

Abstract
mTORC1-dependent translational control plays a key role in several enduring forms of synaptic plasticity such as long term potentiation (LTP) and mGluR-dependent long term depression. Recent evidence demonstrates an additional role in regulating synaptic homeostasis in response to inactivity, where dendritic mTORC1 serves to modulate presynaptic function via retrograde signaling. Presently, it is unclear whether LTP and homeostatic plasticity use a common route to mTORC1-dependent signaling or whether each engage mTORC1 through distinct pathways. Here, we report a unique signaling pathway that specifically couples homeostatic signaling to postsynaptic mTORC1 after loss of excitatory synaptic input. We find that AMPAR blockade, but not LTP-inducing stimulation, induces phospholipase D (PLD)-dependent synthesis of the lipid second messenger phosphatidic acid (PA) in rat cultured hippocampal neurons of either sex. Pharmacological blockade of PLD1/2 or pharmacogenetic disruption of PA interactions with mTOR eliminates mTORC1 signaling and presynaptic compensation driven by AMPAR blockade, but does not alter mTORC1 activation or functional changes during chemical LTP (cLTP). Overexpression of PLD1, but not PLD2, recapitulates both functional synaptic changes as well as signature cellular adaptations associated with homeostatic plasticity. Finally, transient application of exogenous PA is sufficient to drive rapid presynaptic compensation requiring mTORC1-dependent translation of BDNF in the postsynaptic compartment. These results thus define a unique homeostatic signaling pathway coupling mTORC1 activation to changes in excitatory synaptic drive. Our results further imply that more than one canonical mTORC1 activation pathway may be relevant for the design of novel therapeutic approaches against neurodevelopmental disorders associated with mTORC1 dysregulation.
SIGNIFICANCE STATEMENT Homeostatic and Hebbian forms of synaptic plasticity are thought to play complementary roles in regulating neural circuit function, but we know little about how these forms of plasticity are distinguished at the single neuron level. Here, we define a signaling pathway that uniquely links mTORC1 with homeostatic signaling in neurons.
Keywords: BDNF, homeostatic synaptic plasticity, long-term potentiation, mTOR, phosphatidic acid, phospholipase D
Introduction
Plasticity in the strength of excitatory synapses is believed to be a central mechanism by which neuronal networks relay and encode information. A supply of newly synthesized proteins is now known to be vital for maintaining changes in synapse function in an enduring form (Sutton and Schuman, 2006). The mechanistic target of rapamycin complex 1 (mTORC1) is a critical regulator of translation initiation (Ma and Blenis, 2009) and is critical for several distinct forms of synaptic plasticity, including long-term potentiation (LTP; Tang et al., 2002; Cammalleri et al., 2003; Vickers et al., 2005), mGluR-dependent long term depression (LTD) in the hippocampus (Hou and Klann, 2004) and VTA (Mameli et al., 2007), homeostatic adaptation to synaptic inactivity (Henry et al., 2012; Penney et al., 2012), and the antidepressant action of acute NMDAR blockade (Li et al., 2010). Despite the pervasive role of mTORC1 signaling in many forms of synaptic plasticity, our understanding of activity-dependent mTORC1 signaling in neurons remains poorly understood. Since mTORC1 activation accompanies both increases and decreases in excitatory synaptic drive, a question of particular interest is whether LTP and homeostatic plasticity use common or unique pathways to mTORC1 activation.
mTOR signaling is embedded in an extremely complex network of interacting signaling components (Zoncu et al., 2011) and our understanding of this multifaceted pathway is under constant revision (Lipton and Sahin, 2014). The past decade has witnessed growing interest in a unique mechanism for regulating mTORC1 activity via internal synthesis of phosphatidic acid (Chen and Fang, 2002; Foster, 2009). Phosphatidic acid (PA) is a lipid second messenger that was originally discovered to play an important role in relaying mitogenic signals to mTORC1 during cell growth and proliferation (Fang et al., 2001). PA binds directly to mTOR on its FKBP12-Rapamycin Binding (FRB) domain and is thought to strengthen its association with its binding partner raptor, thereby increasing mTOR's catalytic activity (Veverka et al., 2008; Toschi et al., 2009).
Here, we identify a unique mTORC1 signaling pathway in neurons specific to homeostatic signaling. We find that intracellular synthesis of phosphatidic acid (PA) through phospholipase-D (PLD) signaling is an integral step in mTORC1 activation in response to loss of excitatory input, but not in response to multiple forms of chemically-induced LTP (cLTP). Inhibiting PA synthesis or its interaction with mTOR specifically disrupts mTORC1 activation and changes in synaptic function accompanying homeostatic plasticity, but not LTP. These homeostatic effects, in turn, are recapitulated by direct PA treatment of neurons in an mTORC1-dependent manner. Defining unique homeostatic and LTP-associated pathways to mTORC1 activation is important, given that several monogenic disorders resulting in intellectual disability and Autism-like phenotypes share a common feature of dysregulated mTORC1 activation at the synapse (Hoeffer and Klann, 2010). These results further establish mTORC1 as a central hub which meditates multiple forms of synaptic plasticity, and identifies PLD1/PA signaling as a unique entry point in the context of homeostatic adaptation to synaptic inactivity.
Materials and Methods
Animals.
All experimental protocols were approved by the University Committee on the Use and Care of Animals (UCUCA), University of Michigan. Sprague Dawley rats of either sex (Charles River Laboratories, Wilmington, MA) age P1–P3 were used.
BDNF shRNA Transfection.
U6 promotor-driven scrambled and BDNF shRNA-expressing plasmids were obtained from OriGene Technologies; BDNF shRNA 1: 5′-TGTTCCACCAGGTGAGAAGAGTGATGACC-3, BDNF shRNA 2: 5′-GTGATGCTCAGCAGTCAAGTGCCTTTGGA-3′, scrambled: 5′-GCACTACCAGAGCTAACTCAGATAGTACT-3′. Each plasmid additionally contains a tRFP expression cassette driven by a distinct (pCMV) promoter. Neurons were transfected with 0.5 μg of total DNA with the CalPhos Transfection kit (ClonTech;) according to the manufacturer's protocol. All experiments were performed 24 h after transfection.
Cell culture.
Dissociated hippocampal neuron cultures were prepared according to previously published methods (Banker and Goslin, 1990). Briefly, hippocampi from Sprague Dawley rat pups (P1–P3) were dissected in cold dissociation media (DM; 82 mm Na2SO4, 30 mm K2SO4, 5.8 mm MgCl2–6H2O, 252 μm CaCl2–2H2O, 1 mm HEPES, 200 mm glucose, 0.001% w/v phenol red), and transferred to a 15 ml conical tube. Dissociation media (DM) was gently removed (save ∼500 μl covering the tissue itself) and replaced with 5 ml of prewarmed (37°C) cysteine-activated papain solution (3.2 mg l-cysteine (Sigma-Aldrich, Saint Louis, MO) with 500 μl papain (Sigma-Aldrich, Saint Louis, MO) in 10 ml DM, pH ∼7.2). The tissue was then incubated in activated papain solution for 15 min at 37°C to allow for tissue digestion. The tube was inverted ∼2–3 times after 7 min of incubation. Papain inactivation was achieved via two washes in ice-cold DM containing 12.5% v/v fetal bovine serum, followed by two washes in DM alone. Dissociated cells were then washed 2× in chilled normal growth medium [NGM; Neurobasal A (Invitrogen, Grand Island, NY) supplemented with 2% v/v B27 (Invitrogen) and 1% v/v Glutamax (Invitrogen)], then titurated ∼10–15 times in 5 ml NGM to obtain a single cell suspension. This single cell suspension was incubated on ice for ∼3–5 min. 4.5 ml of the cell suspension was then transferred to a new 15 ml tube and centrifuged at 67 ×g (0.5 × 1000 rcf) at 4°C. For plating, 50–70 K cells (in a volume of 150 μl) were dispensed onto poly-d-lysine-coated glass-bottom Petri dishes (Mattek, Ashland, MA) and maintained at 5% CO2/37°C. Approximately 4 h after plating, cells were supplied with 2 ml of NGM-GC (NGM supplemented with 15% v/v glial conditioned media and 10% v/v cortical conditioned media). 24Hr after plating, cells were fed by replacing 50% of the total volume with fresh NGM-GC and every 4 d thereafter by replacing 25% of the total volume with fresh media. Cells were maintained for 14 d with NGM-GC supplementation, then fed every 4 d thereafter with NGM alone. All neurons used for experiments were allowed to mature to an age of ≥21 DIV, unless otherwise noted.
Chemically-induced long-term potentiation (cLTP) protocol.
Pharmacological induction of LTP in cultured hippocampal neurons was achieved using one of two stimulus solutions, designated cLTP1 or cLTP2. cLTP1 stimulus consisted of the following: 0.2 mm Glycine, 0.02 mm Bicuculline, and 0.003 mm Strychnine in a Mg2+-free extracellular solution. The cLTP2 stimulus also used Mg2+-free extracellular solution as a base but contained 50 μm Forskolin and 0.1 μm Rolipram. Both means of cLTP induction followed a similar protocol: Dissociated cultures (DIV 21 or older) were removed from the incubator and place on bench top for 10 min to acclimate to room temperature. Neuronal media from then removed from each dish and replaced with 2mls cLTP1 or cLTP2 solution. Neurons were incubated in cLTP solution for 10 min at room temp. After 10 min, cLTP solution was removed and cells were washed 2× with neuronal media. Dishes were then allowed to recover at 37oC for 2 Hr before use in subsequent electrophysiology or imaging experiments.
Electrophysiology.
Whole-cell patch-clamp recordings were made with an Axopatch 200B amplifier from cultured hippocampal neurons bathed in HEPES-buffered saline (HBS) containing (in mm) the following: 119 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 30 Glucose, 10 HEPES, pH 7.4. Whole-cell pipette had resistances ranging from 3 to 6 MΩ. Internal solution contained (in mm) the following: 100 cesium gluconate, 0.2 EGTA, 5 MgCl2, 2 adenosine triphosphate, 0.3 guanosine triphosphate, 40 HEPES, pH 7.2. mEPSCs were recorded at −70 mV from neurons with a pyramidal-like morphology in the presence of 1 μm TTX and 10 μm bicuculline and analyzed off-line using Synaptosoft minianalysis software. sEPSCs in active networks were recorded at −70 mV in modified HBS (as above, except with 0.5 mm CaCl2 and 3.5 mm MgCl2) without TTX or bicuculline. mIPSCs were recorded at −70 mV in the presence of 1 μm TTX, 10 μm CNQX and 50 μm DL-APV. Internal solution contained (in mm) the following: 135 CsCl, 1 EGTA, 10 HEPES, 5 MgCl2, 4 Mg-ATP, and 1 Li-GTP, pH 7.2. During loose-patch recordings, neurons were bathed in HBS lacking pharmacological inhibitors to permit recording of background spike activity. Pipette solution contained extracellular solution.
For current-clamp recordings, changes in intrinsic excitability were assessed in neurons subject to pharmacological isolation via 10 μm CNQX, 50 μm DL-APV, and 10 μm bicuculline. Internal solution contained (in mm) the following: 120 K-MeSO4, 20 KCl, 0.2 EGTA, 10 HEPES, 2 MgCl2, 4 Mg-ATP, 0.3 Na-GTP, and 7 phosphocreatine, pH 7.2. A series of 500 ms current injections were used to depolarize the soma from resting membrane potential in stepwise increments of 100 pA. Series resistance was compensated for by >80%. Neurons which had a resting membrane potential above −55 mV were not included in further analysis. Membrane hyperpolarization in response to a −25 pA current step was used to measure input resistance. Action potential properties and spike frequency data were analyzed using Clampfit 10.0 (Molecular Devices). Statistical differences between experimental conditions were determined by ANOVA followed by Fisher's LSD post hoc tests
Ca2+ phosphate transfection.
To achieve sparse expression, neurons were transfected with 0.5 μg of total DNA using the Ca2+ phosphate CalPhos Transfection kit (ClonTech) according to the manufacturer's protocol. Unless otherwise indicated, all experiments were performed 24 h post-transfection.
DNA Constructs.
Human HA-PLD1, human HA-PLD2, rat HA-PLD2, and pEGFP-Spo20PABD-WT were provided by Dr. Michael Frohman (SUNY Stony Brook). GFP-Rap1-PABD was provided by Dr. Frank Schmitz (Universität des Saarlandes Medizinische Fakultät). Rapamycin-resistant (S2035T) and Rapamycin/Phosphatic acid resistant (32035T/R2109A) mTOR point mutants were provided by Dr. Jie Chen (University of Illinois at Urbana-Champaign).
Immunocytochemisty and microscopy.
All imaging was performed on an inverted Olympus FV1000 laser-scanning confocal microscope using a Plan-Apochromat 63×/1.4 oil objective with 1× or 2× digital zoom. Alexa 488 was excited with the 488 nm line of an argon ion laser and emitted light was typically collected between 500 and 530 nm with a tunable emission filter. Alexa 555/568 were excited with a 559 nm diode laser and emitted light was typically collected between 570 and 670 nm. Before image collection, the acquisition parameters for each channel were optimized to ensure a dynamic signal range and to ensure no signal bleed-through between detection channels. For every experiment, identical parameters were used for each treatment option. We verified that no detectable staining was observed in control samples incubated in the absence of primary antibody (but otherwise processed identically).
For BDNF and phospho-S6 (PS6) staining, cells were treated in conditioned media as indicated, then fixed at room temperature for 20 min with 4% paraformaldehyde (PFA)/4% sucrose in PBS with 1 mm MgCl2 and 0.1 mm CaCl2 (PBS-MC). After washing in PBS-MC (5 min), cells were permeabilized (0.1% Triton X in PBS-MC, 5 min), blocked with 2% bovine serum albumin in PBS-MC for 30 min, and labeled with a rabbit polyclonal antibody against BDNF (Santa Cruz Biotechnology, 1:100), or a rabbit polyclonal S235/236 phosphospecific antibody (Cell Signaling Technology; 1:50); for each, colabeling and a mouse monoclonal antibody against MAP2 (Sigma-Aldrich, 1:5000) for either 60 min at room temperature or overnight at 4°C. Alexa 555-conjugated goat anti-rabbit (1:500) and Alexa 635-conjugated goat anti-mouse (1:500) secondary antibodies (each 60 min at room temperature) were used to visualize BDNF/PS6 and MAP2 staining, respectively. To ensure blind sampling of BDNF/PS6 expression, neurons with a pyramidal-like morphology were selected for imaging by epifluorescent visualization of MAP2 staining. Analysis of expression in somatic or dendritic neuronal compartments was performed on maximal intensity z-compressed image stacks. Dendrites were linearized and extracted from the full-frame image using the straighten plug-in for ImageJ. For both somatic and dendritic signal, expression was estimated by the average nonzero pixel intensity for each compartment. To analyze dendritic BDNF expression, dendrites were linearized and extracted from the full-frame image using the straighten plug-in for ImageJ. To combine data across multiple experimental runs of the same experiment, expression in each image was normalized against the average nonzero pixel intensity for the respective control group. In experiments where BDNF or PS6 were assessed in transfected cells, neurons were also colabeled with a chicken polyclonal antibody against GFP (Millipore, 1:1000) and an Alexa 488-conjugated goat anti-chicken secondary antibody. Statistical differences were assessed by ANOVA, followed by Fisher's LSD post hoc tests.
Immunocytochemisty and microscopy.
All imaging was performed on an inverted Olympus FV1000 laser-scanning confocal microscope using a Plan-Apochromat 63×/1.4 oil objective with 1× or 2× digital zoom. Alexa 488 was excited with the 488 nm line of an argon ion laser and emitted light was typically collected between 500 and 530 nm with a tunable emission filter. Alexa 555/568 were excited with a 559 nm diode laser and emitted light was typically collected between 570 and 670 nm. Before image collection, the acquisition parameters for each channel were optimized to ensure a dynamic signal range and to ensure no signal bleed-through between detection channels. For every experiment, identical parameters were used for each treatment option. We verified that no detectable staining was observed in control samples incubated in the absence of primary antibody (but otherwise processed identically).
For BDNF and phospho-S6 (PS6) staining, cells were treated in conditioned media as indicated, then fixed at room temperature for 20 min with 4% paraformaldehyde (PFA)/4% sucrose in PBS with 1 mm MgCl2 and 0.1 mm CaCl2 (PBS-MC). After washing in PBS-MC (5 min), cells were permeabilized (0.1% Triton X in PBS-MC, 5 min), blocked with 2% bovine serum albumin in PBS-MC for 30 min, and labeled with a rabbit polyclonal antibody against BDNF (Santa Cruz Biotechnology, 1:100), or a rabbit polyclonal S235/236 phosphospecific antibody (Cell Signaling Technology; 1:50); for each, colabeling and a mouse monoclonal antibody against MAP2 (Sigma-Aldrich, 1:5000) for either 60 min at room temperature or overnight at 4°C. Alexa 555-conjugated goat anti-rabbit (1:500) and Alexa 635-conjugated goat anti-mouse (1:500) secondary antibodies (each 60 min at room temperature) were used to visualize BDNF/PS6 and MAP2 staining, respectively. To ensure blind sampling of BDNF/PS6 expression, neurons with a pyramidal-like morphology were selected for imaging by epifluorescent visualization of MAP2 staining. Analysis of expression in somatic or dendritic neuronal compartments was performed on maximal intensity z-compressed image stacks. Dendrites were linearized and extracted from the full-frame image using the straighten plug-in for ImageJ. For both somatic and dendritic signal, expression was estimated by the average nonzero pixel intensity for each compartment. To analyze dendritic BDNF expression, dendrites were linearized and extracted from the full-frame image using the straighten plug-in for ImageJ. To combine data across multiple experimental runs of the same experiment, expression in each image was normalized against the average nonzero pixel intensity for the respective control group. In experiments where BDNF or PS6 were assessed in transfected cells, neurons were also colabeled with a chicken polyclonal antibody against GFP (Millipore, 1:1000) and an Alexa 488-conjugated goat anti-chicken secondary antibody. Statistical differences were assessed by ANOVA, followed by Fisher's LSD post hoc tests.
Optical analysis of presynaptic function used live-labeling with an Oyster 550-conjugated rabbit polyclonal antibody against the lumenal domain of synaptotagmin 1 (syt-lum; 1:100, Synaptic Systems). Before labeling, neurons were treated with 2 μm TTX for 15 min to isolate spontaneous neurotransmitter release. Neurons were then labeled with anti-syt-lum for 5 min at room temperature, washed, fixed with 4% PFA/4% sucrose in PBS-MC, permeabilized, and blocked as above, then labeled with a guinea pig polyclonal anti-vglut1 antibody (1: 2500, Millipore Bioscience Research Reagents). The intrinsic fluorescent signal of the anti-syt-lum at synaptic sites was amplified by an Alexa 555-conjugated goat anti-rabbit (1:500) secondary antibody. An Alexa 488-conjugated goat anti-guinea pig (1:250) secondary antibody (each for 60 min at room temperature) was used to visualize vglut1 staining. Neurons were selected based on vglut1 epiflourescence to ensure blind sampling of syt-lum expression. For analysis, a “synaptic” syt-lum particle was defined as a particle that occupied >10% of the area defined by a vglut1 particle, and the proportion of vglut1 particles containing synaptic syt-lum particles was calculated using custom written analysis routines for ImageJ. Statistical differences were assessed by ANOVA, followed by Fisher's LSD post hoc tests.
Magnetofection-based siRNA delivery.
BDNF siRNA or nontargeting control siRNA were purchased from Dharmacon (siGENOME SMARTpool, M-080046, Thermo Fisher Scientific., Lafayette, CO.). To favor rapid translational knockdown, a combination of four BDNF-targeting siRNAs were pooled: (1): Sense = 5′-UCGAAGAGCUGCUGGAUGAUU-3′, Antisense = 5′-UCAUCCAGCAGCUCUUCGAUU-3′; (2): Sense = 5′UAUGUACACUGACCAUUAAUU-3′, Antisense: 5′-UUAAUGGUCAGUGUACAUAUU-3′; (3): Sense: 5′-GAGCGUGUGUGACAGUAUUUU-3′, Antisense: 5′-AAUACUGUCACACACGCUCUU-3′; (4): Sense: 5′-GAACUACCCAAUCGUAUGUUU-3′, Antisense: 5′-ACAUACGAUUGGGUAGUUCUU-3′; Control siRNA: Sense: 5′-UAAGGCUAUGAAGAGAUACUU-3′; Antisense: 5′-GUAUCUCUUCAUAGCCUUAUU-3′. Each annealed double-stranded RNA oligonucleotide contained 3′-UU overhangs, a single isomer 6-FAM on the 5′-end of the sense strand, and a 5′-phosphate on the antisense strand. For rapid siRNA transfection, siRNA (67.5 μg in 100 μl media) was combined with 3.5 μl neuromag transfection reagent (Oz Biosciences, France). After a 20 min incubation period, cultured neurons were bathed in the Neuromag/siRNA mix, and then placed on the manufacturer supplied magnet for 15 min at 37°C. After magnetofection, cells were immediately treated with PA or vehicle, and used in experiments as described.
Preparation of PA vesicles.
The lipid second messenger phosphatidic acid (PA) was acquired from Avanti Polar Lipids (Alabaster, Alabama), and was prepared according to previously described methods (Yoon et al., 2011a). PA lipids in chloroform were dried under a stream of nitrogen gas in a 20 ml glass scintillation vial. Dried lipids were reconstituted in a buffer of 150 mm NaCl and 10 mm Tris-Cl (pH8.0), to a concentration of 6 mm. The solution was briefly vortexed, then subjected to bath sonication for a period of 5 min, resulting in a homogenous solution of unilaminar vesicles (15–50 nm in diameter). PA vesicles were added directly to cell media to produce a concentration of 100 μm. Lipid vesicles were made fresh at the start of each experiment.
vglut1-pHluorin imaging.
Cultured hippocampal neurons were transfected at DIV 14–16 with vglut-pHluorin as well as mCherry-synaptophysin to aid in visual identification of terminal boutons. Experiments were conducted 2–3 d post-transfection in HBS containing 10 μm CNQX and 50 μm DL-APV. Action potentials were evoked via a Grass stimulator unit using a custom fabricated electric field stimulator, which was inserted into each dish just before start of an experiment. Each stimulus consisted of a 2 ms 10 V potential difference between platinum-iridium wires 6 mm apart. To observe changes in evoked release, we recorded the change in vglut-pHluorin fluorescence intensity elicited by delivering stimulus repetitions at 10 Hz for 10 s, which has been shown to provide a measure of exocytosis undiluted by signal degradation due to endocytosis or reacidification (Kim and Ryan, 2010). Each stimulus train was delivered three times for each set of synapses, with a 5 min interval separating each train. Images were taken using a Hammamatsu EM-CCD camera mounted on an inverted microscope (Olympus IX8) using a 40× 1.3 NA oil-immersion objective (Olympus). The filter set consisted of a 472/30 excitation filter, a 495 edge dichroic mirror, and a 520/30 emission filter. Illumination was achieved via a Sutter Lambda LS/OR30 300 W xenon bulb control by a Sutter Lambda 10 shutter (Sutter Instrument). All images were captured at a depth of 16 bits to a Dell Optiplex computer using MetaMorph software (Molecular Devices). Full frame (512 × 512, 16 μm pixels) imaging sequences were acquired at a rate of 5 Hz using 50 ms exposure for vglut-pHluorin. Image sequences were analyzed using the “time series” plug-in for ImageJ (http://rsb.info.nih.gov/ij/plugins/time-series.html). Active synapses were identified for each experiment by subtracting the signal before the initiation of each stimulus train from the peak increase in vglut-pHluorin signal elicited by the train. Rectangular ROIs (1.55 × 1.55 μm) were placed over each of the resulting puncta in the “difference image.” The time-lapse plug-in was then used to measure average fluorescence for each ROI over time. To correct for photobleaching during image series acquisition, an identically sized ROI was positioned in a nonsynaptic region adjacent to an identified release site. Measurements obtained from this nonsynaptic ROI were fit with a biexponential decay function, and these values were then subtracted from the data traces associated with active release sites. Automated image analysis and photobleaching correction was performed in MATLB. Fluorescence signals were quantified as ΔF/F0, where F0 is defined as the average of five sequential images recorded before the onset of stimulation for each ROI.
Western blotting.
Samples were collected in lysis buffer containing 100 mm NaCl, 10 mm NaPO4, 10 mm Na4P2O7, 10 mm lysine, 5 mm EDTA, 5 mm EGTA, 50 mm NaF, 1 mm NaVO3, 1% Triton-X, 0.1% SDS, and 1 tablet Complete Mini protease inhibitor mixture (Roche)/7 ml, pH 7.4. Equal amounts of protein for each sample were loaded and separated on 12% polyacrylamide gels, then transferred to PVDF membranes. Blots were blocked with Tris-buffered saline containing 0.1% Triton-X (TBST) and 5% nonfat milk for 60 min at room temperature, and incubated with a rabbit polyclonal primary antibody against BDNF (Santa Cruz Biotechnology, 1: 200) for either 60 min at room temperature or overnight at 4°C. After washing with TBST, blots were incubated with HRP-conjugated anti-rabbit or anti-mouse secondary antibody (1:5000; Jackson Immunoresearch), followed by enhance enhanced chemiluminescent detection (GE Healthcare). All blots were probed with a mouse monoclonal antibody against α-tubulin (1:5000, Sigma-Aldrich) to confirm equal loading. Band intensity was quantified with densitometry with NIH ImageJ and expressed relative to the matched control sample. Statistical differences between treatment conditions and control were assessed first by an overall nonparametric ANOVA (Kruskal–Wallis test) followed by a post hoc Wilcoxon signed rank test for individual comparisons.
Results
Intracellular synthesis of PA is critical for mTOR signaling during HSP
Intracellular synthesis of the lipid second-messenger phosphatidic acid (PA) has emerged as an important signal in mitogenic activation of mTORC1 signaling (Foster, 2009), but little is known about a potential role for PA in relaying synaptic signals to mTOR in mammalian central neurons. To visualize PA synthesis in dissociated hippocampal neurons, we transfected cells with a reporter of endogenous PA synthesis, GFP-Spo20-PABD, at DIV 16–18 and performed live-imaging experiments 2–3 d later. The GFP-Spo20-PABD plasmid contains the PA-binding domain of the yeast N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein Spo20 fused to EGFP (Zeniou-Meyer et al., 2007). Similar to previous findings in COS-7 (Frondorf et al., 2010) and PC12 cells (Zeniou-Meyer et al., 2007), we find that GFP-Spo20-PABD fluorescence is largely limited to the cell nucleus under basal conditions owing to the NLS present in the sequence (Fig. 1A). However, after subjecting neurons to AMPAR blockade (CNQX, 40 μm) we observed a steady, time-dependent increase in PA-related GFP clustering in discrete regions around the soma and extending into the distal regions of the dendrites by 2 Hrs post-treatment (Fig. 1A). Additional experiments using a different fluorescence-based sensor of intracellular PA synthesis produced comparable results (Fig. 1B). This reporter contains the C-terminal PA-binding domain of human Raf1 fused to EGFP (GFP-Raf1-PABD) and displays a strong bias for binding to PA (Schwarz et al., 2011). Similar to our findings using GFP-Spo20-PABD, we observed discrete regions of intracellular clustering of the GFP-Raf1-PABD reporter after subjecting neurons to AMPAR blockade (CNQX, 40 μm, 3 h). Clustering of this sort did not occur in vehicle-treated control cells (Fig. 1B).
Figure 1.
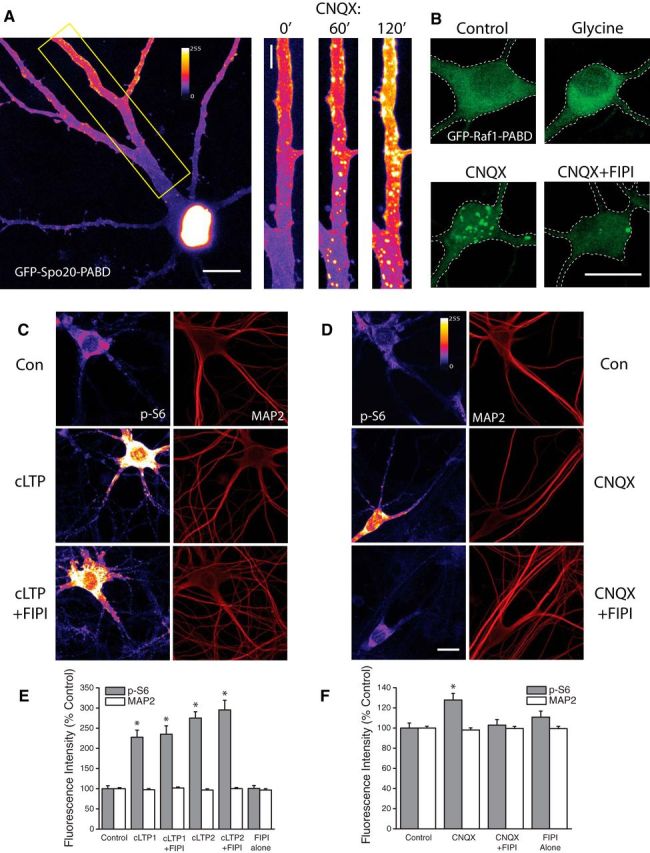
PLD-dependent generation of Phosphatidic Acid during homeostatic plasticity but not cLTP. A, Example of hippocampal neuron expressing the fluorescent PA reporter GFP-Spo20-PABD in culture (left). Right, expanded section of dendrite highlighted in left panel under baseline conditions, and after 60 or 120 min of exposure to CNQX (40 μm). B, Example images of neurons expressing an alternative reporter of intracellular PA synthesis, GFP-Raf1-PABD, under control conditions or after treatment with glycine, CNQX, or CNQX + FIPI. Intracellular accumulation of PA-associated GFP fluorescence was regularly observed after CNQX, but not glycine treatment. FIPI significantly reduced the number of PA-positive puncta after CNQX treatment. Representative images (C, D) and mean (+ SEM) fluorescence intensity (E, F) of MAP2 and phosphorylated ribosomal protein S6 (p-S6) signal in somatic regions under baseline conditions, or under conditions of cLTP or AMPAR blockade with or without concurrent inhibition of PLD signaling (FIPI, 100 nm, 30 min pretreatment). cLTP1= glycine-based protocol, n = 39 cells. cLTP2= forskolin-based protocol, n = 40 cells. CNQX, n = 73 cells. cLTP1+FIPI, n = 40 cells; cLTP2+FIPI, n = 37 cells. CNQX+FIPI, n = 71 cells. FIPI blocks enhanced mTORC1 phosphorylation of downstream target ribosomal protein S6 after AMPAR blockade but not during cLTP. Scale bar, 10 μm in A–C. *p < 0.5 compared to vehicle treated controls.
These data suggest that intracellular synthesis of PA may be a mechanism by which mTORC1 signaling is activated in the context of homeostatic adaptation to synaptic inactivity. Given previous findings that mTORC1 activity is required during other well studied forms of plasticity such as long-term potentiation (Tang et al., 2002; Cammalleri et al., 2003), we also examined a potential role for intracellular PA synthesis in LTP. After stimulation of cultured neurons using a glycine-based chemical LTP (cLTP) paradigm, previously shown to induce long-lasting, mTOR-dependent changes in synaptic strength in dissociated cultures (Lu et al., 2001), we found no difference in the distribution of the GFP-Raf1-PABD reporter (Fig. 1B). The unique responses of the GFP-Raf1-PABD reporter to cLTP and AMPAR blockade raised the possibility that these two forms of plasticity differ in the means by which mTORC1 signaling is engaged, with intracellular synthesis of PA acting as an important point of divergence. Intracellular PA can arise from multiple sources, including lysophosphatidic acid (LPA), diacylglycerol (DG) and phosphatidylcholine (Foster, 2009). Previous reports suggest that the primary source of intracellularly generated PA in cancerous cells is via hydrolysis of phosphatidylcholine by phospholipase-D (Jenkins and Frohman, 2005). To explore a role for phospholipase-D (PLD) signaling in the activation of mTORC1 during either LTP or synaptic homeostasis, we used a small molecule inhibitor of PLD, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI), which inhibits both PLD1 and PLD2 activity and blocks intracellular PA accumulation at subnanomolar concentrations (Su et al., 2009). We found that pretreatment with FIPI severely inhibited the clustering of GFP-Raf1-PABD reporter after treatment with CNQX (Fig. 1B).
To examine the effect of PLD signaling on mTORC1 activation during different forms of synaptic plasticity, we performed immunocytochemical analysis of phosphorylated ribosomal protein S6 (p-S6) at S235/236. We observed significant increases in somatic p-S6 intensity after either glycine or forskolin-based cLTP induction paradigms (Fig. 1C+E). Excitatory synaptic deprivation via AMPAR blockade (CNQX, 40 μm, 3Hr) also produced a significant increase in S6 phosphorylation (Fig. 1D+F), as has been reported previously (Henry et al., 2012). Importantly, coapplication of the PLD1/2 inhibitor FIPI (100 nm, 30 min pretreatment) had no effect on increased p-S6 levels induced by glycine (cLTP1+FIPI) or forskolin (cLTP2+FIPI), but eliminated CNQX-induced increases in mTORC1 activity (CNQX+FIP). Neurons treated with FIPI alone showed no significant difference in p-S6 levels compared with controls, suggesting minimal contribution of steady-state PLD signaling to baseline levels of mTORC1 activity. Together these data suggest that PLD/PA signaling is uniquely engaged by loss of excitatory synaptic drive and is an essential signaling component leading to mTORC1 activation during homeostatic plasticity, but not during the activation of mTORC1 in the context of long-term potentiation.
PLD signaling mediates functional changes during HSP
Previous work has identified BDNF as a unique target of mTORC1-dependent translational regulation in dendrites (Henry et al., 2012; Liao et al., 2012). In hippocampal neurons, this dendritic synthesis of BDNF plays a vital role in the homeostatic regulation of excitatory synapses, acting as a retrograde signal to enhance the efficacy of presynaptic terminals (Jakawich et al., 2010; Lindskog et al., 2010; Henry et al., 2012). In line with a role for PLD/PA signaling operating upstream of mTORC1 in mediating this effect, immunocytochemical analysis revealed enhanced BDNF intensity in dendrites after AMPAR blockade (Fig. 2A,B), which was blocked by coapplication of FIPI. We next used whole-cell patch-clamp electrophysiology to examine a potential role for PLD signaling in the functional changes that emerge at excitatory synapses during homeostatic plasticity or LTP. At both mammalian central synapses and the Drosophila neuromuscular junction, mTORC1-dependent increases in presynaptic function emerge following postsynaptic deprivation of AMPAR-mediated inputs (Penney et al., 2012, Henry et al., 2012). Thus, AMPAR blockade (CNQX, 40 μm, 3 h) elicits a homeostatic increase in mEPSC frequency in hippocampal neurons reflecting enhanced presynaptic release, as well as an increase in mEPSC amplitude that reflects accumulation of new AMPARs at synapses (Aoto et al., 2008; Jakawich et al., 2010; Henry et al., 2012). Notably, coapplication of the PLD1/2 inhibitor FIPI completely suppressed the homeostatic increase in mEPSC frequency induced by AMPAR blockade (Fig. 2E), but did not prevent homeostatic changes in mEPSC amplitude (Fig. 2D) which are mTORC1-independent (Henry et al., 2012). This suggests that the PLD/PA pathway has a unique role in adapting presynaptic function during loss of excitatory input.
Figure 2.
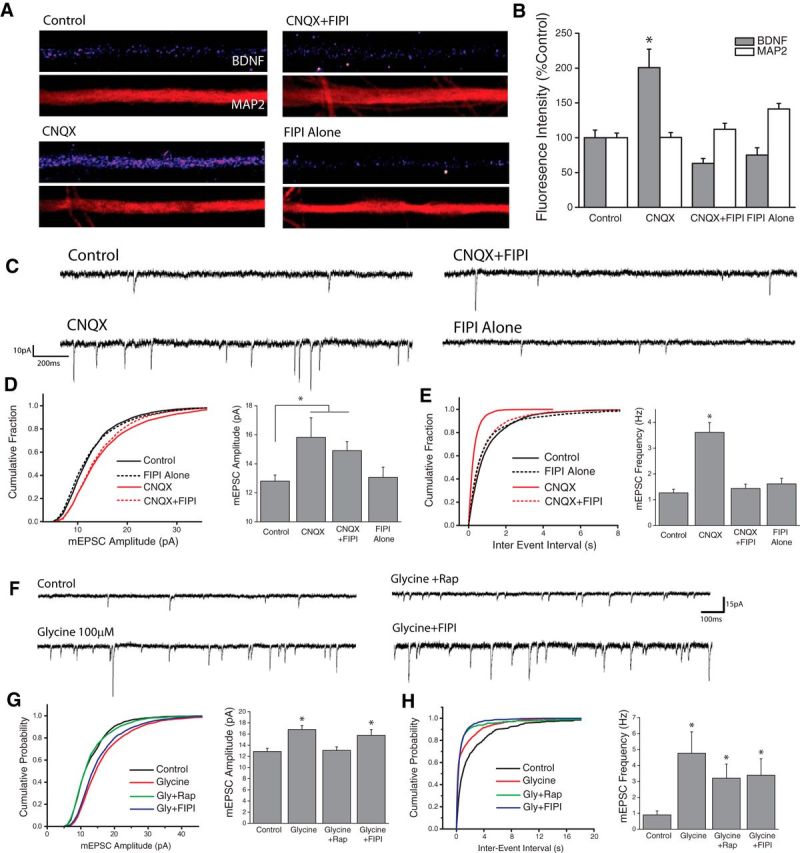
PLD signaling is required for adaptive presynaptic compensation. A, B, Examples images of BDNF and MAP2 expression in linearized dendritic segments (A), and mean (+SEM) expression (B) of MAP2 and BDNF in dendritic compartments normalized to average control values, in neurons treated with CNQX (40 mm, 3Hr) with or without the PLD1/2 inhibitor FIPI (100 nm, 30 min pretreatment). Control, n = 63; cells; CNQX, n = 75 cells; CNQX+FIPI, n = 70 cells; FIPI Alone, n = 56 cells. BDNF expression in dendrites was significantly enhanced by AMPAR blockade compared with control neurons. This effect was completely blocked by concurrent inhibition of PLD signaling. (*p < 0.05, relative to controls). C–E, Representative recordings (C) and mean (+SEM) mEPSC amplitude (D) and frequency (E) recorded from cells treated with vehicle control or CNQX (40 μm, 3Hr) with or without the PLD1/2 inhibitor FIPI (100 nm, 30 m pretreatment). Summary graphs presented on right and cumulative probability distribution shown on left. (*p < 0.05 relative to vehicle controls). Control, n = 22; CNQX, n = 9; CNQX+FIPI, n = 14; FIPI Alone, n = 18. PLD is required for the mTORC1-dependent (Henry et al., 2012) increases in mEPSC frequency observed after blockade of excitatory synaptic input. F–H, Representative recordings (F), and mean (+SEM) mEPSC amplitude (G) and frequency (H) recorded from cells treated with vehicle control or Glycine-based cLTP stimulus (100 μm glycine) with or without the PLD1/2 inhibitor FIPI (100 nm, 30 m pretreatment) *p < 0.05 relative to vehicle controls. Control, n = 12; Glycine, n = 11; Glycine+Rap, n = 8; Glycine+FIPI, n = 11. p < 0.05 compared with control. PLD signaling is not necessary for the mTORC1-dependent changes in mPESC Amplitude observed during chemically-induced LTP.
Consistent with previous reports (Lu et al., 2001), we found that cLTP induction with glycine stimulation produced a significant increase in both the amplitude and frequency of mEPSCs in hippocampal neurons when assessed 2 h after stimulation (Fig. 2F-H). Unlike homeostatic changes in mEPSC frequency, the increase in mEPSC frequency following cLTP remained elevated in the presence of both mTORC1 (100 nm rapamycin, 30 min pretreatment) and PLD (100 nm FIPI, 30 min pretreatment) inhibitors (Fig. 2H). By contrast, glycine-induced increases in mEPSC amplitude were blocked by coapplication of rapamycin (Fig. 2G), which is in line with a role for de novo translation in this form of plasticity. Interestingly, whereas inhibiting PLD1/2 signaling with FIPI prevents mTORC1-dependent presynaptic compensation after AMPAR blockade (Fig. 2E), it had no effect on glycine-induced increases in mEPSC amplitude (Fig. 2G), suggesting that PLD1/2 signaling is not necessary for mTORC1-dependent changes in postsynaptic function at excitatory synapses during cLTP.
PLD1 contributes to PA-induced activation of mTORC1
There are two PLD genes expressed in the mammalian brain, PLD1 and PLD2, each of which exhibits unique cellular compartmentalization. PA is a transient messenger that signals within a limited spatial range of the membrane compartment from which it is generated (Cockcroft, 2001). Thus, unique sites of PLD-mediated PA synthesis could feasibly exert markedly different effects on cellular function. To examine whether PLD1 and PLD2 exhibit unique subcellular localization patterns in neurons, we expressed HA-tagged human PLD1 (hPLD1) or PLD2 (hPLD2) in dissociated hippocampal cultures and assessed HA localization via immunocytochemistry 24–48 Hrs posttransfection (Fig. 3A). In previously published reports using non-neuronal cells, overexpressed HA-PLD1 was primarily localized to late endosomes or the golgi complex, while overexpressed HA-PLD2 was found mainly along the plasma membrane (Du et al., 2004). We observed a similar pattern of HA staining in neurons expressing hPLD1 or hPLD2, with HA-tagged hPLD1 localized to discrete intracellular puncta, and hPLD2 primarily localized to the plasma membrane (Fig. 3A).
Figure 3.
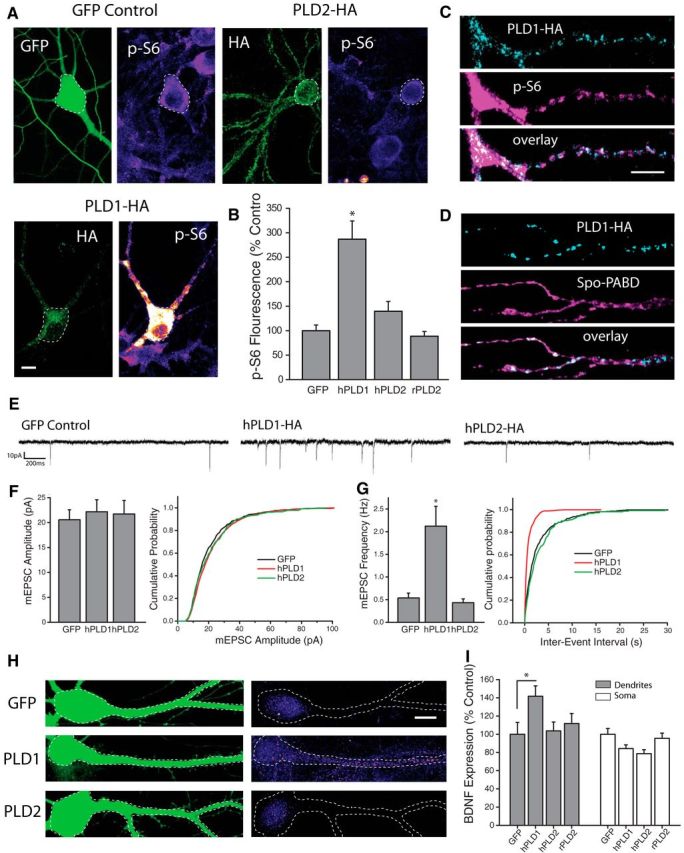
PLD1 overexpression is sufficient to activate mTORC1 and drive changes in synapse function. A, B, Representative images (A) and mean intensity (+SEM,B) of somatic p-S6 staining in neurons expressing HA-tagged hPLD1, hPLD2, or GFP control. GFP control, n = 29 cells; human PLD1-HA, n = 19 cells; human PLD2-HA, n = 26 cells; rat PLD2-HA, n = 25 cells. Expression of hPLD1, but not hPLD2 or rPLD2, significantly (*p < 0.05 relative to GFP-expressing control neurons) increases p-S6 staining. Scale bar represents 20 μm. C, Representative dendritic subregions of cultured hippocampal neurons expressing HA-tagged hPLD1 and stained with an antibody and phosphorylated S6. D, Representative dendritic sections of cells coexpressing HA-tagged hPLD1 as well as the PA reporter Spo-PABD-GFP. Scale bar, 5 μm in C. E–G, Representative recordings (E) and mean (+SEM) mEPSC amplitude (F) and frequency (G) recorded from cells expressing hPLD1, hPLD2 or GFP control. Summary graphs presented on left and cumulative probability distribution shown on right. GFP control, n = 13; hPLD1, n = 11; hPLD2, n = 8. *p < 0.05 relative to vehicle controls. Overexpression of hPLD1, but not hPLD2, is sufficient to elevate mEPSC frequency after 24 Hrs compared with GFP-expressing controls. H, I, GFP and BDNF expression in somatodendritic segments (H), and mean (+SEM) expression (I) of BDNF in somatic and dendritic compartments normalized to average GFP control values, in neurons cotransfected with GFP as well as hPLD1 or hPLD2. GFP control, n = 23 cells; hPLD1, n = 38 cells; hPLD2, n = 26 cells; rPLD2, n = 25 cells. BDNF expression in dendrites was significantly (*p < 0.05, relative to GFP alone) enhanced by expression of hPLD1 compared with control neurons expressing GFP alone, while neurons expressing hPLD2 showed no difference in dendritic BDNF levels compared with control. hPLD1 or hPLD2 overexpression did not increase somatic BDNF intensity compared with controls. Scale bar in H = 10 μm.
Using phosphorylated ribosomal protein S6 as a readout of mTORC1 activity, we found significantly elevated levels of somatic p-S6 in cells expressing hPLD1, compared with cells expressing GFP as a control, indicating potent activation of mTORC1 signaling via PLD1 overexpression (Fig. 3A,B). In a second set of experiments, we found that rapamycin (100 nm) treatment prevents the increase in somatic p-S6 from PLD1 overexpression [Mean +/− SEM somatic p-S6 intensity, relative to GFP control = 225 +/− 50% PLD1 neurons, n = 15; 40 +/− 9.8% in PLD1+Rap neurons, n = 15; 55 +/− 19.2% in GFP+Rap neurons, n = 12; PLD1 vs PLD1+rap, p < 0.05 by t test, data not shown]. By contrast, neurons expressing either human or rat versions of PLD2 showed no significant change in somatic p-S6 levels compared with GFP-expressing controls (Fig. 3B). While more difficult to quantify, distinct patterns of elevated pS6 staining was also commonly observed in distal dendritic regions of cells overexpressing hPLD1 (Fig. 3C). Patches of localized pS6 fluorescence in these cells displayed a remarkable degree of overlap with HA-tagged PLD1 in these same dendritic regions (Fig. 3C). This correspondence was not unique to pS6 staining, however, as GFP signals associated with the Spo-PABD phosphatidic acid reporter also exhibited substantial overlap with HA fluorescence in cells expressing HA-tagged hPLD1 (Fig. 3D). Together, these results suggest that dendritically localized PLD1 can drive synthesis of intracellular PA and recruit enhanced mTORC1 activity in spatially restricted regions. Given reports that PLD1 signaling influences dendrite arborization during neuronal development (Zhu et al., 2012) we also assessed potential changes to excitatory synapse morphology that result from PLD manipulations in mature cells, but found that dendritic spine density was unaffected as a result of 24 h of overexpression of PLD1 or PLD2 (data not shown). To characterize the effects of PLD overexpression on excitatory synapse function, we recorded mEPSCs from dissociated hippocampal neurons expressing hPLD1 or hPLD2 at DIV21–23 (Fig. 3E–G). Similar to postsynaptic mTORC1 activation via overexpression of RhebGTPase (Henry et al., 2012), neurons expressing hPLD1 displayed a significant increase in mEPSC frequency, but no change in mEPSC amplitude, compared with cells expressing GFP alone or those expressing hPLD2 (Fig. 3E–G). Moreover, compared with GFP-expressing control cells, neurons expressing hPLD1 exhibited significantly higher BDNF expression in the dendritic, but not somatic, compartment (Fig. 3H,I). No such change in dendritic BDNF expression was observed in neurons expressing human or rat PLD2. Collectively, these results suggest that overexpression of PLD1, but not PLD2, is sufficient to activate mTORC1 signaling, enhance dendritic BDNF expression, and drive retrograde enhancement of transmitter release from apposed presynaptic terminals.
Of note, whereas PLD1 activation potently drives mTORC1 signaling and enhances neurotransmitter release, inhibiting basal PLD signaling in the absence of AMPAR blockade has little impact on mTORC1 activation or baseline synaptic strength. This pattern of results is consistent with low levels of basal PA synthesis in the absence of activity blockade, an idea that is supported by PA reporter imaging in neurons (Fig. 1).
PA binding to mTOR is essential for homeostatic adaptation
Prior studies have established that the positively charged Arg2109 residue in the FRB domain of mTOR is vital for its interaction with PA (Fang et al., 2001). This site exhibits a high degree of evolutionary conservation (Rodriguez Camargo et al., 2012), suggesting an important role for PA/mTOR signaling across multiple species. Substituting this positively charged Arginine residue with Alanine severely impairs the hydrophobic interaction between mTOR and PA. We took advantage of this fact to develop a pharmacogenetic approach to specifically test the role of PA in driving mTORC1 activation during homeostatic signaling. Neurons were transfected with a variant of mTOR carrying a point mutation in the rapamycin binding domain (S2035T), which eliminates binding between mTOR and the rapamycin-FKB12 complex, thus rendering neurons expressing this mutant insensitive to rapamycin-induced inhibition of mTORC1 function (Brown et al., 1995). A separate group of cells were made to express a mutant form of mTOR carrying both the S2035T and R2109A point mutations, which renders mTORC1 insensitive to both rapamycin and PA. All neurons were cotransfected with GFP as a cell fill and treated with rapamycin (100 nm) to eliminate endogenous mTOR signaling. This scheme (Fig. 4A) results in blanket mTORC1 inhibition across the network, with mTORC1 signaling spared in isolated neurons transfected with the rapamycin-resistant S2035T mutant. Neurons that express the S2035T/R2109A double mutant have intact mTORC1 signaling, but mTORC1 is no longer sensitive to PA binding. Using levels of p-S6 staining intensity to assess mTORC1 activity, we found that disruption of PA-mTOR binding had no impact on mTORC1 activation during cLTP (Fig. 4B,C). In contrast, while neurons expressing the rapamycin-resistant (S2035T) mTOR point mutant showed significant increases in somatic p-S6 intensity after AMPAR blockade (CNQX, 40 μm, 3Hrs), cells in which PA/mTOR binding was disrupted showed no increase in somatic p-S6 intensity after loss of excitatory input compared with controls (Fig. 4B,C). This pattern of results indicates that binding of PA to mTOR's FRB domain is critical for increases in mTORC1 activity engaged during homeostatic adaptation to synaptic inactivity, but not during chemically-induced LTP.
Figure 4.
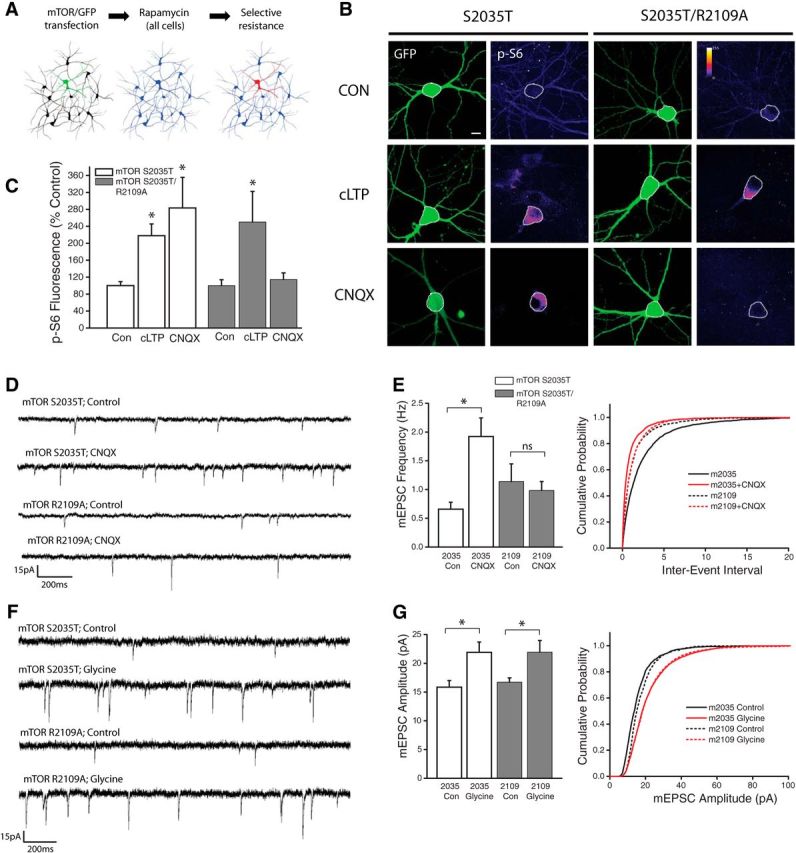
PA binding to mTOR is necessary for enhanced mTORC1 kinase activity during synaptic homeostasis but not cLTP. A, Schematic of experimental design: Neurons were transfected with GFP as well as one of two mutant versions of mTOR (S2035T = Rapamycin resistant; S2035T/R2109A = Rapamycin and PA resistant). Cultures were then treated with rapamycin (100 nm) to eliminate endogenous mTORC1 activity. B, Full frame example images of GFP and p-S6 staining in neurons expressing GFP alongside either mTOR point mutant, under conditions of AMPAR blockade (CNQX, 40 μm, 3Hr), cLTP (Forskolin-based stimulus, 50 μm), or vehicle control. C, Mean (+SEM) somatic intensity of p-S6 staining in groups as indicated. Somatic ps6 in S2035T Control n = 37 cells; S2035T cLTP, n = 22 cells; S2035T CNQX, n = 15 cells; S2035T/R2109A Control, n = 26 cells; S2035T/R2109A cLTP, n = 22 cells; S2035T/R2109A CNQX, n = 16 cells. Inhibition of PA/mTOR interaction (via R2109A mutation) eliminates increased mTORC1 kinase activity in the context of AMPAR blockade, but not during cLTP. Scale bar, 10 μm in B. p < 0.05 compared with vehicle controls in each group. D, E, Representative recordings (D) and mean (+SEM) mEPSC frequency (E) recorded from cells expressing GFP as well as Rap-resistant (mTOR S2035T) or PA-resistant (mTOR R2109A) mTOR point mutants under treatment conditions as indicated. Summary graphs presented on left and cumulative probability distribution shown on right. (*p < 0.05 relative to vehicle controls). S2035T Control, n = 26; S2035 CNQX, n = 14; S2035T/R2109A Control, n = 9; S2035T/R2109A CNQX, n = 16. Inhibition of PA/mTOR interaction (via R2109A mutation) eliminates the mTORC1-dependent increase in mEPSC frequency after AMPAR blockade. F, G, Representative recordings (F) and mean (+SEM) mEPSC amplitude (G) recorded from cells expressing GFP as well as Rap-resistant (mTOR S2035T) or PA-resistant (mTOR R2109A) mTOR point mutants under treatment conditions as indicated. Summary graphs presented on left and cumulative probability distribution shown on right. S2035T Control, n = 12; S2035T cLTP, n = 9; S2035T/R2109A Control, n = 10; S2035T/R2109A cLTP, n = 8. (*p < 0.05 relative to vehicle controls). Inhibition of PA/mTOR interaction (via R2109A mutation) has no effect on the mTORC1-dependent increase in mEPSC amplitude after cLTP induction.
To assess the impact of reduced PA/mTOR binding on synaptic plasticity, we continued the strategy described above (Fig. 4A), in which rapamycin was used to eliminate endogenous mTORC1 function under conditions of rapamycin and/or PA resistant mTOR mutant expression. Whereas rapamycin blocks increases in presynaptic function induced by AMPAR blockade in untransfected neurons (Henry et al., 2012), neurons expressing the S2035T mutant exhibit a robust increase in mEPSC frequency in response to AMPAR blockade (Fig. 4D,E). By contrast, neurons expressing the PA-resistant R2109A mTOR mutant failed to exhibit any change in mEPSC frequency during AMPAR blockade. While PA/mTOR binding appears to be essential for homeostatic increases in presynaptic function during AMPAR blockade, the same was not true for cLTP-induced increases in postsynaptic function (Fig. 4F,G). Indeed, we found largely identical levels of postsynaptic potentiation when assessed 2 h postcLTP stimulation in cells with or without impaired postsynaptic PA/mTOR binding. Together, these data suggest that PLD/PA signaling contributes to a novel mTORC1 activation pathway uniquely engaged by homeostatic signaling but dispensable for other forms of plasticity such as LTP.
Acute activation of mTORC1 and BDNF synthesis with exogenous PA
Having established a potential role for intracellular synthesis of PA in the activation mTORC1 after blockade of excitatory input, but not during chemically-induced LTP, we next sought to directly assess the physiological consequences of PA stimulation. Consistent with experiments using non-neuronal cell lines (Fang et al., 2001; Yoon et al., 2011a; You et al., 2012), we found that exogenous application of PA (100 μm) induced rapid mTORC1 activation in cultured hippocampal neurons. Following PA treatment, Western blots of neuron lysates revealed phosphorylation of the immediate downstream mTORC1 effectors 4E-BP1 and p70S6K within 15 min (Fig. 5A). Similarly, 30 min PA application induced robust phosphorylation of ribosomal protein S6 at S235/236 in the cell bodies and dendrites of hippocampal neurons as assessed via immunocytochemistry (Fig. 5B). Expression of tubulin (Fig. 5A) and microtubule associated protein 2 (MAP2) did not change significantly as a consequence of exposure to PA (Fig. 5B).
Figure 5.

Fast activation of neuronal mTORC1 via exogenous phosphatidic acid. A, Representative Western blots depicting phosphorylated p70S6K (Thr389) and 4EBP1 (Thr37/46) following treatment with PA (100 μm, n = 3 exps) or vehicle for the indicated times; the bands shown are from different blots. Mean (SEM) expression of p-p70S6K and p-4EBP1in neurons subject to treatment with PA. Exogenous PA rapidly enhanced mTORC1 activity, as evidence by significant, time-dependent increases in phosphorylation of its immediate downstream targets p70S6K and 4EBP. 15 min post-PA, n = 3; 30 min post-PA, n = 3; p-p70S6K 15 min post-PA, n = 3; p-p70S6K 30 min post-PA, n = 3. B, Full-frame examples of MAP2 and phosphorylated ribosomal protein S6 (Thr 235/236) staining in neurons treated with vehicle (n = 13) or phosphatidic acid (PA; 100 μm) for 60 min (n = 19). PS6 fluorescence intensity indicated by color look-up table; scale bar equals 40 μm. Mean (+SEM) normalized MAP2 and PS6 expression in neurons treated as indicated. Application of PA activates mTORC1 signaling as indicated by enhanced PS6 staining. *p < 0.05 (t test), relative to vehicle-treated controls. C, BDNF and MAP2 expression in linearized dendritic segments (top), and mean (+ SEM) expression of MAP2 and BDNF in dendritic compartments (bottom) normalized to average control values, in neurons treated with PA (100 μm, 60 min; n = 25) or vehicle (n = 25). BDNF expression in dendrites was significantly (*p < 0.05, relative to vehicle controls) enhanced by treatment with PA compared with vehicle-treated controls. This effect was blockaded by pretreatment with anisomycin (40 μm, 30 min before PA; n = 25) or rapamycin (200 nm, 30 min before PA; n = 25). MAP2 expression did not differ between groups. Scale bar represents 10 μm in A. D, Representative Western blots depicting changes in expression of BDNF or tubulin after treatment with PA (100 μm) ± Rapamycin (300 nm) for the indicated times; the band in the BDNF blot is mature BDNF. Mean (SEM) expression of BDNF in cultured neurons subject to treatment with PA (n = 3 experiments). Exogenous PA rapidly enhanced BDNF expression in an mTORC1-dependent manner, as the increased expression was blocked by rapamycin. E, Representative images of hippocampal neurons expressing myr-d1GFP-nls-A*B treated with either vehicle or PA (100 μm, 45 min). Scale bar, 10 μm. Mean (+SEM) GFP fluorescence in dendrites of hippocampal neurons treated with PA (n = 16) or vehicle (n = 21). Sampled dendritic regions were located 100–200 μm away from the cell body. *p < 0.05 (t test), relative to vehicle-treated controls.
Using PA as a tool to acutely activate mTORC1 signaling in neurons, we next examined the consequences of such activation on dendritic BDNF expression. We found that acute mTORC1 activation with PA (60 min, 100 μm) enhanced dendritic BDNF expression in hippocampal neurons, while having no effect on dendritic MAP2 expression in these same neurons (Fig. 5C). Coincident blockade of protein synthesis (with 40 μm anisomycin, 30 min before PA) or mTORC1 (with 300 nm rapamycin, 30 min before PA) completely suppressed this effect, suggesting that treatment with PA drives new synthesis of BDNF via activation of mTORC1. Similar effects were observed in Western blots of lysates from hippocampal neurons treated with PA alone or in combination with rapamycin (Fig. 5D). PA elicited a time-dependent increase in BDNF expression, with an initial peak at 15 min postPA treatment and persisting for at least 60 min (Fig. 5D). This increase in BDNF expression was also mTORC1-dependent, as concurrent rapamycin (300 nm) treatment completely eliminated PA-induced changes in BDNF expression. To establish that these changes in BDNF expression reflect de novo synthesis, we took advantage of a fluorescent BDNF synthesis reporter to gauge local BDNF synthesis in dendrites (Fig. 5E). The reporter consists of the entire long Bdnf 3′ UTR (sequence 'A*B'), alongside a myristoylation peptide (myr), destabilized d1GFP protein, and a nuclear localization sequence (nls), which in total prevents substantial intracellular movement of reporter-based fluorescence away from the area in which it was synthesized (Liao et al., 2012). BDNF transcripts with long 3′UTR have been previously shown to display preferential localization to the distal regions of dendrites (An et al., 2008), and as such, the myr-d1GFP-nls–A*B reporter has been used as an effective readout of dendritic BDNF synthesis (Liao et al., 2012). We found that application of PA to neuronal cultures transfected with the BDNF-A*B reporter showed enhanced GFP fluorescence intensity in the distal regions of dendrites (located 100–200 μm away from cell bodies), compared with transfected neurons treated with vehicle. Collectively, these experiments reveal that acute mTORC1 activation with exogenous PA drives rapid synthesis of BDNF in hippocampal neurons.
Physiological effects of exogenous PA
We next examined the impact of acute mTORC1 activation on hippocampal neuron activity. To assess the effects on overall network activity levels, we performed loose patch recordings from hippocampal neurons in culture. Recordings performed after a brief (45 min) treatment with PA revealed a profound impact on cell spiking behavior, specifically in the frequency of multispike burst events commonly observed in dissociated neuronal cultures (Fig. 6A). To determine whether this increase in network activity is accompanied by changes in excitatory synaptic function, we examine mEPSCs following acute mTORC1 activation with PA for 45 or 90 min. We found no change in the amplitude of mEPSCs after either 45 or 90 min treatment with PA, suggesting that acute mTORC1 activation is not sufficient to alter excitatory postsynaptic strength (Fig. 6B). In contrast, PA treatment induced a rapid increase in mEPSC frequency after 45 min, which became even more robust following 90 min exposure (Fig. 6C). This time-dependent increase in mEPSC frequency induced by PA was dependent on protein synthesis, mTORC1 activation, and BDNF release, as the increase in mEPSC frequency was blocked by concurrent application of anisomycin (40 μm), Rapamycin (300 nm) and an extracellular scavenger of released BDNF, TrkB-fc (1 μg/ml) (Fig. 6D,E). While previous reports have indicated that increases in network activity in mouse models featuring enhanced mTORC1 activity may reflect changes in inhibitory synapse function (Bateup et al., 2013), we found no changes in the frequency or amplitude of spontaneous mIPSCs in hippocampal cultures after treatment with PA (Fig. 6F), suggesting that altered inhibitory neurotransmission likely emerges following more sustained alterations in mTORC1 signaling. In addition, whole-cell current-clamp recordings revealed no difference between control and PA-treated neurons in a number of measures of intrinsic excitability, including capacitance and resistance (Fig. 6K) or the number of action potentials elicited by a series of depolarizing current steps (Fig. 6G).
Figure 6.
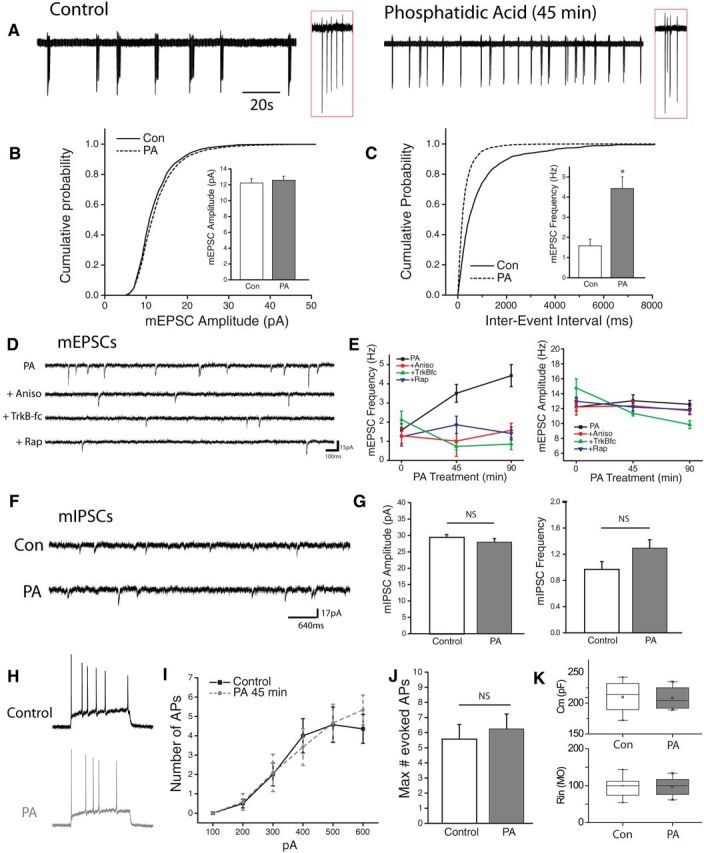
PA enhances network activity via increases in excitatory neurotransmission. A, Cell attached recordings of hippocampal neurons in dissociated cultured treated with vehicle or PA (100 μm, 45 min). Inset: enlarged region of recording (red box) showing individual spikes in a burst. B, C, Cumulative histogram of amplitude (pA) and interevent interval (msec) of mEPSCs recorded from neurons treated with vehicle or 100 μm PA for 45 or 90 min. Inset, mean (+ SEM) mEPSC amplitude and frequency in neurons after treatment with PA (100 μm) or vehicle. *p < 0.5 compared to vehicle treated controls. D, Representative recordings in neurons treated with PA (100 mm, 45 min) with or without 30 min pretreatment with rapamycin (200 nm), anisomysin (40 μm) or TrkB-fc (1 μg/ml). E, Time course of mean (+SEM) mEPSC amplitude and frequency of spontaneous mEPSCs at 45 m or 90 m exposure to PA with or without pretreatment, as indicated above. Sample sizes for the indicated groups: Vehicle (n = 7), PA 45 min (n = 9), PA45 min + rapamycin (n = 9), PA45 min + anisomycin (n = 5), PA45 min +TrKB-fc (n = 6), PA90 min (n = 6), PA90 min + rapamycin (n = 6), PA90 min + anisomycin (n = 9), PA90 min+ TrkBfc (n = 3), rapamycin alone (n = 7), anisomycin alone (n = 7), TrkB-fc alone (n = 6). Acute activation of mTORC1 signaling with PA enhances mEPSC frequency in a protein synthesis, mTORC1, and TrkB-dependent manner. F, Representative recordings of pharmacologically isolated (TTX 1 μm, CNQX 10 μm, APV 50 μm) spontaneous miniature inhibitory currents (mIPSCs) in cells treated with vehicle or PA. G, Mean (+SEM) mIPSC amplitude and frequency of cultured neurons treated with vehicle or PA (100 μm, 45 min). mIPSC control, n = 16; PA 45 min, n = 20. ns=not significant. H, Whole-cell current-clamp recordings showing example action potential traces from vehicle-treated control neurons (black) or cell treated with PA (red,). APs were evoked by injecting 1 s depolarizing current steps in the presence of excitatory and inhibitory synaptic blockers (CNQX 10 μm, APV 50 μm, Bicuculine 10 μm). I, J, Mean (± SEM) and maximum number of action potentials evoked by a series of depolarizing current steps in vehicle-treated (n = 14) or PA-treated (n = 12) cells. K, Box plots showing measures of capacitance (top) and resistance (bottom) for groups as indicated.
Exogenous PA enhances presynaptic release
To confirm that the observed changes in mEPSC frequency following acute mTORC1 activation reflect bona fide changes in presynaptic function, we examined the effect of PA on synaptic uptake of an antibody against the lumenal domain of synaptotagmin 1 (syt-lum uptake) at vglut1-positive excitatory synapses (Fig. 7A,B). The results of these experiments mirrored the observed changes in synaptic physiology, as transient mTORC1 activation induced a significant increase in syt-lum uptake that was blocked by anisomycin, rapamycin, and TrkB-Fc. We next assessed the impact of mTORC1-dependent retrograde signaling on evoked glutamatergic neurotransmission. First, we directly visualized evoked glutamate release by imaging activity-dependent changes in vglut1-pHluorin fluorescence at presynaptic terminals identified by coexpression of mCherry-tagged synaptophysin (mCh-Syn). As previously reported (Voglmaier et al., 2006), basal vglut1-pHluorin fluorescence at presynaptic terminals was low due to effective quenching by the acidic environment of synaptic vesicles, allowing for optical detection of action potential-triggered synaptic vesicle fusion when the lumen of the vesicle is exposed to the neutral extracellular space. To induce action potentials across the network, we applied field stimulation via parallel platinum-iridium electrodes under conditions where individual pulses each faithfully produced an action potential, verified by Ca2+ imaging (data not shown). In control (vehicle-treated) neurons, a 10 s train of action potentials delivered at 10 Hz induced a clear increase in vglut1-pHluorin fluorescence at presynaptic terminals, but following acute mTORC1 activation with PA (100 μm; 45 min), this evoked vesicle fusion was markedly enhanced (Fig. 7C–F). Scavenging extracellular BDNF with TrkB-fc (1 μg/ml) during PA application prevented this enhancement of evoked release, suggesting that acute mTORC1 activation drives increases in evoked glutamate release in a manner dependent on BDNF release. As another means of assessing evoked neurotransmitter release, and to examine whether mTORC1-dependent changes in evoked release are evident in a preparation where intrinsic hippocampal circuitry is preserved, we examined paired-pulse facilitation (PPF, negatively correlated with release probability) at CA3-CA1 synapses in acute hippocampal slices prepared from P18–P24 rats. Following preparation and recovery, slices were treated with PA (100 μm; 45 min), either alone or coincident with the protein synthesis inhibitor anisomycin (40 μm), the mTORC1 inhibitor rapamycin (300 nm), or the Trk receptor inhibitor k252a (300 nm). Relative to control (vehicle-treated) slices, acute mTORC1 activation with PA resulted in significantly diminished PPF (Fig. 7G), consistent with an increase in release probability at these synapses. This effect of PA required new protein synthesis, mTORC1 activation, and Trk receptor activation (Fig. 7G), suggesting that the diminished PPF that accompanies mTORC1 activation in slices, shares core mechanistic features with the retrograde regulation of presynaptic function by dendritic mTORC1 observed in cultured hippocampal neurons. Importantly, these inhibitors have no intrinsic effect on PPF when applied on their own (Fig. 7H). Together, these results demonstrate that acute activation of mTORC1 signaling via phosphatidic acid exerts a strong increase in presynaptic release at hippocampal synapses, largely via synthesis of BDNF.
Figure 7.
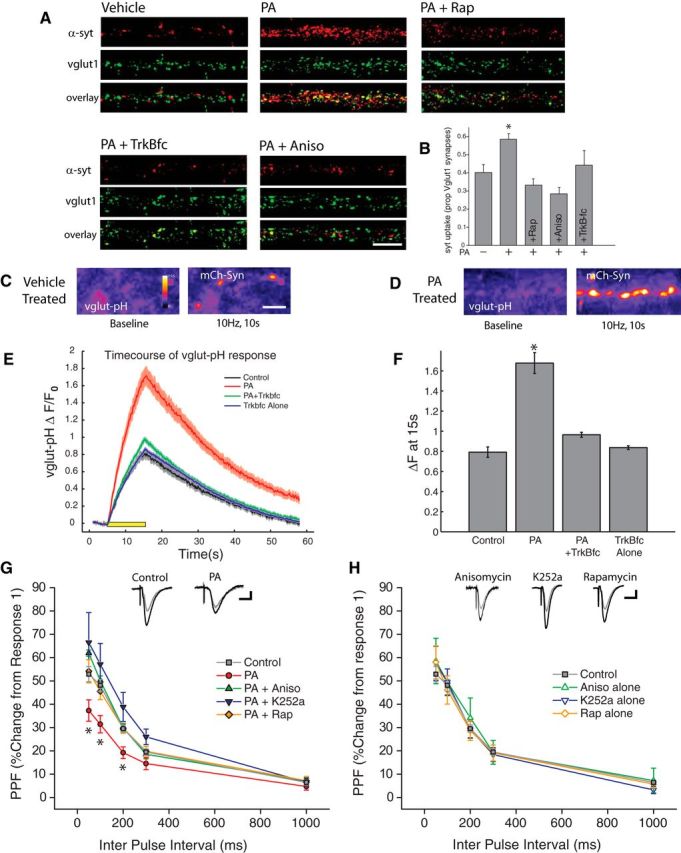
Enhanced presynaptic release emerges quickly after mTORC1 activation. A, B, Representative examples (A) and mean (+ SEM) syt-lum uptake (B) from experiments where the indicated groups were treated with PA (100 μm, 90 min) in the presence or absence of rapamycin (200 nm, 30 min prior), anisomycin (40 μm, 30 min prior) or TrkB-Fc (1 μg/ml, 30 min prior). Sample sizes for the indicated groups: vehicle control (n = 52 images), PA alone (n = 91 images), PA+rapamycin (n = 34 images), PA+anisomycin (n = 41 images), or PA+TrkB-Fc (n = 30 images images). PA treatment induced a significant (*p < 0.05, relative to vehicle control) increase in syt-lum uptake that was blocked by inhibiting protein synthesis, mTORC1, or by scavenging extracellular BDNF. Scale bar represents 10 μm in A. C, D, Example images of synapses expressing vglut1-pHluorin (vglut-pH) at baseline (left panel) as well as immediately after termination of a 10 Hz, 10 s stimulus train (right panel) for vehicle treated control (C) as well and neurons treated with PA (D). Treatment with phosphatidic acid enhanced the changes in vglut-pHluorin fluorescence intensity, compared with cells treated with vehicle in response to an identical stimulus train. Scale bar represents 10 mm in (C). E, Time course of relative change in vglut-pHluorin fluorescence in response to a 10 Hz 10 s stimulus train, recorded from cells treated with vehicle, PA, TrkB-fc or TrkBfc + PA. Black bar represents onset of 10 s stimulus train. Gray: Vehicle control (n = 566 synapses). Red: PA (n = 802 synapses). Green: PA+TrkB-fc (n = 652 synapses). Blue: TrkB-fc alone (n = 750 synapses). F, Mean (± SEM) change in vlgut-pHluorin fluorescence intensity from baseline recorded at the termination of a 10 Hz, 10 s AP train evoked by field stimulus. Evoked changes in vglut-pHluorin fluorescence intensity were significantly enhanced by treatment with PA, and this effect was blocked by pretreatment with the BDNF scavenger TrkB-fc. (*p < 0.05, one-way ANOVA, Tukey–Kramer post hoc). G, H, Paired-pulse facilitation (PPF) in acute hippocampal slices. Treatment with phosphatidic acid (100 μm, 45 min) significantly diminished (*p < 0.05, relative to vehicle controls) the degree of facilitation induced by paired pulses at 50, 100 and 200 ms intervals. Pretreatment with anisomycin, rapamycin, as well as the Trk receptor antagonist K252a, blocked this effect. H, Inhibitors have no effect on their own relative to vehicle-treated controls. Gray Circles: Control (n = 38 slices); Red Circles: PA (n = 10); Green closed Triangles: PA+Aniso (n = 7 slices); Blue closed triangles: PA+ K252a (n = 8 slices); Orange closed diamonds: PA+ Rap (n = 6 slices); Green open Triangles: Aniso Alone (n = 3 slices); Blue open Triangles: K252a alone (n = 9 slices); Orange open Diamonds: Rap alone (n = 8 slices). Scale bar represents 10 ms and 100 mV.
De novo synthesis of BDNF is necessary
The preceding results demonstrate that acute mTORC1 activation drives rapid protein synthesis-dependent increases in dendritic BDNF expression and rapid translation-dependent enhancement of presynaptic function that requires BDNF, suggesting a potential causal relationship between the two. To evaluate this possibility, we examined the specific role of postsynaptic BDNF release using sparse transfection of BDNF shRNA, which has been previously demonstrated to effectively knock down BDNF expression in dissociated hippocampal cultures (Jakawich et al., 2010). 24 h post-transfection, neurons were exposed to PA for 45 min, and postsynaptic mEPSC recordings were made from shRNA-expressing neurons (identified by RFP, expressed by an independent cassette in each shRNA plasmid) or untransfected control neurons (Fig. 8A). We found that whereas acute mTORC1 activation with PA resulted in robust increases in mEPSC frequency, but not mEPSC amplitude, in nontransfected neurons or neurons transfected with a scrambled control shRNA (Fig. 8B–D), PA had no effect in BDNF shRNA-expressing neurons. These results indicate that a postsynaptic source of BDNF is required for the retrograde enhancement of presynaptic function driven by acute mTORC1 activation. To verify a role for de novo synthesis of BDNF in PA-induced enhancement of presynaptic function, we used magnetofection to rapidly deliver siRNA targeting BDNF to specifically block new BDNF synthesis without loss of existing BDNF expression (Fig. 8E–I). BDNF siRNA was visible inside neurons immediately following magnetofection (data not shown), and as expected, a time-dependent decrease in basal levels of BDNF expression ensued over the next 48 h (Fig. 8E). Importantly, however, we found that the existing pool of dendritic BDNF was largely unaffected early after magnetofection, as expression was similar between neurons receiving BDNF siRNA and those receiving a nontargeting control siRNA 90 min following magnetofection. In contrast, BDNF siRNA magnetofection completely suppressed the increase in dendritic BDNF expression induced by PA treatment, suggesting that rapid siRNA delivery can inhibit de novo BDNF synthesis without dramatically impacting the existing BDNF pool due to limited BDNF turnover over this interval (Fig. 8F,G). Thus, we used this 90 min time-point to explore the functional consequences of suppressing new BDNF synthesis. Whereas PA treatment induced robust increases in mEPSC frequency in neurons receiving control siRNA magnetofection (Fig. 8H,I), this effect was completely blocked in neurons receiving BDNF siRNA magnetofection. These results indicate that de novo BDNF synthesis is required for retrograde changes in presynaptic function driven by mTORC1 activation. Together, our findings reveal that dendritic mTORC1 locally regulates the function of opposed presynaptic terminals by controlling BDNF synthesis in dendrites.
Figure 8.
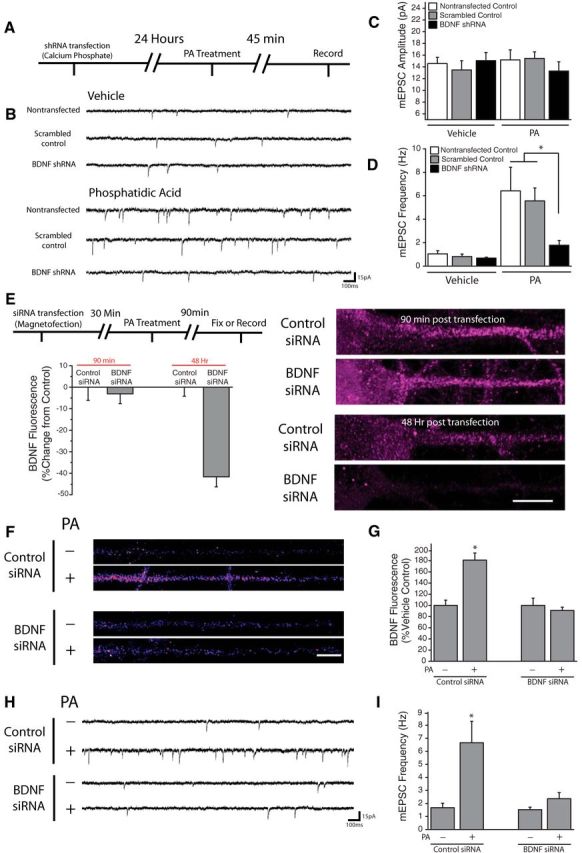
Postsynaptic BDNF expression is necessary for PA-induced changes in synaptic function. A, Experimental timeline: Dissociated hippocampal cultures were transfected with shRNA against BDNF or scrambled control via calcium phosphate. 24 h posttransfection, cells were treated with 100 μm PA for 45 min before recording. B–D, Representative recordings (B) and mean (+SEM) mEPSC amplitude (C) and frequency (D) in control nontransfected neurons (n = 7), as well as cells expressing BDNF shRNA (n = 8), or scrambled control shRNA (n = 4). The top panel in (B) contains representative traces from vehicle-treated group, while the bottom contains traces from cells subject to treatment with PA (100 μm, 45 min; n = 6, 9, 3). Cells were transfected at DIV 15 then used for recordings 24 h later. Postsynaptic BDNF knockdown blocked the increase in mEPSC frequency observed after PA treatment *p < 0.05 versus nontransfected cells and neurons transfected with scrambled shRNA. E, Experimental timeline: Dissociated hippocampal cultures were transfected with siRNA against BDNF or nontargeting control via magnetofection. 30 min posttransfection, cells were treated with phosphatidic acid for 90 min then either used for whole-cell recordings or fixed and processed for subsequent immunocytochemistry. (right) Representative images of straightened somadendritic regions of cultured neurons subject to magnetofection with control or anti BDNF siRNA. Scale bar represents 10 mm. Mean (+ SEM) expression of BDNF, normalized to average control values at identical time points, in neurons treated as indicated. While BDNF fluorescence was unaffected by magnetofection of anti-BDNF siRNA at 90 min (n = 21) compared with control siRNA transfected cells (n = 33), BDNF levels were significantly (*p < 0.05, relative to control) reduced by anti-BDNF siRNA magnetofection after 48 Hrs (n = 21) relative to neurons transfected with control siRNA (n = 32). F, G, BDNF expression in linearized dendritic segments, and mean (+ SEM) expression BDNF in dendritic compartments normalized to average control values, in neurons subject to magnetofection plus or minus PA (100 μm, 90 min). In neurons transfected with control siRNA, treatment with PA (n = 62) significantly enhanced BDNF expression in dendrites (*p < 0.05) compared with vehicle-treated cells (n = 55). In neurons transfected with BDNF siRNA, PA application (n = 60) failed to alter dendritic BDNF expression relative to vehicle (n = 49). Scale bar (in F): 10 μm. H, I, Representative recordings and mean (+ SEM) mEPSC frequency (H) in neurons subject to magnetofection plus or minus PA (100 μm, 90 min). Treatment with PA significantly enhanced mEPSC frequency in neurons transfected with the nontargeting control siRNA (*p < 0.05, t test; n = 6) compared with vehicle-treated neurons (n = 8). In neurons transfected with BDNF siRNA, no differences were observed between cells treated with PA (n = 7) or vehicle (n = 5).
Discussion
Here we report that PLD-mediated hydrolysis of the lipid second messenger PA is a crucial component of the signaling pathway which relays homeostatic signals to postsynaptic mTORC1 after loss of excitatory input. Critically, our data reveal that this PLD/PA-mediated activation of mTORC1 is a point of divergence between homeostatic and Hebbian forms of synaptic plasticity. We find that PLD signaling is necessary for mTORC1 activation after AMPAR blockade but not during LTP, which mirrors the pattern of intracellular PA synthesis detected under each condition (Fig. 1). This dissociation between LTP and HSP also holds true at the functional level as well, as inhibition of PLD signaling, or genetic disruption of PA/mTOR interaction eliminates the well defined presynaptic enhancement observed after AMPAR blockade but not the postsynaptic potentiation that is a central feature of LTP.
How is PLD/PA signaling engaged?
Steady-state levels of PA are believed to be quite low under normal conditions; with some estimates showing baseline levels of intracellular PA ∼5% of that shown for its precursor, phosphatidylcholine (Fang et al., 2003). While there is evidence to support a role for constitutive regulation of mTORC1 and mTORC2 complex integrity by steady-state levels of PA (Toschi et al., 2009), more recent work suggests that actively synthesized PA may directly stimulate mTORC1 kinase activity by displacing the inhibitory FK506 binding protein 38, leading to altered mTOR autophosphorylation (Yoon et al., 2011a). In non-neuronal cells, PLD/PA signaling has been shown to play an important role in signaling nutrient availability to mTOR. Specifically, amino acid availability is believed to act as a signal which causes the Rag-dependent translocation of mTOR (Sancak et al., 2008) and hVps34-dependent translocation of PLD1 (Yoon et al., 2011b) to the late endosomal/lysosomal membrane. It is after this amino-acid-induced translocation occurs that Rheb is thought to activate PLD1-induced synthesis of PA to activate mTORC1 signaling. In addition to Rheb, PLD1 can also be activated by protein kinase C, select Rho-GTPases, and members of the ADP-ribosylation factor (Arf) family (Ktistakis et al., 2003). Interestingly, recent work has implicated postsynaptic action of the RhoGTPase Cdc42 in the expression of presynaptic homeostasis at the drosophila NMJ (Pilgram et al., 2011). Additional experiments are needed to explore the possibility that neurons use PLD/PA signaling as a mechanism similar to that implemented by mitotic cells in relaying nutrient signals to mTOR, or if instead an alternative manner of activating PLD signaling is used, perhaps involving a member of the RhoGTPase family, during synaptic homeostasis.
How do unique types of activation lead to different function outcomes?
The increased postsynaptic strength observed during LTP and presynaptic enhancement that emerges after AMPAR blockade are both critically dependent on mTORC1 signaling, though it is important to appreciate that much of this evidence is based on the use of rapamycin as a specific mTORC1 inhibitor. Given mTOR's prominent role in orchestrating protein translation necessary for many forms of synapse plasticity and learning (Hoeffer and Klann, 2010), we were interested to uncover an upstream point of divergence by which the mTOR pathway is engaged in these two distinct forms of plasticity. Our data suggest that PLD/PA signaling is activated during AMPAR blockade, but not by the strong synaptic stimulation that leads to LTP. Moreover, upregulation of PLD/PA signaling is sufficient to drive mTORC1 signaling, BDNF synthesis, and transsynaptic control of presynaptic neurotransmitter release, yet this signaling axis is completely dispensable for LTP. It remains unclear how the route to mTORC1 activation culminates in the unique synaptic phenotypes underlying LTP (postsynaptic) and homeostasis (presynaptic). One possibility is that mTORC1 acts to regulate the translation of mRNAs in close proximity to the stimulated complex, and the spatial locale of cLTP or PLD/PA-induced mTOR activation occurs in subcellular regions with unique compositions of potential mRNA targets. Previous work has identified mTOR distribution at the cell nucleus (Kim and Chen, 2000), at late endosomes/lysosomes (Sancak et al., 2008; Flinn et al., 2010), along the golgi apparatus (Narita et al., 2011) as well as in the endoplasmic reticulum (Drenan et al., 2004). It is conceivable that stimulation of mTORC1 signaling at the late endosome/lysosome would potentially lead to the translation of specific mRNA transcripts, and hence produce different functional outcomes, than would be observed when mTOR is activated in another subcellular compartment. Support for this type of spatially segregated regulation of mTORC1 signaling in neurons comes from recent work describing differential activation of mTORC1 at the plasma membrane or lysosome by Amyloid-β oligomers or amino acids, respectively (Norambuena et al., 2016). Further experiments are necessary to determine whether activation of mTORC1 at distinct subcellular compartments can explain the unique aspects of PLD/PA-mediated activation we observe in the context of synaptic homeostasis.
Isoform-specific effects on mTORC1 activation and synapse function
Our experiments involving overexpression of either PLD1 or PLD2 reveal an interesting degree of specificity with respect to the influence each of these isoforms exerts over neuronal mTORC1 activation and synapse function. The finding that overexpression of PLD1, but not PLD2, causes an increase in mTORC1 activation (Fig. 3A,B) was somewhat surprising given previous work showing that overexpression of either PLD isoform is sufficient to activate mTORC1 signaling in non-neuronal cell lines (Chen et al., 2003; Fang et al., 2003). Part of this discrepancy could be a result of the different ways in which postmitotic cells such as neurons make use of signaling pathways which normally serve a role in mitogenic signaling. Additionally, PLD1 and PLD2 are known to exhibit markedly different patterns of subcellular localization, with PLD1 present at late endosomes/ lysosomes (Toda et al., 1999) or the golgi apparatus (Freyberg et al., 2001), and PLD2 occupying lipid raft sites along the plasma membrane (Colley et al., 1997). Our finding that PLD1 overexpression dramatically increases dendritic BDNF expression and mEPSC frequency (Fig. 7) mirrors our previously reported effects on BDNF expression and presynaptic function after overexpression of a constitutively active version of Rheb GTPase (Henry et al., 2012). This is perhaps not surprising, given previous reports that Rheb, mTOR, and PLD1 share a similar subcellular localization (Toda et al., 1999; Sancak et al., 2008) and that Rheb directly binds to and activates PLD1 (Sun et al., 2008). If PLD1 is directly implicated in certain aspects of Rheb-induced changes in synaptic function, it could serve as an important point of entry of future therapeutic interventions for neurodevelopmental disorders which share upregulated mTORC1 signaling as a common phenotype, including PTEN hamartoma syndrome and Tuberous sclerosis.
Chronic vs acute mTOR activation
Though the effects of chronic mTORC1 upregulation on basal characteristics of synapse form and function have been extremely varied, our findings mirror that of several recent publications. Specifically, we find that acute activation of mTORC1 signaling after PA treatment mirrors the effects of more prolonged activation reported previously with respect to increases in mEPSC frequency (Bateup et al., 2011; Luikart et al., 2011), and increases in presynaptic release probability (Weston et al., 2012). Additionally, we found no change in inhibitory neurotransmission, mEPSC amplitude, or dendritic spine morphology, similar to effects seen after hyperactivation of mTORC1 signaling resulting from mutations in the inhibitory mTOR regulators PTEN or TSC1/2 (Luikart et al., 2011; Bateup et al., 2011; Sperow et al., 2012). Our results differ from previous reports describing changes in inhibitory synapse connectivity (Bateup et al., 2013), increases in spine density (Fraser et al., 2008), and increases in mEPSC amplitude (Tavazoie et al., 2005). One potential source of this discrepancy may be the amount of time during which mTORC1 activity is upregulated. For instance, our previous work demonstrated that somatic hypertrophy in cultured hippocampal neurons is observed after 5 d, but not after 1 d of genetic mTORC1 hyperactivation (Henry et al., 2012). This may also suggest that the synaptic functional abnormalities we report here actually precede changes in cell morphology. Given the essential role played by synaptic activity in shaping neuronal connectivity during development (Bleckert and Wong, 2011), it is conceivable that early shifts in excitatory synapse function resulting from mTORC1 hyperactivation may drive the subsequent changes in spine density and circuit structure that have been observed after longer periods of mTORC1 hyperactivation (Nie et al., 2010; Luikart et al., 2011; Weston et al., 2012; Normand et al., 2013). With its common role in several monogenic neurodevelopmental disorders, additional insight into the consequences of mTORC1 upregulation on synapse form and function will provide a crucial link between cellular and behavioral abnormalities observed in patients with Autism and Intellectual Disability.
Footnotes
This work was supported by NIH Grants F31MH093112 (F.E.H.), RO1MH085798 (M.A.S.), and RO1NS097498 (M.A.S.). We thank Hisashi Umemori and members of the Sutton laboratory for many helpful discussions. Human HA-PLD1, human HA-PLD2, rat HA-PLD2, and pEGFP-Spo20PABD-WT were generously provided by Dr. Michael Frohman (SUNY Stony Brook); GFP-Rap1-PABD was provided by Dr. Frank Schmitz (Universität des Saarlandes Medizinische Fakultät); and rapamycin-resistant (S2035T) and rapamycin/phosphatic acid resistant (32035T/R2109A) mTOR point mutants were provided by Dr. Jie Chen (University of Illinois at Urbana-Champaign).
The authors declare no competing financial interests.
References
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, Xu B (2008) Distinct role of long 3' UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 134:175–187. 10.1016/j.cell.2008.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, Chen L (2008) Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60:308–320. 10.1016/j.neuron.2008.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker GA, Goslin K (1990) Culturing nerve cells. Cambridge, MA: MIT. [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL (2011) Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J Neurosci 31:8862–8869. 10.1523/jneurosci.1617-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL (2013) Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78:510–522. 10.1016/j.neuron.2013.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleckert A, Wong RO (2011) Identifying roles for neurotransmission in circuit assembly: insights gained from multiple model systems and experimental approaches. Bioessays 33:61–72. 201000095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL (1995) Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature 377:441–446. 10.1038/377441a0 [DOI] [PubMed] [Google Scholar]
- Cammalleri M, Lütjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP (2003) Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci U S A 100:14368–14373. 10.1073/pnas.2336098100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Fang Y (2002) A novel pathway regulating the mammalian target of rapamycin (mTOR) signaling. Biochem Pharmacol 64:1071–1077. 10.1016/s0006-2952(02)01263-7 [DOI] [PubMed] [Google Scholar]
- Chen Y, Zheng Y, Foster DA (2003) Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 22:3937–3942. 10.1038/sj.onc.1206565 [DOI] [PubMed] [Google Scholar]
- Cockcroft S. (2001) Signalling roles of mammalian phospholipase D1 and D2. Cell Mol Life Sci 58:1674–1687. 10.1007/PL00000805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA (1997) Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol 7:191–201. 10.1016/S0960-9822(97)70090-3 [DOI] [PubMed] [Google Scholar]
- Drenan RM, Liu X, Bertram PG, Zheng XF (2004) FKBP12-rapamycin-associated protein or mammalian target of rapamycin (FRAP/mTOR) localization in the endoplasmic reticulum and the golgi apparatus. J Biol Chem 279:772–778. 10.1074/jbc.M305912200 [DOI] [PubMed] [Google Scholar]
- Du G, Huang P, Liang BT, Frohman MA (2004) Phospholipase D2 localizes to the plasma membrane and regulates angiotensin II receptor endocytosis. Mol Biol Cell 15:1024–1030. 10.1091/mbc.E03-09-0673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294:1942–1945. 10.1126/science.1066015 [DOI] [PubMed] [Google Scholar]
- Fang Y, Park IH, Wu AL, Du G, Huang P, Frohman MA, Walker SJ, Brown HA, Chen J (2003) PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr Biol 13:2037–2044. 10.1016/j.cub.2003.11.021 [DOI] [PubMed] [Google Scholar]
- Flinn RJ, Yan Y, Goswami S, Parker PJ, Backer JM (2010) The late endosome is essential for mTORC1 signaling. Mol Biol Cell 21:833–841. 10.1091/mbc.E09-09-0756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DA. (2009) Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim Biophys Acta 1791:949–955. 10.1016/j.bbalip.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ (2008) Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 151:476–488. 10.1016/j.neuroscience.2007.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyberg Z, Sweeney D, Siddhanta A, Bourgoin S, Frohman M, Shields D (2001) Intracellular localization of phospholipase D1 in mammalian cells. Mol Biol Cell 12:943–955. 10.1091/mbc.12.4.943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frondorf K, Henkels KM, Frohman MA, Gomez-Cambronero J (2010) Phosphatidic acid is a leukocyte chemoattractant that acts through S6 kinase signaling. J Biol Chem 285:15837–15847. 10.1074/jbc.M109.070524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry FE, McCartney AJ, Neely R, Perez AS, Carruthers CJ, Stuenkel EL, Inoki K, Sutton MA (2012) Retrograde changes in presynaptic function driven by dendritic mTORC1. J Neurosci 32:17128–17142. 10.1523/JNEUROSCI.2149-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E (2010) mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 33:67–75. 10.1016/j.tins.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Klann E (2004) Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci 24:6352–6361. 10.1523/JNEUROSCI.0995-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, Sutton MA (2010) Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 68:1143–1158. 10.1016/j.neuron.2010.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM, Frohman MA (2005) Phospholipase D: a lipid centric review. Cell Mol Life Sci 62:2305–2316. 10.1007/s00018-005-5195-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Chen J (2000) Cytoplasmic-nuclear shuttling of FKBP12-rapamycin-associated protein is involved in rapamycin-sensitive signaling and translation initiation. Proc Natl Acad Sci U S A 97:14340–14345. 10.1073/pnas.011511898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA (2010) CDK5 serves as a major control point in neurotransmitter release. Neuron 67:797–809. 10.1016/j.neuron.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ktistakis NT, Delon C, Manifava M, Wood E, Ganley I, Sugars JM (2003) Phospholipase D1 and potential targets of its hydrolysis product, phosphatidic acid. Biochem Soc Trans 31:94–97. 10.1042/bst0310094 [DOI] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS (2010) mTOR dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964. 10.1126/science.1190287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao GY, An JJ, Gharami K, Waterhouse EG, Vanevski F, Jones KR, Xu B (2012) Dendritically targeted Bdnf mRNA is essential for energy balance and response to leptin. Nat Med 18:564–571. 10.1038/nm.2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog M, Li L, Groth RD, Poburko D, Thiagarajan TC, Han X, Tsien RW (2010) Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci U S A 107:21806–21811. 10.1073/pnas.1016399107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JO, Sahin M (2014) The neurology of mTOR. Neuron 84:275–291. 10.1016/j.neuron.2014.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29:243–254. 10.1016/S0896-6273(01)00194-5 [DOI] [PubMed] [Google Scholar]
- Luikart BW, Schnell E, Washburn EK, Bensen AL, Tovar KR, Westbrook GL (2011) Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J. Neurosci. 31:4345–4354. 10.1523/jneurosci.0061-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J (2009) Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10:307–318. 10.1038/nrm2672 [DOI] [PubMed] [Google Scholar]
- Mameli M, Balland B, Luján R, Lüscher C (2007) Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317:530–533. 10.1126/science.1142365 [DOI] [PubMed] [Google Scholar]
- Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, Tavaré S, Inoki K, Shimizu S, Narita M (2011) Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332:966–970. 10.1126/science.1205407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie D, Di Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, Dabora S, Codeluppi S, Pandolfi PP, Pasquale EB, Sahin M (2011) Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci 13:163–172. 10.1038/nn.2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norambuena A, Wallrabe H, McMahon L, Silva A, Swanson E, Khan SS, Baerthlein D, Kodis E, Oddo S, Mandell JW, Bloom GS (2017) mTOR and neuronal cell cycle reentry: how impaired brain insulin signaling promotes Alzheimer's disease. Alzheimers Dement 13:152–167. 10.1016/j.jalz.2016.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normand EA, Crandall SR, Thorn CA, Murphy EM, Voelcker B, Browning C, Machan JT, Moore CI, Connors BW, Zervas M (2013) Temporal and mosaic Tsc1 deletion in the developing thalamus disrupts thalamocortical circuitry, neural function, and behavior. Neuron 78:895–909. 10.1016/j.neuron.2013.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney J, Tsurudome K, Liao EH, Elazzouzi F, Livingstone M, Gonzalez M, Sonenberg N, Haghighi AP (2012) TOR is required for the retrograde regulation of synaptic homeostasis at the Drosophila neuromuscular junction. Neuron 74:166–178. 10.1016/j.neuron.2012.01.030 [DOI] [PubMed] [Google Scholar]
- Pilgram GS, Potikanond S, van der Plas MC, Fradkin LG, Noordermeer JN (2011) The RhoGAP crossveinless-c interacts with dystrophin and is required for synaptic homeostasis at the Drosophila neuromuscular junction. J Neurosci 31:492–500. 10.1523/JNEUROSCI.4732-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Camargo DC, Link NM, Dames SA (2012) The FKBP-rapamycin binding domain of human TOR undergoes strong conformational changes in the presence of membrane mimetics with and without the regulator phosphatidic acid. Biochemistry 51:4909–4921. 10.1021/bi3002133 [DOI] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (2008) The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501. 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz K, Natarajan S, Kassas N, Vitale N, Schmitz F (2011) The synaptic ribbon is a site of phosphatidic acid generation in ribbon synapses. J Neurosci 31:15996–16011. 10.1523/JNEUROSCI.2965-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperow M., Berry RB, Bayazitov IT, Zhu G, Baker SJ, Zakharenko SS (2012) Phosphatase and tensin homologue (PTEN) regulates synaptic plasticity independently of its effect on neuronal morphology and migration. J Physiol 590:777–792. 10.1113/jphysiol.2011.220236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Yeku O, Olepu S, Genna A, Park JS, Ren H, Du G, Gelb MH, Morris AJ, Frohman MA (2009) 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol 75:437–446. 10.1124/mol.108.053298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, Armstrong M, Brown HA, Chen J (2008) Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc Natl Acad Sci U S A 105:8286–8291. 10.1073/pnas.0712268105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM (2006) Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127:49–58. 10.1016/j.cell.2006.09.014 [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A 99:467–472. 10.1073/pnas.012605299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005) Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734. 10.1038/nn1566 [DOI] [PubMed] [Google Scholar]
- Toda K, Nogami M, Murakami K, Kanaho Y, Nakayama K (1999) Colocalization of phospholipase D1 and GTP-binding-defective mutant of ADP-ribosylation factor 6 to endosomes and lysosomes. FEBS Lett 442:221–225. 10.1016/S0014-5793(98)01646-9 [DOI] [PubMed] [Google Scholar]
- Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA (2009) Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol 29:1411–1420. 10.1128/MCB.00782-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veverka V, Crabbe T, Bird I, Lennie G, Muskett FW, Taylor RJ, Carr MD (2008) Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene 27:585–595. 10.1038/sj.onc.1210693 [DOI] [PubMed] [Google Scholar]
- Vickers CA, Dickson KS, Wyllie DJ (2005) Induction and maintenance of late-phase long-term potentiation in isolated dendrites of rat hippocampal CA1 pyramidal neurones. J Physiol 568:803–813. 10.1113/jphysiol.2005.092924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH (2006) Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron 51:71–84. 10.1016/j.neuron.2006.05.027 [DOI] [PubMed] [Google Scholar]
- Weston MC, Chen H, Swann JW (2012) Multiple roles for mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. J. Neurosci. 32:11441–11452. 10.1523/jneurosci.1283-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Sun Y, Arauz E, Jiang Y, Chen J (2011a) Phosphatidic acid activates mammalian target of rapamycin complex 1 (mTORC1) kinase by displacing FK506 binding protein 38 (FKBP38) and exerting an allosteric effect. J Biol Chem 286:29568–29574. 10.1074/jbc.M111.262816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Du G, Backer JM, Frohman MA, Chen J (2011b) Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol 195:435–447. 10.1083/jcb.201107033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JS, Frey JW, Hornberger TA (2012) Mechanical stimulation induces mTOR signaling via an ERK-independent mechanism: implications for a direct activation of mTOR by phosphatidic acid. PLoS One 7:e47258 10.1371/journal.pone.0047258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeniou-Meyer M, Zabari N, Ashery U, Chasserot-Golaz S, Haeberlé AM, Demais V, Bailly Y, Gottfried I, Nakanishi H, Neiman AM, Du G, Frohman MA, Bader MF, Vitale N (2007) Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J Biol Chem 282:21746–21757. 10.1074/jbc.M702968200 [DOI] [PubMed] [Google Scholar]
- Zhu YB, Kang K, Zhang Y, Qi C, Li G, Yin DM, Wang Y (2012) PLD1 negatively regulates dendritic branching. J Neurosci 32:7960–7969. 10.1523/JNEUROSCI.5378-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12:21–35. 10.1038/nrm3025 [DOI] [PMC free article] [PubMed] [Google Scholar]