

Abstract
Background
Epilepsy regroups a common and diverse set of chronic neurological disorders that are characterized by spontaneous, unprovoked, and recurrent epileptic seizures. Epilepsies have a highly heterogeneous background with a strong genetic contribution and various mode of inheritance. X-linked epilepsy usually manifests as part of a syndrome or epileptic encephalopathy. The variability of clinical manifestations of X-linked epilepsy may be attributed to several factors including the causal genetic mutation, making diagnosis, genetic counseling and treatment decisions difficult. We report the description of a Moroccan family referred to our genetic department with X-linked epileptic seizures as the only initial diagnosis.
Case presentation
Knowing the new contribution of Next-Generation Sequencing (NGS) for clinical investigation, and given the heterogeneity of this group of disorders we performed a Whole-Exome Sequencing (WES) analysis and co-segregation study in several members of this large family. We detected a novel pathogenic PLP1 missense mutation c.251C > A (p.Ala84Asp) allowing us to make a diagnosis of Pelizaeus-Merzbacher Disease for this family.
Conclusion
This report extends the spectrum of PLP1 mutations and highlights the diagnostic utility of NGS to investigate this group of heterogeneous disorders.
Keywords: Missense mutation, PLP1 gene, Connatal Pelizaeus-Merzbacher disease
Background
Epilepsy regroups a large group of highly heterogeneous disorders that have in common an abnormally increased predisposition to seizures. Epilepsy is defined as a common and diverse set of neurologic disorders characterized by spontaneous, unprovoked and recurrent epileptic seizures [1].
Multiple genetic factors were identified to be involved in primary epilepsy syndromes for which epileptic seizures are the predominant clinical feature; the same hold true in brain developmental disorders for which epileptic seizures are secondary clinical features. Recent whole-exome sequencing (WES) data suggest that mutations causing epileptic encephalopathies are often sporadic. These mutations are typically de novo dominant variations that affect a single autosomal gene but autosomal recessive and X-linked inheritance were also reported [2].
Generally the X-linked epileptic forms orient to a group of disorders which, among others, consist of: Epilepsy, X-linked, with Variable Learning Disabilities and Behavior Disorders (OMIM #300491), Epileptic Encephalopathy, Early Infantile, type 1 (EIEE1; OMIM #308350) and type 2 (EIEE2; OMIM #300672), and rarely Pelizaeus-Merzbacher Disease (PMD; OMIM #312080) [3–5].
We report here a family addressed to our department with an initial diagnosis of X-linked epileptic seizures. Given the heterogeneity of this group of diseases we used a whole-exome sequencing approach that allowed us to detect a novel PLP1 missense mutation p.Ala84Asp related to PMD. This diagnosis was consistent with the phenotype of the affected patients, and confirmed by segregation analysis of the mutation in the family.
Case presentation
The propositus (V-4) is a Moroccan 5 years old boy, non-consanguineous, last child of three siblings, and was addressed for familial epilepsy (Fig. 1A). His parents and older sister appear to be healthy and have been free of epilepsy seizure in the past. He had neonatal hypotonia, psychomotor delay, and no seating position with the support of the head at 2 years old. Daily-generalized seizures started at 8 months old and treated by valproate of sodium, but without any response. At clinical examination, he had stature and weight delay at <3rd percentile, microcephaly at -2SD, severe hypotonia, with no facial dysmorphia nor nystagmus. The computerized tomography (CT) scan was normal, but the electroencephalogram (EEG) showed abnormal waves at right fronto-temporal region. Unfortunately, MRI could not be available for the patient.
Fig. 1.
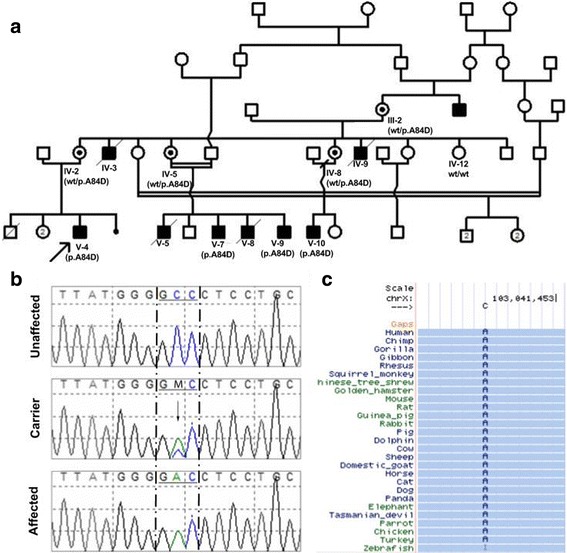
a Pedigree of the studied Moroccan family with X-linked epileptic seizures, presenting the co-segregation of the c.251C > A mutation. The patient above the arrow indicates the proband. b Chromatograms by Sanger sequencing of family members who are hemizygous, heterozygous and wild-type for the PLP1 variant. c Amino acid conservation map across species demonstrating a highly conserved residues position 84 (based on Multiz Alignments Track of UCSC Genome Browser and The human genome reference hg19/GRCch37)
Five cousins (V-5, V-7, V-8, V-9, V-10) and maternal uncles (IV-3, IV-9) were also followed for epilepsy. IV-3, IV-9, V-5 and V-8 died respectively at four, seven, three and two years. They had all neonatal hypotonia, psychomotor delay and epilepsy beginning at 6 months old.
In an effort to elucidate the causal mutation, we undertook a broader approach by WES on two family members, the propositus V-4 and his cousin V-7. Genomic DNA was isolated from peripheral blood leukocytes using the salting-out method.
The WES was performed at McGill University and Genome Québec Innovation Centre (MUGQIC) on a HiSeq 2000 sequencing instrument (Illumina Inc.). The WES capture was done using SureSelect Human All Exon V4 in solution capture kit (Agilent Technologies) according to the manufacturer’s instructions.
The well-validated GATK software (Genome Analysis ToolKit (GATK) [6] has been used in conjunction with the gold-standard aligner BWA (Burrow-Wheeler Alignment) [7] for assembly. Prior to the alignment, each read was properly assessed and trimmed using various tools from the Picard suite (http://broadinstitute.github.io/picard). Once the alignment was performed, quality of each base has been normalized for each sample, and local realignment for suspected indels region was performed. Variant calling has been performed jointly on all samples processed together using UnifiedGenotyper from the GATK suite. We also used the ANNOVAR software to annotate variants using the most up-to-date public databases (dnSNP, REFSEQ) [8]. The potential functional impact of mutations was predicted using Sorting Intolerant From Tolerant (SIFT), at http://sift.bii.a-star.edu.sg, and Polymorphism Phenotyping 2 (PolyPhen2), at http://genetics.bwh.harvard.edu/pph2/.
The putative mutation was validated via Sanger, sequencing the DNA of 9 family members with the following primers: Forward (AGCCTTGTTAAGGTGCTCGCT) and Reverse (GCTTGATGTTGGCCTCTGGA). Sequencing was performed using BigDye Terminator v3.1 cycle sequencing, and the obtained sequences were analyzed on an ABI 3000 DNA Analyzer (Applied Biosystems).
Informed consent for DNA analysis was obtained from all participants and parents’ patients.
A total of 188,043 variants were detected. The WES data covered all exons of known X-Linked Epilepsy related genes, and no known pathogenic mutations were identified in these genes. Nevertheless, we identified a single hemizygous base substitution in coding exon3, c.251C > A of the PLP1 gene (based on GRCh37/hg19, and transcript ID NM_000533), resulting in substitution of alanine by an aspartic acid at amino acid 84. This mutation was found in hemizygous state in the propositus V-4 and in his cousin V-7. Sanger sequencing confirmed this mutation in hemizygous state in the affected subjects (V-9, V-10) and in heterozygous state in the obligatory carrier mothers, showing co-segregation of the mutation with the disease phenotype (Fig. 1A). The non-affected IV-12 was wild type for this mutation (Fig. 1B).
Mutation c.251C > A (p.Ala84Asp) located within exon 3, results in changes of a non-polar alanine to a charged aspartic acid at position 84, it was predicted by Polyphen2 to be probably damaging with a score of 0.989 (sensitivity: 0.72, specificity: 0.97). The Mutation was also predicted to be damaging by SIFT with a score of 0. As shown in Fig. 1C, this amino acid change affects highly conserved residues.
Discussion
The advent and increasingly decreasing cost of NGS and WES has opened up vast new opportunities for its application as a clinical tool to identify mutations in genes established to cause disorders, or in novel genes; altogether facilitating both discovery efforts and conclusive diagnoses [9]. Many studies have demonstrated that systematic application of WES in specific clinical situations has been more accurate, faster, and less expensive than conventional diagnostic procedures [10].
In this study, we report a family showing X-linked connatal severe neurological disorder with epilepsy. WES allowed us to have a conclusive diagnosis identifying a novel missense PLP1 mutation, suggesting a diagnosis of Pelizaeus-Merzbacher Disease (PMD).
PMD is caused by mutations involving the PLP1 gene that encodes the myelin protein proteolipid protein 1 (PLP1) and the spliced variant DM20. The c.251C > A mutation, located within the second hydrophobic transmembrane domain of PLP1, has not been previously described. Mutations in the same region of the protein, including c.247G > A and c.254 T > C mutations causing a p.Gly83Arg and p.Leu85Arg amino acid substitutions respectively, have been shown to cause connatal form of PMD [11, 12].
There is a spectrum of PLP1-related disorders with some genotype–phenotype correlations [4, 11–13]. Generally, patients with PLP1 missense mutations show the most severe form of PMD (connatal form), the most common PLP1 duplications result in the classical PMD, whereas deletions and null mutations in mild form of PMD and SPG2 [4, 11, 13, 14]. Epilepsy, major hypotonia, and severe psychomotor delay were constant features in our affected children and they have the same clinical history. Thus, epilepsy, not being a frequent sign in PMD, represents the specificity of this family. Considering that the majority of PLP1 point mutations cause more severe dysmyelinating diseases than null mutations, Inoue speculated that the profound dysmyelination resulting from PLP1 point mutations probably arises not from the absence of functional protein, but rather from the cytotoxic effect of mutant protein [13].
Conclusion
Our study expands the mutation spectrum of PLP1 gene and shows that whole-exome analysis in a patient from a family exhibiting X-linked epilepsy could be used efficiently to identify causative mutations, thus providing help to clinicians in making a definitive diagnosis.
Acknowledgements
The authors would like to gratefully acknowledge the patients and his parents for their collaboration.
Funding
None.
Availability of data and materials
The datasets generated will be available from the corresponding author on reasonable request.
Abbreviations
- PLP1
Proteolipid protein 1
- PMD
Pelizaeus-Merzbacher Disease
- WES
Whole-exome sequencing
Authors’ contributions
JL: data analysis, literature search, manuscript preparation; SE and MM: clinical assessments and preparation of the manuscript; BO and LB: carried out the Molecular genetic studies; AD and DS: Computational analysis; PD, PC, GR, AS: participated in the design of the study and manuscript review. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Signed informed consent from adult participants, and patient’s parents under 18 years of age, were obtained to perform DNA analysis.
Consent for publication
The Consent to publish the case report was obtained from the patient’s parent under 18 years of age: Propositus (V-4). The consent for the use and publication of medical information of the deceased individuals was provided from their respective mothers. It was discussed with the family that the details of the case were described including the pedigree in a completely anonymous way.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jaber Lyahyai, Email: jaber.lyahyai@gmail.com.
Bouchra Oulad Amar Bencheikh, Email: bouchra.ouledabencheikh@mail.mcgill.ca.
Siham C. Elalaoui, Email: sihamgen@yahoo.fr
Maria Mansouri, Email: mariamansourig@hotmail.fr.
Lamia Boualla, Email: lamiae_boualla@yahoo.fr.
Alexandre DIonne-Laporte, Email: alexandre.dionne-laporte@mcgill.ca.
Dan Spiegelman, Email: dan.spiegelman@crchum.qc.ca.
Patrick A. Dion, Email: patrick.a.dion@umontreal.ca
Patrick Cossette, Email: patrick.cossette@umontreal.ca.
Guy A. Rouleau, Email: guy.rouleau@mcgill.ca
Abdelaziz Sefiani, Email: sefianigen@hotmail.com.
References
- 1.Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J., Jr Epileptic seizures and epilepsy: definitions proposed by the international league against epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46(4):470–472. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- 2.Nieh SE, Sherr EH. Epileptic encephalopathies: new genes and new pathways. Neurotherapeutics. 2014;11(4):796–806. doi: 10.1007/s13311-014-0301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukumura S, Adachi N, Nagao M, Tsutsumi H. A novel proteolipid protein 1 gene mutation causing classical type Pelizaeus-Merzbacher disease. Brain and Development. 2011;33(8):697–699. doi: 10.1016/j.braindev.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Shiihara T, Watanabe M, Moriyama K, Uematsu M, Sameshima K. A novel PLP1 frameshift mutation causing a milder form of Pelizaeus-Merzbacher disease. Brain and Development. 2015;37(4):455–458. doi: 10.1016/j.braindev.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Xie H, Feng H, Ji J, Wu Y, Kou L, Li D, Ji H, Wu X, Niu Z, Wang J, et al. Identification and functional study of novel PLP1 mutations in Chinese patients with Pelizaeus-Merzbacher disease. Brain and Development. 2015;37(8):797–802. doi: 10.1016/j.braindev.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 6.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulkarni S, Pfeifer J. Clinical genomics: a guide to clinical next-generation sequencing. London: Academic Press; 2015. [Google Scholar]
- 11.Cailloux F, Gauthier-Barichard F, Mimault C, Isabelle V, Courtois V, Giraud G, Dastugue B, Boespflug-Tanguy O. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European network on brain Dysmyelinating disease. Eur J Hum Genet. 2000;8(11):837–845. doi: 10.1038/sj.ejhg.5200537. [DOI] [PubMed] [Google Scholar]
- 12.Grossi S, Regis S, Biancheri R, Mort M, Lualdi S, Bertini E, Uziel G, Boespflug-Tanguy O, Simonati A, Corsolini F, et al. Molecular genetic analysis of the PLP1 gene in 38 families with PLP1-related disorders: identification and functional characterization of 11 novel PLP1 mutations. Orphanet J Rare Dis. 2011;6:40. doi: 10.1186/1750-1172-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005;6(1):1–16. doi: 10.1007/s10048-004-0207-y. [DOI] [PubMed] [Google Scholar]
- 14.Osaka H, Koizume S, Aoyama H, Iwamoto H, Kimura S, Nagai J, Kurosawa K, Yamashita S. Mild phenotype in Pelizaeus-Merzbacher disease caused by a PLP1-specific mutation. Brain and Development. 2010;32(9):703–707. doi: 10.1016/j.braindev.2009.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated will be available from the corresponding author on reasonable request.