

Abstract
Familial myelodysplastic syndromes arise from haploinsufficiency of genes involved in hematopoiesis and are primarily associated with early-onset disease. Here we describe a familial syndrome in seven patients from four unrelated pedigrees presenting with myelodysplastic syndrome and loss of chromosome 7/7q. Their median age at diagnosis was 2.1 years (range, 1–42). All patients presented with thrombocytopenia with or without additional cytopenias and a hypocellular marrow without an increase of blasts. Genomic studies identified constitutional mutations (p.H880Q, p.R986H, p.R986C and p.V1512M) in the SAMD9L gene on 7q21, with decreased allele frequency in hematopoiesis. The non-random loss of mutated SAMD9L alleles was attained via monosomy 7, deletion 7q, UPD7q, or acquired truncating SAMD9L variants p.R1188X and p.S1317RfsX21. Incomplete penetrance was noted in 30% (3/10) of mutation carriers. Long-term observation revealed divergent outcomes with either progression to leukemia and/or accumulation of driver mutations (n=2), persistent monosomy 7 (n=4), and transient monosomy 7 followed by spontaneous recovery with SAMD9L-wildtype UPD7q (n=2). Dysmorphic features or neurological symptoms were absent in our patients, pointing to the notion that myelodysplasia with monosomy 7 can be a sole manifestation of SAMD9L disease. Collectively, our results define a new subtype of familial myelodysplastic syndrome and provide an explanation for the phenomenon of transient monosomy 7. Registered at: www.clinicaltrials.gov; #NCT00047268.
Introduction
Germline predisposition has been recognized as an underlying cause for the development of myelodysplastic syndromes (MDS) in children. Recently, it has also been gaining importance in the etiology of adult myeloid neoplasia, particularly in cases with a positive family history. Various genes are known to be associated with heritable forms of MDS and acute myeloid leukemia,1,2 e.g., GATA2,3 CEBPA,4 RUNX1,5 ANKRD26,6 ETV67 and DDX41,8 in addition to inherited bone marrow failure syndromes. Germline mutations in DDX41 can result in adult-onset myeloid neoplasia, while aberrations in RUNX1 and GATA2 are associated with myeloid neoplasia in younger individuals. We recently reported that GATA2 deficiency is the most common genetic cause of primary childhood MDS, accounting for 15% of all cases of advanced MDS, and 37% of MDS with monosomy 7 (MDS/-7).9 However, in the majority of cases of pediatric MDS, and also in a considerable number of cases of familial myeloid neoplasia, the presumed germline cause has not yet been discovered.10,11
Monosomy 7 is the most frequent cytogenetic lesion in children with MDS and, unlike in adults, it often occurs as the sole cytogenetic abnormality.12 Due to the rapid and progressive course of the disease, it is considered an urgent indication for hematopoietic stem cell transplantation.13 However, transient monosomy 7 has occasionally been documented in childhood MDS.14–16 Considering that complete (−7) and partial [del(7q)] deletion of chromosome 7 are common aberrations in MDS, extensive efforts have been undertaken to discern causative tumor suppressor genes located on chromosome 7. Asou and colleagues identified SAMD9 (Sterile Alpha Motif Domain-containing 9), its paralogue SAMD9L (SAMD9-like), and Miki/HEPACAM2 as commonly deleted genes within a 7q21 cluster in patients with myeloid neoplasia.17 Notably Samd9l-haploinsufficient mice were shown to develop myeloid malignancies characterized by different cytopenias and mimicking human disease with monosomy 7.18
In line with these findings, germline heterozygous gain-of-function SAMD9L mutations p.H880Q, p.I891T, p.R986C, and p.C1196S were recently discovered in four pedigrees with variable degrees of neurological symptoms (ataxia, balance impairment, nystagmus, hyperreflexia, dysmetria, dysarthria) and hematologic abnormalities (single to tri-lineage cytopenias, MDS/-7). For most carriers, the clinical presentation was compatible with the diagnosis of ataxia-pancytopenia syndrome.19,20 Similarly, in two recent studies, we and others reported de novo gain-of-function mutations in SAMD9 in a total of 18 patients with MIRAGE syndrome (myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy), of whom four notably also developed MDS/-7.21,22 However, not all patients develop the full MIRAGE disease spectrum, as documented in one family with SAMD9-related MDS.23 The SAMD9 and SAMD9L genes share 62% sequence identity and apart from their putative role as myeloid tumor suppressors, their general function and their specific effect pertaining to hematopoiesis are not well-understood.18
In this study we aimed to identify the genetic cause in pedigrees with non-syndromic familial MDS. We discovered constitutional SAMD9L mutations associated with non-random patterns of clonal escape leading to loss of the mutant allele. We further demonstrate in two cases that SAMD9L–related disease can be associated with transient −7, occurring as a one-time clonal event followed by somatic correction of hematopoiesis achieved by UPD7q with double wild-type SAMD9L.
Methods
Patients
The diagnosis of MDS was established according to World Health Organization criteria.24,25 Patients 1 (P1), 2 (P2), 5 (P5), and 7 (P7) were enrolled in prospective study 98 of the European Working Group of MDS in Childhood (EWOG-MDS) (www.clin-icaltrials.gov; #NCT00047268). Patient 6 (P6) was the father of P5 (family III). Family II (P3 and P4) was referred for evaluation of familial MDS from Phoenix Childreńs Hospital, USA. The study had been approved by an institutional ethics committee (University of Freiburg, CPMP/ICH/135/95 and 430/16). Written informed consent to participation had been obtained from patients and parents.
Genomic studies and bioinformatics
Exploratory whole exome sequencing was performed in bone marrow granulocytes in P1 and P2, as outlined in the Online Supplementary Material. Targeted deep sequencing for SAMD9/SAMD9L and genes related to MDS/inherited bone marrow failure syndromes was performed in other patients, except for P3 and P6 due to unavailability of material. All relevant variants were validated using Sanger sequencing. For germline confirmation, DNA was extracted from skin fibroblasts and/or hair follicles, and targets were amplified and sequenced as previously described.9 The degree of deleteriousness was calculated using the combined annotation-dependent depletion scoring system (CADD-score).26 The variants with CADD-scores higher than 20 were further evaluated for their role in hematologic disease or cancer, thereby focusing on the top 1% most deleterious variants in the human genome. In addition, pathogenicity calculations were performed using standard prediction tools. The evolutionary conservation across species and the physicochemical difference between amino acids were estimated by PhyloP, PhastCons and the Grantham score, respectively.27 Mutant clonal size was inferred from allelic frequencies and the total number of next-generation sequencing reads normalized to the ploidy level. Further details are provided in the Online Supplementary Methods.
Evaluation of variant allelic configuration
Genomic DNA of P1 collected at the time of progress to chronic myelomonocytic leukemia (CMML) was amplified to obtain a 1333 bp region encompassing both SAMD9L mutations: p.V1512M (germline) and p.R1188X (acquired). Polymerase chain reaction products were TA-cloned and sequenced as previously reported.28 Sequences of 170 colonies were evaluated for the presence of SAMD9L mutations.
Cellular and functional studies
Metaphase karyotyping and interphase fluorescence in situ hybridization were performed using bone marrow specimens according to standard procedures.12 Human colony-forming cell assays were performed in P1 (at CMML disease stage) and in P7 (at diagnosis) as previously described.29 Furthermore, to evaluate the effect of the patient-derived SAMD9L p.V1512M and p.R986C mutations on cellular proliferation, 293FT cells were dye-labeled and consequently transfected with wild-type or mutant teal fluorescent protein (TFP)-SAMD9L as previously described.20 The transfected cells were tracked by flow cytometry for TFP-SAMD9L expression and dye dilution as an indicator of cell division. SAMD9L variant p.T233N, recently reported as “disease-protective”,20 was used as a control.
Results
Clinical phenotype of patients
Patients P1-P6 (4 males and 2 females) belong to three unrelated families of German descent and were diagnosed with bona fide familial MDS after known inherited bone marrow failure syndromes had been excluded by targeted sequencing and functional tests (Figure 1, Table 1). Index patient P7 is the only child of a non-consanguineous Swedish family. Detailed clinical descriptions of all the patients are given in the Online Supplementary Material. All affected individuals had normal measurements without dysmorphic stigmata at birth and at last follow-up (Table 1). Psychomotor development and neurocognitive function were normal and, in particular, no ataxia or movement disorders were diagnosed in the ten mutation carriers (7 patients and 3 silent carriers with SAMD9L mutations). Previous family histories were unremarkable for cytopenias, neurological disease, malignancies, or stillbirths, with the exception of the father of P7 who at the last follow-up presented with unclear ataxia. Prior non-invasive recurrent respiratory tract infections were noted in three of the seven patients (P1, P2, and P4) and endogenous eczema in two (P1 and P3). Moreover, P1 developed transient pancytopenia during infancy and suffered from self-limiting seizures during infancy with no structural brain abnormalities or neurological deficits identified. Peripheral blood findings at diagnosis included isolated thrombocytopenia in one (P2), thrombocytopenia with neutropenia in two (P1, P3) and pancytopenia in four patients (P4-7). Mean corpuscular volume was increased in five of the seven patients at diagnosis, and HbF was elevated in two out of three tested patients. MDS manifested at the median age of 2.1 (range, 1.0–42) years as hypocellular refractory cytopenia of childhood (RCC) or in the adult (P6) as refractory cytopenia with multilineage dysplasia (Table 1). Severe dysplasia with vacuolization was observed in two patients (P1, P2). A common cytogenetic feature in all patients was the complete or partial loss of chromosome 7 (Table 1).
Figure 1.

Germline SAMD9L mutations in pedigrees with familial myelodysplastic syndrome. (A) Identification of four pedigrees with MDS and monosomy 7 harboring germline heterozygous SAMD9L mutations: p.V1512M (pedigree I), p.R986H (pedigree II) and p.R986C (pedigree III), p.H880Q (pedigree IV), and somatic mutations: p.R1188X (P1) and p.S1317RfsX21 (P7). Dotted symbols indicate healthy mutation carriers. Sanger sequencing on DNA extracted from hair follicles (HR) confirmed the germline status of mutations as visualized in electropherograms. Sequencing in P1 was performed on peripheral blood granulocytes (GR) revealing a minor mutational peak, corresponding to a variant allelic frequency of 8.3% by whole exome sequencing. In pedigree III, the mutation in P5 was confirmed in fibroblast (FB) DNA, while for P6 and remaining family members whole blood (WB) was analyzed. In pedigree IV other family members were not tested (n.t.). (B) SAMD9L and SAMD9 gene orientation on 7q22 in reverse strand direction (3′–5′). The SAMD9L protein is coded by one exon and contains two known functional sites: N-terminal sterile alpha motif (SAM) and nuclear localization sequence (NLS). Four germline and two somatic (*) mutations were identified in SAMD9L. Germline missense mutations are evolutionarily highly conserved. (C) TA cloning of the double mutated SAMD9L region of P1 revealed cis-configuration of mutations p.V1512M (germline) and p.R1188X (somatic) in ten out of 172 clones tested.
Table 1.
Clinical data of SAMD9L-mutated patients.
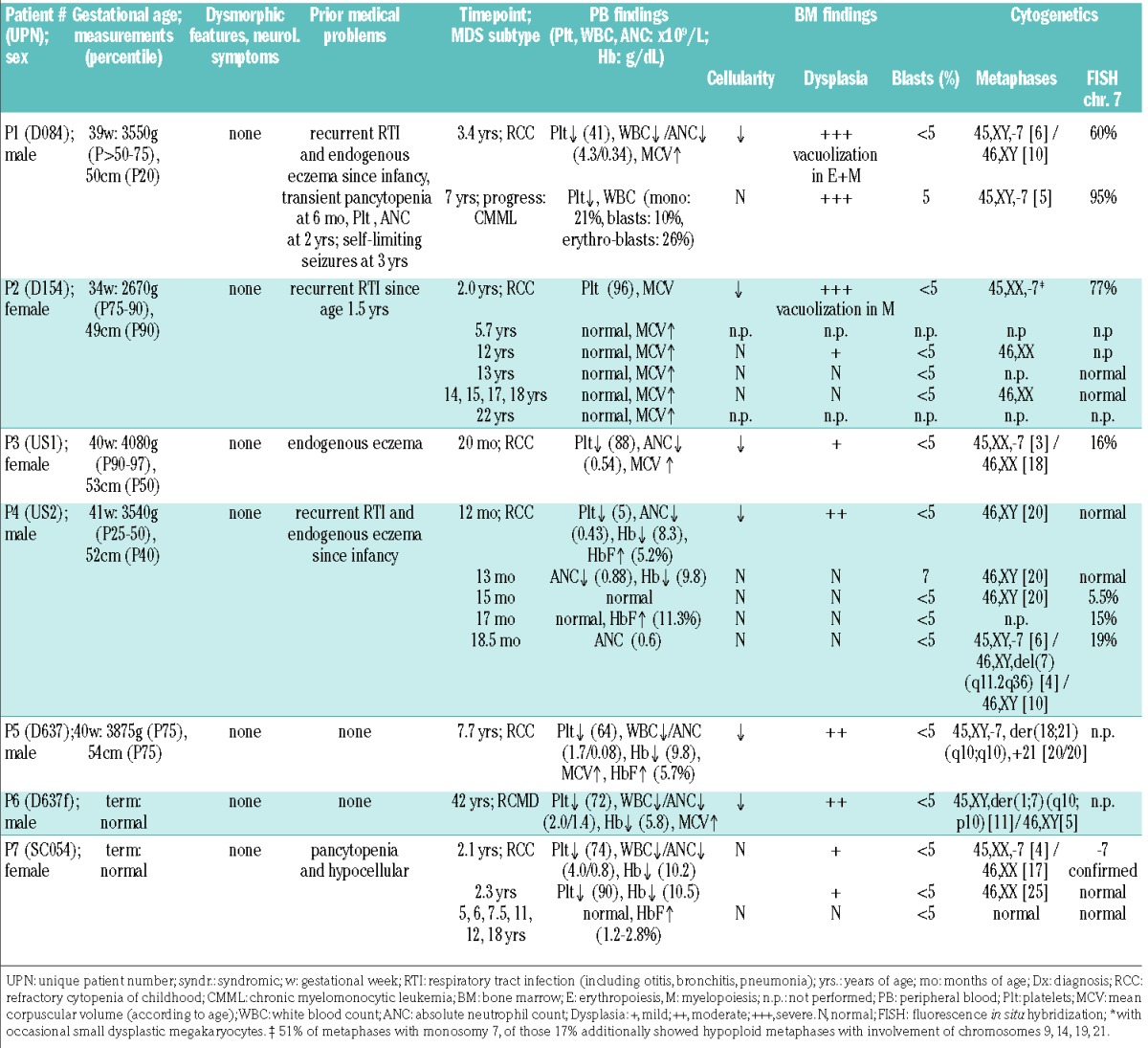
The clinical course was remarkable for several patients. Three years after the initial diagnosis of RCC, P1 developed severe infections and hepatosplenomegaly, and for the first time required platelet transfusions, while his blood smear showed 21% blasts, compatible with the diagnosis of CMML (with no in vitro hypersensitivity to granulocyte-macrophage colony-stimulating factor), (Figure 2). Following hematopoietic stem cell transplantation, he developed late-onset acute graft-versus-host disease and died from cerebral hemorrhage. At the same time, his younger sister (P2) was diagnosed with RCC/-7; however, the parents decided against follow-up in the hematology clinic. Unexpectedly, her complete blood count normalized 3.7 years later and remained stable until the last follow-up 20 years after the initial diagnosis. She achieved a spontaneous remission as indicated by normalization of bone marrow morphology/cellularity and cytogenetics (Figure 2, Table 1). A similar clinical picture was seen in P7 who was diagnosed with RCC with a −7 clone at the age of 2.1 years, and experienced rapid cytogenetic remission with normal marrow and blood counts until the last follow-up, 16 years after diagnosis (Table 1). Hematologic findings also normalized in P4, shortly after the initial manifestation; however, fluorescence in situ hybridization revealed chromosome 7 loss in bone marrow, which gradually worsened and culminated, after 3.5 months, in the emergence of two independent clones with −7 and del(7q) (Table 1).
Figure 2.

Bone marrow findings in P1 and P2 at different timepoints during the course of the disease. Hematoxylin and eosin staining of bone marrow (BM) at diagnosis of RCC in P1 showing dysplastic granulopoiesis with hypergranulation and a pseudo-Pelger cell (top left), myeloblast and dysplastic eosinophil (top right). BM at diagnosis in P2 (synchronous with monosomy 7) showing hypergranulation and vacuolization in myelocytes, and dysplastic erythropoiesis with double nuclei (bottom left). Normal BM morphology in P2, 15 years after initial BM confirming spontaneous phenotype reversion (bottom right).
Due to high-risk cytogenetics and disease progression, five of the seven patients (P1, P3-6) underwent hematopoietic stem cell transplantation after myeloablative conditioning. At last follow-up, six of the seven patients were alive, four after transplantation, and two without therapy.
Constitutional and acquired SAMD9L mutations
Exploratory whole exome sequencing performed in family I identified two shared candidate variants in P1 and P2 evaluated as highly conserved and deleterious by in silico prediction: SAMD9L (p.V1512M) and PTEN (p.Y188C) (Table 2, Online Supplementary Figure S1). Sequencing of DNA from hair follicles confirmed the constitutional nature of both novel mutations. The SAMD9L p.V1512M variant was inherited from the mother (Figure 1A) whereas PTEN p.Y188C was of paternal origin; both parents were asymptomatic and had normal complete blood counts at the time of testing. Finally, truncating acquired SAMD9L mutation p.R1188X (VAF 5.9%) was identified in P1 in hematopoiesis (Table 2).
Table 2.
Overview of germline and somatic mutations.
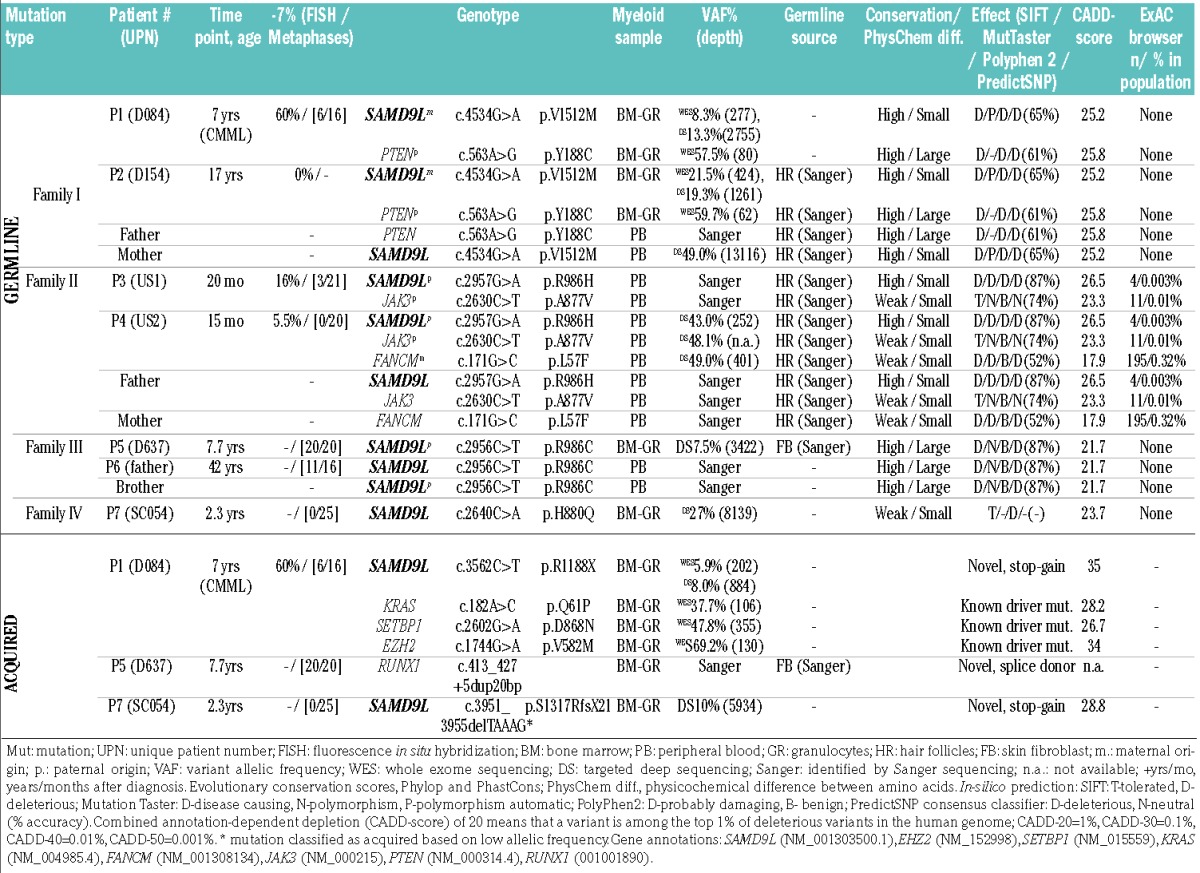
In pedigree II, targeted next-generation sequencing in P4 revealed SAMD9L p.R986H as the most plausible candidate constitutional mutation predicted to be highly conserved and deleterious (Table 2, Figure 1A,B). This mutation was found in four individuals in ExAC (out of 120976 alleles). Additional missense variants in JAK3 p.A877V (ExAC: 11 individuals, 121372 alleles), and FANCM p.L57F (ExAC: 195 individuals, 121190 alleles) had lower and moderate pathogenicity scores, respectively (Table 2). Chromosomal breakage studies on P4 were negative thus arguing against a pathogenic role of the heterozygous FANCM variant. Germline analysis revealed SAMD9L and JAK3 variants in P3, P4, and their father, while the FANCM variant was transmitted from the mother only to P4. Both parents were asymptomatic.
In pedigree III, the SAMD9L p.R986C mutation was identified in P5 and the affected father, P6 (Figure 1A). This mutation has been reported in a family with ataxia-pancytopenia phenotype, with one of three carriers developing MDS/-7 at the age of 18 months.20 The HLA-identical brother of P5 was thoroughly evaluated as a potential bone marrow donor. He was clinically healthy and had a normal complete blood count, but he did not qualify as a donor because of hypocellular bone marrow with mild dysplastic features. He was also a carrier of the p.R986C mutation.
In P7 of pedigree IV, targeted next-generation sequencing identified two SAMD9L mutations (Table 2): missense p.H880Q with a variant allele frequency of 27% out of 8139 reads (likely constitutional; this mutation was reported in multiple individuals within a family with ataxia pancytopenia but no MDS phenotype) and nonsense p.S1317RfsX21 likely acquired in a subclone as inferred from the much lower variant allele frequency of 10% (5934 reads). In summary, inherited SAMD9L mutations p.V1512M, p.R986H, and p.R986C were identified in three families (each with 2 individuals diagnosed with MDS/-7 and 1 healthy carrier Figure 1A,B), and p.H880Q in P7 who presented with transient monosomy 7.
Acquired mutations in known oncogenes
All patients with exception of P3 and P6 were evaluated for the presence of somatic mutations in leukemia-associated genes using whole exome sequencing or targeted next-generation sequencing. In P1 previously reported leukemia driver mutations SETBP1 p.D868N, EZH2 p.V582M, and KRAS p.Q61P were identified as somatic (Table 2, Online Supplementary Figure S2). Similarly, in P5 a somatic RUNX1 mutation c.413_427+5dup20bp was detected (Table 2). This is a novel splice-donor site mutation not present in databases. No additional mutations in leukemia driver genes were observed in other affected cases.
Clonal escape mechanisms from SAMD9L mutations are not random
In comparison with other constitutional variants, SAMD9L missense mutations showed significantly lower median allelic frequencies across all patients (53% versus 20%, P<0.05) (Table 2). This finding was corroborated by the consistent partial or complete loss of chromosome 7 (Figure 3, Table 1). In P1, −7 progressively expanded from 60% at the time that RCC was diagnosed to 95% at the time of progression to CMML. In addition, P1 and P7 harbored subclones with acquired stop-gain SAMD9L mutations located upstream of the constitutional missense substitutions (Figure 3). TA cloning confirmed the cis orientation of both mutations on the same chromosome in P1 (Figure 1C). Western blot of 293T cells transiently transfected with double-mutated (p.V1512M/p.R1188X) SAMD9L plasmid revealed stable expression of the truncated protein (data not shown). In P2 and P7, the initial −7 disappeared and was replaced by a large somatic clone with uniparental disomy (UPD) 7q that duplicated the paternal wild-type SAMD9L allele (Figures 3 and 4).
Figure 3.

Mechanisms of clonal escape from SAMD9L germline mutations. Multiple mechanisms of clonal escape from damaging germline missense SAMD9L mutations are observed and lead to complete (monosomy 7) or partial (deletion 7q) loss of chromosome 7 with decreasing mutant SAMD9L allele (red circles), both situations can lead to MDS development; UPD7q and truncating somatic SAMD9L mutations (green circles), which have a benign outcome and contribute to normal hematopoiesis. Multiple clonal outcomes can occur in a single patient.
Figure 4.

Loss of mutated SAMD9L allele due to genomic deletion or mitotic recombination. Variant allelic frequency (VAF) scores for chromosome 7 in P1 and P2. single nucleotide polymorphisms and Indels detected using whole exome sequencing (~4000 variants with a VAF score >5% and <95%), show a complete loss of chromosome 7 in P1, as the VAF scores are either low or high. P2, unlike P1 demonstrates a partial loss of the chromosome 7 after position 7q11.22 towards the q terminal site. The read depth of the single nucleotide polymorphisms for P2 was maintained throughout for chromosome 7 with no loss thus confirming that loss of heterozygosity is due to UPD and not −7q. Whole exome sequencing VAF values are marked by a star within the graph. VAF: variant allelic frequency; UPD: uniparental isodisomy. Blue line: centromere; red line: SAMD9L gene position; yellow dotted line: start of UPD. For P7, targeted next-generation sequencing identified 14 informative (heterozygous) polymorphisms located on chromosome 7q with an average depth of 1036 reads (Online Supplementary Table S1). Single nucleotide polymorphisms are represented in a VAF graph depicting the skewing of heterozytosity towards one allelle occurring after position g.66098482 (rs3764903).
The natural history of SAMD9L-related myelodysplastic syndrome reveals divergent clinical outcomes
We next studied in detail the disease course in patients who were left untreated for longer periods of time. In P4, the evolution from normal karyotype to independent clones with −7 and del(7q) was rapid and occurred within a few months (Table 1). Cytogenetic progress was associated with partial recovery of complete blood counts and normalization of bone marrow cellularity, pointing to the possibility of hematopoietic stem cell niche repopulation by −7/del(7q) retaining only the wild-type SAMD9L allele. P1 developed CMML 3.6 years after the initial diagnosis of MDS/-7. This patient carried somatic driver mutations representing major subclones co-existing on the −7 background (Table 2). An additional somatic SAMD9L mutation p.R1188X (co-occurring in cis with p.V1512M) was found in a minor clonal fraction of approximately 6%.
In contrast, P2 and P7 showed an unexpected clinical course with spontaneous hematologic recovery, disappearing monosomy 7 and the presence of a large double wild-type UPD7q clone in the bone marrow (Figures 4 and 5). Both patients remained healthy, had normal follow-up bone marrow examinations with no signs of dysplasia (Table 1), and normal compete blood counts until their last follow-ups, 20 (P1) and 16 (P7) years after initial diagnosis.
Figure 5.

Clonal evolution and spontaneous reversion due to UPD7q. Clonal evolution model in P2 (D154) depicting disease history during an observation period of 20 years. At diagnosis, initial bone marrow harbored monosomy 7 (77% by fluorescence in situ hybridization and 51% by metaphase karyotyping). Blood counts normalized 3.7 years later and since then P2 maintained normal complete blood counts until last follow-up at the age of 22 years. From the age of 12 years, repeated yearly bone marrow examinations revealed normocellular hematopoiesis with no dysplasia and normal cytogenetics. Bone marrow collected at the age of 17 years (*) was subjected to whole exome and targeted deep sequencing. Germline heterozygous SAMD9L mutation p.V1512M was detected at a variant allelic frequency (VAF) of ~20%, corresponding to a clonal size of ~40% in a diploid chromosome 7 background. Concurrently, a spontaneous genetic correction of the SAMD9L locus occurred resulting from uniparental isodisomy (UPD)7q of paternal origin. This self-corrected clone occurred either initially (dotted line) or after termination of monosomy 7 and contributed to normal hematopoiesis. Abbreviations: Dx, diagnosis; pat, paternal origin; mat, maternal origin; UPD; uniparental isodisomy, LFU; last follow-up.
SAMD9L mutations inhibit cell proliferation
An inhibitory effect on cell proliferation reported for SAMD9L mutants overexpressed in 293FT cells in vitro was termed as gain-of-function. In contrast, ectopic expression of p.T233N was shown to mitigate cell proliferation to a lesser extent in comparison to wild-type SAMD9L, and was categorized as a disease-protective or loss-of-function variant.20 SAMD9L p.R986H was previously functionally studied and shown to be gain-of-function,20 while p.H880Q was shown to induce loss of heterozygosity by −7/del(7q) or UPD7q in an Epstein-Barr virus-transformed cell line in vitro.19 To determine the effect of the SAMD9L mutation identified in families I and III, we transiently transfected 293FT cells with vectors containing disease-associated mutations p.V1512M and p.R986C respectively, along with the loss-of-function variant p.T233N. Cell proliferation was assessed in dye dilution assays. Both SAMD9L p.V1512M and p.R986C decreased dye dilution in comparison to wild-type SAMD9L and p.T233N (Figure 6A,B), pointing to an amplified growth restrictive effect of the disease-associated variants (Figure 6B).
Figure 6.

Functional evaluation of SAMD9L mutations. (A,B) The effect of SAMD9L mutations on cell proliferation was assessed by dye dilution assays. 293FT cells were transiently transfected with TFP-SAMD9L wild type (WT), the disease-associated mutations p.R986C and p.V1512M, and the protective variant p.T233N previously reported by Tesi et al.20 (A) Histograms depict the dye levels in transfected cells. Dye levels were monitored in TFP-transfected cells (filled gray histograms) and compared to cells expressing uniformly intermediate levels of TFP-SAMD9L wild-type (blue histograms) or variants (red/orange lines), as indicated. A single representative experiment is shown. (B) Cumulative summary of three independent experiments on inhibition of cell proliferation associated with indicated TFP-SAMD9L mutations. Values (mean ± SD) are calculated based on a scale defined by 0 (dye levels in TFP-transfected cells) and −1 (dye levels in cells transfected with TFP-SAMD9L wild-type). Unpaired t-test, two tailed: *P<0.05; ** P<0.005; ***P<0.001.
Discussion
In this study, we describe a familial MDS syndrome caused by heterozygous missense mutations in the SAMD9L gene located on chromosome 7q21. We present seven individuals from four unrelated pedigrees who developed MDS/-7 from the age of 1 to 42 years without any neurological involvement. Unlike other reported SAMD9L mutation carriers,19,20 the sole clinical manifestation in our cases was hematologic. Other novel findings outlined here are the description of the somatic mutational landscape likely contributing to the progression of MDS, the observation of transient monosomy 7, and finally the occurrence of non-random revertant mosaicism leading to complete hematologic recovery.
The SAMD9L mutations p.R986H, p.R986C, and p.V1512M identified in this cohort affect evolutionarily highly conserved amino acid residues and are assessed as pathogenic by in silico prediction. The mutation p.H880Q (P7) shows a weak conservation score; however, this mutation had already been reported as causative for the ataxia-pancytopenia phenotype.19 We were not able to test SAMD9L genetics in P7, however the unclear ataxia that this patient had been evaluated for at his last visit points to a carrier status and indicates that there must be an overlap between sole hematologic and ataxia phenotypes in SAMD9L disease. Summarizing all SAMD9L mutations recently reported or identified in our cohort, a total of six germline mutations can be discerned (p.H880Q, p.I981T, p.R986H, p.R986C, p.C1196S, and p.V1512M). Of note, all these mutations cluster exclusively to the C-terminal half of the protein. Further, upon comparing reported mutations in the paralogue gene SAMD9 (p.R982H/C)22 with that of the present study in SAMD9L (p.R986H/C), we identified a potential mutational hotspot affecting highly conserved regions in both SAMD9L (p.984-989: GVRIIH) and SAMD9 (p980-985: GVRIIH) proteins.
The reported constitutional variants in both SAMD9/SAMD9L were classified as gain-of-function based on the observation of decreased cell proliferation in a 293T cell line.20–22 Similarly, we observed growth deficiency in 293T cells harboring SAMD9L p.V1512M and p.R986C. Based on these findings, one cautious speculation hints at a gain-of-function effect that is toxic to cells. This is supported by the discovery of an acquired stopgain SAMD9L mutation in P1 and P7 that likely “eliminates” germline missense mutations. In the cases studied here, complete or partial deletion of chromosome 7 and also UPD7q was non-random and each time resulted in loss of the germline-mutated SAMD9L gene copy. Additional studies, which are essential to further define the effect of SAMD9L variants, might be challenging due to the growth inhibitory effect of the alterations. It also remains to be determined whether SAMD9L missense mutations lead to increased protein stability, alter protein structure, enhance an unknown functional domain, or exert a completely neomorphic effect.
We describe three silent mutation carriers from separate families demonstrating no previous relevant medical history. Despite normal complete blood counts and mean corpuscular volume, the brother of P5 had a hypocellular marrow with mild dysplasia, evidently attributed to the identical pathogenic SAMD9L mutation. This finding emphasizes the need for thorough hematologic workup, including marrow studies, in potential sibling donors especially when they lack a genetic marker for familial disease. The intrafamiliar heterogeneity regarding the hematologic presentation remains elusive; one can speculate that other yet unknown genetic or epigenetic mechanisms might act as modifiers.
Thus far there is only limited knowledge about the regulation and cellular functions of SAMD9L. It has been postulated that both SAMD9L and the adjacent paralogous SAMD9 gene share functional redundancy, shaped by a long-term, possibly virus-induced selective pressure.30 Both genes can be upregulated by type I and II interfer-ons20,31–33 and by this might suppress inflammatory pathways and exert anti-viral properties.31,34 Although there was no evidence for a defined immunodeficiency in our cohort, three of the seven patients experienced recurrent respiratory infections with or without cytopenias before they developed RCC/-7.
Further evidence supports that SAMD9/SAMD9L genes act as tumor suppressors as their inactivation is associated with increased cellular proliferation, i.e. in normophosphatemic familiar tumoral calcinosis (SAMD9), in hepatitis B virus-associated hepatocellular carcinoma (SAMD9L), in MDS/acute myeloid leukemia with microdeletion in 7q21 (both genes), and in Samd9l-haploinsufficient mice. Based on the observations in this murine model, the cytokine-receptor complexes cannot be properly degraded due to impaired endosomal function in SAMD9L-haploinsufficient cells, which results in constitutive intracellular signaling with prolonged cell survival.18 Moreover, Samd9l+/− and Samd9l−/− mice develop MDS with normo/hypercellular bone marrow, drawing a parallel to human SAMD9L-related MDS in which the initial marrow hypocellularity associated with the “toxic” mutation is restored upon the loss of the mutant SAMD9L allele. This loss is accomplished through −7, del(7q) or UPD7q, and leads to a proliferative advantage with clonal expansion. In our cohort, although initially all patients presented with hypocellular marrow, longer observation periods in P1, P2, P4, and P7 revealed successive normalization of marrow cell content associated with increasing −7/del(7q) clone or the appearance of UPD7q. In P1, the leukemic progression was further aggravated by the accumulation of typical MDS driver mutations in SETBP1, KRAS and EZH2 within the −7 clone. P5 also demonstrated an acquired splice site mutation in RUNX1, a known leukemic driver gene. Notably, typical adult MDS driver mutations (i.e., TET2, DNMT3A, and IDH1/2) were not encountered in our cohort. This is in line with previously published findings discussing SETBP1, RAS pathway mutations and RUNX1 (identified in our SAMD9L-mutated patients) as recurrent drivers of pediatric MDS.10 Building on our observations we propose the following mechanism of MDS evolution in SAMD9L disease: the bone marrow attempts to circumvent the toxicity of the constitutional SAMD9L mutation and selects for fitter, yet premalignant −7/del(7q) clones (with only one wild-type SAMD9L copy), or benign clones with truncated SAMD9L (Figure 3). Over time the resulting haploinsufficiency of tumor supressor genes on 7q (e.g. EZH2 or CUX1) in all patients likely provides the first step towards progression. Finally, additional driver somatic mutations might be encountered in some but not all patients.
Somatic revertant mosaicism has been reported in inherited bone marrow failure syndromes with hypocellular bone marrow, including telomeropathy with germline mutations in TERT,35 and Fanconi anemia in which mosaicism in blood occurs at rates of up to 140 times higher than in the general population.36,37 However, in general, revertant mosaicism is a rare facet to clonal hematopoiesis because spontaneous correction of the pathogenic allele is a random event. In our study, we report two patients (P2 and P7) who presented with RCC/-7 at young age and demonstrated complete hematopoietic remission with normal cytogenetics throughout an observation period ranging from 16 to 20 years. The clinical picture of these patients fits the previously described transient monosomy 7 syndrome; to our knowledge eight patients with primary MDS with transient −7 or del(7q) have been reported in the literature.14–16,38–40 Their ages at diagnosis ranged from 8 months to 3 years, and spontaneous remission was achieved within 1–20 months from diagnosis. It seems that the −7 clones in our patients either vanished spontaneously or were outcompeted by “fitter” UPD7q-corrected clones with a diploid copy of the wild-type SAMD9L allele. Based on these observations, a watch-and-wait strategy might be proposed for younger patients with RCC/-7 who have no additional somatic driver mutations and are clinically stable. However, prolonged “watchful waiting” poses the risk of progression as witnessed in P1, who developed CMML and acquired oncogenic mutations 3.6 years after he was diagnosed with RCC.
In conclusion, our observations establish the molecular basis of a distinct subtype of familial MDS and point to the notion that MDS with chromosome 7 loss can be the sole and common manifestation of SAMD9L-related disease. The negative mutational effect leads to escape and outgrowth of clones carrying −7/del(7q) with only wild-type SAMD9L allele, which might spontaneously dissapear or persist and provide the first step towards disease progression. Finally, this is the first description of long-term revertant mosaicism due to non-random UPD7q in SAMD9L disease, and a plausible explanation for transient monosomy 7 syndrome.
Supplementary Material
Acknowledgments
We are grateful to Sophia Hollander, Christina Jäger, Yahaira Pastor, Alexandra Fischer, Wilfried Truckenmüller (Freiburg), Tamara Szattler (Stockholm) and Bart Przychodzien (USA) for their excellent laboratory assistance and data management, and to Dr. Anett Schmidt, Dr. Dagmar Möbius, and Dr. Elisabeth Holfeld (Cottbus) for clinical care. The authors are grateful to the Genomics Core Facility at the German Cancer Research Center/DKFZ, Heidelberg, Germany for performing whole exome sequencing and Claritas Genomics (Cambridge, MA, USA) for targeted sequencing in P4.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/3/427
Funding
Deutsche Krebshilfe (Max Eder grant #109005) to MWW, BMBF (DKTK German Cancer Consortium, topic molecular diagnostics of pediatric malignancies) to CMN and MWW, BMBF (e:Med FKZ 01ZX1409B) to MB, the German Science Foundation (DFG, SFB 850, to MB and EXE306 to HB), the Research Council, Swedish Cancer Society, and Foundation for Strategic Research to YTB. MWW and ME are past trainees of EHA-ASH translational research training in hematology. MV is an EMBO Long-term Fellow (ALTF 206-2015 co-funded by the European Commission (LTFCOFUND2013, GA-2013-609409). JCA is a Wellcome Trust Senior Research Fellow in Clinical Science (grant 098513/Z/12/Z) with support from the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children, NHS Foundation Trust and UCL, and Great Ormond Street Children’s Charity. This study was supported in part by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School).
References
- 1.Churpek JE, Godley LA. How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood. 2016;128(14):1800–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43(5):598–608. [DOI] [PubMed] [Google Scholar]
- 3.Hahn CN, Chong CE, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351(23):2403–2407. [DOI] [PubMed] [Google Scholar]
- 5.Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175. [DOI] [PubMed] [Google Scholar]
- 6.Pippucci T, Savoia A, Perrotta S, et al. Mutations in the 5′ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet. 2011;88(1):115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polprasert C, Schulze I, Sekeres MA, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell. 2015;27(5):658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wlodarski MW, Hirabayashi S, Pastor V, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–1397. [DOI] [PubMed] [Google Scholar]
- 10.Pastor V, Hirabayashi S, Karow A, et al. Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia. 2017;31(3):759–762. [DOI] [PubMed] [Google Scholar]
- 11.Churpek JE, Pyrtel K, Kanchi KL, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015;126(22):2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gohring G, Michalova K, Beverloo HB, et al. Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood. 2010;116(19):3766–3769. [DOI] [PubMed] [Google Scholar]
- 13.Kardos G, Baumann I, Passmore SJ, et al. Refractory anemia in childhood: a retrospective analysis of 67 patients with particular reference to monosomy 7. Blood. 2003;102(6):1997–2003. [DOI] [PubMed] [Google Scholar]
- 14.Mantadakis E, Shannon KM, Singer DA, et al. Transient monosomy 7 - A case series in children and review of the literature. Cancer. 1999;85(12):2655–2661. [DOI] [PubMed] [Google Scholar]
- 15.Parker TM, Klaassen RJ, Johnston DL. Spontaneous remission of myelodysplastic syndrome with monosomy 7 in a young boy. Cancer Genet Cytogenet. 2008;182(2):122–125. [DOI] [PubMed] [Google Scholar]
- 16.Leung EW, Woodman RC, Roland B, Abdelhaleem M, Freedman MH, Dror Y. Transient myelodysplastic syndrome associated with isochromosome 7q abnormality. Pediatr Hematol Oncol. 2003;20(7):539–545. [DOI] [PubMed] [Google Scholar]
- 17.Asou H, Matsui H, Ozaki Y, et al. Identification of a common microdeletion cluster in 7q21.3 subband among patients with myeloid leukemia and myelodysplastic syndrome. Biochem Biophys Res Commun. 2009;383(2):245–251. [DOI] [PubMed] [Google Scholar]
- 18.Nagamachi A, Matsui H, Asou H, et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell. 2013;24(3):305–317. [DOI] [PubMed] [Google Scholar]
- 19.Chen DH, Below JE, Shimamura A, et al. Ataxia-pancytopenia syndrome is caused by missense mutations in SAMD9L. Am J Hum Genet. 2016;98(6):1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tesi B, Davidsson J, Voss M, et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS and neurological symptoms. Blood. 2017;129(16):2266–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Narumi S, Amano N, Ishii T, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48(7):792–797. [DOI] [PubMed] [Google Scholar]
- 22.Buonocore F, Kuhnen P, Suntharalingham JP, et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest. 2017;127(5):1700–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz JR, Wang S, Ma J, et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia. 2017;31(8):1827–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumann I, Niemeyer CMBJ, Shannon K. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008, 2008:104–107. [Google Scholar]
- 25.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classifi cation of the myeloid neoplasms. Blood. 2002;100(7):2292–2302. [DOI] [PubMed] [Google Scholar]
- 26.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862–864. [DOI] [PubMed] [Google Scholar]
- 28.Wlodarski MW, O’Keefe C, Howe EC, et al. Pathologic clonal cytotoxic T-cell responses: nonrandom nature of the T-cell-receptor restriction in large granular lymphocyte leukemia. Blood. 2005;106(8):2769–2780. [DOI] [PubMed] [Google Scholar]
- 29.Vraetz T, Emanuel PD, Niemeyer CM. In vitro regulation of colony stimulating factor-mediated hematopoiesis in healthy individuals and patients with different types of myeloproliferative disease. Methods Mol Biol. 2003;215:293–309. [DOI] [PubMed] [Google Scholar]
- 30.Lemos de Matos A, Liu J, McFadden G, Esteves PJ. Evolution and divergence of the mammalian SAMD9/SAMD9L gene family. BMC Evol Biol. 2013;13:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chefetz I, Ben Amitai D, Browning S, et al. Normophosphatemic familial tumoral calcinosis is caused by deleterious mutations in SAMD9, encoding a TNF-alpha responsive protein. J Invest Dermatol. 2008;128(6):1423–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka M, Shimbo T, Kikuchi Y, Matsuda M, Kaneda Y. Sterile alpha motif containing domain 9 is involved in death signaling of malignant glioma treated with inactivated Sendai virus particle (HVJ-E) or type I interferon. Int J Cancer. 2010;126(8):1982–1991. [DOI] [PubMed] [Google Scholar]
- 33.Hershkovitz D, Gross Y, Nahum S, et al. Functional characterization of SAMD9, a protein deficient in normophosphatemic familial tumoral calcinosis. J Invest Dermatol. 2011;131(3):662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Wennier S, Zhang L, McFadden G. M062 is a host range factor essential for myxoma virus pathogenesis and functions as an antagonist of host SAMD9 in human cells. J Virol. 2011;85(7):3270–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jongmans MCJ, Verwiel ETP, Heijdra Y, et al. Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet. 2012;90(3):426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waisfisz Q, Morgan NV, Savino M, et al. Spontaneous functional correction of homozygous fanconi anaemia alleles reveals novel mechanistic basis for reverse mosaicism. Nat Genet. 1999;22(4):379–383. [DOI] [PubMed] [Google Scholar]
- 37.Reina-Castillon J, Pujol R, Lopez-Sanchez M, et al. Detectable clonal mosaicism in blood as a biomarker of cancer risk in Fanconi anemia. Blood Advances. 2017;1(5):319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagasawa M, Tomizawa D, Tsuji Y, et al. Pancytopenia presenting with monosomy 7 which disappeared after immunosuppressive therapy. Leuk Res. 2004;28(3):315–319. [DOI] [PubMed] [Google Scholar]
- 39.Benaim E, Hvizdala EV, Papenhausen P, Moscinski LC. Spontaneous remission in monosomy 7 myelodysplastic syndrome. Br J Haematol. 1995;89(4):947–948. [DOI] [PubMed] [Google Scholar]
- 40.Scheurlen W, Borkhardt A, Ritterbach J, Huppertz HI. Spontaneous hematological remission in a boy with myelodysplastic syndrome and monosomy 7. Leukemia. 1994;8(8):1435–1438. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.