5:1290–1293. doi: 10.1002/cbic.200400077. [DOI] [PubMed] [Google Scholar]
- 34.Kalaitzis JA, Cheng Q, Meluzzi D, Xiang L, Izumikawa M, Dorrestein PC, Moore BS. Policing starter unit selection of the enterocin type II polyketide synthase by the type II thioesterase EncL. Bioorg Med Chem. 2011;19:6633–6638. doi: 10.1016/j.bmc.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rui Z, Petříčková K, Škanta F, Pospíšil S, Yang Y, Chen CY, Tsai SF, Floss HG, Petříček M, Yu TW. Biochemical and genetic insights into asukamycin biosynthesis. J Biol Chem. 2010;285:24915–24924. doi: 10.1074/jbc.M110.128850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Claxton HB, Akey DL, Silver MK, Admiraal SJ, Smith JL. Structure and functional analysis of RifR, the type II thioesterase from the rifamycin biosynthetic pathway. J Biol Chem. 2009;284:5021–5029. doi: 10.1074/jbc.M808604200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harvey BM, Hong H, Jones MA, Hughes-Thomas ZA, Goss RM, Heathcote ML, Bolanos-Garcia VM, Kroutil W, Staunton J, Leadlay PF, Spencer JB. Evidence that a novel thioesterase is responsible for polyketide chain release during biosynthesis of the polyether ionophore monensin. ChemBioChem. 2006;7:1435–1442. doi: 10.1002/cbic.200500474. [DOI] [PubMed] [Google Scholar]
- 38.Liu T, You D, Valenzano C, Sun Y, Li J, Yu Q, Zhou X, Cane DE, Deng Z. Identification of NanE as the Thioesterase for Polyether Chain Release in Nanchangmycin Biosynthesis. Chem Biol. 2006;13:945–955. doi: 10.1016/j.chembiol.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Whicher JR, Florova G, Sydor PK, Singh R, Alhamadsheh M, Challis GL, Reynolds KA, Smith JL. Structure and function of the RedJ protein, a thioesterase from the prodiginine biosynthetic pathway in Streptomyces coelicolor. J Biol Chem. 2011;286:22558–22569. doi: 10.1074/jbc.M110.213512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heathcote ML, Staunton J, Leadlay PF. Role of type II thioesterases: Evidence for removal of short acyl chains produced by aberrant decarboxylation of chain extender units. Chem Biol. 2001;8:207–220. doi: 10.1016/s1074-5521(01)00002-3. [DOI] [PubMed] [Google Scholar]
- 41.Schneider A, Marahiel MA. Genetic evidence for a role of thioesterase domains, integrated in or associated with peptide synthetases, in non-ribosomal peptide biosynthesis in Bacillus subtilis. Arch Microbiol. 1998;169:404–410. doi: 10.1007/s002030050590. [DOI] [PubMed] [Google Scholar]
- 42.Koglin A, Löhr F, Bernhard F, Rogov VV, Frueh DP, Strieter ER, Mofid MR, Güntert P, Wagner G, Walsh CT, Marahiel MA, Dötsch V. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature. 2008;454:907–911. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritchie MK, Johnson LC, Clodfelter JE, Pemble CW, Fulp BE, Furdui CM, Kridel SJ, Lowther WT. Crystal structure and substrate specificity of human thioesterase 2: Insights into the molecular basis for the modulation of fatty acid synthase. J Biol Chem. 2016;291:3520–3530. doi: 10.1074/jbc.M115.702597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raisch J, Rolhion N, Dubois A, Darfeuille-Michaud A, Bringer M-A. Intracellular colon cancer-associated Escherichia coli promote protumoral activities of human macrophages by inducing sustained COX-2 expression. Lab Invest. 2015;95:296–307. doi: 10.1038/labinvest.2014.161. [DOI] [PubMed] [Google Scholar]
- 45.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A. The α/ β hydrolase fold. Protein Eng, Des Sel. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 46.Holm L, Rosenström P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Zheng T, Bruner SD. Structural basis for phosphopantetheinyl carrier domain interactions in the terminal module of nonribosomal peptide synthetases. Chem Biol. 2011;18:1482–1488. doi: 10.1016/j.chembiol.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Healy AR, Vizcaino MI, Crawford JM, Herzon SB. Convergent and Modular Synthesis of Candidate Precolibactins. Structural Revision of Precolibactin A. J Am Chem Soc. 2016;138:5426–5432. doi: 10.1021/jacs.6b02276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dimise EJ, Widboom PF, Bruner SD. Structure elucidation and biosynthesis of fuscachelins, peptide siderophores from the moderate thermophile Thermobifida fusca. Proc Natl Acad Sci U S A. 2008;105:15311–15316. doi: 10.1073/pnas.0805451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruner SD, Weber T, Kohli RM, Schwarzer D, Marahiel MA, Walsh CT, Stubbs MT. Structural basis for the cyclization of the lipopeptide antibiotic surfactin by the thioesterase domain SrfTE. Structure. 2002;10:301–310. doi: 10.1016/s0969-2126(02)00716-5. [DOI] [PubMed] [Google Scholar]
- 52.Healy AR, Nikolayevskiy H, Patel JR, Crawford JM, Herzon SB. A Mechanistic Model for Colibactin-Induced Genotoxicity. J Am Chem Soc. 2016;138:15563–15570. doi: 10.1021/jacs.6b10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–95. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 54.Yin J, Lin AJ, Golan DE, Walsh CT. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat Protoc. 2006;1:280–285. doi: 10.1038/nprot.2006.43. [DOI] [PubMed] [Google Scholar]
- 55.Kabsch W. XDS. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr, Sect D: Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.