

Optimal management of central nervous system (CNS) infiltration is a key remaining challenge in delivering precision therapy for childhood acute lymphoblastic leukemia (ALL).1 Most CNS relapses occur in children without high-risk features, and minimal residual disease (MRD) does not reliably predict CNS relapse.1,2 Consequently, all children receive intensive CNS-directed therapy, which is potentially toxic to the developing brain.3 The nature of cells infiltrating the CNS is a subject of much debate, with some believing that a subset of cells selectively traffic to the CNS compartment whilst others believe that all ALL cells are capable of entering the CNS. The majority of mechanistic work addressing this question has been performed in patient-derived xenograft (PDX) models. Herein, we report the first description of the clonal architecture of CNS and paired bone marrow (BM) compartments in patients with ALL, using high-throughput sequencing (HTS) of primary patient BM and cerebrospinal fluid (CSF) samples taken at diagnosis and relapse. This addresses a significant gap in our understanding of how faithfully PDX models recapitulate clonal architecture in patients. Our data support our hypothesis that CNS and BM ALL share common characteristics, and after their development in the BM almost all B-cell precursor ALL clones are capable of disseminating to the CNS. In addition, they suggest that in most cases CNS and accompanying BM relapses are not intrinsically different biologically, often arising from the same subclones at the two sites.
Tailored CNS-directed therapy requires an improved understanding of the mechanisms of CNS relapse, and the development of more sensitive assays to track CNS response during therapy. CNS leukemia is usually detected by CSF cytology. CSF flow cytometry or polymerase chain reaction (PCR) are more sensitive,1,4,5 but increased detection at diagnosis has not improved prediction of CNS relapse, possibly because it measures disease load at baseline rather than response to therapy.6 PDX models suggest that up to 80% of diagnostic BM samples can engraft in the CNS,7 and cellular barcoding experiments show the clonal composition within murine CNS and BM compartments are similar.7 This suggests the ability to enter the CNS compartment is a generic property of ALL. However, these results contrast with other studies indicating that CNS leukemic blasts may selectively express genes associated with trafficking.2,8,9 The interpretation of PDX data has caveats, since CNS infiltration is measured without selective pressures of CNS-directed therapy or immunosurveillance and species differences may make microenvironments more or less permissive for expansion.1
New techniques using HTS of immunoglobulin gene rearrangements allow detailed interrogation of ALL subclonal composition within individual patients.10 Unlike conventional PCR, using allele-specific primers, HTS is able to visualize all gene rearrangements simultaneously, and therefore uncover the extent of initial clonal diversity and the influence of selective pressures over time.11 Herein, we use HTS to investigate clonal relationships between leukemic cells directly isolated from patients CSF and BM in childhood ALL. This provides an important test of the validity of PDX models and insights into CNS and BM clonal selection during therapy.
Following ethical approval (REC 13/LO/1262), BM and CSF were obtained from patients with CNS involvement. Clinical details and samples available for analysis are given in Table 1. Samples were processed as previously described.11 Briefly, DNA was amplified by multiplex-PCR of rearranged variable, diverse, joining (VDJ) segments of the immunoglobulin heavy chain (IGH) genes, which encode the hypervariable CDR3 domain, and sequenced on a MiSeq (Illumina, San Diego, CA, USA). IGH was chosen due to sample constraints, as this was the dominant clone by screening at diagnosis in all the patients and was used as the MRD marker. Further experimental details are described in Online Supplementary Methods and Online Supplementary Table S1. Rearrangements were annotated with reference to ImMunoGeneTics germline sequences12 using Vidjil.13 The ultrasensitivity of HTS detecting VDJ rearrangements comes with the problem of distinguishing leukemia-specific from normal background repertoire. The frequency of most common IGH gene rearrangements from BM aspirates of healthy individuals has been shown to average 0.04 – 0.08%.14 For the study herein, a neoplastic clone was defined as rearrangements with frequencies >1% in an individual sample, or where the identical rearrangement (at level >1%) is seen in a paired sample at any level.
Table 1.
Patient characteristics of ALL samples with CNS involvement.
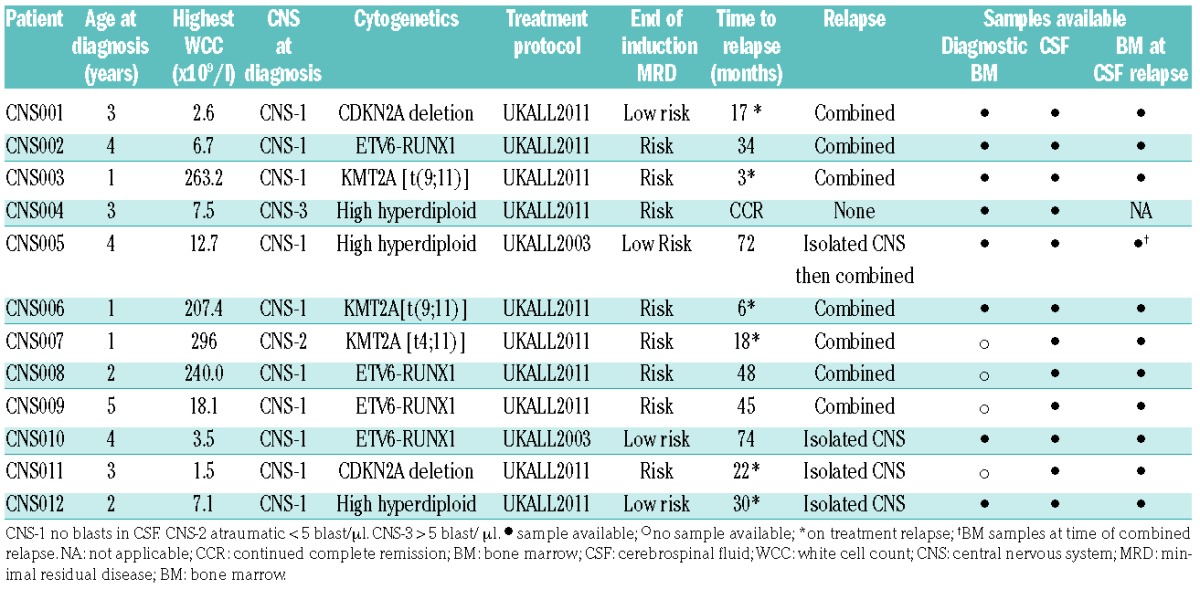
We analyzed 12 consecutive patients with B-cell precursor ALL and CNS involvement. High- and low-risk patients were represented, and four cases had a clinically isolated CNS relapse (Table 1). Criteria for CNS relapse was defined as ≥ 5 white blood cells per ml of CSF with morphological evidence of lymphoblasts. A combined BM/CNS relapse was defined as presence of CNS disease with ≥ 5% blasts in a concomitant BM aspirate. One patient had CNS involvement at diagnosis, in eight patients, material was available from diagnosis and relapse, and three patients had samples from CNS relapse but no original diagnostic material. Combined relapses are shown in Figure 1 and isolated CNS relapses in Figure 2, detailed information on the identity and percentages of clones in different compartments is given in Online Supplementary Table S2.
Figure 1.

Combined relapses, CNS disease at presentation and schematic presentation of the types of relapse seen. (A) CNS004. CNS involvement at diagnosis, showing that both compartments share the two dominant clones, albeit at reversed frequencies. (B) CNS001. The dominant diagnostic BM clone is IGHV4-34*01/IGHD2-2*01/IGHJ5*02 at 85%; present at 0.002% in BM relapse and undetectable at CNS relapse. The dominant clone in BM and CNS relapse samples is IGHV6-1*01/IGHJ5*02 at 78% and 88%, respectively (only 4% in original diagnostic BM). (C) CNS008. The identical dominant clone IGHV3-30-3*01/IGHD3-9*01/IGHJ6*03 at 95% and 64% in BM relapse and CNS relapse, respectively. (D) CNS002. The dominant diagnostic BM clones are 36% IGHV1-69*06/IGHD5-12*01/IGHJ4*02, 11% IGHV3-72*01/IGHD2-8*02/IGHJ4*02 and 10% IGHV1-18*01/IGHD2-2*02/IGHJ6*02. The dominant clone in the relapse BM sample is also IGHV1-69*06/IGHD5-12*01/IGHJ4*02 at 45%, however, this is only present at 5% in the CNS relapse sample. The second dominant clone in the relapse BM sample, IGHV3-74*01/IGHD5-12*01/IGHJ4*02 at 34%, is only present at 0.1% in diagnostic BM and 0.1% in CNS relapse. The CNS relapse dominant clone is IGHV3-74*01/IGHD7-27*01/IGHJ6*02 at 43%, which is absent in the original diagnostic BM, and only 0.6% in BM relapse. The second dominant clone in the CNS is IGHV3-74*01/IGHJ6*02 at 14% (which is again absent in the original diagnostic, and only 0.06% in BM relapse). The dominant clones in the CNS (IGHV3-74*01/IGHD7-27*01/IGHJ6*02 and IGHV3-74*01/IGHJ6*02) share the same IGHV and IGHJ gene usage, but the CDR3 regions are different and unrelated, as illustrated in Online Supplementary Figure S1. (E) CNS009. The identical dominant clones IGHV1-46*01/IGHD5-12*01/IGHJ4*02 at 34% and 48%, and IGHV3-71*02/IGHD6-6*01/IGHJ4*02 at 31% and 40% in BM and CNS, respectively. (F) CNS003. The dominant clone in all three samples is IGHV1-46*01/IGHD3-3*01/IGHJ4*02 at 91%, 50% and 94% in diagnosis BM, BM relapse and CNS relapse, respectively. (G and H) Schematic presentation of the two patterns of relapse seen. Left panel (“Pattern 1”) shows the same ALL clones in BM and CNS at relapse with no evidence for selection of “CNS-tropic” subclones, which could be due to relapse occurring at one or other site and then free trafficking between the CNS and BM, or to the same clones surviving treatment independently at the two sites. The right panel (“Pattern 2”) shows the alternative pattern, where there is evidence for some separate clonal evolution at the two sites, although many clones are also present in both compartments. (I) CNS006. Shows change in dominant clones from diagnosis through to combined BM and CNS relapse. The dominant diagnostic BM clone is IGHV6-1*01/IGHD3-10*01/IGHJ6*02 26% (this is at a very low level of 0.9% in relapse BM, and undetectable in CNS relapse). IGHV3-30-3*01/IGHJ4*02 was only 2% at diagnosis but 10% in relapse BM and 1% in CNS relapse. The dominant clone in the relapse BM, IGHV3-11*06/IGHJ4*02 at 41% was not detectable in the diagnostic sample or in CNS relapse samples. Other dominant relapse BM clones were 11% IGHV1-3*01/IGHD2-8*01/IGHJ4*02 (2% in original diagnostic BM and 1% in CNS relapse), and 10% IGHV3-30-3*01/IGHD2-8*01/IGHJ4*02 (2% in original diagnostic BM and 1% in CNS relapse). The dominant clone in CNS relapse, IGHV6-1*01/IGHD2-8*01/IGHJ4*02 at 26% was only low level in original diagnostic and relapse BM samples at 0.4% and 0.5%, respectively. The other dominant clones in the CNS were 21% IGHV3-30*18/IGHJ4*02 (0.01% in original diagnostic and relapse BM, respectively) and 16% IGHV7-4-1*01/IGHJ6*02 (0.02% in original diagnostic and 0.1% in relapse BM). (J) CNS007. The identical dominant clone IGHD6-13*01/IGHJ4*02 at 37% and 45% in the BM relapse and CNS relapse samples, respectively. However there is a smaller clone (15%) IGHD2-2*02/IGHJ4*02 in BM, not detectable in the CNS sample. In contrast, there is 11% IGHD2-2*02/IGHJ4*02 in CNS relapse not detectable in the BM relapse sample. CSF: cerebrospinal fluid; BM: bone marrow.
Figure 2.

Isolated CNS relapses. (A) CNS005. The dominant diagnostic BM clone is IGHV1-8*01/IGHD2-2*02/IGHJ6*03 at 55%. At relapse, the dominant clones are IGHV3-23/IGHD2-2*02/IGHJ6*03 and IGHV1-2*04/IGHD3-22*01/IGHJ6*03 at 40% and 48% in BM, and 39% and 45% in CNS, respectively. In original diagnostic BM, IGHV3-23/IGHD2-2*02/IGHJ6*03 represents 0.005% and IGHV1-2*04/IGHD3-22*01/IGHJ6*03 represents 0.01%. The dominant clone in the diagnostic BM, IGHV1-8*01/IGHD2-2*02/IGHJ6*03 at 55% is not detectable at relapse, but further analysis showed that this has been V-V replaced, whereby IGHV1-8*01 was replaced to create the IGHV3-23/IGHD2-2*02/IGHJ6*03 clone at relapse (Online Supplementary Figure S2). The presence of the IGHV3-23/IGHD2-2*02/IGHJ6*03 clone in diagnostic BM, albeit at a very low level, also demonstrates there is V-V replacement at diagnosis. (B) CNS010. The dominant clone in diagnostic BM is IGHV3-15*01/IGHD6-19*01/IGHJ6*02 at 31%. There are three other subclones, 24% IGHV4-59*01/IGHD3-10*01/IGHJH3*02, 12% IGHV3-33*01/IGHD6-6*01/IGHJ2*01 and 6% IGHV1-46*01/IGHJ4*02. At time of CNS relapse, the dominant clone is also IGHV3-15*01/IGHD6-19*01/IGHJ6*02 at 41%, however, the three other dominant clones present in diagnostic BM are undetectable in the CSF sample. The other dominant clone in CNS relapse is IGHV4-59*01/IGHD3-10*01/IGHJH3*02 at 46%, which is only present at a level of 0.0001% in diagnostic BM. The BM in morphological remission at time of CNS relapse shows a very low level of the CNS dominant clone IGHV4-34*01/IGHJ2*01 at a level of 0.12%, likewise the original diagnostic BM clone IGHV3-15*01/IGHD6-19*01/IGHJ6*02 at 0.05%. (C) CNS011. The dominant clones in CSF at CNS relapse; IGHV4-34/IGHD3-9*01/IGHJ6*02 at 54% and IGHV3-33*01/IGHD3-10*02/IGHJ3*02 at 34%. These clones are detected at a low level of 0.54% and 0.33%, respectively, in BM at time of CNS relapse, with no additional dominant clones detected. (D) CNS012. The dominant clones in diagnostic BM are IGHV1-2*04/IGHD7-27*01/IGHJ2*01 and IGHV7-4-1*02/IGHD5-12*01/IGHJ6*02 at 32.4% and 31.7%, respectively. In the CNS relapse CSF sample, the same two dominant clones are present at comparable levels. The BM at time of CNS relapse was morphologically negative, however, there was low-level positivity for both clones by both real-time PCR and HTS. CSF: cerebrospinal fluid; BM: bone marrow.
Firstly, we examined CNS and BM clones in the patient with overt CNS infiltration at original diagnosis (CNS004, Figure 1A). The CNS and BM compartments share identical dominant clones, albeit with reversed proportions between compartments. Next, we compared BM and CSF at the time of combined relapse; these results fell into two distinct patterns. In the majority of patients, (5/7: CNS001, CNS002, CNS003, CNS008, CNS009) (Figure 1B–H and Online Supplementary Table S2), BM and CSF clonal composition are remarkably similar at the time of relapse, in terms of both the extent of clonal diversity and the dominant clones (“pattern 1” Figure 1G). In the remaining patients there was evidence-for some separate clonal evolution at the two sites (“pattern 2” Figure 1H), although notably CNS clones are detectable in the BM, albeit sometimes at very low frequencies (patients CNS006, CNS007, Figure 1I,J).
We then examined the four patients with apparently isolated CNS relapse (Figure 2A–D). In cases CNS010, CNS011 and CNS012, examination of the paired (apparently uninvolved) BM at the time of relapse using the HTS assay identified low-level BM involvement with all CNS clones also detected in the BM compartment, although at very low frequencies. The fourth patient (CNS005) initially had an isolated CNS relapse at 72 months (CSF sample analyzed), then achieved remission before combined relapse at 100 months (BM sample analyzed). Again the two dominant clones at CNS relapse were the dominant clones at BM relapse. Therefore, these cases of clinical CNS involvement with subclinical or later BM relapse also fit into pattern 1 (Figure 1G). In the seven patients with diagnostic BM available (CNS001 [Figure 1B], CNS002 [Figure 1D], CNS003 [Figure 1F], CNS005 [Figure 2A], CNS006 [Figure 1I], CNS010 [Figure 2B] and CNS012 [Figure 2D]), three patients showed persistence of the original dominant clone, two showed falling levels of the dominant diagnostic clone with a rise in minor subclones whilst the other two showed loss of the dominant clone and selection of minor subclones at relapse. This rise and fall of subclones between diagnosis and relapse is a phenomenon widely reported in the literature.15
These data represent the first description of the clonal architecture of CNS and paired BM compartments in patients with ALL, and extends our understanding of the mechanisms of CNS leukemia. However, several limitations should be noted when interpreting our results. Patient numbers are small due to the difficulty of obtaining these rare samples. Additionally, in order to complete the full clonal picture, other immunoglobulin and T-cell receptor gene loci could be assessed if DNA quantity allowed.
Results from the patient with CNS disease at diagnosis are consistent with our hypothesis that ALL cells freely traffic to the CNS with no evidence for selection of “CNS-tropic” subclones. A similar situation is seen at relapse, with nine out of 11 patients sharing subclones between BM and CNS compartments. This supports our previous PDX work showing the polyclonal nature of CNS-infiltrating cells, which share similar clonal architecture with BM and the spleen.7 Interestingly, although most patients showed emergence of minor subclones during treatment, they did not differ between CNS and BM. This is perhaps surprising as selective pressures (chemotherapy exposure, immune surveillance and nutrient availability) vary between the two sites. Two possible explanations can be postulated. Firstly, relapse clones emerge at one site and then freely traffic to the other site prior to overt clinical relapse (akin to free trafficking seen at original diagnosis). This mechanism is further supported by our data showing that clones identified in the CNS at apparently isolated relapse are also detectable in the corresponding BM at low frequency. Alternatively, relapse mechanisms are cell-intrinsic, with certain subclones predetermined to be biologically fitter (presumably due to additional mutations), irrespective of their local microenvironment.
In contrast, two out of 11 patients with CNS relapse had some separate clonal evolution between BM and CNS, although many shared clones were also seen. In these cases, varying selective pressures or local acquisition of advantageous mutations may cause differential expansion in the two compartments. Using the more sensitive HTS assay, BM disease was detectable in all patients with apparently isolated CNS relapse; further evidence that BM infiltration is present at some level, even in apparently isolated CNS relapse.1,5
Overall, our data support the hypothesis that CNS and BM ALL share common characteristics, and that almost all clones can disseminate to the CNS. In addition, they suggest that CNS and BM relapses are not intrinsically biologically different, often arising from the same subclones at the two sites or freely trafficking between compartments at the time of relapse. Thus we provide underpinning science to the long-held belief, derived from clinical observations, that CNS and BM relapses are “competing events” rather than distinct clinical entities; i.e., increased CNS-directed therapy reduces overt CNS relapses but results in late BM relapses and vice versa.5 These observations support the use of at least some CNS-directed therapy for ALL, irrespective of CSF findings at diagnosis. Ultra-sensitive techniques such as HTS may allow for dynamic tracking of CNS and BM response to treatment, permitting more precise risk-adapted therapy tailored to the perils of BM and CNS relapse.
Supplementary Material
Acknowledgments
The authors would like to thank the Smurfit family for the generous support of this research.
Footnotes
Funding: this work was supported by Great Ormond Street Hospital Children’s Charity (W1069) and The Chief Scientist Office (ETM/374).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Frishman-Levy L, Izraeli S. Advances in understanding the pathogenesis of CNS acute lymphoblastic leukaemia and potential for therapy. Br J Haematol. 2017;176(2):157–167. [DOI] [PubMed] [Google Scholar]
- 2.van der Velden VH, de Launaij D, de Vries JF, et al. New cellular markers at diagnosis are associated with isolated central nervous system relapse in paediatric B-cell precursor acute lymphoblastic leukaemia. Br J Haematol. 2016;172(5):769–781. [DOI] [PubMed] [Google Scholar]
- 3.Halsey C, Buck G, Richards S, Vargha-Khadem F, Hill F, Gibson B. The impact of therapy for childhood acute lymphoblastic leukaemia on intelligence quotients; results of the risk-stratified randomized central nervous system treatment trial MRC UKALL XI. J Hematol Oncol. 2011;4:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levinsen M, Marquart HV, Groth-Pedersen L, et al. Leukemic blasts are present at low levels in spinal fluid in one-third of childhood acute lymphoblastic leukemia cases. Pediatr Blood Cancer. 2016;63(11):1935–1942. [DOI] [PubMed] [Google Scholar]
- 5.Pui CH, Howard SC. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol. 2008;9(3):257–268. [DOI] [PubMed] [Google Scholar]
- 6.Martinez-Laperche C, Gomez-Garcia AM, Lassaletta A, et al. Detection of occult cerebrospinal fluid involvement during maintenance therapy identifies a group of children with acute lymphoblastic leukemia at high risk for relapse. Am J Hematol. 2013;88(5):359–364. [DOI] [PubMed] [Google Scholar]
- 7.Williams MT, Yousafzai YM, Elder A, et al. The ability to cross the blood-cerebrospinal fluid barrier is a generic property of acute lymphoblastic leukemia blasts. Blood. 2016;127(16):1998–2006. [DOI] [PubMed] [Google Scholar]
- 8.Gomez AM, Martinez C, Gonzalez M, et al. Chemokines and relapses in childhood acute lymphoblastic leukemia: A role in migration and in resistance to antileukemic drugs. Blood Cells Mol Dis. 2015;55(3):220–227. [DOI] [PubMed] [Google Scholar]
- 9.Munch V, Trentin L, Herzig J, et al. Central nervous system involvement in acute lymphoblastic leukemia is mediated by vascular endothelial growth factor. Blood. 2017;130(5):643–654. [DOI] [PubMed] [Google Scholar]
- 10.Gawad C, Pepin F, Carlton VE, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120(22):4407–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartram J, Mountjoy E, Brooks T, et al. Accurate sample assignment in a multiplexed, ultrasensitive, high-throughput sequencing assay for minimal residual disease. J Mol Diagn. 2016;18(4):494–506. [DOI] [PubMed] [Google Scholar]
- 12.Giudicelli V, Brochet X, Lefranc MP. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc. 2011;2011(6):695–715. [DOI] [PubMed] [Google Scholar]
- 13.Duez M, Giraud M, Herbert R, Rocher T, Salson M, Thonier F. Correction: analysis of high-throughput repertoire sequencing. PLoS One. 2017;12(2):e0172249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu D, Emerson RO, Sherwood A, et al. Detection of minimal residual disease in B lymphoblastic leukemia by high-throughput sequencing of IGH. Clin Cancer Res. 2014;20(17):4540–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bashford-Rogers RJ, Nicolaou KA, Bartram J, et al. Eye on the B-ALL: B-cell receptor repertoires reveal persistence of numerous B-lymphoblastic leukemia subclones from diagnosis to relapse. Leukemia. 2016;30(12):2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.