

Abstract
In patients with dysfunctions of the Ca2+ channel ORAI1, stromal interaction molecule 1 (STIM1) or integrin-regulating kindlin-3 (FERMT3), severe immunodeficiency is frequently linked to abnormal platelet activity. In this paper, we studied platelet responsiveness by multiparameter assessment of whole blood thrombus formation under high-shear flow conditions in 9 patients, including relatives, with confirmed rare genetic mutations of ORAI1, STIM1 or FERMT3. In platelets isolated from 5 out of 6 patients with ORAI1 or STIM1 mutations, store-operated Ca2+ entry (SOCE) was either completely or partially defective compared to control platelets. Parameters of platelet adhesion and aggregation on collagen microspots were impaired for 4 out of 6 patients, in part related to a low platelet count. For 4 patients, platelet adhesion/aggregation and procoagulant activity on von Willebrand Factor (VWF)/rhodocytin and VWF/fibrinogen microspots were impaired independently of platelet count, and were partly correlated with SOCE deficiency. Measurement of thrombus formation at low shear rate confirmed a greater impairment of platelet functionality in the ORAI1 patients than in the STIM1 patient. For 3 patients/relatives with a FERMT3 mutation, all parameters of thrombus formation were strongly reduced regardless of the microspot. Bone marrow transplantation, required by 2 patients, resulted in overall improvement of platelet function. We concluded that multiparameter assessment of whole blood thrombus formation in a surface-dependent way can detect: i) additive effects of low platelet count and impaired platelet functionality; ii) aberrant ORAI1-mediated Ca2+ entry; iii) differences in platelet activation between patients carrying the same ORAI1 mutation; iv) severe platelet function impairment linked to a FERMT3 mutation and bleeding history.
Introduction
Severe, genetically linked immunodeficiency can be accompanied by platelet function defects, especially in cases of rare mutations in the ORAI1, STIM1 and FERMT3 genes on platelet properties, in spite of solid evidence for a role of the mouse orthologs in arterial thrombosis.
In platelets and other blood cells, stromal interaction molecule 1 (STIM1) acts as a major Ca2+ sensor located in the endoplasmic reticulum. When the reticular Ca2+ concentration is lower, it assembles with the ORAI1 Ca2+ (ICRAC) channels in the plasma membrane to mediate store-operated Ca2+ entry (SOCE).1–4 Therefore, the conventional test for SOCE (i.e. for STIM1 and ORAI1 activity) is to provoke depletion of the STIM1-linked Ca2+ store with the endoplasmic Ca2+ -ATPase inhibitor thapsigargin, and then measure Ca2+ entry through ORAI1 channels upon addition of extracellular CaCl2.5,6
Both ORAI1 and STIM1 have non-redundant roles in patroling and defense functions of white blood cells. In platelets, ORAI1 as well as STIM1 is considered to enhance the Ca2+ signal generation, induced in particular by protein tyrosine kinase-linked receptors, such as glycoprotein VI (GPVI). In mouse, both ORAI1 and STIM1 are implicated in hemostasis and arterial thrombus formation.7–10 Murine knockout studies have indicated that the ORAI1-STIM1 Ca2+ signaling contributes to multiple platelet activation processes, such as adhesiveness via integrin activation, granule release, aggregation of platelets, and procoagulant activity.8,11,12 However, in humans, the consequences of defective ORAI1 or STIM1 activity in platelets have not been thoroughly investigated.
In humans, dysfunctional mutations in the ORAI1 or STIM1 genes are very rare.13 The patients described with such mutations usually suffer from severe immunodeficiency, congenital myopathy, ectodermal dysplasia or other Ca2+ -linked abnormalities.14–16 The immune deficiency can be attributed to the loss-of-function of leukocyte and lymphocyte subsets. Given the severity of the symptoms, often already evident at a young age, these patients are commonly treated by allogeneic hematopoietic stem cell transplantation. The few patients described do not have an overt bleeding history, although they may periodically show autoimmune thrombocytopenia.16
Leukocyte adhesion deficiency type III (LAD-III) forms another severe immune disease, in this case accompanied by epistaxis or petechiae. It is associated with dysfunctional mutations in the FERMT3 gene of the integrin-regulation protein, kindlin-3.13 LAD-III patients present with normal platelet count, but impaired platelet adhesion, which may explain the bleeding symptoms.17,18 These patients may require hematopoietic stem cell transplantation during childhood.19
Recently, we developed a microspot-based assay for multiparameter assessment of whole blood thrombus formation under flow conditions.20 This assay proved to be valuable in characterizing platelet count and function abnormalities in patients with a variety of genetic bleeding disorders. Here, we used this integrative method to investigate overall platelet functions in 9 patients with severe immunodeficiencies, including parents, with a confirmed dysfunctional mutation in ORAI1, STIM1 or FERMT3. For 2 patients, we also investigated the effects of bone marrow transplantation. The results suggest a variable phenotypic penetrance on platelet properties in patients and relatives carrying these mutations.
Methods
Patients and controls
Blood was drawn from patients and healthy controls after full informed consent in compliance with the Declaration of Helsinki. The studies were approved by the local medical ethics committees. Healthy controls had normal blood cell counts, had not received anti-platelet medication for at least two weeks, and did not have a known history of bleeding or immunodeficiency.
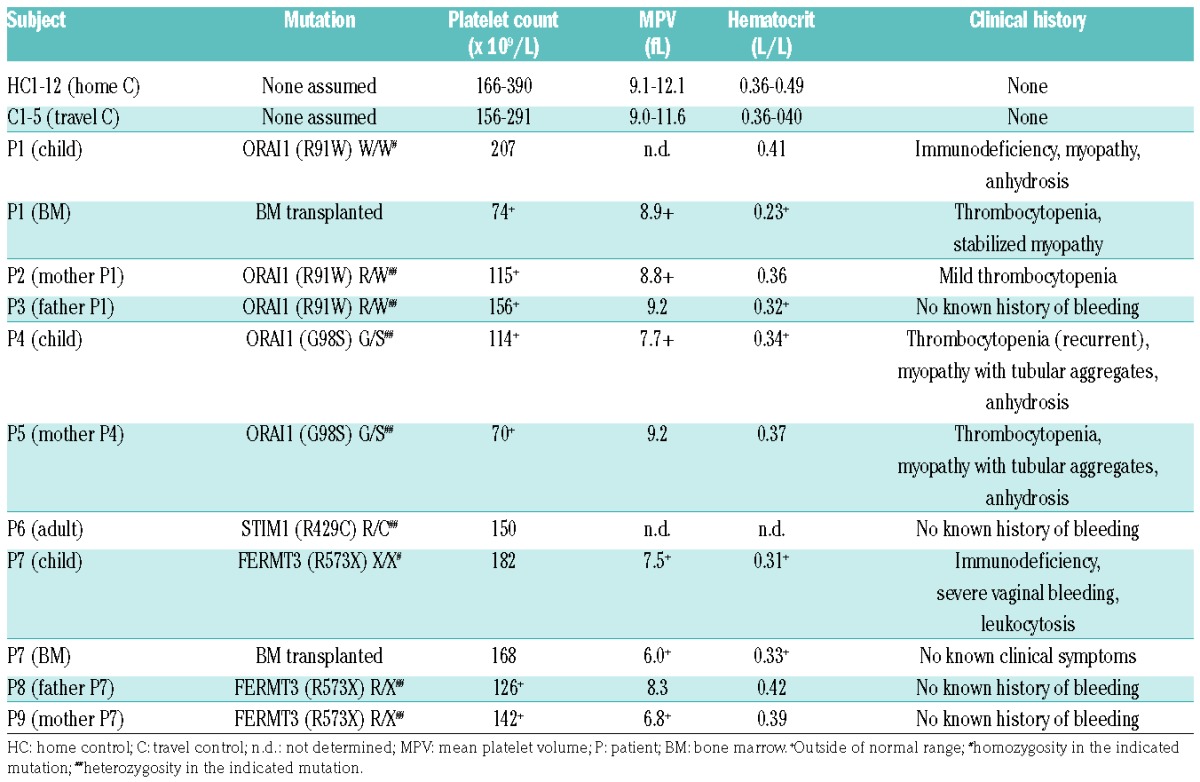
Blood samples from patients with immunodeficiency and their parents (all genotyped) were collected at the Department of Pediatrics and Adolescent Medicine, University Medical Center in Freiburg, and at the Children’s Hospital, University of Ulm, Germany (Table 1). Simultaneously, blood samples were taken from healthy donors serving as daily travel controls. Relevant patients’ characteristics, including identified mutations and clinical history, are summarized in Table 1. Patient P1 showed homozygosity in the R91W mutation in the ORAI1, known to be linked to severe combined immunodeficiency and defective T-cell Ca2+ signaling.1 Heterozygosity in this mutation was confirmed for both parents (P2 and P3). Patient P4 carried a heterozygous G98S mutation in ORAI1, which was confirmed in the mother (P5). This mutation is reported to be linked to tubular aggregated myopathy-2.21 Patient P6 carried a heterozygous R429C mutation of STIM1, which is also linked to T-cell immunity.22 Patient P7 suffered from severe immunodeficiency, linked to a homozygous R573X mutation in kindlin-3 encoded by the FERMT3 gene. The 2 parents (P8 and P9) were heterozygous for this mutation. Two of the patients with severe immunodeficiency, P1 and P7, were eligible for bone marrow transplantation. Blood samples in these cases were obtained before and at two or three months after transplantation, respectively. Only limited volumes of blood were available for investigation due to the young age of the patients.
Table 1.
Subjects’ characteristics.
Platelet Ca2+ responses
Rises in cytosolic [Ca2+]i were measured in platelets after loading with Fura-2 acetoxymethyl ester (2.5 μM) by calibrated ratio fluorometry.23 Data are presented as nM increases in cytosolic [Ca2+].24
Multiparameter thrombus formation on microspots under flow
Whole blood thrombus formation was assessed under flow conditions, basically as described before.20 In brief, blood samples (0.5 mL) were re-calcified in the presence of thrombin inhibitors, and perfused over a coverslip coated with three microspots (spot 1: type I collagen; spot 2: von Willebrand Factor (VWF)/rhodocytin; spot 3: VWF/fibrinogen) in a transparent parallel-plate perfusion chamber. Perfusion was at high wall-shear rate of 1600 s−1 for 3.5 minutes (min), or at a low shear rate of 150 s−1 for 6.0 min. The thrombi formed on the microspots were immediately post stained with FITC-labeled anti-fibrinogen mAb (1:100), FITC-anti-CD62P mAb (25 μg/mL) and AF647-annexin A5 (0.25 μg/mL). Representative brightfield and fluorescence images were captured from each microspot in real-time without fixation. Duplicate runs were performed whenever possible. Analysis of brightfield and fluorescence images, giving 7 parameters per microspot, was performed with pre-defined scripts in Fiji software.25 (Un)supervised heatmaps using scaled parameters (range 0–10) of thrombus formation were constructed using R software.
Statistical analysis
Significance of differences was determined with the paired sample t-test (intervention effects) and by principal component analysis (PCA), using the statistical package for social sciences (SPSS, v.11.0).
A detailed description of the methods used is available in the Online Supplementary Appendix.
Results
Variable aberrances in SOCE in platelets from patients with ORAI1 or STIM1 mutations
Blood samples were obtained from 3 patients with a mutation in the Ca2+ flux-regulating proteins ORAI1 or STIM1, as well as from parents carrying the same mutation. For comparison, blood samples were also taken from two cohorts of healthy subjects, i.e. a group of normal home controls (HC1-12) and a group of normal travel controls (C1-6). Using Fura-2-loaded platelets, we first evaluated the alterations in Ca2+ signaling in response to the GPVI receptor agonist, convulxin, the PAR receptor agonist, thrombin, and the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor, thapsigargin. In each case, the platelets were first stimulated with agonist in Ca2+-free medium, after which extracellular CaCl2 was added to measure secondary Ca2+ entry.
In comparison to control platelets, platelets from patient P1, carrying a homozygous ORAI191W/W mutation, responded normally to each agonist, but showed a substantially reduced Ca2+ entry following stimulation with convulxin but not thrombin (Figure 1A and B). Markedly, in this patient’s platelets, Ca2+ entry after stimulation with thapsigargin (as a default condition for SOCE) was completely abolished. This pointed to a complete absence of the STIM1-ORAI1 pathway, similar to that established for platelets from STIM1- or ORAI1-deficient- mice.7,8 Flow cytometric evaluation indicated that convulxin-stimulated platelets from P1 showed a reduced phosphatidylserine (PS) exposure [7±1% vs. 28±2% for control platelets, mean±SEM, n=3; P<0.05], but were not altered in integrin αIIbβ3 activation or P-selectin expression (P>0.10). Platelets from the patient’s parents (both with confirmed heterozygosity) were diminished in SOCE, but to a different extent (Figure 2). Platelets from the mother (P2) showed a nearly annulled Ca2+ entry, whereas platelets from the father (P3) were less severely reduced when compared to platelets from two cohorts of healthy controls. It was possible to obtain a second blood sample from patient P1 at two months after bone marrow transplantation. The platelets showed a 50% recovery in SOCE signal after thapsigargin stimulation (SOCEP1: 22 nM vs. SOCEP1-BM: 575 nM).
Figure 1.

Defective Ca2+ entry in platelets from a patient with a homozygous R91W mutation in ORAI1. Fura-2-loaded platelets from patient P1 (ORAI1W/W) and travel control C1 were used, suspended in Hepes buffer with 0.1 mM EGTA. Rises in cytosolic Ca2+ were measured in time upon stimulation with convulxin (Cvx, 50 ng/mL), thrombin (Thr, 4 nM) or thapsigargin (Thaps, 1.0 μM). After a defined time, CaCl2 (2 mM) was added to induce Ca2+ entry. Representative traces (A) and quantification (B) of Ca2+ increases in platelets from control subject and patient P1. Note that the maximal Ca2+ rise in response to CaCl2 following thapsigargin was used as a measure of store-operated Ca2+ entry (SOCE) capacity.
Figure 2.

Variably defective Ca2+ entry in platelets from patients with ORAI1W/W, ORAI1G/S or STIM1R/C mutations. Fura-2-loaded platelets in Hepes buffer with 0.1 mM EGTA were stimulated with thapsigargin (1.0 μM), and after a defined time with CaCl2 (2 mM). Platelets were analyzed from 12 healthy home controls (HC1-12), 6 healthy travel controls (C1-6), and the indicated patients/relatives (P1-6). Platelets from patient P1 were also analyzed after bone marrow (BM) transplantation. Shown are maximal increases in Ca2+. Dotted lines indicate range of store-operated Ca2+ entry (SOCE) levels (CaCl2-induced Ca2+ rise after thapsigargin) for platelets from home controls (mean ± SD, for HC1-12).
Platelets from one patient (P4), carrying a heterozygous mutation ORAI189G/S in the same protein region, responded differently. These platelets displayed a high SOCE signal (Ca2+ entry after thapsigargin). In contrast, platelets from parent P5 (also carrying the mutation) were greatly reduced in SOCE (Figure 2). This difference was confirmed for a second set of blood samples (data not shown). A different picture was obtained with patient P6 with a heterozygous mutation in STIM1429R/C.22 In these platelets, increases in Ca2+ evoked by convulxin/CaCl2 or thrombin/CaCl2 were in the normal range, whereas SOCE after thapsigargin was completely abolished (Figure 3A and B).
Figure 3.

Defective Ca2+ entry in platelets from a patient with a heterozygous R429C mutation in STIM1. Fura-2-loaded platelets from patient P6 (STIM1R/C) and travel control C6, suspended in Hepes buffer with 0.1 mM EGTA, were stimulated with convulxin (Cvx, 50 ng/mL), thrombin (Thr, 4 nM) or thapsigargin (Thaps, 1.0 μM), after which CaCl2 (2 mM) was given to induce Ca2 entry. Representative traces (A) and quantification (B) of Ca2+ rises in platelets from control subject and patient P6.
Phenotypic analysis of platelets was also performed of a LAD-III patient (P7) who carried a homozygous FERMT3573X/X mutation, and presented with immunodeficiency and a history of bleeding.26 Flow cytometry indicated an almost complete lack of integrin αIIbβ3 activation in response to receptor agonists, as observed in another patient carrying this mutation.27 In platelets from the 2 heterozygous parents (P8, P9), αIIbβ3 integrin activation was in the normal range (data not shown).
Lower range blood cell counts in patients with ORAI1, STIM1 or FERMT3 mutations
Table 1 provides an overview of the clinical histories of patients P1-9 including hematologic parameters determined in freshly isolated blood samples. In all patients, with the exception of P1 and P7, platelet counts were between 70 and 150×109/L, which are slightly below the normal ranges presented in both control cohorts. In the majority of patients, also hematocrit levels were in the lower range of normal, i.e. between 0.31 and 0.42 L/L. After bone marrow transplantation (2–3 months), platelet count in P1 and P7 had restored to 74 and 168×109/L, respectively.
Different patterns between patients of aberrant whole-blood thrombus formation at high shear rate
To obtain detailed insight into the hemostatic potential of the patients’ platelets, whole blood samples were used for multiparameter assessment of thrombus formation under flow at high wall-shear rate of 1600 s−1. Samples from corresponding control subjects (C1-6) and home control subjects (HC1-12) were again used for comparison. This high-throughput method, previously established,20 allowed simultaneous examination of platelet adhesion, aggregation and activation at three microspots: spot 1 with coated type I collagen (involving platelet receptors: GPIb-V-IX, GPVI and integrin α2β1); spot 2 with coated VWF/rhodocytin (receptors GPIb-V-IX and CLEC-2); and spot 3 with coated VWF/fibrinogen (receptors GPIb-V-IX and αIIbβ3). From each microspot, microscopic brightfield images were recorded to assess platelet adhesion and aggregation (parameters V1-4); in parallel, fluorescence images were recorded in three colors for detection of PS exposure, P-selectin expression and αIIbβ3 activation (parameters V5-7).
Representative examples of brightfield and fluorescence images from spot 1 for blood samples of control C2 and immunodeficient patients with ORAI1 or STIM1 mutation (P1, P4, and P6) are shown in Figure 4. Regarding the patient blood samples, the images show patterns of platelet adhesion, aggregation and activation that vary from from normal to reduced, as was also observed for spot 2 (VWF/rhodocytin) and spot 3 (VWF/fibrinogen) (see below).
Figure 4.

Altered thrombus formation of blood from patients with ORAI1W/W, ORAI1G/S or STIM1R/C mutations. Whole blood from indicated control subjects and patients was perfused over three microspots [(Sp1, collagen type I; Sp2, von Willebrand Factor (VWF)/rhodocytin; Sp3, VWF/fibrinogen; downstream → upstream)] at wall-shear rate of 1600 s−1. After 3.5 minutes of perfusion, brightfield images were taken from thrombi on all microspots, and platelets were stained for phosphatidylserine (PS) exposure (AF568-annexin A5), P-selectin expression (AF647 anti-CD62P mAb), and integrin αIIbβ3 activation (FITC anti-fibrinogen mAb). Shown are representative brightfield and fluorescence images at spot 1, obtained with blood from control C2 and patients P1, P4 and P6 (bars, 50 μm).
A heatmap was constructed with data from all analyzed images (3 spots × 7 parameters) for the cohort of 12 normal home controls (HC1-12), the 6 travel controls (C1-6), and the 9 individual patients/relatives with mutations in ORAI1 (P1-5), STIM1 (P6) or FERMT3 (P7-9), in which all values were normalized to a scale of 0–10 per parameter (Figure 5A). In the derived subtraction heatmap of Figure 5B, the patient data were expressed relative to those from the home controls. This subtraction confirmed an overall high similarity between the values of the two control groups (HC1-12 and C1-6), with the exception of a parameter reflecting platelet deposition on spot 2. Datasets for individual subjects C1-6 were within the normal ranges (data not shown). This confirmed the usefulness of the multiparameter test,20 and underscored the quality of the analyzed blood samples. For each patient/relative, we arbitrarily set a relevant effect threshold, i.e. when outside the range of mean±2 SD of the control group (HC1-12). This filter produced a ‘relevant’ subtraction heatmap, indicating distinct patterns of altered parameters of thrombus formation for individual patients P1-9 (Figure 5C).
Figure 5.

Integrated analysis of thrombus formation for patients with ORAI1, STIM1 or FERMT3 mutations. Thrombus formation on three microspots was measured with blood from home controls (HC1-12), travel controls (C1-6) and indicated patients/relatives with a genetic mutation in ORAI1 (P1-5), STIM1 (P6) or FERMT3 (P7-9), at wall-shear rate of 1600 s−1, as for Figure 4. Coding of microspots: Sp1, collagen type I; Sp2, von Willebrand Factor (VWF)/rhodocytin; Sp3, VWF/fibrinogen. Coding of outcome parameters: V1, thrombus morphological score (scale 0-5); V2, platelet surface area coverage (% SAC); V3, thrombus contraction score (scale 0-3); V4, thrombus multilayer score (scale 0-3); V5, phosphatidylserine (PS) exposure (% SAC); V6, P-selectin expression (% SAC); V7, αIIbβ3 activation (% SAC). Data were scaled per parameter from 0-10. (A) Heatmap of scaled values for control groups HC1-12 and C1-6 (means); and of scaled values for individual patients (*) and relatives. (B) Subtraction heatmap of scaled values, compared to those from HC1-12. (C) Subtraction heatmap after filtering for differences considered to be relevant, i.e. outside the range of mean ± 2 SD (HC1-12).
Figure 5C underlines that, overall, multiple parameters on spot 1 were reduced for patients P2-5, whereas especially parameters on spots 2–3 were reduced for patients P1 and P5. Interestingly, patient P4 carrying the assumed gain-of-function mutation ORAI1G98S (but not relative P5 with low SOCE) showed a typical increase in platelet PS exposure. For the patient with homozygous FERMT3 mutation (P7), a more severe reduction on all three spots was seen in comparison to the 2 heterozygous relatives (P8 and P9).
Effects of low platelet count
Considering that whole blood thrombus formation can be influenced by not only the inherited platelet disorder, but also a low platelet count,28 the present heatmaps may reflect both platelet-related properties. To examine this in more detail, the subtraction heatmap data were extended with values of platelet count and SOCE. Unsupervised clustering of the extended dataset indicated that particularly spot 1, parameters (V1-4) clustered with platelet count, while spot 2 parameters (V3-5) and PS exposure (Sp1V5, Sp2V5) clustered with altered SOCE (Online Supplementary Figure S1).
As an alternative approach, we used the primary (non-normalized) data of thrombus formation (Sp1-3, V1-7), platelet count and SOCE for PCA. This revealed a similar pattern, in that the majority of spot 1 parameters together with platelet count determined component 1, whereas most spot 2 parameters determined component 2 (Online Supplementary Figure S2A). Pearson regression coefficient confirmed a correlation of spot 1 parameters with platelet count, and also a correlation of platelet PS exposure (Sp1V5, Sp3V5) with SOCE (Online Supplementary Figure S2B).
This information was then used to interpret the alterations in thrombus formation for individual patients (see clustered heatmap in Online Supplementary Figure S1). Concerning the patient (P1) with homozygous R91W mutation in ORAI1, with normal platelet count (207×109/L) and near abolished SOCE, thrombus formation was near normal on spot 1 (collagen), but markedly reduced on spot 2 (VWF/rhodocytin) and spot 3 (VWF/fibrinogen). The impaired Ca2+ signal was linked to a low PS exposure on all spots (Online Supplementary Table S1). On the other hand, for the patients P2 and P3 with heterozygous R91W mutation in ORAI1 (relatively low platelet counts of 115–156×109/L, and 20–60% of normal SOCE, respectively), particularly parameters for spot 1 were below normal, with a reduced PS exposure.
Concerning patients P4 and P5, heterozygous for the G98S mutation in ORAI1 (low platelet counts of 70 and 114×109/L; 100% and 30% of normal SOCE, respectively), thrombus formation on spot 1 was reduced, while parameters of thrombus formation on spots 2–3 (including PS exposure) were only reduced for patient P5. In P4 (but not P5), a gain-of-function of Ca2+ channel activity was apparent from an increased PS exposure on spots 2–3. For patient P6, heterozygous for the R429C mutation in STIM1, thrombus formation on spot 1 (collagen) was normal in spite of the abolished SOCE. Together, this reinforced the idea that a low platelet count rather than altered SOCE determines the thrombus formation on collagen, but not on the other surfaces.
Control experiments with reconstituted blood confirmed that, for control donors, lowering of the platelet count to 100×109/L had only a limited effect on particular parameters of thrombus formation limited to spot 1, whereas lowering to 50×109/L affected thrombus formation on all spots 1–3 (Online Supplementary Figure S3). Taken together with the predictive effect of platelet count in the PCA, this suggests that a relatively low count affects thrombus formation under flow conditions more severely when combined with a reduced platelet functionality.
Patterns of aberrant whole-blood thrombus formation at low shear rate
Using remaining samples from patients/relatives P1-6, we went on to perform whole blood flow measurements at a low shear rate of 150 s-1. Subtraction heatmap analysis of the normalized parameter values, in comparison to control data from HC1-12 cohort, indicated for all patients with ORAI1 mutations (P1-5) a consistent pattern of reduced thrombus formation parameters on spot 1 (Online Supplementary Figure S4A–C). In particular for patients P2 and P3, markers of platelet activation appeared to be reduced (PS exposure, P-selectin expression and αIIbβ3 activation). Once again, for patient P6 with STIM1 mutation, most parameters were unchanged.
Principal component analysis of the low shear data again pointed to a linkage of spot 1 parameters with platelet count and a linkage of Sp1V5 (PS exposure) with SOCE activity (Online Supplementary Figure S5A and B).
Partial recovery of whole blood thrombus formation after bone marrow transplantation
Blood samples from 2 patients were also obtained after bone marrow transplantation (P1 after 2 months, P7 after 3 months), and used for platelet activation and thrombus formation studies. Transplantation of patient P1 led to a partially recovered SOCE from 20% to 60% of normal (Figure 2). After transplantation, parameters of thrombus formation were particularly reduced on spot 1 (paralleling a reduction in platelet count from 207 to 74×109/L), but were unchanged for spot 2, and enhanced for spot 3 (Figure 6). Transplantation of patient P7 resulted in an overall improvement of most parameters on all spots (platelet count decreased from 182 to 168×109/L).
Figure 6.

Altered thrombus formation after bone marrow transplantation. Thrombus formation (1600 s−1) on three microspots measured with blood from indicated home controls (HC1-12), travel controls (C1-6) and 2 patients P1 (ORAI1W/W) and P7 (FERMT3R/X), both before (*) and after bone marrow transplantation (BMT). For description of microspots and parameters, see Figure 4. Subtraction heatmap of scaled values after filtering for differences considered to be relevant, i.e. outside the range of mean ± 2 SD (HC1-12).
Discussion
In the present study, we used a multiparameter test of whole blood thrombus formation under flow conditions, as a proxy measurement of hemostatic activity20 to characterize quantitative and qualitative platelet abnormalities in rare patients and their relatives with severe immunodeficiencies, linked to signaling protein defects and mutations in the ORAI1, STIM1 and FERMT3 genes. So far, functional effects of the ORAI1 and STIM1 mutations have only been described for human immune cells or cell lines. Hence, the present data are the first to report on comparative alterations in platelet SOCE and platelet functions in as many as 9 genotyped patients/relatives.
Aberrations in SOCE accompanied by mutations in ORAI1 or STIM1
Earlier work with bone marrow chimeric mice with megakaryocytic deficiency in Orai1 or Stim1 demonstrated a prominent role of these Ca2+-entry regulating proteins in platelet calcium homeostasis and activation, including PS exposure.11,29 Our human data are compatible with these findings in that SOCE after thapsigargin or GPVI stimulation appeared to be impaired in platelets from patient P1 with a homozygous ORAI1W/W mutation. A similar impairment of SOCE has been reported in platelets from Orai1R93W mice, i.e. a loss-of-function mutation orthologous to the human ORAI1 R91W variant.29 Platelets from the latter mice showed a defect in Ca2+ fluxes and other responses, when triggered with low concentrations of (thrombin or collagen) receptor agonists. These platelets displayed normal aggregation under flow conditions, but a decreased procoagulant activity (PS exposure).29
Relatives P2 and P3, heterozygous for the R91W mutation, showed approximately 20–50% residual SOCE activity, which is compatible with co-expression of the non-mutated allele in the platelets. Interestingly, Ca2+ entry after thrombin stimulations remained unaffected in patients/relatives P1-3, which is explained by involvement of the SOCE-independent Ca2+ entry mechanism through TRPC6 channels.30
On the other hand, the R98S mutation in ORAI1 carried by patient P4, with assumed gain-of-function,31 was not accompanied by altered SOCE activity after thapsigargin or GPVI stimulation, whereas SOCE was substantially reduced in the relative P5 (confirmed in two independent blood samples). The difference in SOCE activity between P4 and P5 might be explained by a different ‘penetration’ of the mutated allele in ORAI1 expression in megakaryocytes and platelets. However, an alternative explanation is the presence of other modifying genetic or acquired factors between P4 and P5, with possible effects on platelet SOCE activity, on which we can only speculate.
Concerning patient P6 with a heterozygous R429C mutation in STIM1, Ca2+ entry in platelets was impaired after thapsigargin, but not after GPVI or PAR1/4 stimulation. In mammalian cell lines, the R429 mutation modulates the C-terminal oligomerization and puncta formation of STIM1 with ORAI1.32 Our results suggest that, in platelets, this mutation strongly inhibits the interaction of STIM1 with ORAI1 after full Ca2+ store depletion, such as that provoked by thapsigargin.
Altered platelet functions in thrombus formation accompanied by mutations in ORAI1, STIM1 or FERMT3
Previous murine studies have pointed out that deficiency in platelet ORAI1 or STIM1 led to a moderately reduced collagen-dependent thrombus formation with minimal effect on bleeding,8,11 whereas murine deficiency in FERMT3 (kindlin-3) resulted in impaired thrombus formation and a clear bleeding phenotype.33 Furthermore, both ORAI1-deficient and ORAI1R93W mouse platelets were found to be partly defective in procoagulant activity (PS exposure), along with the annulled SOCE activity.8,11,29
In the present study, we observed even more variable platelet phenotypes in the 9 patients/relatives with a mutation in ORAI1, STIM1 or FERMT3. Heatmap analysis indicated extensive but distinct patterns of reduced thrombus formation between patients, compared to blood from cohorts of healthy control subjects. Blood analysis further indicated that most patients/relatives carrying such a mutation had platelet counts below or near the lower range of normal. Low platelet count was found to correlate with low thrombus formation parameters, especially on spot 1. By comparison with flow studies using blood from healthy controls at lower counts, it appears that mild thrombocytopenia can lead to a more severe reduction in thrombus formation if combined with lower platelet functionality. Mild thrombocytopenia for patients with a loss-of-function mutation in ORAI1 or STIM1 has been reported before.16 So far, published papers on patients with a FERMT3 mutation have not reported on thrombocytopenia.26,27
Because of the relatively large number of patients in our study, we were able to separate the thrombus formation parameters linked to qualitative or quantitative platelet defects. Both unsupervised clustering of the heatmap data and PCA of the raw data indicated that mostly thrombus parameters on spot 1 (collagen; platelet receptors GPIb-V-IX, GPVI, α2β1) showed a dependency on platelet count, whereas those of spot 2 (VWF/rhodocytin; receptors GPIb-V-IX, CLEC-2) and spot 3 (VWF/fibrinogen; receptors GPIb-V-IX, αIIbβ3) were not dependent on platelet count. Markedly, this was true for both the high shear and low shear flow tests. An explanation for this finding is that, with collagen as a relatively strong agonist for GPVI, on spot 1 platelet delivery (thus, count) rather than platelet activation is a limiting factor for thrombus-forming parameters. In contrast, the immobilized ligands of spots 2 and 3, being less platelet-stimulating, may rely more on full platelet activation including normal SOCE and αIIbβ3 integrin activation. Correlation analysis also indicated that PS exposure was a key parameter linked to SOCE, in agreement with earlier mouse data.10,11
Improved platelet functions after bone marrow transplantation
Bone marrow transplantation of patient P1 (ORAI1W/W), two months before, resulted in improved SOCE activity and normalized thrombus formation on spot three but not on spot 1 (linked to a low platelet count). Transplantation of P7 (FERMT3X/X), three months before, led to an overall improvement of thrombus formation parameters (at normal platelet count). The partial restoration of platelet count at two months post transplantation is compatible with reports that a full normalization can take several months.26,34
Based on the relevant differential analysis of thrombus formation parameters (Figure 5C), Table 2 provides a summative overview for each patient of alterations in: platelet adhesion/aggregation, integrin activation/secretion and procoagulant activity (phases 1–3). This approach is based on the rationale that this whole blood flow assay senses additive effects of low platelet count and impaired platelet functionality. Table 2 shows an overall defect in adhesion/aggregation for all patients/relatives with ORAI1 (R91W) mutation, as well as a defect in procoagulant activity. It also underlines the fact that the assumed gain-of-function ORAI1 (G98S) mutation in patient P4 is accompanied by a typical increase in procoagulant activity (but not in relative P5 with low SOCE). Furthermore, in patients carrying the FERMT3 (R573X) mutation, all platelet responses appeared to be more severely reduced in cases of homozygosity than those of heterozygosity. This may be of clinical relevance, since only the homozygous carrier P7 had a history of bleeding (Table 1).
Table 2.
Overall comparison of defective store-operated Ca2+ entry (SOCE) and impairment in thrombus formation of individual patients.

Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/3/540
Funding
Financial support from the Netherlands Centre for Translational Molecular Medicine (CTMM, MICRO-BAT), the Interreg V Euregio Meuse-Rhine program (Poly-Valve), Dutch Heart Foundation (2015T79 to TGM and JMEMC) and the Netherlands Organization for Scientific Research (NWO Vidi 91716421 to JMEMC).
References
- 1.Feske S, Gwack Y, Prakriya M, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–185. [DOI] [PubMed] [Google Scholar]
- 2.Penna A, Demuro A, Yeromin AV, et al. The CRAC channel consists of a tetramer formed by STIM-induced dimerization of Orai dimers. Nature. 2008;456(7218):116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luik RM, Wang B, Prakriya M, Wu M, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454(7203):538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13(9):549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varga-Szabo D, Braun A, Nieswandt B. STIM1 and Orai1 in platelet function. Cell Calcium. 2011;50(3):270–278. [DOI] [PubMed] [Google Scholar]
- 6.Ambily A, Kaiser WJ, Pierro C, et al. The role of plasma membrane STIM1 and Ca2+entry in platelet aggregation. STIM1 binds to novel proteins in human platelets. Cell Signal. 2014;26(3):502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varga-Szabo D, Braun A, Kleinschnitz C, et al. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. 2008;205(7):1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun A, Varga-Szabo D, Kleinschnitz C, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. 2009;113(9):2056–2063. [DOI] [PubMed] [Google Scholar]
- 9.Gilio K, Harper MT, Cosemans JM, et al. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J Biol Chem. 2010;285(30):23410–23419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Kruchten R, Braun A, Feijge MA, et al. Antithrombotic potential of blockers of store-operated calcium channels in platelets. Arterioscler Thromb Vasc Biol. 2012;32(7):1717–1723. [DOI] [PubMed] [Google Scholar]
- 11.Gilio K, van Kruchten R, Braun A, et al. Roles of platelet STIM1 and Orai1 in glycoprotein VI- and thrombin-dependent procoagulant activity and thrombus formation. J Biol Chem. 2010;285(31):23629–23638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heemskerk JW, Mattheij NJ, Cosemans JM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost. 2013;11(1):2–16. [DOI] [PubMed] [Google Scholar]
- 13.Malacards: human disease database. 2016. Available from: www.malacards/org
- 14.Shaw PJ, Feske S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J Physiol. 2012;590(17):4157–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura L, Sandrock-Lang K, Speckmann C, et al. Platelet secretion defect in a patient with stromal interaction molecule 1 deficiency. Blood. 2013;122(22):3696–3698. [DOI] [PubMed] [Google Scholar]
- 16.Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 2015;1356:45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuijpers TW, van de Vijver E, Weterman MAJ, et al. LAD-1/variant syndrome is caused by mutations in FERMT3. Blood. 2009;113(19):4740–4746. [DOI] [PubMed] [Google Scholar]
- 18.Van de Vijver E, De Cuyper IM, Gerrits AJ, et al. Defects in Glanzmann thrombasthenia and LAD-III (LAD-1/v) syndrome: the role of integrin 1 and 3 in platelet adhesion to collagen. Blood. 2012;119(2):583–586. [DOI] [PubMed] [Google Scholar]
- 19.Stepensky PY, Wolach B, Gavrieli R, et al. Leukocyte adhesion deficiency type III: clinical features and treatment with stem cell transplantation. J Pediatr Hematol Oncol. 2015;37(4):264–268. [DOI] [PubMed] [Google Scholar]
- 20.De Witt SM, Lamers MME, Swieringa F, et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat Commun. 2014;5:4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Endo Y, Noguchi S, Hara Y, et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca2+ channels. Hum Mol Genet. 2015;24(3):637–648. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs S, Rensing-Ehl A, Speckmann C, et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J Immunol. 2012;188(3):1523–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feijge MA, van Pampus EC, Lacabaratz-Porret C, et al. Inter-individual varability in Ca2+ signalling in platelets from healthy volunteers, relation with expression of endomembrane Ca2+-ATPases. Br J Haematol. 1998;102(3):850–859. [DOI] [PubMed] [Google Scholar]
- 24.Heemskerk JW, Vis P, Feijge MA, et al. Roles of phospholipase C and Ca2+-ATPase in calcium responses of single, fibrinogen-bound platelets. J Biol Chem. 1993;268(1):356–363. [PubMed] [Google Scholar]
- 25.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crazzolara R, Maurer K, Schulze H, et al. A new mutation in the KINDLIN-3 gene ablates integrin-dependent leukocyte, platelet, and osteoclast function in a patient with leukocyte adhesion deficiency-III. Pediatr Blood Cancer. 2015;62(9):1677–1679. [DOI] [PubMed] [Google Scholar]
- 27.Jurk K, Schulz AS, Kehrel BE, et al. Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3. Thromb Haemost. 2010;103(5):1053–1064. [DOI] [PubMed] [Google Scholar]
- 28.Cauwenberghs S, Feijge MA, Theunissen E, et al. Novel methodology for the assessment of platelet transfusion therapy by measuring increased thrombus formation and thrombin generation. Br J Haematol. 2007;136(3):480–490. [DOI] [PubMed] [Google Scholar]
- 29.Bergmeier W, Oh-Hora M, McCarl CA, et al. R93W mutation in Orai1 causes impaired calcium influx in platelets. Blood. 2009;113(3):675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hassock SR, Zhu MX, Trost C, Flockerzi V, Authi KS. Expression and role of TRPC proteins in human platelets: evidence that TRPC6 forms the store-independent calcium entry channel. Blood. 2002;100(8):2801–2811. [DOI] [PubMed] [Google Scholar]
- 31.Bohm J, Bulla M, Urquhart JE, et al. ORAI1 mutations with distinct channel gating defects in tubular aggregate myopathy. Hum Mutat. 2017;38(4):426–438. [DOI] [PubMed] [Google Scholar]
- 32.Maus M, Jairaman A, Stathopulos PB, et al. Missense mutation in immunodeficient patients shows the multifunctional roles of coiled-coil domain 3 (CC3) in STIM1 activation. Proc Natl Acad Sci USA. 2015;112(19):6206–6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14(3):325–330. [DOI] [PubMed] [Google Scholar]
- 34.Takami A, Shibayama M, Orito M, et al. Immature platelet fraction for prediction of platelet engraftment after allogeneic stem cell transplantation. Bone Marrow Transplant. 2007;39(8):501–507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.