

Abstract
Key points
Neurotransmitter release is inhibited by metabotropic glutamate type 7 (mGlu7) receptors that reduce Ca2+ influx, yet synapses lacking this receptor also produce weaker release, suggesting that mGlu7 receptors may also prime synaptic vesicles for release. Prolonged activation of mGlu7 receptors with the agonist l‐AP4 first reduces and then enhances the amplitude of EPSCs through a presynaptic effect.
The inhibitory response is blocked by pertussis toxin, while the potentiating response is prevented by a phospholipase C inhibitor (U73122) and an inhibitor of diacylglycerol (DAG) binding (calphostin C), suggesting that this receptor also couples to pathways that generate DAG.
Release potentiation is associated with an increase in the number of synaptic vesicles close to the plasma membrane, which was dependent on the Munc13‐2 and RIM1α proteins. The Glu7 receptors activated by the glutamate released following high frequency stimulation provoke a bidirectional modulation of synaptic transmission.
Abstract
Neurotransmitter release is driven by Ca2+ influx at synaptic boutons that acts on synaptic vesicles ready to undergo exocytosis. Neurotransmitter release is inhibited when metabotropic glutamate type 7 (mGlu7) receptors provoke a reduction in Ca2+ influx, although the reduced release from synapses lacking this receptor suggests that they may also prime synaptic vesicles for release. These mGlu7 receptors activate phospholipase C (PLC) and generate inositol trisphosphate, which in turn releases Ca2+ from intracellular stores and produces diacylglycerol (DAG), an activator of proteins containing DAG‐binding domains such as Munc13 and protein kinase C (PKC). However, the full effects of mGlu7 receptor signalling on synaptic transmission are unclear. We found that prolonged activation of mGlu7 receptors with the agonist l‐AP4 first reduces and then enhances the amplitude of EPSCs, a presynaptic effect that changes the frequency but not the amplitude of the mEPSCs and the paired pulse ratio. Pertussis toxin blocks the inhibitory response, while the PLC inhibitor U73122, and the inhibitor of DAG binding calphostin C, prevent receptor mediated potentiation. Moreover, this DAG‐dependent potentiation of the release machinery brings more synaptic vesicles closer to the active zone plasma membrane in a Munc13‐2‐ and RIM1α‐dependent manner. Electrically evoked release of glutamate that activates mGlu7 receptors also bidirectionally modulates synaptic transmission. In these conditions, potentiation now occurs rapidly and it overcomes any inhibition, such that potentiation prevails unless it is suppressed with the PLC inhibitor U73122.
Keywords: PLC, RIM1α KO, Munc13‐2 KO, RRP size, glutamate release
Key points
Neurotransmitter release is inhibited by metabotropic glutamate type 7 (mGlu7) receptors that reduce Ca2+ influx, yet synapses lacking this receptor also produce weaker release, suggesting that mGlu7 receptors may also prime synaptic vesicles for release. Prolonged activation of mGlu7 receptors with the agonist l‐AP4 first reduces and then enhances the amplitude of EPSCs through a presynaptic effect.
The inhibitory response is blocked by pertussis toxin, while the potentiating response is prevented by a phospholipase C inhibitor (U73122) and an inhibitor of diacylglycerol (DAG) binding (calphostin C), suggesting that this receptor also couples to pathways that generate DAG.
Release potentiation is associated with an increase in the number of synaptic vesicles close to the plasma membrane, which was dependent on the Munc13‐2 and RIM1α proteins. The Glu7 receptors activated by the glutamate released following high frequency stimulation provoke a bidirectional modulation of synaptic transmission.
Introduction
Metabotropic glutamate type 7 (mGlu7) receptors belong to group III mGlu receptors (mGluRs), along with mGluR types 4, 6 and 8. These mGlu7 receptors are G protein coupled receptors (GPCRs) that are widely expressed in the brain (Kinzie et al. 1995; Ohishi et al. 1995; Corti et al. 1998) and they localize to the presynaptic active zone of glutamatergic synapses (Shigemoto et al. 1996, 1997; Kinoshita et al. 1998) where they dampen neurotransmitter release (Millán et al. 2002, 2003; Martín et al. 2007). The inhibition of glutamate release mediated by mGlu7 receptors requires activation of a pertussis toxin (PTx) sensitive Gi/o protein and it reduces Ca2+ channel activity (Perroy et al. 2000, 2002; Millán et al. 2002, 2003; Pelkey et al. 2006; Martín et al. 2007). This effect is consistent with the weaker synaptic transmission induced by the group III mGluR agonist l‐2‐amino‐4‐phosphonobutyrate (l‐AP4) (Baskys & Malenka, 1991; Gereau & Conn, 1995; Capogna, 2004; Pelkey et al. 2005; Ayala et al. 2008; Klar et al. 2015). Moreover, prolonged activation of mGlu7 receptors de‐represses excitatory synaptic transmission due to receptor internalization (Pelkey et al. 2005). However, synaptic transmission is depressed in mGlu7 receptor knockout mice when high frequency stimulation (HFS) activates this receptor at Schaffer collateral (SC)–CA1 hippocampal synapses, suggesting that mGlu7 receptors also potentiate neurotransmitter release at these synapses (Bushell et al. 2002).
The mGlu7 receptor can also couple to phosphatidylinositol bisphosphate (PIP2) hydrolysis through the activation of phospholipase C (PLC: Perroy et al. 2000; Martín et al. 2010; Ferrero et al. 2011), although the effects of this signalling on synaptic transmission have not yet been addressed. PLC activation hydrolyses phospholipid PIP2 to generate inositol trisphosphate (IP3) and diacylglycerol (DAG), the former releasing Ca2+ from intracellular stores while the latter activates proteins containing DAG‐binding domains, such as Munc13 and protein kinase C (PKC). Phorbol esters, the stable analogues of DAG, enhance synaptic transmission through PKC (Malenka et al. 1986; Herrero et al. 1992; Parfitt & Madison, 1993) and through the non‐kinase active zone protein Munc13, which is essential for synaptic vesicle (SV) priming (Betz et al. 1998; Wu & Wu, 2001; Rhee et al. 2002; Wierda et al. 2007; Lou et al. 2008). Therefore, it might be anticipated that the coupling of mGlu7 receptors to PLC and the ensuing generation of DAG would potentiate synaptic transmission. However, there is currently no evidence of such PLC‐coupled mGlu7 receptor mediated potentiation of synaptic transmission. Nevertheless, group III mGluRs at the calyx of Held do augment the readily releasable pool (RRP) of SVs, although with little impact on synaptic amplitude since the receptor also decreases the release probability (Billups et al. 2005).
Here we studied how mGlu7 receptors influence synaptic transmission at SC–CA1 hippocampal pyramidal cell synapses where these receptors are expressed strongly (Shigemoto et al. 1997). Indeed, this receptor is the only group III mGluR present at adult SC–CA1 synapses (Baskys & Malenka, 1991; Ayala et al. 2008). We found that mGlu7 receptors bidirectionally modulate synaptic transmission at SC–CA1 hippocampal synapses. Indeed, receptor activation with the agonist l‐AP4 first reduces and then enhances the excitatory postsynaptic current (EPSC) amplitude through a presynaptic effect. The inhibition of release reflects the rapid coupling of the receptor to a PTx sensitive G protein, while mGlu7 mediated potentiation is slower and results from receptor coupling to a PTx resistant pathway. Synaptic potentiation, which is prevented by inhibitors of PLC‐ and DAG‐binding, drives SVs closer to the active zone (AZ) plasma membrane in a Munc13‐2‐ and RIM1α‐dependent manner. Both these responses (release inhibition and potentiation) provoked by mGlu7 receptors occur simultaneously following electrically evoked glutamate release, and while potentiation prevails, inhibition can be revealed by blocking this potentiation.
Methods
Animals
All procedures for handling and killing animals were performed in accordance with European Commission guidelines (2010/63/UE) and they were approved by the Animal Research Committee at the Complutense University. The animal experiments also comply with the policies and regulations of The Journal of Physiology (Grundy, 2015). Hippocampal slices were obtained from C57BL/6 (30‐ to 45‐day‐old) wild‐type (WT) mice and in some experiments, slices from Munc13‐2 and RIM1α knockout (KO) mice were used (generously provided by Dr N. Brose (Varoqueaux et al. 2002) and Dr S. Schoch (Schoch et al. 2002), respectively). In all instances adequate measures were taken to minimize the animal's pain and discomfort.
Hippocampal slice preparation
Animals were anaesthetized with isoflurane (1.5–2% in a mixture of 80% synthetic air–20% oxygen) and decapitated. The brain was quickly removed and placed in ice‐cold artificial CSF (aCSF, in mm: NaCl 124, KCl 2.69, KH2PO4 1.25, MgSO4 2, NaHCO3 26, CaCl2 2, ascorbic acid 0.4, and glucose 10 (pH 7.3)). Coronal vibratome hippocampal slices (325 μm thick: Leica VT 1200S, Leica Microsystems, Wetzlar, Germany) were obtained and maintained at room temperature (21–24°C) in a holding chamber (≥1 h) in aCSF continuously bubbled with carbogen (95% O2 and 5% CO2). The slices were then transferred to an immersion recording chamber and superfused at 1 mL min−1 with gassed aCSF containing 50 μm picrotoxin to block GABAA receptors. To measure miniature EPSCs (mESPCs: Fig. 3 A‐G), tetrodotoxin (TTx, 1 μm) was added to the aCSF. Pyramidal CA1 cells were visualized on an Eclipse FN1 Nikon microscope using a 40× water immersion objective with a Nomarski condenser combined with infrared microscopy (Nikon Instruments, Tokyo, Japan).
Figure 3. The mGlu7 receptors increase the frequency but not the amplitude of mEPSCs.
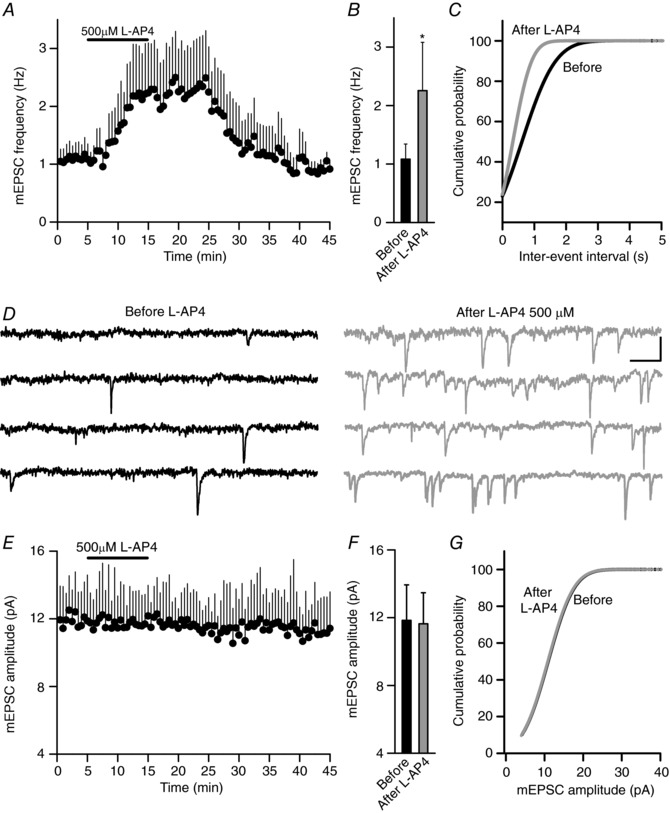
A, l‐AP4 (500 μm, 10 min) increases the mEPSC frequency. B, quantification of the changes in the mEPSC frequency before and after l‐AP4 (500 μm, 10 min, P = 0.0101, n = 21 cells/21 slices/10 mice). C, cumulative frequency plots showing the increase in the mEPSC frequency induced by l‐AP4 (500 μm, 10 min). D, sample traces recorded before and after exposure to l‐AP4 (500 μm, 10 min). E, l‐AP4 (500 μm, 10 min) fails to alter the mEPSC amplitude. F, quantification of the changes in mEPSC amplitude before and after exposure to l‐AP4 (500 μm, 10 min, P = 0.7016). G, cumulative frequency plots show that l‐AP4 has no effect on the mEPSC amplitude. The data were compared to the basal state prior to l‐AP4 addition using an unpaired Student's t test and the values represent the mean ± SD. Scale in D: 10 pA and 10 ms.
Electrophysiology experiments
Pyramidal neurons from the hippocampal CA1 region were identified by their morphology and characteristic electrophysiological properties, and recorded in the whole‐cell configuration. Series and input resistances were monitored throughout the experiment using a −5 mV pulse, and the recordings were considered stable when the series and input resistances, resting membrane potential and stimulus artifact duration did not change >20%. Cells that did not meet these criteria were discarded.
Patch pipettes were pulled from thick‐walled borosilicate glass (1.5 mm outer diameter) on a P‐97 puller (Sutter Instrument, Novato, CA, USA). The pipettes (3–4 MΩ) were filled with the internal solution containing (in mm): potassium gluconate 135, KCl 10, Hepes 10, MgCl2 1, ATP‐Na 2 (pH adjusted to 7.3 with KOH; osmolarity 280–290 mOsm L−1). Recordings were obtained with a PC‐ONE amplifier under voltage‐clamp conditions and the membrane potential was held at −70 mV to record glutamatergic evoked EPSCs (eEPSCs) from SC afferents. Signals were fed to a Pentium‐based PC through a DigiData1322A interface board (Molecular Devises, Sunnyvale, CA, USA). The pCLAMP 10.2 software was used to generate stimuli, as well as for data display, acquisition, storage and analysis. Experiments were performed at 25°C using a temperature controller (Warner Instruments, Hamden, CT, USA).
Theta capillaries (2–5 μm tip) filled with aCSF were used for bipolar stimulation. The electrodes were connected to a stimulator (S38, Grass, Artisan Technology Group, Champaign, IL, USA) through an isolation unit and placed in the stratum radiatum layer at the border of the CA3–CA1 regions to stimulate glutamatergic afferents. Stimuli were delivered at 0.33 Hz and the paired pulse ratio (PPR) was obtained as (2nd EPSC/1st EPSC) after delivering paired pulses (50 ms interval) at 0.33 Hz. The stimulus intensity was adjusted to yield EPSC amplitudes between 20 and 200 pA and it remained unchanged during the experiment. To activate mGlu7 receptors by electrically induced release of glutamate, a HFS protocol was applied that consisted of two (1 s at 50 Hz, every 20 s) or three trains (1 s at 100 Hz, every 20 s).
For statistical analysis, an average of 30 consecutive EPSCs were compared (normalized to the baseline), delivered at 0.33 Hz (90 s) before, during (90 s after the onset of the treatment) and 10 min after l‐AP4 perfusion. In the mEPSC studies (Fig. 3), the frequency and mean amplitude of the events that appeared in the 5 min prior to and after l‐AP4 application were compared. In all the electrophysiology experiments only one neuron was analysed per slice.
Analysis of synaptic vesicle distribution at the active zone by electron microscopy
Hippocampal slices (325 μm thick) obtained as described above for the electrophysiology experiments were transferred to an immersion recording chamber and superfused at 1 mL min−1 with gassed aCSF including 50 μm picrotoxin. In some cases, the slices were also treated with 500 μm l‐AP4 for 10 min. The slices were fixed immediately afterwards by immersion for 45 min at 37°C in 3.5% glutaraldehyde in 0.1 m phosphate buffer (PB, pH 7.4) and they were then left in glutaraldehyde solution for 30 min at room temperature before storing them for 20 h at 4°C. The slices were then rinsed six times with large volumes of 0.1 m PB and the hippocampus was dissected out, post‐fixing it in 1% OsO4–1.5% K3Fe(CN)6 for 1 h at room temperature. After dehydrating through a graded series of ethanol (30, 50, 70, 80, 90, 95 and 100%), the samples were then embedded using the SPURR embedding kit (TAAB, Aldermaston, UK). Ultrathin ultramicrotome sections (70–80 nm thick: Leica EM UC6 Leica Microsystems, Wetzlar, Germany) were stained routinely with uranyl acetate and lead citrate, and images were obtained on a Jeol 1010 transmission electron microscope (Jeol, Tokyo, Japan). Randomly chosen areas of the CA1 striatum radiatum were then photographed at 80,000× magnification and only asymmetric synapses with clearly identifiable electron‐dense postsynaptic densities were analysed by measuring them with ImageJ software. The relative percentage of SVs per AZ was calculated in 10 nm bins at the AZ of the inner layer membrane and the total number of SVs per synaptic terminal was also determined. The data were analysed blind to the genotype and treatment.
Immunohistochemistry for electron microscopy
Electron microscopy examination of immunoreactivity for mGlu7, Munc13‐1 and Munc13‐2 in the hippocampus was performed as described previously, using the pre‐embedding immunoperoxidase and immunogold double‐labelling method (Luján et al. 1996). Briefly, 30‐day‐old C57BL/6J mice were perfused with 4% paraformaldehyde, 0.05% glutaraldehyde and 15% (v/v) saturated picric acid in 0.1 m PB. Vibratome brain sections (60 μm) were processed for immunohistochemical detection of Munc13‐1 or Munc13‐2 using the horseradish peroxide (HRP) reaction, and of mGlu7 using a silver‐enhanced immunogold reaction. Briefly, sections were incubated for 1 h in 10% normal goat serum (NGS) diluted in Tris buffer (TB, 50 mm, pH 7.4) containing 0.9% NaCl (TBS) and 0.2% Triton X‐100. The sections were incubated in a mixture of anti‐mGlu7 (1–2 μg mL−1 diluted in TBS containing 1% NGS) and anti‐Munc13‐1 or anti‐Munc13‐2 (1–2 μg mL−1 diluted in TBS containing 1% NGS). Subsequently, they were incubated overnight at 4°C with a mixture of goat anti‐mouse IgG coupled to 1.4 nm gold (Nanoprobes Inc., Stony Brook, NY, USA) and biotinylated goat anti‐rabbit IgG (Vector Laboratories, Burlingame, CA, USA) in TBS containing 1% NGS. After several washes in TBS, the sections were post‐fixed in 1% (v⁄v) glutaraldehyde and washed in double‐distilled water, prior to performing silver enhancement of the gold particles with an HQ Silver kit (Nanoprobes Inc.). Subsequently, the sections were incubated with an avidin–biotin–peroxidase complex diluted 1:100 (ABC kit, Vector Laboratories). Bound peroxidase enzyme activity was revealed using 3,3′‐diaminobenzidine tetrahydrochloride (DAB: 0.05% in TB, pH 7.4) as the chromogen and 0.01% H2O2 as the substrate. After washing with TB and PB, the sections were then treated with osmium tetroxide (1% in 0.1 m PB), block‐stained with uranyl acetate, dehydrated in a graded series of ethanol and flat‐embedded in Durcupan resin (Fluka). Regions of interest were cut at 70–90 nm on an ultramicrotome (Reichert Ultracut E, Leica, Austria) and collected on single slot pioloform‐coated copper grids. Staining was performed on drops of 1% aqueous uranyl acetate followed by Reynolds's lead citrate, and the ultrastructure was analysed in a Jeol‐1010 electron microscope.
To test the specificity of the electron microscopy staining, the primary antibody was omitted or replaced with 5% (v/v) NGS from the species of the primary antibody. Under these conditions, no selective labelling was observed. In addition, some sections were incubated with only gold‐labelled and biotinylated secondary antibodies, followed by the ABC complex and peroxidase reaction without silver intensification. This resulted in an amorphous HRP end‐product, and no metal particles were detected. When the same protocol was performed but with silver intensification and without the HRP reaction, silver grains were evident but there was no amorphous HRP end‐product. Under these conditions, only infrequent small patches of HRP end‐product were detected and the patches were not selectively associated with any particular cell profile. In addition, the selective location of the signals in structures labelled with only one or the other of the signalling products in the same section, as well as when there were adjacent double‐labelled structures, indicated that the method used does not produce false‐positive double‐labelling results.
Quantitative analysis at EM level
To establish the relative abundance of mGlu7 in hippocampal axon terminals expressing Munc13‐1 and Munc13‐2, immunolabelling of the CA1 stratum radiatum was quantified in 60 μm coronal slices, as described previously (Luján et al. 1996). Briefly, three tissue samples from three animals were embedded in blocks, totalling nine blocks. To minimize false negatives, serial ultrathin electron microscopy sections were cut close to the surface of each block as the immunoreactivity decreased with depth. We estimated the quality of the immunolabelling by always selecting areas with optimal gold labelling at approximately the same distance from the cutting surface, within 5–10 μm of the surface. Randomly selected areas were then photographed from the selected ultrathin sections at a final magnification of 50,000×. To determine the frequency of mGlu7 in axon terminals, immunogold labelling was quantified relative to the total area of ∼5000 μm2. Munc13‐1‐ and Munc13‐2‐positive axon terminals were assessed for the presence of mGlu7 immunoparticles and the percentage of these terminals containing mGlu7 immunoparticles was calculated in the hippocampal stratum radiatum. In total, 75 dendrites were selected and the area of the individual dendritic profiles was measured. The percentage of immunoparticles for HCN1 at postsynaptic and presynaptic sites was calculated in layer III.
Statistical analysis
The data are expressed as the mean ± standard deviation (SD) and the results were compared using an unpaired Student's t test. Statistical differences were established at P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***).
Results
While group III mGluRs with low and high affinity for the l‐AP4 agonist inhibit synaptic transmission at hippocampal SC–CA1 synapses in young animals (Baskys & Malenka, 1991), only mGlu7 receptors inhibit such synaptic transmission in adult mice (Ayala et al. 2008). Thus, adult (>30‐day‐old) SC–CA1 synapses are suitable to study mGlu7 receptor mediated modulation of synaptic transmission without interference from the high l‐AP4‐affinity mGlu4 and mGlu8 receptors. l‐AP4 (20 μm) did not alter the EPSC amplitude at adult (30‐ to 45‐day‐old) SC–CA1 hippocampal synapses (98.0 ± 15.3% of the baseline, n = 9 cells/9 slices/9 mice, P = 0.9013: Fig. 1 A and B). However, short applications (3 min) of l‐AP4 (500 μm) did reversibly reduce the EPSC amplitude (59.3 ± 20.7% of the baseline, n = 9 cells/9 slices/9 mice, P = 0.0004: Fig. 1 A and B). We also found that prolonged application (10 min) of l‐AP4 (500 μm) caused a rapid reduction in the EPSC amplitude (60.5 ± 13.8% of the baseline, n = 12 cells/12 slices/10 mice; P ˂ 0.0001: Fig. 1 C and D) that subsequently faded, resulting in a net increase (139.2 ± 25.2% of the baseline, n = 12 cells/12 slices/10 mice, P = 0.0002: Fig. 1 C and D). The specific group III mGluR antagonist 2‐Methyl‐O‐phosphonoserine (MSOP) completely abolished the responses to l‐AP4 (basal 100.4 ± 12.9%; inhibition 99.1 ± 17.8%, P = 0.8886; potentiation 99.0 ± 13.4%, P = 0.8586, n = 6 cells/6 slices/6 mice: Fig. 1 C and D). This bidirectional modulation of EPSC amplitude by l‐AP4 is associated with contrasting changes in the EPSC2/EPSC1 PPR (basal, 1.42 ± 0.31; inhibition, 2.12 ± 0.27 (P ˂ 0.0001); potentiation, 0.99 ± 0.31, P = 0.0027: Fig. 1 E and F). Hence, this bidirectional modulation of EPSC amplitude by mGlu7 receptors appeared to be due to changes in neurotransmitter release.
Figure 1. The potentiation but not the inhibition of synaptic transmission by mGlu7 receptors requires prolonged activation with l‐AP4.
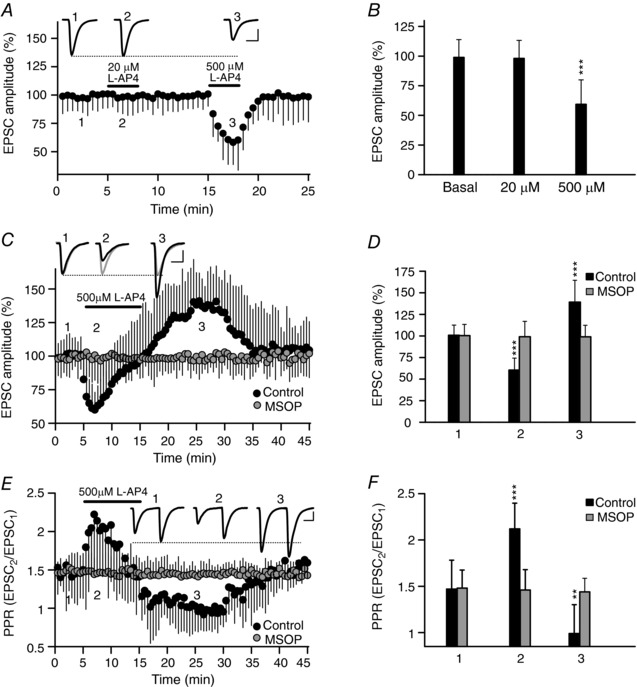
EPSCs were recorded from CA1 pyramidal cells after stimulation of Schaffer collaterals with an electrode placed in the stratum radiatum. A, the application of a low concentration of l‐AP4 (20 μm, 3 min) does not produce any change, whereas a higher concentration of l‐AP4 (500 μm, 3 min) reversibly reduces the EPSC amplitude. EPSC sample traces represent the mean of 30 consecutive EPSCs at 0.33 Hz 90 s before (1) and 90 s after the addition of l‐AP4 at 20 μm (2) or 500 μm (3). B, quantification of the changes in EPSC amplitude with l‐AP4 (20 μm, 3 min, P = 0.9013 or 500 μm, 3 min, P = 0.0004: n = 9 cells/9 slices/9 mice). C, a longer application of l‐AP4 (500 μm, 10 min) first reduces and then increases the amplitude of the EPSC. The group III mGluR antagonist MSOP (200 μm, 30–60 min) blocked the l‐AP4 induced changes in EPSC amplitude. The EPSC sample traces represent the mean of 30 consecutive EPSCs at 0.33 Hz from experiments on control (black) and MSOP treated slices (grey) taken 90 s before (1, basal) or 90 s after the addition of l‐AP4 (2, inhibition), and 10 min after l‐AP4 perfusion (3, potentiation). D, quantification of the changes in EPSC amplitude induced by l‐AP4 (500 μm, 10 min) during inhibition (2) and potentiation (3) in both control (P ˂ 0.0001 and P = 0.0002) and MSOP treated slices (P = 0.8886 and P = 0.8586, respectively): control, n = 12 cells/12 slices/10 mice; MSOP treated slices, n = 6 cells/6 slices/6 mice. E, the data shown in C were used to determine the l‐AP4 induced changes in the paired pulse ratio (PPR: EPSC1/EPSC2) from control and MSOP treated slices. The sample traces are the mean of 30 consecutive EPSCs. F, quantification of the changes in the PPR induced by l‐AP4 (500 μm, 10 min) during inhibition (2) and potentiation (3) in both control (P ˂ 0.0001 and P = 0.0027) and MSOP treated slices (P = 0.8886 and P = 0.8586, respectively). The data were compared to the basal conditions prior to l‐AP4 addition using an unpaired Student's t test and the values represent the mean ± SD. Scale bars in A, C, E: 50 pA and 15 ms.
l‐AP4 is a weak agonist of NMDA receptors (NMDARs: Lafon‐Cazal et al. 1999). However, the l‐AP4 induced bidirectional modulation of synaptic transmission does not involve NMDARs, as l‐AP4‐induced inhibition (63.0 ± 9.8% of the baseline, P = 0.0013) and potentiation (141.3 ± 29.0% of the baseline, P = 0.0381, n = 5 cells/5 slices/4 mice) was unchanged in the presence of the NMDAR antagonist d‐2‐amino‐5‐phosphonovalerate (d‐AP5) (50 μm: Fig. 2 A and B). l‐AP4 can also activate mGlu7 receptors at GABA terminals and reduce GABA release (Klar et al. 2015), thereby attenuating the inhibition of glutamate release via GABAB receptors. However, again l‐AP4‐induced inhibition (61.7 ± 8.6% of the baseline, P = 0.0003) and potentiation of synaptic transmission (135.6 ± 11.3% of the baseline, P = 0.0157, n = 5 cells/5 slices/3 mice) was not altered in the presence of the GABAB antagonist CGP 55845 (5 μm: Fig. 2 C and D). Hence, it appears that neither NMDARs nor GABAB receptors are involved in the bidirectional modulation of synaptic transmission induced by l‐AP4.
Figure 2. There is no change in the inhibition and potentiation of synaptic transmission by l‐AP4 in the presence of the NMDAR and GABAB receptor antagonists d‐AP5 and CGP 55845.
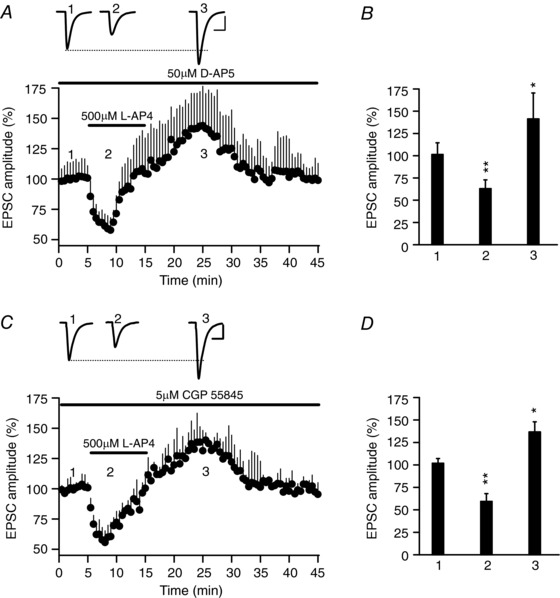
A, l‐AP4 (500 μm, 10 min) first reduces and then increases the amplitude of the EPSC in the continued presence of the NMDAR antagonist d‐AP5 (50 μm). B, quantification of the changes in EPSC amplitude induced by l‐AP4 (500 μm, 10 min) during inhibition (2) and potentiation (3: P = 0.0013 and P = 0.0381, n = 5 cells/5 slices/4 mice). C, l‐AP4 (500 μm, 10 min) first reduces and then increases the amplitude of the EPSC in the continued presence of the GABAB receptor antagonist CGP 55845 (5 μm). D, quantification of the changes in EPSC amplitude induced by l‐AP4 (500 μm, 10 min) during inhibition (2) and potentiation (3: P = 0.0003 and P = 0.0157, n = 5 cells/5 slices/3 mice). In A and C, the EPSC sample traces represent the mean of 30 consecutive EPSCs at 0.33 Hz taken 90 s before (1, basal) or 90 s after the addition of l‐AP4 (2, inhibition), and 10 min after the end of l‐AP4 addition (3, potentiation). The data were compared to the basal state prior to l‐AP4 addition using an unpaired Student's t test and the values represent the mean ± SD.
l‐AP4 increased the frequency of the mEPSCs measured with the Na+ channel blocker tetrodotoxin (TTx, 1 μm; 1.08 ± 0.25 Hz to 2.25 ± 0.82 Hz, n = 21 cells/21 slices/10 mice, P = 0.0101: Fig. 3 A–C) without changing the mEPSC amplitude (before 11.84 ± 2.1 pA, after 11.64 ± 1.8 pA, P = 0.7420: Fig. 3 D–G). Moreover, l‐AP4 did not decrease the mEPSC frequency because voltage dependent Ca2+ channels (VDCCs) that mediate release inhibition by mGlu7 (Millán et al. 2002) are not active in the presence of TTx. Indeed, the VDCC blocker Cd2+ (100 μm) failed to decrease the mEPSC frequency (1.18 ± 0.40 Hz in the presence and 1.16 ± 0.48 Hz in the absence of Cd2+, n = 4 cells/4 slices/2 mice: data not shown: see also Parfitt & Madison, 1993; Gereau & Conn, 1995). Thus, the modulation of synaptic transmission by the mGlu7 receptor has a presynaptic origin.
There was an increase in the amplitude of the second EPSC during potentiation compared to basal conditions in PPR experiments (Fig. 1 E), suggesting that an increase in the RRP size may contribute to synaptic strength. The RRP size was calculated from cumulative amplitude plots as the y‐intercept from a linear fit of the steady state levels attained during the high frequency train (Schneggenburger et al. 2002; Gioia et al. 2016). The RRP size of l‐AP4 (500 μm, 10 min) treated slices stimulated at 40 Hz for 2.5 s increased from 1150.2 (±268.3) to 1589.7 (±309.9) pA, P = 0.0035: Fig. 4 A and B). Hence, an increase in the number of release competent SVs may contribute to the mGlu7 receptor mediated potentiation of synaptic transmission.
Figure 4. The mGlu7 receptor mediated potentiation of synaptic transmission involves an increase in the RRP size.
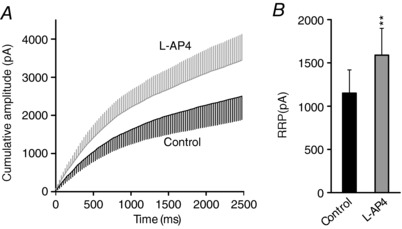
Slices were treated with l‐AP4 (500 μm, 10 min) and after a 5 min wash, they were stimulated at 40 Hz for 2.5 s. The RRP size was calculated from the cumulative amplitude plots as the y‐intercept from a linear fit of the steady state level attained during the high frequency train. A, cumulative amplitude plot of a 2.5 s 40 Hz train in control and l‐AP4 treated cells. B, quantification of the changes in RRP size induced by treating cells with l‐AP4 (500 μm, 10 min, P = 0.0035): control, n = 10 cells/10 slices/4 mice; l‐AP4, n = 10 cells/10 slices/4 mice. The data were compared to the controls using an unpaired Student's t test and the values represent the mean ± SD.
PLC inhibition impairs mGlu7 receptor mediated potentiation of synaptic transmission
While mGlu7 receptors activate PLC (Perroy et al. 2000; Martín et al. 2010), it is not clear how this signalling affects neurotransmitter release as both inhibition (Perroy et al. 2000) and potentiation (Martín et al. 2010) have been reported. l‐AP4 caused a sustained reduction in the EPSC amplitude (62.3 ± 17.0% of the baseline, n = 8 cells/8 slices/8 mice, P ˂ 0.0001: Fig. 5 A and B) and it failed to increase this parameter in slices treated with the PLC inhibitor U73122 (2 μm, 30 min; 97.9 ± 26.8%, n = 8 cells/8 slices/8 mice, P = 0.7689: Fig. 5 A and B). By contrast, l‐AP4 did provoke transient inhibition in the presence of the inactive PLC inhibitor U73343 (60.8 ± 9.5% of baseline, n = 6 cells/6 slices/6 mice, P = 0.011: Fig. 5 A and B) followed by potentiation of the EPSC amplitude (142.5 ± 16.3% of the baseline, n = 6 cells/6 slices/6 mice, P = 0.0434: Fig. 5 A and B). Thus, PLC appeared to be required for mGlu7 receptor mediated potentiation of synaptic transmission at SC–CA1 synapses. Moreover, these results also indicate that the fading of mGlu7 receptor mediated inhibition could be the result of the simultaneous coupling of the receptor to an additional signalling pathway that potentiates synaptic transmission, first counteracting the release inhibition and then resulting in a net potentiation.
Figure 5. The l‐AP4‐induced potentiation is sensitive to the PLC inhibitor U73122 and to the DAG binding inhibitor calphostin C, but not to pertussis toxin.
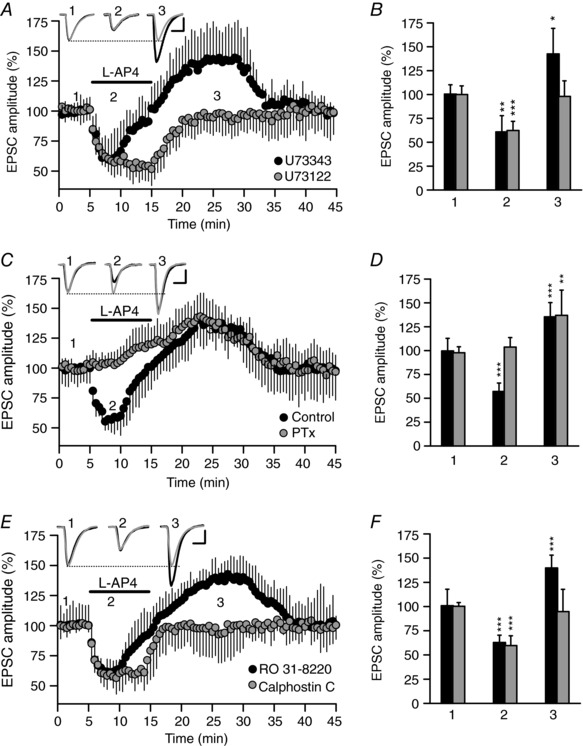
A, in the presence of the PLC inhibitor U73122 (2 μm, 30 min), l‐AP4 (500 μm, 10 min) fails to enhance the EPSC amplitude but, rather, it causes sustained inhibition of this parameter. By contrast, the inactive PLC inhibitor U73343 (2 μm, 30 min) caused a transient inhibition that subsequently faded, resulting in the net potentiation of the EPSC amplitude. B, quantification of the changes in EPSC amplitude induced by l‐AP4 (500 μm, 10 min) during inhibition (2) and potentiation (3) in the presence of U73122 (P ˂ 0.0001 and P = 0.7689, n = 6 cells/6 slices/6 mice) or U73343 (P = 0.011 and P = 0.0434, n = 8 cells/8 slices/8 mice). C, in the presence of PTx (2 μg mL−1 for 2–4 h at 30°C), l‐AP4 (500 μm, 10 min) failed to reduce, but rather it enhanced, the EPSC amplitude, whereas in control slices incubated for 2–4 h at 30°C, l‐AP4 (500 μm, 10 min) caused a transient inhibition that subsequently faded into a net potentiation of the EPSC amplitude. D, quantification of the changes in EPSC amplitude induced by l‐AP4 (500 μm, 10 min) during inhibition (2) and potentiation (3) in both PTx treated (P = 0.1709 and P = 0.0048, n = 8 cells/8 slices/8 mice) and in control slices (P ˂ 0.0001 and P = 0.0002, n = 8 cells/8 slices/8 mice). E, in the presence of the DAG binding inhibitor calphostin C (0.1 μm, 30 min), l‐AP4 (500 μm, 10 min) induced sustained inhibition of the EPSC amplitude and it failed to enhance the EPSC amplitude. By contrast, the ATP binding antagonist and PKC inhibitor RO 31‐8220 (1 μm, 30 min) did not alter the l‐AP4 responses, as the transient inhibition was followed by a net potentiation of the EPSC amplitude. F, quantification of the changes in EPSC amplitude induced by l‐AP4 (500 μm, 10 min) during inhibition (2) and potentiation (3) in the presence of calphostin C (P ˂ 0.0001 and P = 0.4741, n = 10 cells/10 slices/10 mice) or RO 31‐8220 (P = 0.0001 and P ˂ 0.0001, n = 9 cells/9 slices/6 mice). In A, C and E, the EPSC sample traces represent the mean of 30 consecutive EPSCs at 0.33 Hz taken 90 s before (1, basal) or 90 s after the addition of l‐AP4 (2, inhibition), and 10 min after l‐AP4 perfusion (3, potentiation). The data were compared to the basal state (1) using an unpaired Student's t test and the values represent the mean ± SD. Scale in A, C and E: 50 pA and 15 ms.
Pertussis toxin impairs inhibition but not mGlu7 receptor mediated potentiation of synaptic transmission
As mGlu7 receptors coupled to Gi/o proteins inhibit Ca2+ channels and adenylyl cyclase (Perroy et al. 2000; Millán et al. 2002, 2003; Capogna, 2004; Pelkey et al. 2005; Martín et al. 2007, 2010; Kammermeier, 2015), we tested whether blocking Gi/o proteins with PTx alters mGlu7 receptor mediated potentiation of synaptic transmission. In hippocampal slices incubated with PTx (2 μg mL−1 for 2–4 h at 30°C), l‐AP4 failed to reduce (103.8 ± 9.9% of the baseline, n = 8 cells/8 slices/8 mice, P = 0.1709: Fig. 5 C and D), while the increase of EPSC amplitude was not altered (137.0 ± 26.4% of the baseline, n = 8 cells/8 slices/8 mice, P = 0.0048: Fig. 5 C and D). By contrast, l‐AP4 first inhibited (57.1 ± 8.9% of the baseline, n = 8 cells/8 slices/8 mice, P ˂ 0.0001: Fig. 5 C and D) and then potentiated the amplitude of the EPSCs in control slices that were not exposed to PTx (135.4 ± 14.9% of the baseline, n = 8 cells/8 slices/8 mice, P = 0.0002: Fig. 5 C and D). Therefore, the modulation of EPSCs by mGlu7 receptors is the result of the temporal summation of PTx‐sensitive and PTx‐resistant components of synaptic transmission. The PTx‐sensitive component develops rapidly upon exposure to l‐AP4 as a consequence of the well‐established inhibition of Ca2+ channels by mGlu7 receptors that activate Gi/o proteins (Okamoto et al. 1994; Millán et al. 2002, 2003; Perroy et al. 2002; Martín et al. 2007, 2010; Kammermeier, 2015). By contrast, the PTx‐resistant component of synaptic transmission develops more slowly, probably reflecting the mGlu7 receptor signalling pathway described in synaptosomes, a pathway targeting the release machinery through PIP2 hydrolysis coupled to the generation of IP3 and DAG (Martín et al. 2010).
The inhibitor of DAG binding, calphostin C, prevents mGlu7 receptor mediated potentiation of synaptic transmission
To further understand the mechanism underlying mGlu7 receptor mediated potentiation of synaptic transmission, we distinguished between PKC and Munc13 (Lou et al. 2008) as targets of the DAG generated by mGlu7 receptors. To study these targets, we used the PKC inhibitor RO 31‐8220, which prevents ATP binding, and calphostin C, which prevents DAG binding and therefore also inhibits non‐kinase DAG‐binding proteins. Slices exposed to RO 31‐8220 (1 μm, 30 min) responded to l‐AP4 with a transient inhibition of the EPSC amplitude (62.9 ± 7.6% of the baseline, n = 9 cells/9 slices/6 mice, P = 0.0001: Fig. 5 E and F), which then faded into net potentiation (139.8 ± 13.3% of the baseline, n = 9 cells/9 slices/6 mice, P ˂ 0.0001: Fig. 5 E and F). By contrast, the inhibition of the EPSC amplitude in hippocampal slices exposed to calphostin C (0.1 μm, 30 min) was sustained until the agonist was removed (59.7 ± 10.1% of the baseline, n = 10 cells/10 slices/10 mice, P ˂ 0.0001: Fig. 5 E and F), with no potentiation of the EPSC amplitude (94.7 ± 23.1% of the baseline, n = 10 cells/10 slices/10 mice, P = 0.4741: Fig. 5 E and F). These data suggest that Munc13 proteins and not PKC participate in mGlu7 receptor‐mediated potentiation of synaptic transmission at SC–CA1 hippocampal synapses.
Lack of mGlu7 receptor mediated potentiation in Munc13‐2 KO mice
The AZ protein Munc13‐1 is a phorbol ester receptor essential for SV priming that plays an important role in neurotransmitter release (Betz et al. 1998; Rhee et al. 2002). There are three Munc13 isoforms expressed in the brain (Munc13‐1–3), although only Munc13‐1 and Munc13‐2 are found in the hippocampus (Augustin et al. 1999b). However, Munc13‐1 KO mice (Augustin et al. 1999a) are not viable and thus they cannot be analysed by electrophysiology. Munc13‐2 KO mice are viable and basal synaptic transmission at SC–CA1 hippocampal synapses is unaffected by the absence of Munc13‐2 (Varoqueaux et al. 2002; Breustedt et al. 2010). Thus, we tested whether mGlu7 receptor modulation of synaptic transmission is maintained in slices lacking Munc13‐2. In hippocampal slices from Munc13‐2 KO mice there was a sustained dampening of the EPSC amplitude in the presence of l‐AP4 (65.5 ± 6.5% of the baseline, n = 11 cells/11 slices/4 mice, P < 0.0001: Fig. 6 A and B) and no subsequent potentiation of the EPSC amplitude (97.7 ± 13.3% of the baseline, n = 11 cells/11 slices/4 mice, P = 0.5488: Fig. 6 A and B). By contrast, WT littermates showed bidirectional modulation of the EPSC amplitude (Fig. 6 A and B). We also assessed whether mGlu7 receptor mediated potentiation of synaptic transmission was related to changes in SV distribution. In control slices from WT littermates, l‐AP4 increased the number of SVs close to the plasma membrane (from 6.8 ± 1.5%, n = 86 synapses/2 control mice, to 10.9 ± 1.2%, 90 synapses/2 l‐AP4 mice, P < 0.0001: Fig. 6 C–E). Conversely, in slices from Munc13‐2 KO mice there was a similar number of SVs close to the AZ plasma membrane to those in the WT (6.6 ± 1.2%, n = 145 synapses/3 mice, P = 0.7551: Fig. 6 G and I) but l‐AP4 did not increase the number of SVs close to the plasma membrane (6.9 ± 1.2%, n = 147 synapses/3 mice, P = 0.5815: Fig. 6 H and I). Interestingly, l‐AP4 did not alter the number of SVs at the AZ in WT (38.2 ± 4.8 and 34.5 ± 3.9, P = 0.0745 before and after exposure to l‐AP4: Fig. 6 F) or in the Munc13‐2 KO synapses (36.8 ± 3.9 and 34.6 ± 3.6, P = 0.2147 before and after l‐AP4: Fig. 6 J). Thus, mGlu7 receptor mediated potentiation of synaptic transmission requires an increase in the size of the RRP of SVs that does not occur in cells lacking Munc13‐2.
Figure 6. The lack of mGlu7 receptor mediated potentiation of synaptic transmission in Munc13‐2 knockout mice is associated with the failure of l‐AP4 to increase the number of synaptic vesicles close to the active zone plasma membrane.
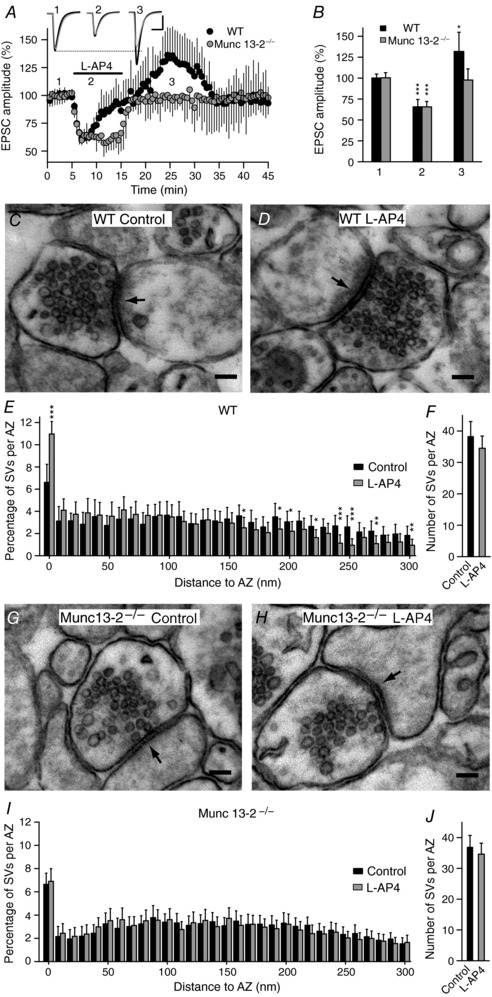
A, l‐AP4 fails to enhance the EPSC amplitude in Munc13‐2 KO mice. The EPSC sample traces represent the mean of 30 consecutive EPSCs at 0.33 Hz in slices from wild‐type (WT) and Munc13‐2 KO mice taken 90 s prior to (1, basal), 90 s after (2, inhibition) and 10 min after l‐AP4 perfusion (3, potentiation). B, quantification of the changes in the EPSC amplitude induced by l‐AP4 (500 μm, 10 min) in WT (n = 5 cells/5 slices/3 mice) and Munc13‐2 KO mice (n = 11 cells/11 slices/4 mice), during inhibition (2, P = 0.0002 and P = 0.0397) and potentiation (3, P < 0.0001 and P = 0.5488). The data were compared to the basal state (1) using an unpaired Student's t test. C and D, l‐AP4 treatment brings SVs close to the plasma membrane in WT mice. E, quantification of the l‐AP4 induced increase (P < 0.0001) in SV distribution less than 10 nm from the AZ membrane. F, l‐AP4 does not change (P = 0.0745, n = 86 synapses/2 mice) the total number of SVs per AZ in WT mice compared to the controls (n = 90 synapses/2 mice). The data from l‐AP4 treated slices were compared with the control slices (untreated) using an unpaired Student's t test. G and H, l‐AP4 treatment fails to bring SVs less than 10 nm from AZ plasma membrane in Munc13‐2 KO mice. I, quantification of the l‐AP4 induced effect on SV distribution in Munc13‐2 KO mice. l‐AP4 fails (P = 0.5815) to bring SVs to less than 10 nm from the AZ membrane. J, l‐AP4 does not change (P = 0.2147, n = 145 synapses/3 mice) the total number of SVs per AZ in Munc13‐2 KO mice (n = 147 synapses/3 mice). The values represent the mean ± SD and the data from l‐AP4 treated slices were compared with control (untreated) slices using an unpaired Student's t test. Scale in A: 50 pA and 15 ms. Scale bar in C, D, G and H: 100 nm.
The mGlu7 receptors co‐localize with Munc13‐2 and Munc13‐1 at glutamatergic nerve terminals
The precise subcellular localization of mGlu7 receptors in axon terminals expressing Munc13‐1 or Munc13‐2 was studied by double labelling pre‐embedding immunogold/HRP electron microscopy. Munc13‐1, Munc13‐2 and mGlu7 immunoreactivity was found at presynaptic sites (Fig. 7 A and B) and when dual mGlu7/Munc13‐1 labelling was quantified in the neuropil of the stratum radiatum, 128 (38.55%) of the 332 axon terminals labelled for mGlu7 were also labelled for Munc13‐1. In terms of Munc13‐2, this proportion increased to 201 (63.21%) double‐labelled terminals from a total of 318 axon terminals labelled for mGlu7.
Figure 7. Nerve terminals containing the mGlu7 receptor also express the Munc13‐1 or Munc13‐2 proteins.
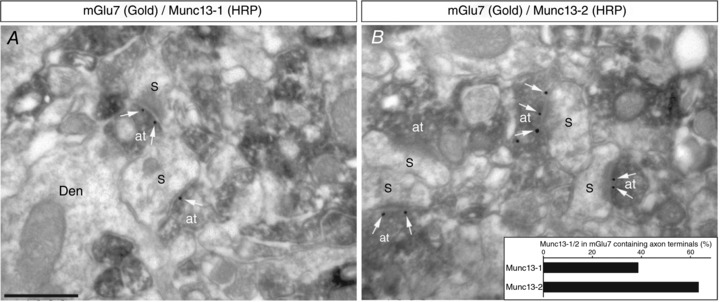
A and B, double‐labelling pre‐embedding immunogold/HRP electron microscopy demonstrates mGlu7 immunolabelling (gold/silver‐intensified immunogold reaction) in the presynaptic AZ (white arrows) of axon terminals (at) immunopositive (HRP reaction) for Munc13‐1 (A) and Munc13‐2 (B) that make synapses with dendritic spines (S) of pyramidal cells. Scale bar: 0.5 μm. Den, dendrite.
Lack of mGlu7 receptor mediated potentiation in RIM1α KO mice
RIMs are multidomain proteins that play a central role in the organization of the AZ. RIM proteins tether Ca2+ channels to the presynaptic AZ (Kaeser et al. 2011) and they activate SV priming by reversing the auto‐inhibitory homodimerization of Munc13 (Deng et al. 2011). In addition, RIM1α also recruits and stabilizes Munc13 proteins at the AZ (Betz et al. 2001; Schoch et al. 2002), and it drives the distribution of SVs (Fernández‐Busnadiego et al. 2013). Given the physical and functional association between the Munc13 and RIM1α proteins, we studied whether mGlu7 receptor modulation of synaptic transmission is altered in slices from RIM1α KO mice. Inhibition of EPSC amplitude was sustained in hippocampal slices lacking RIM1α (66.4 ± 14.3% of the baseline, n = 10 cells/10 slices/10 mice, P = 0.0024: Fig. 8 A and B), which reversed upon l‐AP4 washout, although there was no subsequent potentiation of the EPSC amplitude (97.4 ± 16.2% of the baseline, n = 10 cells/10 slices/10 mice, P = 0.8095: Fig. 8 A and B). l‐AP4 also failed to promote the accumulation of SVs close to the plasma membrane of RIM1α KO synapses (4.3 ± 1.2%, n = 152 synapses/3 mice in the presence of l‐AP4 and 5.0 ± 1.2%, n = 147 synapses/3 mice in the absence of l‐AP4, P = 0.2456: Fig. 8 G–I). Thus, mGlu7 mediated potentiation of synaptic transmission is supported by the recruitment of SVs to the plasma membrane, which requires the Munc13‐2 and RIM1α proteins. By contrast, the number of SVs close to the AZ plasma membrane (Fig. 8 G and I) was similar in control slices from RIM1α KO and their WT littermates (Fig. 8 C and E), supporting basic synaptic transmission and its inhibition by l‐AP4 (Fig. 8 A and B).
Figure 8. The lack of mGlu7 receptor mediated potentiation of synaptic transmission in RIM1α knockout mice is associated with the failure of l‐AP4 to increase the number of synaptic vesicles close to the active zone plasma membrane.
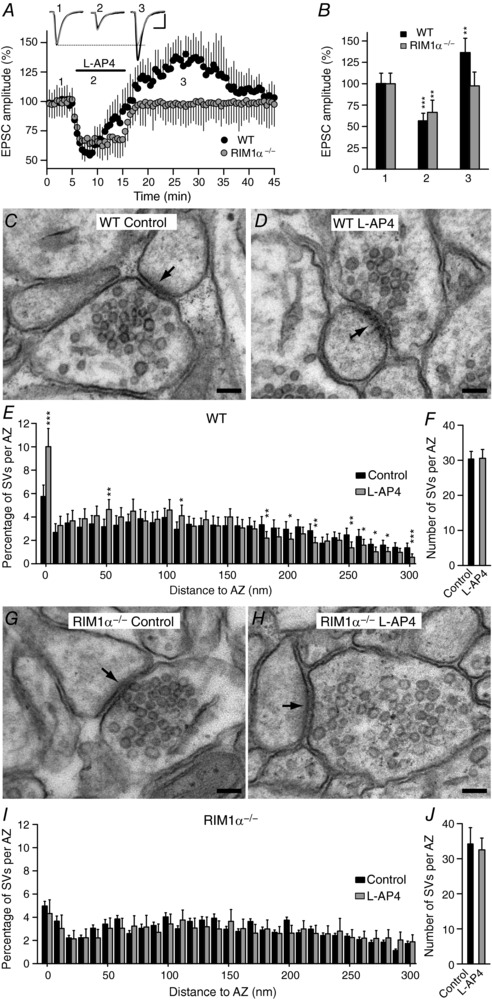
A, l‐AP4 fails to potentiate the EPSC amplitude in RIM1α KO mice. EPSC sample traces represent the mean of 30 consecutive EPSCs at 0.33 Hz from experiments in wild‐type (WT) and RIM1α KO mice taken 90 s prior to (1, basal), 90 s after (2, inhibition) and 10 min after l‐AP4 perfusion (3, potentiation). B, quantification of the changes in the EPSC amplitude induced by l‐AP4 (500 μm, 10 min) in WT (n = 5 cells/5 slices/5 mice) and RIM1α KO mice (n = 10 cells/10 slices/10 mice) during both inhibition (2, P = 0.0004 and P = 0.0061) and potentiation (3, P = 0.0024 and P = 0.8095). The data were compared to the basal state (1) using an unpaired Student's t test. C and D, l‐AP4 treatment brings SVs closer to the plasma membrane in WT mice. E, quantification of the l‐AP4 induced increase (P = 0.0013) in the SVs at least 10 nm from the AZ membrane. F, l‐AP4 does not change (P = 0.944, n = 112 synapses/3 mice) the total number of SVs per AZ in WT mice. The data from l‐AP4 treated slices were compared with the control slices (untreated) using an unpaired Student's t test. G and H, l‐AP4 treatment fails to bring SVs close to the plasma membrane in RIM1α KO mice. I, quantification of the l‐AP4 induced change in SV distribution in RIM1α KO mice. l‐AP4 fails (P = 0.2456) to bring SVs at least 10 nm from the AZ membrane. J, l‐AP4 does not change (P = 0.3616, n = 152 synapses/3 mice) the total number of SVs per AZ in RIM1α KO mice (n = 147 synapses/3 mice). The data from l‐AP4 treated slices were compared with control (untreated) slices using an unpaired Student's t test and the values represent the mean ± SD. Scale in A: 50 pA and 15 ms. Scale bar in C, D, G and H: 100 nm.
Electrically evoked release of glutamate also activates mGlu7 receptors
We tested whether mGlu7 receptors activated by electrically evoked release of glutamate also modulate excitatory synaptic transmission bidirectionally. As mGlu7 receptors have a low affinity for both l‐AP4 and glutamate agonists (Okamoto et al. 1994), we used three‐train HFS (1 s at 100 Hz, every 20 s) to increase the synaptic glutamate. Accordingly, a MSOP‐sensitive component of synaptic potentiation (50.7 ± 24.2%, P = 0.0052) that lasted several minutes after HFS was unmasked (10 cells/10 slices/5 mice: Fig. 9 A and B). Nevertheless, as only a rapidly developing potentiation was observed, activation of mGlu7 receptors in these conditions did not result in the bidirectional modulation of synaptic transmission seen with the exogenous agonist l‐AP4. While the PLC inhibitor U73122 prevented mGlu7 induced potentiation, it did not unravel any inhibitory response as it completely eliminated the MSOP‐sensitive component of the EPSC (6.6 ± 24.4%, P = 0.7464, n = 8 cells/8 slices/4 mice: Fig. 9 A and B).
Figure 9. HFS‐induced activation of mGlu7 receptors bidirectionally modulates synaptic transmission, although a potentiating response prevails.
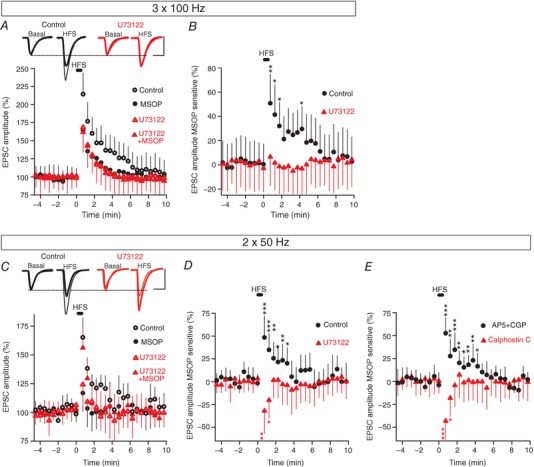
A, changes in EPSC amplitude induced by three trains (1 s at 100 Hz, every 20 s) in control slices (untreated: n = 10 cells/10 slices/5 mice) or in slices exposed to: MSOP (200 μm, 30–60 min, n = 10 cells/10 slices/5 mice), U73122 (2 μm, 30 min, n = 8 cells/8 slices/4 mice), or MSOP plus U73122 (n = 8 cells/8 slices/4 mice). B, quantification of the MSOP sensitive component of the EPSC normalized to the basal values prior to HFS. C, changes in EPSC amplitude induced by two trains (1 s at 50 Hz, every 20 s) in control slices (untreated, n = 9 cells/9 slices/3 mice) or slices exposed to: MSOP (200 μm, 30–60 min, n = 9 cells/9 slices/ 3 mice), U73122 (2 μm, 30 min, n = 9 cells/9 slices/3 mice) or MSOP plus U73122 (n = 9 cells/9 slices/3 mice). D, quantification of the MSOP sensitive component of the EPSC normalized to the basal values prior to HFS. E, quantification of the MSOP sensitive component of the EPSC normalized to the basal values prior to HFS in d‐AP5 (50 μm) plus CGP 55845 (5 μm) treated slices (n = 10 cells/10 slices/3 mice) and in calphostin C (0.1 μm, 30 min) treated slices (n = 10 cells/10 slices/3 mice). Unpaired Student's t test compared to basal values. Scale bars in A and C: 50 pA and 15 ms. EPSC sample traces in A and C represent the mean of 10 consecutive EPSCs at 0.33 Hz 90 s before (basal) and 20 s after HFS. Thin and thick traces indicate in the absence and presence of MSOP, respectively.
The effects of the inhibitory response might be counteracted by the strong Ca2+ influx during HFS, therefore we used a weaker stimulation with only two trains (1 s at 50 Hz, every 20 s). With this protocol, a MSOP‐sensitive component of synaptic potentiation was evident (48.8 ± 23.5%, P = 0.0003) (n = 9 cells/9 slices/3 mice: Fig. 9 C and D), indicating that potentiation of synaptic transmission by mGlu7 receptors also dominates under these conditions. However, U73122 now revealed an inhibitory effect of mGlu7 (32.0 ± 24.9%, P = 0.0055, n = 9 cells/9 slices/3 mice: Fig. 9 D) that lasted less than the potentiating one.
We assessed the presynaptic origin of this MSOP‐sensitive component of synaptic transmission by measuring the corresponding changes in the PPR. The PPR decreased to −0.29 ± 0.21 (P = 0.0098) after HFS compared with basal (−0.03 ± 0.16, n = 9 cells/9 slices/3 mice) in the control slices, whereas the PPR increased to 0.25 ± 0.17 (P = 0.0011) after HFS compared to basal (−0.03 ± 0.11%, n = 9 cells/9 slices/3 mice) in U73122 treated slices (data not shown). We also confirmed that the modulation of synaptic transmission by HFS‐induced activation of mGlu7 receptors was independent of NMDARs and GABAB receptors, as the NMDAR antagonist d‐AP5 (50 μm) and the GABAB antagonist CGP 55845 (5 μm), added together, did not alter the MSOP component of synaptic transmission (52.5 ± 17.2%, P = 0.0001, n = 10 cells/10 slices/3 mice, Fig. 9 E). We also tested that the mGlu7 receptor dependent potentiation was abolished in the presence of calphostin C, revealing an inhibitory response (43.2 ± 20.5%, P = 0.0001, n = 10 cells/10 slices/3 mice: Fig. 9 E).
The bidirectional modulation of synaptic transmission by the HFS‐induced activation of mGlu7 receptors was tested for its sensitivity to the lack of Munc13‐2 and RIM1α proteins. In Munc13‐2 KO the potentiation was abolished, although the inhibitory response was preserved (27.0 ± 24.7%, P = 0.0075, n = 11 cells/11 slices/4 mice). In contrast, WT littermates showed potentiation (54.5 ± 24.9%, P = 0.0001, n = 11 cells/11 slices/3 mice, Fig. 10 A and B). Similarly, in RIM1α KO mice the potentiation was abolished and unchanged inhibition was observed (30.3 ± 26.0%, P = 0.0043, n = 11 cells/11 slices/4 mice) while WT littermates showed potentiation (50.3 ± 29.1%, P = 0.0012, Fig. 10 C and D). Thus, HFS‐induced activation of mGlu7 receptors provokes the bidirectional modulation of synaptic transmission. However, as both responses occur simultaneously, the potentiating response alone prevails while the inhibitory one is only evident when potentiation is blocked with calphostin C, or when there is no Munc13‐2 or RIM1α protein.
Figure 10. The mGlu7 dependent HFS‐induced potentiation is abolished by the lack of Munc13‐2 or RIM1α proteins.
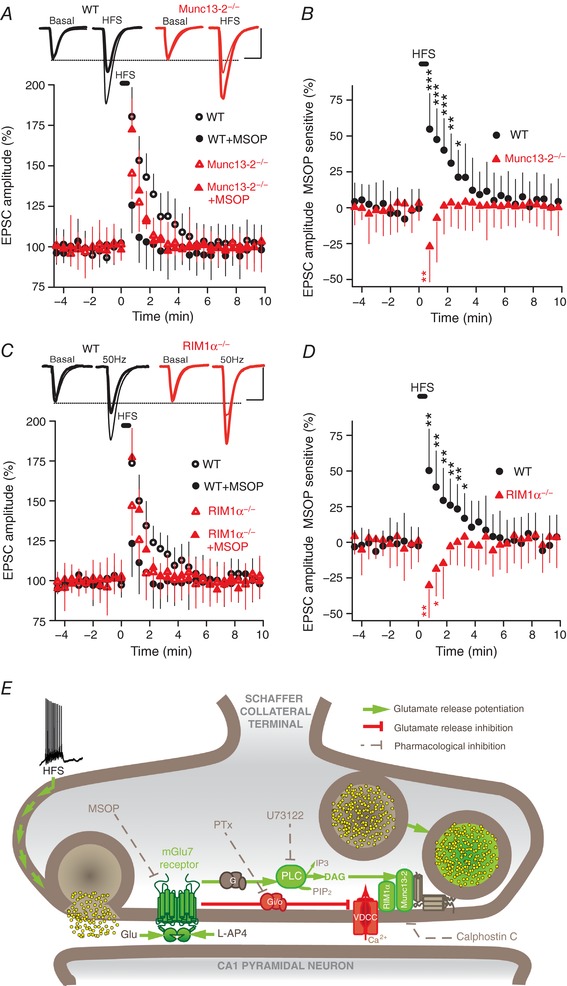
A, changes in the EPSC amplitude induced by two trains (1 s at 50 Hz, every 20 s) in control (untreated) and MSOP (200 μm, 30–60 min) treated slices from Munc13 KO (n = 11 cells/11 slices/4 mice) and WT littermates (n = 11 cells/11 mice/3 mice). B, quantification of the MSOP sensitive component of the EPSC normalized to the basal values prior to HFS. C, changes in EPSC amplitude induced by two trains (1 s at 50 Hz, every 20 s) in control (untreated) and MSOP (200 μm, 30–60 min) treated slices from RIM1α KO (n = 11 cells/11 slices/4 mice) and WT littermates (n = 9 cells/9 mice/3 mice). D, quantification of the MSOP sensitive component of the EPSC normalized to the basal values prior to HFS. Unpaired Student's t test compared to basal values. Scale bars in A and C: 50 pA and 15 ms. EPSC sample traces in A and C represent the mean of 10 consecutive EPSCs at 0.33 Hz 90 s before (basal) and 20 s after HFS (thin and thick traces in the absence and presence of MSOP, respectively). E, scheme of the proposed signalling for the bidirectional modulation of synaptic transmission by mGlu7 receptors. The mGlu7 receptor stimulates a PTx‐sensitive G protein that inhibits voltage dependent Ca2+ channels and decreases neurotransmitter release. The mGlu7 receptors also activate a PTx‐resistant G protein to enhance PLC activity, thereby increasing PIP2 hydrolysis and the generation of DAG. This DAG binds to and activates/translocates the non‐kinase DAG‐binding protein Munc13‐2 to promote glutamate release with the help of RIM1α. DAG, diacylglycerol; HFS, high frequency stimulation; PIP2, phosphatidylinositol bisphosphate; PLC, phospholipase C; PTx, pertussis toxin; VDCC, voltage dependent Ca2+ channel. The blockade/inhibition (dashed line) of Gi/o proteins by PTx, mGlu7 receptors by MSOP, PLC by U73122 and Munc13 by calphostin C is also indicated, as well as the activation (arrows) of mGlu7 receptors with the agonist l‐AP4 or by HFS induced glutamate release.
Discussion
Activation of mGlu7 receptors with the agonist l‐AP4 or following glutamate release induced by HFS provokes a bidirectional modulation of excitatory synaptic transmission at SC–CA1 synapses. This inhibition is sensitive to the blockage of Gi/o proteins with PTx, while potentiation is prevented by the PLC inhibitor U73122 and by the inhibitor of DAG binding, calphostin C. This DAG‐dependent modulation of the release machinery requires the Munc13‐2 or RIM1α proteins. Both inhibition and potentiation occur simultaneously when mGlu7 receptors are activated by the glutamate released in response to HFS, although potentiation prevails and the inhibitory response can only be revealed after the blockade of potentiation.
We found that mGlu7 receptors bidirectionally modulate synaptic transmission through PTx‐sensitive and PTx‐resistant signalling pathways, which inhibit and enhance neurotransmitter release, respectively. The inhibition of synaptic transmission mediated by mGlu7 (Baskys & Malenka, 1991; Gereau & Conn, 1995; Ayala et al. 2008; Klar et al. 2015) is the result of Ca2+ channel inhibition (Millán et al. 2002, 2003; Perroy et al. 2000, 2002; Pelkey et al. 2006), yet it is independent of changes in cAMP unless adenylyl cyclase is activated (Millán et al. 2002; Martín et al. 2007; Ferrero et al. 2016). Direct evidence that mGlu7 receptors potentiate neurotransmitter release was obtained from synaptosomes (Martín et al. 2010), and our electrophysiological data are consistent with that biochemical study. First, the potentiating response is resistant to PTx but dependent on PLC, and second, the potentiating response is resistant to PKC inhibitors but blocked by the DAG‐binding antagonist calphostin C. Finally, the potentiation of neurotransmitter release does not involve a switch in receptor signalling from inhibition to potentiation but, rather, the coupling of the receptor to an additional PLC‐dependent signalling cascade. This is consistent with the capacity of the mGlu7 receptor to simultaneously activate several signalling pathways (Martín et al. 2010; Ferrero et al. 2016). Thus, mGlu7 receptor coupling to PLC‐dependent pathways in synaptosomes results in the net potentiation of release, despite the ongoing inhibition of Ca2+ channels (Martín et al. 2010).
The mGlu7 receptor mediated potentiation of synaptic transmission requires the AZ protein Munc13‐2, consistent with the central role of Munc13 proteins in neurotransmitter release. The hippocampal CA regions express Munc13‐1 and Munc13‐2 (Augustin et al. 1999a), which are essential for evoked and spontaneous release, and to maintain the RRP (Varoqueaux et al. 2002; Imig et al. 2014). We could not test the role of Munc13‐1 in mGlu7 modulation of synaptic transmission because Munc13‐1 KO mice are not viable (Augustin et al. 1999a). By contrast, mice lacking Munc13‐2 (Varoqueaux et al. 2002) maintain basal synaptic transmission and the number of SVs close to the AZ plasma membrane, and mGlu7 receptor mediated inhibition persists in these mice, although they failed to potentiate synaptic transmission because no further SVs are recruited to the AZ plasma membrane. This is consistent with the fact that these receptors are distributed in both Munc13‐1‐ and Munc13‐2‐containing synapses. Munc13 proteins have a DAG binding site (Betz et al. 1998), through which PLC‐coupled GPCRs activate/translocate these proteins (Bauer et al. 2007; Martín et al. 2010; Ferrero et al. 2013) and initiate the PLC dependent potentiation of synaptic transmission. Munc13‐2 is more abundant in cytosolic fractions than Munc13‐1 (Augustin et al. 1999a) and thus, Munc13‐2 could provide extra priming of SVs in response to DAG generation by mGlu7 receptors. In addition, Munc13‐2 proteins have been associated with the potentiation of synaptic transmission at several synapses (Gioia et al. 2016; Kawabe et al. 2017) in a PLC dependent manner (Rosenmund et al. 2002). The mGlu7 receptor mediated potentiation of both glutamate release (Martín et al. 2010) and synaptic transmission is also PLC dependent, suggesting that mGlu7 receptors are involved in this form of synaptic plasticity. In fact, short‐term potentiation is attenuated in mGlu7 KO mice (Bushell et al. 2002).
The mGlu7 receptor mediated potentiation of synaptic transmission requires RIM1α proteins, consistent with its essential role in transmitter release and synaptic plasticity (Castillo et al. 2002; Schoch et al. 2002; Kaeser et al. 2011). RIM1α supports the function of Munc13 proteins by disrupting inactive Munc13 homodimers and stabilizing the protein (Schoch et al. 2002; Deng et al. 2011). In addition, RIM1α organizes AZ proteins and recruits Ca2+ channels to the AZ (Kaeser et al. 2011). Finally, the dependence of mGlu7 receptor mediated potentiation on RIM1α is also consistent with the co‐immunoprecipitation of these two proteins in the hippocampus (Pelkey et al. 2008).
The mGlu7 receptor mediated potentiation of synaptic transmission is faster when glutamate is released by HFS than by the agonist l‐AP4. The activation of mGlu7 receptors also depends on Ca2+/calmodulin binding to the intracellular C domain (O'Connor et al. 1999; Caldwell et al. 2008; Scheschonka et al. 2008). Thus, the Ca2+ requirements for optimal receptor activation can be better fulfilled during HFS than with agonist activation (0.33 Hz). Strikingly, mGlu7 receptors respond to much lower l‐AP4 concentrations after light‐induced channel rhodopsin‐2 mediated Ca2+ influx (Caldwell et al. 2008). Therefore, AZ mGlu7 receptors (Shigemoto et al. 1996, 1997) exposed to high concentrations of extracellular glutamate and intracellular Ca2+ during HFS may be more active than when exposed to l‐AP4. High Ca2+ levels may also favour the activation of other Ca2+ dependent proteins in the mGlu7 receptor signalling cascade that are involved in the potentiation of neurotransmitter release (Martín et al. 2011), such as Munc13 (Junge et al. 2004).
The HFS‐induced release of glutamate activates mGlu7 receptors that signal simultaneously through pathways leading to the potentiation and inhibition of neurotransmitter release. As the potentiating effect is stronger and lasts longer than the inhibitory one, the potentiation of synaptic transmission prevails, consistent with the role of these receptors in short term potentiation (Bushell et al. 2002). The duration of the inhibitory response is probably related to the time during which synaptic glutamate persists above the threshold for mGlu7 receptor activation, which exhibits a low affinity for glutamate. By contrast, mGlu7 receptor potentiation lasts longer because a more complex signalling cascade is associated with this response. Thus, mGlu7 receptors activate a PTx resistant G protein that enhances PLC activity and generates DAG, which in turn translocates Munc13‐2 proteins to plasma membrane and recruits SVs to the AZ, with the help of RIM1α (see Fig. 10 E and Martín et al. 2010). Accordingly, the duration of the potentiating response could be dependent on the DAG levels and/or on the time that Munc13 priming lasts. The rapid potentiation of synaptic transmission by mGlu7 may have important implications for the short‐term plasticity of excitatory synapses. Moreover, mGlu7 receptors are also present at GABAergic terminals, although it is not known if these receptors exert bidirectional control of inhibitory synaptic transmission. Additional studies will be required to address these issues.
Additional information
Competing interests
None declared.
Author contributions
R.M., M.T. and J.S.‐P. conceived and designed the experiments with input from all the co‐authors; R.M. performed and analysed the electrophysiological recordings; R.M., J.J.F. and A.C.‐A. performed and analysed the electron microscopy experiments at Universidad Complutense; C.A. and R.L. performed and analysed the immunohistochemistry for electron microscopy at Universidad Castilla‐La Mancha. J.S.‐P. wrote the paper with input from all the co‐authors. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the “Spanish Ministry for Economy and Competitivity” MINECO (grant numbers BFU 2013‐43163R to J.S.‐P. and BFU 2012‐32105 to M.T., and BFU 2015‐63769‐R to R.L.) and the “Junta de Comunidades de Castilla‐La Mancha” (PPII‐2014‐005‐P to R.L.).
Acknowledgements
The Munc13‐2 and RIM1α KO mice were a generous gift from Dr Nils Brose (Max Planck Institute for Experimental Medicine, Gottingen, Germany) and Dr Susanne Schoch (University of Bonn Medical Centre, Bonn, Germany), respectively. We also thank Agustin Fernandez and Marisa Garcia from the electron microscopy facility at the Universidad Complutense Madrid for their technical support, and María del Carmen Zamora for her excellent technical assistance. We are grateful to Dr David Bartolomé‐Martín for installing the electrophysiology recording system and to Dr Mark Sefton for editorial assistance.
Edited by: Yoshihiro Kubo & Michisuke Yuzaki
References
- Augustin I, Betz A, Herrmann C, Jo T & Brose N (1999a). Differential expression of two novel Munc13 proteins in rat brain. Biochem J 337, 363–371. [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Sudhof TC & Brose N (1999b). Munc13‐1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Niswender CM, Luo Q, Banko JL & Conn PJ (2008). Group III mGluR regulation of synaptic transmission at the SC‐CA1 synapse is developmentally regulated. Neuropharmacology 54, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskys A & Malenka RC (1991). Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol 444, 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Woolley RJ, Teschemacher AG & Seward EP (2007). Potentiation of exocytosis by phospholipase C‐coupled G‐protein‐coupled receptors requires the priming protein Munc13‐1. J Neurosci 27, 212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J & Brose N (1998). Munc13‐1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron 21, 123–136. [DOI] [PubMed] [Google Scholar]
- Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J & Brose N (2001). Functional interaction of the active zone proteins Munc13‐1 and RIM1 in synaptic vesicle priming. Neuron 30, 183–196. [DOI] [PubMed] [Google Scholar]
- Billups B, Graham BP, Wong AY & Forsythe ID (2005). Unmasking group III metabotropic glutamate autoreceptor function at excitatory synapses in the rat CNS. J Physiol 565, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breustedt J, Gundlfinger A, Varoqueaux F, Reim K, Brose N & Schmitz D (2010). Munc13‐2 differentially affects hippocampal synaptic transmission and plasticity. Cereb Cortex 20, 1109–1120. [DOI] [PubMed] [Google Scholar]
- Bushell TJ, Sansig G, Collett VJ, van der Putten H & Collingridge GL (2002). Altered short‐term synaptic plasticity in mice lacking the metabotropic glutamate receptor mGlu7. ScientificWorldJournal 2, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell JH, Herin GA, Nagel G, Bamberg E, Scheschonka A & Betz H (2008). Increases in intracellular calcium triggered by channelrhodopsin‐2 potentiate the response of metabotropic glutamate receptor mGluR7. J Biol Chem 283, 24300–24307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M (2004). Distinct properties of presynaptic group II and III metabotropic glutamate receptor‐mediated inhibition of perforant pathway‐CA1 EPSCs. Eur J Neurosci 19, 2847–2858. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Sudhof TC & Malenka RC (2002). RIM1α is required for presynaptic long‐term potentiation. Nature 415, 327–330. [DOI] [PubMed] [Google Scholar]
- Corti C, Restituito S, Rimland JM, Brabet I, Corsi M, Pin JP & Ferraguti F (1998). Cloning and characterization of alternative mRNA forms for the rat metabotropic glutamate receptors mGluR7 and mGluR8. Eur J Neurosci 10, 3629–3641. [DOI] [PubMed] [Google Scholar]
- Deng L, Kaeser PS, Xu W & Sudhof TC (2011). RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 69, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Busnadiego R, Asano S, Oprisoreanu AM, Sakata E, Doengi M, Kochovski Z, Zurner M, Stein V, Schoch S, Baumeister W & Lucic V (2013). Cryo‐electron tomography reveals a critical role of RIM1α in synaptic vesicle tethering. J Cell Biol 201, 725–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero JJ, Alvarez AM, Ramírez‐Franco J, Godino MC, Bartolomé‐Martín D, Aguado C, Torres M, Luján R, Ciruela F & Sánchez‐Prieto J (2013). β‐Adrenergic receptors activate exchange protein directly activated by cAMP (Epac), translocate Munc13‐1, and enhance the Rab3A‐RIM1α interaction to potentiate glutamate release at cerebrocortical nerve terminals. J Biol Chem 288, 31370–31385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero JJ, Ramírez‐Franco J, Martín R, Bartolomé‐Martín D, Torres M & Sánchez‐Prieto J (2016). Cross‐talk between metabotropic glutamate receptor 7 and beta‐adrenergic receptor signaling at cerebrocortical nerve terminals. Neuropharmacology 101, 412–425. [DOI] [PubMed] [Google Scholar]
- Ferrero JJ, Torres M & Sánchez‐Prieto J (2011). Inhibitors of diacylglycerol metabolism reduce time to the onset of glutamate release potentiation by mGlu7 receptors. Neurosci Lett 500, 144–147. [DOI] [PubMed] [Google Scholar]
- Gereau RW Jr & Conn PJ (1995). Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci 15, 6879–6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioia DA, Alexander NJ & McCool BA (2016). Differential expression of Munc13‐2 produces unique synaptic phenotypes in the basolateral amygdala of C57BL/6J and DBA/2J mice. J Neurosci 36, 10964–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero I, Miras‐Portugal MT & Sánchez‐Prieto J (1992). Activation of protein kinase C by phorbol esters and arachidonic acid required for the optimal potentiation of glutamate exocytosis. J Neurochem 59, 1574–1577. [DOI] [PubMed] [Google Scholar]
- Imig C, Min SW, Krinner S, Arancillo M, Rosenmund C, Sudhof TC, Rhee J, Brose N & Cooper BH (2014). The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 84, 416–431. [DOI] [PubMed] [Google Scholar]
- Junge HJ, Rhee J‐S, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C & Brose N (2004). Calmodulin and Munc13 form a Ca2+ sensor/effector complex that control short‐term synaptic plasticity. Cell 118, 389–401. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J & Sudhof TC (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ‐domain interaction. Cell 144, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammermeier PJ (2015). Constitutive activity of metabotropic glutamate receptor 7. BMC Neurosci 16, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe H, Mitkovski M, Kaeser PS, Hirrlinger J, Opazo F, Nestvogel D, Kalla S, Fejtova A, Verrier SE, Bungers SR, Cooper BH, Varoqueaux F, Wang Y, Nehring RB, Gundelfinger ED, Rosenmund C, Rizzoli SO, Südhof TC, Rhee JS & Brose N (2017). ELKS1 localizes the synaptic vesicle priming protein bMunc13‐2 to a specific subset of active zones. J Cell Biol 216, 1143–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A, Shigemoto R, Ohishi H, van der Putten H & Mizuno N (1998). Immunohistochemical localization of metabotropic glutamate receptors, mGluR7a and mGluR7b, in the central nervous system of the adult rat and mouse: a light and electron microscopic study. J Comp Neurol 393, 332–352. [PubMed] [Google Scholar]
- Kinzie JM, Saugstad JA, Westbrook GL & Segerson TP (1995). Distribution of metabotropic glutamate receptor 7 messenger RNA in the developing and adult rat brain. Neuroscience 69, 167–176. [DOI] [PubMed] [Google Scholar]
- Klar R, Walker AG, Ghose D, Grueter BA, Engers DW, Hopkins CR, Lindsley CW, Xiang Z, Conn PJ & Niswender CM (2015). Activation of metabotropic glutamate receptor 7 is required for induction of long‐term potentiation at SC‐CA1 synapses in the hippocampus. J Neurosci 35, 7600–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon‐Cazal M, Fagni L, Guiraud M‐J, Mary S, Lerner‐Natoli M, Pin J‐P, Shigemoto R & Bockaert J (1999). mGluR7‐like metabotropic glutamate receptors inhibit NMDA‐mediated excitotoxicity in cultured mouse cerebellar granule neuron. Eur J Neurosci 11, 663–672. [DOI] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N & Schneggenburger R (2008). Phorbol esters modulate spontaneous and Ca2+‐evoked transmitter release via acting on both Munc13 and protein kinase C. J Neurosci 28, 8257–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luján R, Nusser Z, Roberts JD, Shigemoto R & Somogyi P (1996). Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci 8, 1488–1500. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Madison DV & Nicoll RA (1986). Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321, 175–177. [DOI] [PubMed] [Google Scholar]
- Martín R, Bartolomé‐Martín D, Torres M & Sánchez‐Prieto J (2011). Non‐additive potentiation of glutamate release by phorbol esters and metabotropic mGlu7 receptor in cerebrocortical nerve terminals. J Neurochem 116, 476–485. [DOI] [PubMed] [Google Scholar]
- Martín R, Durroux T, Ciruela F, Torres M, Pin JP & Sánchez‐Prieto J (2010). The metabotropic glutamate receptor mGlu7 activates phospholipase C, translocates munc‐13‐1 protein, and potentiates glutamate release at cerebrocortical nerve terminals. J Biol Chem 285, 17907–17917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín R, Torres M & Sánchez‐Prieto J (2007). mGluR7 inhibits glutamate release through a PKC‐independent decrease in the activity of P/Q‐type Ca2+ channels and by diminishing cAMP in hippocampal nerve terminals. Eur J Neurosci 26, 312–322. [DOI] [PubMed] [Google Scholar]
- Millán C, Castro E, Torres M, Shigemoto R & Sánchez‐Prieto J (2003). Co‐expression of metabotropic glutamate receptor 7 and N‐type Ca2+ channels in single cerebrocortical nerve terminals of adult rats. J Biol Chem 278, 23955–23962. [DOI] [PubMed] [Google Scholar]
- Millán C, Luján R, Shigemoto R & Sánchez‐Prieto J (2002). The inhibition of glutamate release by metabotropic glutamate receptor 7 affects both [Ca2+]c and cAMP: evidence for a strong reduction of Ca2+ entry in single nerve terminals. J Biol Chem 277, 14092–14101. [DOI] [PubMed] [Google Scholar]
- O'Connor V, El Far O, Bofill‐Cardona E, Nanoff C, Freissmuth M, Karschin A, Airas JM, Betz H & Boehm S (1999). Calmodulin dependence of presynaptic metabotropic glutamate receptor signaling. Science 286, 1180–1184. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Akazawa C, Shigemoto R, Nakanishi S & Mizuno N (1995). Distributions of the mRNAs for L‐2‐amino‐4‐phosphonobutyrate‐sensitive metabotropic glutamate receptors, mGluR4 and mGluR7, in the rat brain. J Comp Neurol 360, 555–570. [DOI] [PubMed] [Google Scholar]
- Okamoto N, Hori S, Akazawa C, Hayashi Y, Shigemoto R, Mizuno N & Nakanishi S (1994). Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J Biol Chem 269, 1231–1236. [PubMed] [Google Scholar]
- Parfitt KD & Madison DV (1993). Phorbol esters enhance synaptic transmission by a presynaptic, calcium‐dependent mechanism in rat hippocampus. J Physiol 471, 245–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW & McBain CJ (2005). mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron 46, 89–102. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Lacaille JC & McBain CJ (2006). Compartmentalized Ca2+ channel regulation at divergent mossy‐fiber release sites underlies target cell‐dependent plasticity. Neuron 52, 497–510. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Yuan XQ, Lacaille JC & McBain CJ (2008). State‐dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron 60, 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroy J, El Far O, Bertaso F, Pin JP, Betz H, Bockaert J & Fagni L (2002). PICK1 is required for the control of synaptic transmission by the metabotropic glutamate receptor 7. EMBO J 21, 2990–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroy J, Prezeau L, De Waard M, Shigemoto R, Bockaert J & Fagni L (2000). Selective blockade of P/Q‐type calcium channels by the metabotropic glutamate receptor type 7 involves a phospholipase C pathway in neurons. J Neurosci 20, 7896–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Sudhof TC, Takahashi M, Rosenmund C & Brose N (2002). β phorbol ester‐ and diacylglycerol‐induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108, 121–133. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N & Rhee JS (2002). Differential control of vesicle priming and short‐term plasticity by Munc13 isoforms. Neuron 33, 411–424. [DOI] [PubMed] [Google Scholar]
- Scheschonka A, Findlow S, Schemm R, El Far O, Caldwell JH, Crump MP, Holden‐Dye K, O'Connor V, Betz H & Werner JM (2008). Structural determinants of calmodulin binding to the intracellular C‐terminal domain of the metabotropic glutamate receptor 7A. J Biol Chem 283, 5577–5588. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T & Neher E (2002). Vesicle pools and short term synaptic depression: lessons from a large synapse. Trends Neurosci 25, 206–212. [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC & Sudhof TC (2002). RIM1α forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415, 321–326. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S & Mizuno N (1997). Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci 17, 7503–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T & Somogyi P (1996). Target‐cell‐specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature 381, 523–525. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K & Rosenmund C (2002). Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13‐mediated vesicle priming. Proc Natl Acad Sci USA 99, 9037–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB & Verhage M (2007). Interdependence of PKC‐dependent and PKC‐independent pathways for presynaptic plasticity. Neuron 54, 275–290. [DOI] [PubMed] [Google Scholar]
- Wu XS & Wu LG (2001). Protein kinase C increases the apparent affinity of the release machinery to Ca2+ by enhancing the release machinery downstream of the Ca2+ sensor. J Neurosci 21, 7928–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]