

Abstract
Fanconi anemia is a rare disease characterized by congenital malformations, aplastic anemia, and predisposition to cancer. Despite the consolidated role of the Fanconi anemia proteins in DNA repair, their involvement in mitochondrial function is emerging. The purpose of this work was to assess whether the mitochondrial phenotype, independent of genomic integrity, could correlate with patient phenotype. We evaluated mitochondrial and clinical features of 11 affected individuals homozygous or compound heterozygous for p.His913Pro and p.Arg951Gln/Trp, the two residues of FANCA that are more frequently affected in our cohort of patients. Although p.His913Pro and p.Arg951Gln proteins are stably expressed in cytoplasm, they are unable to migrate in the nucleus, preventing cells from repairing DNA. In these cells, the electron transfer between respiring complex I–III is reduced and the ATP/AMP ratio is impaired with defective ATP production and AMP accumulation. These activities are intermediate between those observed in wild-type and FANCA−/− cells, suggesting that the variants at residues His913 and Arg951 are hypomorphic mutations. Consistent with these findings, the clinical phenotype of most of the patients carrying these mutations is mild. These data further support the recent finding that the Fanconi anemia proteins play a role in mitochondria, and open up possibilities for genotype/phenotype studies based on novel mitochondrial criteria.
Introduction
Fanconi anemia (FA) is a rare autosomal or X-linked recessive disease characterized by congenital abnormalities, bone marrow failure, and predisposition to cancer (mainly leukemia and squamous cell carcinomas). FA is caused by mutations in at least 22 genes. The FA proteins co-operate in a pathway whose role in maintaining the genome integrity is well known. In the presence of DNA damage, eight of the FA proteins, including FANCA, assemble in a nuclear ‘core complex’ responsible for monoubiquitination of FANCD2, an FA player that localizes in foci where it interacts with other FA components for DNA repair.1 FA cells are consistently hypersensitive to interstrand cross-link inducing agents, such as diepoxybutane (DEB) or mitomycin C (MMC).
In addition to their nuclear localization, the FA proteins are also localized in the cytoplasm, where they are likely to be involved in different processes that have yet to be defined, such as maintenance of mitochondrial aerobic metabolism, suppression of intracellular reactive oxygen species (ROS) levels, and protection from proinflammatory cytokine-induced apoptosis.2 Indeed, FA cells have structurally abnormal mitochondria, as they appear swollen with matrix rarefaction, altered cristae and reticulum fragmentation.3,4 All these features further affect the mitochondrial functions, suppressing cell respiration, perpetuating ROS production and switching respiration from oxidative phosphorylation (OXPHOS) to aerobic glycolysis.2–5
An important milestone in unraveling the role of the FA proteins in the cytoplasm is the discovery of their involvement in selective autophagy.6 They are fundamental for removal of damaged mitochondria probably modulating mitochondrial fission-fusion balance, explaining why mutations of the FA genes result in accumulation of morphological defective mitochondria and inbalance of the cellular redox status.6,7
The clinical expressivity in FA is extremely variable from severe to mild, sometimes without clinical signs. Except for mutations of the FANCD2 and BRCA2 genes, no strong association has been found between the clinical phenotype with the genotype, mainly because of the wide genetic heterogeneity and the limited number of patients who belong to rare complementation groups.8,9 Even considering FANCA, the most frequently mutated gene accounting for approximately two-thirds of the cases,10 the spectrum of mutation is extremly heterogeneous with private mutations and large intragenic deletions.11,12
We report functional studies of three missense FANCA mutations (p.His913Pro, p.Arg951Gln, and p.Arg951Gln), whose pathogenic effect in DNA repair is distinct from that in mitochondrial activity. The mutant proteins are stably expressed but localized only in the cytoplasm, suggesting that they are functionally inactive at least for the DNA repair. They are instead functionally hypomorphic for the mitochondrial function. Consistent with the hypothesis of a residual activity of p.His913Pro, p.Arg951Gln, and p.Arg951Trp, the clinical phenotype of patients carrying these mutations is characterized by late onset of mild cytopenia, which tends to remain stable during follow up.
Methods
Biological samples
Eleven FA probands with positive chromosomal breakage test were included in this study. The institutional review board of the “G. Gaslini” Hospital, Genoa, Italy, approved the study, and all the subjects or their legal guardians gave written informed consent to the investigation according to the Declaration of Helsinki. In 10 cases (F2-F11), mutations had been previously reported.13 Patient P1 was a novel case analyzed by next generation sequencing.14
Complementation assay
Lymphoblast (LFB) cells were transduced with retroviral vectors expressing the cDNAs for FANCA, as previously reported.15,16 Mitomycin C (MMC) survival assay and cell cycle evaluation were performed as previously described.15
Western blot and immunofluorescence assay
A full-length FANCA sequence was amplified and cloned into the pcDNA3.1-Flag tagged expression vector. The mutant FANCA cDNAs (p.His913Pro and p.Arg951Gln/Trp) were generated by site-directed mutagenesis using specific primers (available upon request) and transfected, using calcium phosphate, in 293T cells treated with 2 mM hydroxyurea (HU) for 24 hours.17 Protein whole and fractionated cell extracts were prepared using M-PER™ Mammalian Protein Extraction Reagent and NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific), respectively. Primary antibodies were used as follows: anti-FANCA (Savino et al.,18 1:500), anti-FANCD2 (Santa Cruz, sc-20022, 1:500), add anti-HSP90 (Santa Cruz, sc-7947, 1:4000), anti-ORC2 (Abcam, ab68348; 1:500), anti-FLAG (OctA Probe H5, Santa Cruz, 1:500), and anti-α Tubulin (Santa Cruz, sc-5286, 1:2000). Immuno-reactivity was visualized using the Enhanced Chemiluminescent SuperSignalTm West Femto Maximum Sensitivity Substrate (Pierce).
For immunofluorescence assay, anti-FLAG antibody was used with anti-mouse FITC secondary antibody (F0479, DakoCytomation), while nuclei were stained with 1 ug/mL propidium iodide solution (Sigma Aldrich).
Biochemical assays
FANCA pcDNA3.1 constructs were transfected in LFB cells not expressing the FANCA protein (FANCA−/−) using Lipofectamine (Thermo Fisher Scientific). Biochemical assays (ATP/AMP evaluation, electron transfer between complex I to complex III and Oxygen consumption measurements) were performed as described in Columbaro et al.19
Fo-F1 ATP synthase activity assay
Evaluation of the Fo-F1 ATP synthase activity was performed as previously described.20 Briefly, 200,000 cells were incubated for 10 minutes (min) in a proper medium and ATP synthesis was induced by the addition of 0.1 mM ADP. The reaction was monitored for 2 min, every 30 seconds (sec) in a luminometer (GloMax® 20/20n Luminometer, Promega Italia, Milan, Italy), by the luciferin/luciferase chemiluminescent method, with ATP standard solutions between 10-8 and 10-5 M (luciferin/luciferase ATP bioluminescence assay kit CLSII, Roche, Basel, Switzerland). Data were expressed as nmol ATP produced/min/106 cells.
The oxidative phosphorylation efficiency (P/O ratio) was calculated as the ratio between the concentration of the produced ATP and the amount of consumed oxygen in the presence of respiring substrate and ADP.21
Clinical data
Clinical information, phenotypic score and grade of cytopenia was defined as described in Svahn et al.8 The hematologic condition was evaluated by scores grouped in three categories, from 1 to 3, from > 3 to 7, and > 7, representing mild, moderate, and severe phenotypes, respectively. Regarding the malformation features, a score was assigned to every patient, dividing the population into three groups: mild (<6), moderate (6–15), and severe (>15).
Results
Missense p.His913Pro and p.Arg951Gln mutants of FANCA are stably expressed with loss-of-function effect on DNA repair
In our cohort of FA individuals, there are relatively recurrent missense mutations of the FANCA gene, such as c.2738A>C (p.His913Pro) accounting for 9 alleles from six different families (Table 1). Three patients were homozygous (F1, F2, and F3) and another 3 (F4, F5 and F6) were compound heterozygous for the mutation with the second allele being a small in-frame p.Glu1239_Arg1243del or large intragenic deletion, and another missense p.Arg951Gln mutation, respectively. All the six families were from Sicily, suggesting a founder effect of the c.2738A>C variant. Indeed, genotyping four polymorphic loci closed to FANCA, we found that the variant was associated with the same haplotype in all the families (data not shown). Bioinformatics tools showed low pathogenicity scores for p.His913Pro.13 Consistently, the CADD score (score 13.25) is lower than the threshold (score 15.00) commonly used to predict pathogenicity of missense variants.22
Table 1.
Molecular and clinical data of Fanconi anemia individuals with biallelic or monoallelic p.His913Proand p.Arg951Gln alleles of FANCA.
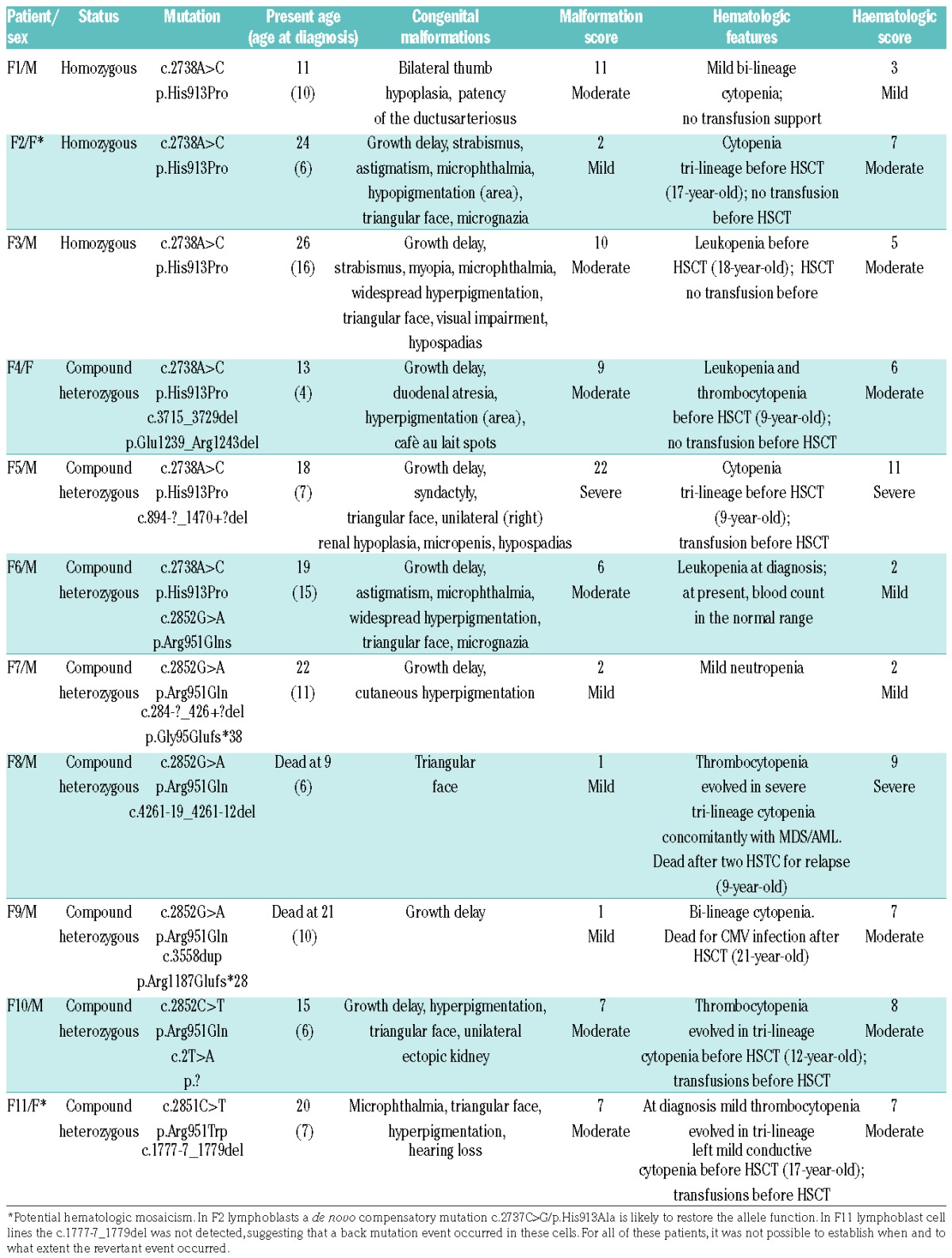
To exclude the possibility that another FA gene or another unknown FANCA variant in linkage disequilibrium with c.2738A>C could be responsible for the disease, we further investigated the role of FANCA as a disease-causing gene. First of all, complementation analysis of FANCA restored the MMC sensitivity and/or G2 arrest induced by melphalan in LFB homozygous for p.His913Pro (F3) (Figure 1A and B). Then, to investigate the functional impact of p.His913Pro on protein stability and localization, we performed Western blot analysis using LFB from 4 affected individuals (F3-F6). In all the four cell lines, FANCA was not degraded and was expressed at similar level as in wild-type cells (FANCA+/+) (Figure 2A). The protein was instead undetectable, even at different exposures, in LFB (FANCA−/−) from an FA patient homozygous for a FANCA large intragenic deletion (exons 4 to 20) resulting in a frameshift mutation (p.Gly95Glyfs*31). Finally, in order to evaluate whether the mutant p.His913Pro protein could play a role in nuclear DNA repair, we analyzed fractionated cellular lysates. As expected, FANCA was detected in both cytoplasm and nucleus of the wild-type LFB cells. Instead, the protein was present only in the cytoplasm of cells from F3 (Figure 2B). Immunofluorescence and Western blot of fractionated cellular lysate from the 293T cells over-expressing the wild-type or the mutated FANCA tagged with FLAG confirmed that the mutant protein was retained in the cytoplasm (Figure 2C and D). Taken together, these data indicated that the p.His913Pro mutant form of FANCA is stably expressed but is unable to enter the nucleus and play its role in the DNA repair pathway.
Figure 1.

Functional studies of the p.His913Pro and p.Arg951Gln variants. (A) Complementation analysis determined as cell survival after mitomycin C (MMC) treatment of lymphoblast (LFB) cells from patients F3 (homozygous for p.His913Pro) and F7 (compound heterozygous for p.Arg951Gln and p.Gly95Glufs*38) transduced with retroviral vector expressing the wild-type FANCA cDNA. (B and C) Complementation of the G2 cell cycle arrest after melphalan exposure of F3 and F7 LFB. (D and E) Comparison of the cell survival and cell cycle analysis in LFB cells of patients carrying the p.His913Pro (F3, F4, F5 and F6) and the p.Arg951Gln (F7) mutations and in control cells: wild-type (+/+) LFB cells and LFB cells not expressing the protein (−/−) due to a homozygous large intragenic deletion (c.284-?_1826+?del) of FANCA. There is no significant difference between LFB−/− cells and LFB carrying the missense mutations.
Figure 2.

Expression of p.His913Pro and p.Arg951Gln FANCA proteins. (A) Western blot of total lysates from lymphoblast (LFB) cells of patients carrying the p.His913Pro (F3, F4, F5 and F6) and the p.Arg951Gln (F7 and F10) mutations, showing that the mutant FANCA proteins are expressed. The controls are wild-type (+/+) LFB cells and LFB cells not expressing the FANCA protein (−/−) due to a homozygous large intragenic deletion (c.284-?_1826+?del) of FANCA. (B) Western blot of cytoplasmic (C) and nuclear (N) fractionated cellular lysates after 2 mM hydroxyurea treatment (24 hours) of LFB from patients F3 and F7, showing that the endogenous p.His913Pro and p.Arg951Gln proteins do not translocate to the nucleus. Controls +/+ and −/−, as indicated in (A). (C) Western blot of cytoplasmic (C), nuclear (N) fractionated, or total (TL) cellular lysates from 293T cells transfected with the wild-type (wt) or the mutant (H913P and R951Q) forms of FANCA tagged with FLAG, confirming the exogenous p.His913Pro and p.Arg951Gln FANCA proteins are retained in the cytoplasm. (D) Immunofluorescence analyses on 293T cells transfected as indicated in (C). Nuclei are stained with propidium iodide (PI). (E) Western blot of different LFB cells exposed to 2 mM hydroxyurea (24 hours) showing no monoubiquitination of the FANCD2 protein. Control +/+, as indicated in (A).
Of note, patient F6 is a compound heterozygous for the p.His913Pro and p.Arg951Gln mutations. The latter was identified in another 4 probands (F7, F8, F9, and F10) who are compound heterozygous for deleterious frameshift, splicing or start-loss mutation of FANCA (Table 1). Consistent with haplotype analysis (data not shown), the families carrying p.Arg951Gln come from different geographical areas, suggesting that the mutation originated from independent de novo events and that the presence of another disease-causing mutation in linkage disequilibrium was unlikely. In the LFB cells of F7, the sensitivity to MMC and the G2 arrest were restored by complementation analysis (Figure 1A and C). Like the p.His913Pro substitution, p.Arg951Gln did not affect FANCA stability, being the protein expressed in LFB of patients F7 and F10 (Figure 2A). Moreover, neither the endogenous nor the over-expressed mutant protein migrated into the nucleus (Figure 2B and C).
Taken together, these data suggest that p.His913Pro and p.Arg951Gln are loss-of-function mutations for the role of FANCA in controlling the genomic integrity. Consistent with this hypothesis, FANCD2 was not monoubiquitinated in any of the six cell lines available for the analysis (Figure 2E). Moreover, in the same cells, the sensitivity to MMC and the percentage of cells blocked in the G2 phase are comparable with those of the FANCA−/− cells (Figure 1D and E).
Finally, at position 951 an additional substitution (c.2851C>T/p.Arg951Trp) was identified in one allele of patient F11. However, the LFB cell line or other biological samples were not available for investigations to confirm that the mutation is associated with protein stability and lack of FANCD2 monoubiquitination, as reported by Karras et al.23
p.His913Pro and p.Arg951Gln are hypomorphic mutations for mitochondrial activity
In order to evaluate whether FANCA could affect the mitochondrial function, we investigated the electron transport chain functionality and the cellular energy status in LFB from patients with mild (F6 and F7), moderate (F3 and F4), and severe (F5, the only LFB cell line available within this group) hematologic score. Consistent with the FA mitochondrial phenotype,2–4 the cells showed an altered ATP/AMP ratio with reduced ATP production and AMP accumulation, and impaired electron transfer between complexes I and III (Figure 3A). Of note, the biochemical values were intermediate between those obtained in FANCA+/+ and FANCA−/− cells, suggesting that p.His913Pro and p.Arg951Gln are hypomorphic alleles for the mitochondrial activity.
Figure 3.

Hypomorphic effect of missense mutations on mitochondrial activity. (A) Intracellular concentrations of ATP and AMP, ATP/AMP ratio, and electron transfer between complex I and III were determined in lymphoblasts (LFB) from patients with mild (F6, F7), moderate (F3, F4) and severe (F6) hematologic scores. LFB from 3 Fanconi anemia (FA) patients not expressing FANCA (−/−) and 5 healthy individuals (+/+) have been used as controls. The three −/− cell lines are compound heterozygous (p.Arg18Profs*19/p.Asn1221Thrfs*26 and p.Ser175Leufs*5/p.Trp183*) and homozygous (Gly95Glufs*31) for nonsense or frameshift mutations. Each graph is representative of three experiments carried out in each cell line and data are expressed as mean±Standard Deviation (SD). t-test indicates a significant difference of P<0.05 (*) and P<0.01 (**) between the +/+ cells and the other samples, while # and ## indicate a significant difference of P<0.05 and P<0.01, respectively, between the −/− LFB cells and the other samples. (B) The same parameters reported in (A) were evaluated in an LFB −/− cell line transfected with the wild-type (wt) or mutant (H913P, R951Q and R951W) forms of FANCA tagged with FLAG, showing that the mitochondrial activity is intermediate between that of LFB +/+ and LFB −/−cells transfected or untransfected (+/+ or −/− vectors) with the empty vector. Each graph is representative of three experiments carried out in each cell line and data are expressed as mean±Standard Deviation (SD). t-test indicates a significant difference of P<0.05 (* or #) and P<0.01 (** or ##) between LFB cells −/− transfected with empty vector or expressing the three missense mutant forms of FANCA and LFB cells +/+ (* and **) or LFB cells −/− (# and ##).
In order to demonstrate that these alleles are directly involved in determining a mild mitochondrial phenotype, we over-expressed the p.His913Pro and p.Arg951Gln, as well as p.Arg951Trp, FANCA proteins in FANCA−/− cells. Whereas the ATP and AMP concentrations, the ATP/AMP ratio, and the electron transport were complemented by transfection of the wild-type cDNA, the same phenotypes were only partially restored by expression of the three mutant cDNAs. Of note, the biochemical measurements are significantly different when FANCA−/− cells are compared with those over-expressing the mutant forms of FANCA (Figure 3B).
The defect in the electron transfer between complexes I and III determines an impaired oxygen consumption and ATP synthesis in FANCA−/− cells, while in the samples expressing His913Pro, Arg951Gln and Arg951Trp, this activity is partially recovered, although with values lower than those obtained in cells expressing the wild-type FANCA protein (Figure 4A and B). Conversely, respiration was observed in all samples after stimulation with succinate, indicating that the electron transfer through complex II, III and IV pathways is not impaired (Figure 4A). This implies that the FA mutations may have a specific interaction with the complexes I, III and IV pathway. However, similarly to that reported in literature,21 the P/O ratio in the presence of pyruvate/malate is around 2.5 in all samples, suggesting that, despite the impaired mitochondrial function in FANCA−/− and in cells expressing the three mutant forms of FANCA, the residual oxygen consumption is devoted to ATP synthesis (Figure 4C).
Figure 4.

Oxymetric measures and ATP synthesis. (A) Amperometric traces of the oxygen consumption for LFB FANCA−/− cells transfected with wild-type (+/+), p.His913Pro, p.Arg951Gln, and p.Arg951Trp mutant forms of FANCA. (B) ATP synthesis rate in the same samples as in (A). The data in (A) and (B) are also depicted as histograms. Each bar graph is representative of three experiments and data are expressed as mean±Standard Deviation (SD). Anova test indicates a significant difference of P<0.05 (*) and P<0.01 (**) between the +/+ sample and the other samples, while ## indicates a significant difference of P<0.01 between LFB −/−transfected with the p.His913Pro, p.Arg951Gln and p.Arg951Trp mutant forms of FANCA and LFB −/−.
Moderate clinical phenotype in patients with missense mutations affecting residues His913Pro and Arg951
The clinical data of the affected individuals carrying the p.His913Pro, p.Arg951Gln, and p.Arg951Trp mutations are reported in Table 1. Of the 11 patients, 8 were male and 3 females. Mean age at diagnosis was 8.9 years (range 4–16 years). Ten had mild or moderate somatic phenotype (range 1–11). Nine of these individuals had concordant mild or moderate hematologic scores ranging from 2 to 8. Three individuals (F1, F6, and F7) had mild cytopenia that did not require transfusions. Of note, F6 and F7 had a mild leukopenia/neutropenia at diagnosis; at present, blood count is in the normal range in F6 and neutropenia is stable in F7 after 4 and 11 years, respectively, from the first abnormal blood count. The remaining 6 probands underwent HSCT. The first abnormal blood count was a mild tri-lineage cytopenia in F2 and F9 or thrombocytopenia, as in F10 and F11. They all underwent HSCT for progression to aplastic anemia, which occurred 10–11 years after diagnosis in F2, F9, and F11. Even F3 and F4 did receive HSCT after two and five years, respectively, of stable moderate cytopenia because a matched healthy sibling brother was available. Except for F9, who died for HSCT complications due to cytomegalovirus infection, the other patients are all alive after a follow up ranging from 1 to 18 years (mean 9.5 years) and none have developed hematologic adverse events (myelodysplastic syndrome, acute myelogenous leukemia or cytogenetic alterations) and/or solid tumor.
Patients F5 and F8 were the 2 affected individuals with severe hematologic scores. In addition to having multiple congenital malformations (score 22), F5 was characterized by severe tri-lineage requiring blood transfusions. He successfully underwent HSCT two years after diagnosis. At diagnosis, F8 had no congenital malformation except for growth delay and mild thrombocytopenia. However, the mono-lineage cytopenia evolved in severe tri-lineage cytopenia concomitantly with myelodysplastic syndrome transformed in acute myelogenous leukemia. He died three years later for relapse after two HSCT.
Discussion
Mutations of the FA genes are almost private, though common founder mutations have been reported in few populations, including Spanish Gypsies, the Afrikaner population of South Africa, Ashkenazi Jews, and people from some Italian geographical regions.24–27 This is the case of p.His913Pro, a mutation of the FANCA gene that is relatively frequent, even as a homozygous condition, in patients from Sicily. Indeed, the analysis of microsatellite markers in the six families revealed the presence of a common haplotype compatible with a founder effect. In contrast, p.Arg951Gln has been identified, never as a homozygous mutation, in individuals from different geographical areas, suggesting that it occurred as independent de novo mutational events. At the same residue, an additional rare pathogenic mutant (p.Arg951Trp) affects one allele of our cohort of patients.23
Since these alterations are missense mutations, their pathogenic effect on protein function has been ascertained by considering several aspects. Unlike p.Arg951Gln/Trp, p.His913Pro was classified as a variant of uncertain significance, as the predictive bioinformatic tools indicated that this amino acid substitution has no or mild effect on protein function.13 However, genetic studies support their pathogenic role, as they are present in FA patients and only rarely in controls (ExAC or other databases).13 Consistent with this hypothesis, we found that the mutant FANCA proteins (both endogenous and over-expressed) localize only in the cytoplasm, where they are stably expressed at similar levels to those observed in wild-type cells. As a consequence, the FA/BRCA pathway remains inactive because FANCD2 is not monoubiquitinated, as demonstrated by Western blot analysis.
These data are consistent with those reported by Castella et al.,24 who showed that all the FANCA missense mutations are stably expressed. Like p.His913Pro and p.Arg951Gln, their products do not enter the nucleus, preventing FANCD2 being monoubiquitinated. On the contrary, the same authors demonstrated that null mutations, such as nonsense or frameshift alterations, are unstable and not detectable within cells.24 Therefore, our data confirm the hypothesis of a correlation between type of mutation and expression level of mutant FANCA, but as to why these mutant proteins are retained in the cytoplasm without any apparent function remains a subject of debate.
In trying to unravel any role of the FA proteins in other cell compartments, we investigated the mitochondria OXPHOS function and the cellular energy status whose alteration may lead to an impaired mitophagy process.28 As demonstrated by biochemical assays, expression of the p.His913Pro and p.Arg951Gln/Trp proteins are associated with a mild mitochondrial function impairment. In particular, the electron transport between complexes I and III, the ATP production, and the O2 consumption appear defective but not to the same extent as in FA cells homozygous for null mutations. Interestingly, the defective OXPHOS is limited to the pathway composed by complexes I, III and IV, considering that the oxygen consumption and the relative ATP synthesis is similar in control and FA samples when induced by succinate. This suggests that the potential function of FANCA, as well as other FA proteins, on mitochondria is confined to the OXPHOS led by complex I. However, the evaluation of P/O ratio shows that, in all samples, the oxygen consumption, even when very low, is finalized to the ATP synthesis, indicating that the mitochondria are always in a coupled status.
In accordance with the recent discovery that the FA proteins are required for clearance of damaged mitochondria and to decrease the mitochondrial reactive oxygen species,6 our finding supports the role of FANCA in these organelles. Considering that small interfering RNA of the FA genes is associated with increased puncta, which is a marker for defects in clearance of damaged mitochondria,6 we can speculate that mutations of FANCA could negatively influence the efficiency of the mitophagy, causing an accumulation of damaged mitochondria. This may determine not only a decrement in the energy production, but also the increment of oxidative stress, which could induce damage of other mitochondria. However, we cannot exclude the possibility that the FANCA protein may have a more direct role in the modulation of the mitochondria structure and function, i.e. influencing the organization of inner mitochondrial membrane, whose integrity is essential for the proper functioning of the OXPHOS.
Of interest, our data allow us to dissect the activity of FANCA in DNA repair from that in preserving the mitochondrial function, as has previously been hypothesized for FANCC.6 Indeed, one mutation of this gene (c.67delG), resulting in an N-terminus truncated but stable mutant form of FANCC, is unable to repair DNA but is able to restore the mitochondrial function. Therefore, we regard the p.His913Pro and p.Arg951Gln/Trp as variants with a hypomorphic effect of FANCA in maintaining the mitochondrial activity. Even mutations of the FANCD2 gene have been regarded as hypomorphic.29 However, their ‘hypomorphic’ effect is associated with the role of the protein in controlling genomic integrity. In FANCD2 individuals, at least one allele is always associated with expression of a low amount of FANCD2, which is detectable in both the non- and monoubiquitinated form, suggesting that residual activity of FANCD2 for the DNA repair processes is essential for life. On the contrary, biallelic null mutations of FANCA and FANCC are relatively common in FA and are always associated with lack of FANCD2 monoubiquitination.24
Considering that the c.67delG mutation of FANCC, which is able to restore the mitochondrial function,6 is associated with mild clinical phenotypes,9,30 we evaluated whether we could reach the same conclusion in our cohort of 11 patients. The mean age (8.9 years) at diagnosis was higher than that reported in the FA Italian cohort (6.8 years).8 Nine of them had concordant mild/moderate hematologic and somatic phenotypes. Regarding malformations, 4, 6, and one patients were classified as mild, moderate and severe, respectively, and percentages [0.36 vs. 0.43 (mild), 0.55 vs. 0.37 (moderate), and 0.09 vs. 0.20 (severe)] are not significantly different from those of the entire Italian cohort, although only in one case were the congenital malformations severe. For the hematologic features, 3 (0.30), 6 (0.50) and 2 (0.20) of patients received mild, moderate and severe scores, respectively, compared with 16% (mild), 27% (moderate), and 49% (severe) of the FA Italian population, suggesting that the missense mutations at residues His913 and Arg951 are associated with better hematologic prognosis.8
Two patients had severe hematologic score and underwent HSCT (together with another 6 affected individuals). However, HSCT per se should not be regarded as a marker of hematologic severity. Indeed, despite their moderate hematologic condition, 2 patients took advantage of having a suitable healthy matched sibling donor, which is known to provide the best outcome.31 Another 4 underwent familiar or matched unrelated donor HSCT even more than ten years after the first abnormal blood count, when cytopenia dropped from mild/moderate to severe. They are alive and well at 3–7 years after HSCT, except for F9 and F8 who died for cytomegalovirus infection after HSCT and relapse after two HSCT, respectively. Three probands (F2, F6, and F7) with mild hematologic score did not undergo HSCT and their hematologic condition appears to be stable even 11 years (F7) after diagnosis.
Taken together, these data suggest that FA individuals carrying the p.His913Pro and p.Arg951Gln/Trp mutations at one or both the FANCA alleles have a relatively mild phenotype. This conclusion is consistent with data of Faivre et al.,9 showing that patients with altered FANCA protein have milder phenotypes than those with a complete loss of FANCA. However, the clinical phenotype depends on many factors, including different genetic and environmental factors, preventing us from confirming any clear correlations. Although FA affected individuals carrying the same disease-causing mutation (even among siblings) could have different outcomes, the 3 patients homozygous for p.His913Pro have mild/moderate clinical phenotype. Of note, our cohort numbers were limited, as the number of cases studied is relatively low. In order to assign statistical significance to the correlation, we should explore whether other FA mutations are hypomorphic for the mitochondrial activity and eventually correlate this with mild clinical outcome.
Finally, another factor that could modulate the clinical phenotype in FA is the occurrence of hematopoietic mosaicism, in which cells have lost mitomycin C sensitivity due to back mutations or other genetic mechanisms, as it occurs in almost 20% of cases.32 In consideration of this, F2 and F11 are 2 individuals with potential hematopoietic mosaicism, since one of the two FA mutations was reverted to wild-type allele in their LFB cell lines.13 However, none of them were further investigated, preventing us from ascertaining whether and to what extent the mosaicism was present in the bone marrow and blood cells.
In conclusion, at least for the mitochondrial activity, p.His913Pro and p.Arg951Gln/Trp are hypomorphic mutations, which are associated with a mild/moderate phenotype characterized by late onset of the disease and slow hematologic progression. Since the hematopoietic stem cells are relatively sensitive to their redox status,33 a residual activity of mitochondria in cells expressing these, and maybe other missense mutations, could help patients avoid experiencing worsening cytopenia and postponing their eligibility for HSCT. Therefore, we should explore whether other FA mutations with hypomorphic effect could explain the clinical variability in FA.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/3/417
Funding
This study was supported by Telethon Foundation (grant GGP11076), Cariplo Foundation (2012-0529), Italian Ministry of Health (RF-2010-2309222), AIRFA (Italian Association for Research in Fanconi Anemia), ERG S.p.A., Cambiaso Risso Group, Rimorchiatori Riuniti S.p.A., and Saar Depositi Oleari Portuali S.p.A. We are grateful to the “Cell Line and DNA Biobank from Patients affected by Genetic Diseases” (“G. Gaslini” Institute) and Telethon Genetic Biobank Network (project n. GTB07001) for the sample providing. MF is supported by a fellowship (ID 19432) from AIRC (Italian Association of Cancer Research) and RB from “Umberto Veronesi” Foundation.
References
- 1.Longerich S, Li J, Xiong Y, Sung P, Kupfer GM. Stress and DNA repair biology of the Fanconi anemia pathway. Blood. 2014;124(18):2812–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumari U, Ya Jun W, Huat Bay B, Lyakhovich A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene. 2014;33(2):165–172. [DOI] [PubMed] [Google Scholar]
- 3.Cappelli E, Cuccarolo P, Stroppiana G, et al. Defects in mitochondrial energetic function compels Fanconi Anaemia cells to glycolytic metabolism. Biochim Biophys Acta. 2017;1863(6):1214–1221. [DOI] [PubMed] [Google Scholar]
- 4.Ravera S, Vaccaro D, Cuccarolo P, et al. Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie. 2013;95(10):1828–1837. [DOI] [PubMed] [Google Scholar]
- 5.Capanni C, Bruschi M, Columbaro M, et al. Changes in vimentin, lamin A/C and mitofilin induce aberrant cell organization in fibroblasts from Fanconi anemia complementation group A (FA-A) patients. Biochimie. 2013;95(10):1838–1847. [DOI] [PubMed] [Google Scholar]
- 6.Sumpter R, Levine B. Novel functions of Fanconi anemia proteins in selective autophagy and inflammation. Oncotarget. 2016;7(32):50820–50821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shyamsunder P, Esner M, Barvalia M, et al. Impaired mitophagy in Fanconi anemia is dependent on mitochondrial fission. Oncotarget. 2016;7(36):58065–58074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Svahn J, Bagnasco F, Cappelli E, et al. Somatic, hematologic phenotype, long-term outcome, and effect of hematopoietic stem cell transplantation. An analysis of 97 Fanconi anemia patients from the Italian national database on behalf of the Marrow Failure Study Group of the AIEOP (Italian Association of Pediatric Hematology-Oncology). Am J Hematol. 2016;91(7):666–671. [DOI] [PubMed] [Google Scholar]
- 9.Faivre L, Guardiola P, Lewis C, et al. Association of complementation group and mutation type with clinical outcome in fanconi anemia. European Fanconi Anemia Research Group. Blood. 2000;96(13):4064–4070. [PubMed] [Google Scholar]
- 10.Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest. 2012;122(11):3799–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levran O, Diotti R, Pujara K, Batish SD, Hanenberg H, Auerbach AD. Spectrum of sequence variations in the FANCA gene: an International Fanconi Anemia Registry (IFAR) study. Hum Mutat. 2005;25(2):142–149. [DOI] [PubMed] [Google Scholar]
- 12.Morgan NV, Tipping AJ, Joenje H, Mathew CG. High frequency of large intragenic deletions in the Fanconi anemia group A gene. Am J Hum Genet. 1999;65(5):1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Rocco D, Bottega R, Cappelli E, et al. Molecular analysis of Fanconi anemia: the experience of the Bone Marrow Failure Study Group of the Italian Association of Pediatric Onco-Hematology. Haematologica. 2014;99(6):1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicchia E, Greco C, De Rocco D, et al. Identification of point mutations and large intragenic deletions in Fanconi anemia using next-generation sequencing technology. Mol Genet Genomic Med. 2015;3(6):500–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antonio Casado J, Callén E, Jacome A, et al. A comprehensive strategy for the subtyping of patients with Fanconi anaemia: conclusions from the Spanish Fanconi Anemia Research Network. J Med Genet. 2007;44(4):241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanenberg H, Batish SD, Pollok KE, et al. Phenotypic correction of primary Fanconi anemia T cells with retroviral vectors as a diagnostic tool. Exp Hematol. 2002;30(5):410–420. [DOI] [PubMed] [Google Scholar]
- 17.Molina B, Marchetti F, Gómez L, et al. Hydroxyurea induces chromosomal damage in G2 and enhances the clastogenic effect of mitomycin C in Fanconi anemia cells. Environ Mol Mutagen. 2015;56(5):457–467. [DOI] [PubMed] [Google Scholar]
- 18.Savino M, Borriello A, D’Apolito M, et al. Spectrum of FANCA mutations in Italian Fanconi anemia patients: identification of six novel alleles and phenotypic characterization of the S858R variant. Hum Mutat. 2003;22(4):338–339. [DOI] [PubMed] [Google Scholar]
- 19.Columbaro M, Ravera S, Capanni C, et al. Treatment of FANCA cells with resveratrol and N-acetylcysteine: a comparative study. PLoS One. 2014;9(7):e104857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravera S, Dufour C, Cesaro S, et al. Evaluation of energy metabolism and calcium homeostasis in cells affected by Shwachman-Diamond syndrome. Sci Rep. 2016;6:25441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinkle PC. P/O ratios of mitochondrial oxidative phosphorylation. Biochim Biophys Acta. 2005;1706(1–2):1–11. [DOI] [PubMed] [Google Scholar]
- 22.Easton DF, Deffenbaugh AM, Pruss D, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81(5):873–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karras GI, Yi S, Sahni N, et al. HSP90 Shapes the Consequences of Human Genetic Variation. Cell. 2017;168(5):856–866.e812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castella M, Pujol R, Callén E, et al. Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood. 2011;117(14):3759–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tipping AJ, Pearson T, Morgan NV, et al. Molecular and genealogical evidence for a founder effect in Fanconi anemia families of the Afrikaner population of South Africa. Proc Natl Acad Sci USA. 2001;98(10):5734–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kutler DI, Auerbach AD. Fanconi anemia in Ashkenazi Jews. Fam Cancer. 2004;3(3–4):241–248. [DOI] [PubMed] [Google Scholar]
- 27.Savino M, Ianzano L, Strippoli P, et al. Mutations of the Fanconi anemia group A gene (FAA) in Italian patients. Am J Hum Genet. 1997;61(6):1246–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacVicar TD, Lane JD. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J Cell Sci. 2014;127(Pt 10):2313–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalb R, Neveling K, Hoehn H, et al. Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FAD2 patients with severe phenotype. Am J Hum Genet. 2007;80(5):895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashita T, Wu N, Kupfer G, et al. Clinical variability of Fanconi anemia (type C) results from expression of an amino terminal truncated Fanconi anemia complementation group C polypeptide with partial activity. Blood. 1996;87(10):4424–4432. [PubMed] [Google Scholar]
- 31.MacMillan ML, DeFor TE, Young JA, et al. Alternative donor hematopoietic cell transplantation for Fanconi anemia. Blood. 2015;125(24):3798–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo Ten Foe JR, Kwee ML, Rooimans MA, et al. Somatic mosaicism in Fanconi anemia: molecular basis and clinical significance. Eur J Hum Genet. 1997;5(3):137–148. [PubMed] [Google Scholar]
- 33.Du W, Adam Z, Rani R, Zhang X, Pang Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid Redox Signal. 2008;10(11):1909–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.