

Abstract
Systemic fentanyl induces hyperalgesic priming, long-lasting neuroplasticity in nociceptor function characterized by prolongation of inflammatory mediator hyperalgesia. To evaluate priming at both nociceptor terminals, we studied, in male Sprague Dawley rats, the effect of local administration of agents that reverse type I (protein translation) or type II [combination of Src and mitogen-activated protein kinase (MAPK)] priming. At the central terminal, priming induced by systemic, intradermal, or intrathecal fentanyl was reversed by the combination of Src and MAPK inhibitors, but at the peripheral terminal, it was reversed by the protein translation inhibitor. Mu-opioid receptor (MOR) antisense prevented fentanyl hyperalgesia and priming. To determine whether type I and II priming occur in the same population of neurons, we used isolectin B4–saporin or [Sar9, Met(O2)11]-substance P–saporin to deplete nonpeptidergic or peptidergic nociceptors, respectively. Following intrathecal fentanyl, central terminal priming was prevented by both saporins, whereas that in peripheral terminal was not attenuated even by their combination. However, after intradermal fentanyl, priming in the peripheral terminal requires both peptidergic and nonpeptidergic nociceptors, whereas that in the central terminal is dependent only on peptidergic nociceptors. Pretreatment with dantrolene at either terminal prevented fentanyl-induced priming in both terminals, suggesting communication between central and peripheral terminals mediated by intracellular Ca2+ signaling. In vitro application of fentanyl increased cytoplasmic Ca2+ concentration in dorsal root ganglion neurons, which was prevented by pretreatment with dantrolene and naloxone. Therefore, acting at MOR in the nociceptor, fentanyl induces hyperalgesia and priming rapidly at both the central (type II) and peripheral (type I) terminal and this is mediated by Ca2+ signaling.
SIGNIFICANCE STATEMENT Fentanyl, acting at the μ-opioid receptor (MOR), induces hyperalgesia and hyperalgesic priming at both the central and peripheral terminal of nociceptors and this is mediated by endoplasmic reticulum Ca2+ signaling. Priming in the central terminal is type II, whereas that in the peripheral terminal is type I. Our findings may provide useful information for the design of drugs with improved therapeutic profiles, selectively disrupting individual MOR signaling pathways, to maintain an adequate long-lasting control of pain.
Keywords: calcium, endoplasmic reticulum, fentanyl, hyperalgesia, hyperalgesic priming, μ-opioid receptor (MOR)
Introduction
Chronic pain is a major health issue that affects quality of life markedly (Aronoff, 2016; Jackson et al., 2016; Maixner et al., 2016; Roeckel et al., 2016; Turk et al., 2016). As much as 30–40% of the United States population suffers from chronic pain at an estimated cost of 560–635 billion dollars annually (Renfrey et al., 2003; Johannes et al., 2010; Roeckel et al., 2016). Although opioids remain the most potent treatment for moderate to severe pain, the increase in opioid prescriptions in the United States has been accompanied by a sharp rise in the incidence of addiction and opioid-related mortality and has produced an “opioid epidemic” (Volkow and McLellan, 2016). Opioids induce several adverse effects such as analgesic tolerance, addiction, opioid-induced hyperalgesia (OIH), and hyperalgesic priming (Chu et al., 2008; Joseph et al., 2010; Lee et al., 2011; Araldi et al., 2015, 2017a, 2018; Trang et al., 2015; Roeckel et al., 2016).
Hyperalgesic priming, here referred as priming, is a form of neuroplasticity in primary afferent nociceptors that has been characterized by marked prolongation of prostaglandin E2 (PGE2)-induced mechanical hyperalgesia (Aley et al., 2000; Reichling and Levine, 2009; Bogen et al., 2012; Ferrari et al., 2013, 2015; Araldi et al., 2015, 2016a,b, 2017a, 2018; Khomula et al., 2017). We have recently demonstrated a novel form of priming (type II), induced by repeated exposure to the μ-opioid receptor (MOR) selective agonist, DAMGO ([D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin acetate salt) (Joseph et al., 2010; Araldi et al., 2015, 2017a, 2018) that occurs in isolectin B4 (IB4)-negative (peptidergic) nociceptors (Araldi et al., 2018); this contrasts with type I priming, which occurs in IB4-positive (nonpeptidergic) (Joseph and Levine, 2010) nociceptors. Another distinguishing feature of type II priming is that MOR agonist signaling switches from being antihyperalgesic to inducing hyperalgesia, a model of OIH (Joseph et al., 2010; Araldi et al., 2015, 2017a, 2018). OIH has been demonstrated, not only in humans suffering from different types of pain, but also in healthy volunteers and opioid addicts (Lee and Yeomans, 2014; Stoicea et al., 2015; Mauermann et al., 2016). Because the fentanyl class of opioids are the ones most frequently reported to induce OIH and can elicit OIH after acute administration (Célèrier et al., 2000; Richebé et al., 2005; Célèrier et al., 2006; Waxman et al., 2009), we studied the neurobiological basis of fentanyl-induced hyperalgesia and priming. In contrast to DAMGO, acute administration of fentanyl induced hyperalgesia and priming.
It has been demonstrated that activation of the ryanodine receptor, which releases Ca2+ from the endoplasmic reticulum (ER) (Sutko et al., 1985; Fill and Copello, 2002; Khomula et al., 2017) and induces Ca2+ signaling (Stutzmann and Mattson, 2011; Adasme et al., 2015; Futagi and Kitano, 2015; Evans et al., 2016), is associated with neuroplasticity, including induction of type I priming (Chen et al., 2015; Futagi and Kitano, 2015; Khomula et al., 2017). Because MOR activation may also cause ER stress and alter signal transduction (Aoe, 2015), we also evaluated the involvement of Ca2+ signaling in fentanyl-induced priming at the terminal opposite from fentanyl administration and determined whether it was type I or II and if it occurred in nonpeptidergic or peptidergic nociceptors.
Materials and Methods
Animals
All experiments were performed on 220–420 g adult male Sprague Dawley rats (Charles River Laboratories). Animals were housed three per cage under a 12 h light/dark cycle in a temperature- and humidity-controlled animal care facility at the University of California–San Francisco. Food and water were available ad libitum. Nociceptive testing was performed between 9:00 A.M. and 5:00 P.M. Experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of California at San Francisco and adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Effort was made to minimize the number of animals used and their suffering.
Testing mechanical nociceptive threshold
Mechanical nociceptive threshold was quantified using an Ugo Basile Analgesymeter® [Randall-Selitto paw-withdrawal device (Stoelting)], which applies a linearly increasing mechanical force to the dorsum of the rat's hindpaw, as described previously (Taiwo and Levine, 1989; Taiwo et al., 1989; Araldi et al., 2015, 2017a, 2018; Ferrari and Levine, 2015). Rats were placed in cylindrical acrylic restrainers designed to provide ventilation, allow extension of the hind leg from lateral ports in the cylinder during the assessment of nociceptive threshold, and minimize restraint stress. To acclimatize rats to the testing procedure, they were placed in a restrainer for 1 h before starting each training session (3 consecutive days of training, once a day) and for 30 min before experimental manipulations. The nociceptive threshold was defined as the force, in grams, at which the rat withdrew its paw. Baseline paw-pressure nociceptive threshold was defined as the mean of the three readings taken before test agents were injected. To minimize experimenter bias, individuals conducting the behavioral experiments (D.A. and L.F.F) were blinded to experimental interventions.
Drugs and their administration
The following compounds were used in this study: cordycepin 5′-triphosphate sodium salt (a protein translation inhibitor), dantrolene sodium salt (a ryanodine receptor blocker that prevents calcium release from the endoplasmic reticulum), fentanyl citrate salt (a MOR agonist), naloxone (a opioid receptor antagonist), PGE2 (a direct-acting hyperalgesic agent that sensitizes nociceptors), SU 6656 (a Src family kinase inhibitor), and U0126 (a MAPK/ERK inhibitor), all of which were from Sigma-Aldrich.
The stock solution of PGE2 (1 μg/μl) was prepared in 10% ethanol and additional dilutions made with physiological saline (0.9% NaCl), yielding a final ethanol concentration <1%. Fentanyl, cordycepin, and naloxone were dissolved in saline. All other drugs were dissolved in 100% DMSO (Sigma-Aldrich) and further diluted in saline containing 2% Tween 80 (Sigma-Aldrich). The final concentration of DMSO and Tween 80 was ∼2%.
Intradermal drug administration was performed on the dorsum of the hindpaw using a 30-gauge hypodermic needle adapted to a 50 μl Hamilton syringe by a segment of PE/10 polyethylene tubing (Becton Dickinson). The combination of SU 6656 and U0126 was diluted to a concentration of 1 μg/2 μl each and the combination injected by adding 2 μl into a syringe separated by an air bubble to avoid mixing in the syringe. The intradermal administration of all drugs except PGE2, naloxone, and fentanyl was preceded by a hypotonic shock to facilitate the permeability of the cell membrane to these agents (1 μl of distilled water separated by an bubble to avoid mixing in the same syringe) to enhance entry into the nerve terminal (Borle and Snowdowne, 1982; Burch and Axelrod, 1987). Importantly, in vivo control experiments have shown previously that the final concentration of ethanol (2%) used to prepare the solution of PGE2 had no effect on the mechanical threshold per se; DMSO, which was used to dissolve dantrolene, SU 6656, and U0126, did not produce any effect on the mechanical nociceptor threshold (Ferrari et al., 2016; Araldi et al., 2017a).
Intrathecal administration of fentanyl, PGE2, dantrolene, cordycepin, and the combination of SU 6656 and U0126 was performed in rats briefly anesthetized with 2.5% isoflurane (Phoenix Pharmaceuticals) in 97.5% O2 using a 29-gauge hypodermic needle (300 units/μl syringe) inserted into the subarachnoid space between the L4 and L5 vertebrae. The maximum volume injected into the spinal cord was 20 μl. The combination of SU 6656 (10 μg/5 μl) and U0126 (10 μg/5 μl) was injected in a final volume of 10 μl (these drugs were mixed in the syringe at the moment of injection), followed by an injection of PGE2 (400 ng/10 μl) at the same site. The intrathecal site of injection was confirmed by a sudden flick of the rat's tail, a reflex that is evoked by subarachnoid space access and bolus injection (Mestre et al., 1994).
PGE2 was injected intrathecally (400 ng) or intradermally (100 ng) after intrathecal or intradermal administration of fentanyl and/or inhibitors. Mechanical nociceptive threshold was evaluated 30 min and 4 h after the injection of PGE2.
MOR oligodeoxynucleotide antisense
To investigate the role of MOR in the hyperalgesia and priming induced by intradermal and intrathecal fentanyl, oligodeoxynucleotides (ODN) antisense (AS) for MOR mRNA (Khasar et al., 1996; Sanchez-Blazquez et al., 1997; Araldi et al., 2017a, 2018) was used. The AS-ODN sequence for MOR, 5′-CGC-CCC-AGC-CTC-TTC-CTC-T-3′, was directed against a unique region of rat MOR (UniProtKB database entry P33535 [OPRM_RAT] antisense sequence to block translation and downregulate the gene expression of all eight known isoforms [MOR]). The ODN mismatch (MM) sequence, 5′-CGC-CCC-GAC-CTC-TTC-CCT-T-3′ for MOR, was a scrambled version of the antisense sequence that has the same base pairs and GC ratio, with little or no homology to any mRNA sequences posted at GeneBank with four mismatched bases (denoted by bold letters). A nucleotide BLAST search was performed to confirm that the mRNA sequence targeted by the AS-ODN, or its MM-ODN control, were not homologous to any other sequences in the rat database. The oligodeoxynucleotides were synthesized by Invitrogen Life Technologies.
Before use, lyophilized ODNs were reconstituted in nuclease-free 0.9% NaCl and then administered intrathecally at a dose of 6 μg/μl in a volume of 20 μl (120 μg/20 μl). MM-ODNs or AS-ODNs were injected for 3 consecutive days, once a day and, on the fourth day, fentanyl (100 ng) was injected intrathecally (20 μl) or intradermally (5 μl). When fentanyl was injected intrathecally, PGE2 was injected 12 hours (intrathecal) or 24 hours (intradermal) after fentanyl. However, when fentanyl was injected intradermally, PGE2 was injected intradermally or intrathecally 3 h after fentanyl. The mechanical nociceptive threshold was evaluated 1 h after the injection of fentanyl and 30 min and 4 h after the injection of PGE2. As described previously (Alessandri-Haber et al., 2003), rats were anesthetized with isoflurane (2.5% in O2) and ODN was injected using a syringe (300 units/μl) with a 29-gauge needle inserted into the subarachnoid space between the L4 and L5 vertebrae. A total of 120 μg of ODN, in a volume of 20 μl, was then injected. When anesthesia was stopped, rats regained consciousness ∼2 min after the injection. Use of AS-ODN to attenuate the expression of proteins, which is essential for their role in nociceptor sensitization, is well supported by previous studies (Song et al., 2009; Su et al., 2011; Bogen et al., 2012; Quanhong et al., 2012; Sun et al., 2013; Araldi et al., 2015, 2016b; 2017a; Ferrari et al., 2016; Oliveira-Fusaro et al., 2017).
Intrathecal administration of saporins
IB4-saporin.
IB4-saporin, an IB4-positive nociceptor neurotoxin (Advanced Targeting Systems), was diluted in saline and a dose of 3.2 μg in a volume of 20 μl administered intrathecally 14 d before experiments. The dose and timing of IB4-saporin administration were chosen based on previous reports from our group and others (Vulchanova et al., 2001; Nishiguchi et al., 2004; Joseph et al., 2008; Joseph and Levine, 2010; Araldi et al., 2015, 2016b, 2017b).
[Sar9, Met(O2)11]-substance P-saporin (SSP-saporin).
SSP-saporin, an SP-positive nociceptor neurotoxin (Advanced Targeting Systems), was diluted in saline and a dose of 100 ng in a volume of 20 μl was administered intrathecally 14 d before priming experiments. The addition of [Sar9, Met(O2)11] to the substance P-conjugated to saporin makes the agent more stable and potent than when substance P alone is bound to saporin. The dose and pretreatment interval was based on the studies of Wiley et al. (2007) and Choi et al. (2012), who observed no loss of intrinsic lumbar dorsal horn neurons expressing the neurokinin 1 (NK1) receptor in deeper laminae and prominent loss of NK1 receptor in laminae I, and studies by others (Khasabov et al., 2002; Vierck et al., 2003; Wiley et al., 2007; Choi et al., 2012; Weisshaar and Winkelstein, 2014; Kras et al., 2015; Araldi et al., 2016a, 2017b, 2018).
To administer IB4-saporin, SSP-saporin, or their combination, rats were briefly anesthetized with 2.5% isoflurane (Phoenix Pharmaceuticals) in 97.5% O2 and then a 29-gauge hypodermic needle was inserted, on the midline, into the subarachnoid space, between the L4 and L5 vertebrae. The control treatment consisted of intrathecal injection of the same volume of vehicle (saline). Rats regained consciousness ∼2 min after stopping anesthesia. There was no effect of IB4-saporin, SSP-saporin, or their combination on the mechanical nociceptive threshold per se. The group that was treated intrathecally with the combination of saporins received IB4-saporin (3.2 μg/10 μl) in the morning and SSP-saporin (100 ng/10 μl) in the afternoon.
Protocol for induction of priming by fentanyl
Whereas a single injection of the selective MOR agonist DAMGO alone had no effect on nociceptive threshold and attenuates the mechanical hyperalgesia induced by PGE2 (Levine and Taiwo, 1989; Taiwo and Levine, 1990), when injected repeatedly, it produces changes in nociceptor function such as OIH, no longer producing an antihyperalgesic effect and by itself producing mechanical hyperalgesia (Aley et al., 1995; Aley and Levine, 1997; Araldi et al., 2015, 2017a, 2018). In contrast, a single intrathecal or intradermal injection of fentanyl (100 ng), another MOR agonist, induced acute hyperalgesia (see Fig. 1). Repeated injections of DAMGO are required to induce a latent state of hyperresponsiveness to the subsequent injection of proalgesic mediators, prototypically PGE2 (Joseph et al., 2010; Araldi et al., 2015, 2017a, 2018), referred to as type II priming (Araldi et al., 2015, 2017a, 2018). Priming (Aley et al., 2000; Reichling and Levine, 2009; Ferrari et al., 2014; Araldi et al., 2015, 2017a, 2018), a model of neuroplasticity, is expressed as prolongation of the mechanical hyperalgesia produced by PGE2, lasting at least 4 h, as opposed to the injection of PGE2 in naive paws, in which hyperalgesia fully dissipated by 2 h (Aley and Levine, 1999). In preliminary experiments, we observed that a single injection of intrathecal or intradermal fentanyl (100 ng) was able to induce hyperalgesia and priming at both the central and peripheral terminal of the nociceptor. To study the mechanisms involved, intrathecal or intradermal injections of fentanyl were performed. Changes in the mechanical nociceptive threshold, induced by intrathecal or intradermal fentanyl, were evaluated 1 h after its injection. To investigate the changes in nociceptor function produced by a previous injection of fentanyl (intrathecal or intradermal), measured as prolonged response to a hyperalgesic mediator at a point in time (>3 h) when the mechanical nociceptive threshold was not different from pre-fentanyl baseline levels, PGE2 was injected at the same site or at the opposite terminal and hyperalgesia was evaluated after 30 min and again at 4 h. The continued presence of hyperalgesia at the fourth hour is characteristic of priming (Aley et al., 2000; Ferrari et al., 2014; Araldi et al., 2015, 2017a, 2018). To elucidate the contribution of intracellular signaling pathways involved in hyperalgesia and priming induced by fentanyl and to investigate the mechanisms that play a role in the induction of the changes in nociceptor function, pharmacological agents were injected intrathecally or intradermally before the administration of fentanyl (prevention protocol). To investigate the second messengers involved in the expression of the neuroplasticity, inhibitors were administered intrathecally or intradermally before the injection of PGE2 in fentanyl-primed rats (reversal protocol).
Figure 1.
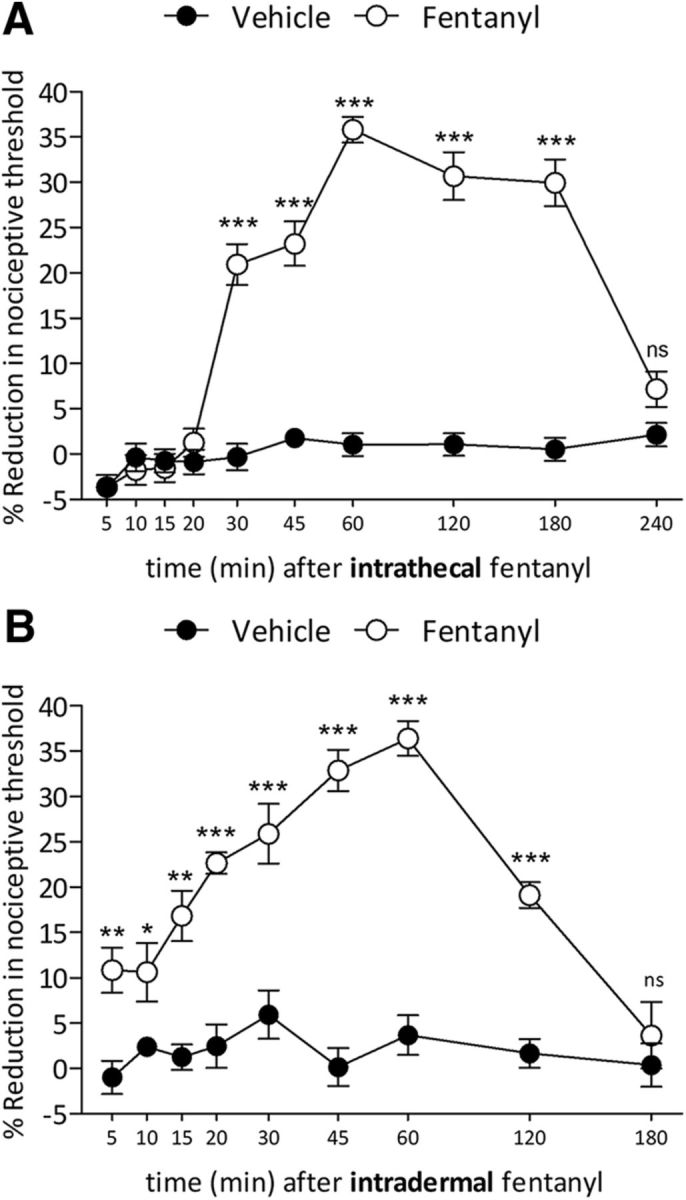
Intrathecal and intradermal fentanyl-induced hyperalgesia. A, Mechanical nociceptive threshold was evaluated 5, 10, 15, 20, 30, 45, 60, 120, 180, and 240 min after intrathecal injection of vehicle (saline, 20 μl; black circles) or fentanyl (100 ng/20 μl; white circles). Significant hyperalgesia was first observed by 30 min and persisted unattenuated to 180 min (F(1,90) = 756.59, ***p < 0.0001, when fentanyl-treated group was compared with saline, at 30, 45, 60, 120, and 180 min after the intrathecal; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). At 240 min, mechanical nociceptive threshold had returned to baseline. (n = 10 paws/10 rats per group). B, Five, 10, 15, 20, 30, 45, 60, 120, and 180 min after intradermal injection of vehicle (saline, 5 μl; black circles) or fentanyl (100 ng/5 μl; white circles), the mechanical nociceptive threshold was evaluated. Hyperalgesia was first observed 5 min after injection of fentanyl and persisted to 120 min (F(1,80) = 118.84, *p < 0.05, **p < 0.001 and ***p < 0.0001; when fentanyl-treated group is compared with saline; two-way repeated-measures ANOVA followed by Bonferroni post hoc test; n = 8 paws/8 rats per group).
Repeated subcutaneous administration of fentanyl
Priming was also induced by systemic (subcutaneous [s.c.]; performed over the rat's shoulders into the loose skin over the neck) administration of fentanyl. Rats received four injections of fentanyl (20 μg/kg per injection, s.c.) 15 min apart, resulting in a cumulative dose of 80 μg/kg/rat (Célèrier et al., 2000; Laulin et al., 2002). Mechanical nociceptive threshold was evaluated before the first injection of fentanyl and 48 h later. Fentanyl was dissolved in physiologic saline (0.9%) and administered subcutaneously (100 μl/100 g body weight). Using this protocol, we evaluated whether systemic fentanyl induces type I or II priming in the central and peripheral nociceptor terminal and if its induction is dependent on ER Ca2+ signaling.
DRG neuron culture
Primary cultures of rat dorsal root ganglia (DRG) sensory neurons were obtained from adult male Sprague Dawley rats (220–235 g) and prepared as described previously (Ferrari et al., 2016; Khomula et al., 2017). In brief, under isoflurane anesthesia, rats were decapitated, the dorsum of the vertebral column was opened, and L4 and L5 DRGs were removed rapidly, chilled in Hanks' balanced salt solution (HBSS) on ice, and desheathed. Ganglia were treated with 0.125% collagenase P (Worthington Biochemical) in HBSS for 90 min at 37°C and then treated with 0.25% trypsin (Worthington Biochemical) in calcium- and magnesium-free PBS (Invitrogen Life Technologies) for 10 min, followed by 3× washout and trituration in Neurobasal A medium (Invitrogen Life Technologies) to produce a single-cell suspension. The suspension was centrifuged at 1000 RPM for 3 min and resuspended in Neurobasal A medium supplemented with 50 ng/ml nerve growth factor, 100 U/ml penicillin/streptomycin, and B-27 (Invitrogen Life Technologies). Cells were then plated on coverslips and incubated at 37°C in 5% CO2 for at least 24 h before use in experiments.
Calcium imaging
Cultured rat DRG neurons were used for in vitro experiments between 24 and 96 h after dissociation and plating. At least three rats/culture preparation were used for each experimental series. Within the text, “n” refers to the number of neurons. Cells were identified as neurons by having double birefringent plasma membranes (Cohen et al., 1968; Landowne, 1993). Although small, medium, and large neurons were routinely observed in the same preparation, this study focused only on cells with a cell body diameter <30 μm (small DRG neurons, predominantly representing the C-type nociceptor subpopulation). After mounting a coverslip to a recording chamber, the culture medium was replaced with Tyrode's solution containing the following (in mm): 140 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES; adjusted to pH 7.4 with NaOH as previously described (Ferrari et al., 2016, 2017; Khomula et al., 2017). Tyrode's solution was used in the in vitro experiments as an external perfusion solution. To eliminate extracellular Ca2+ entry into cells, in some experiments, Tyrode's solution was replaced with nominally calcium-free solution containing the following (in mm): 140 NaCl, 4 KCl, 4 MgCl2, 2 EGTA, 10 glucose, and 10 HEPES; adjusted to pH 7.4 with NaOH (Khomula and Voitenko, 2006; Shutov et al., 2006). The volume of the recording chamber was 150 μl. The perfusion system was gravity driven at a flow rate of 1–2 ml/min. All experiments were performed at room temperature (20–23°C).
Our bright-field imaging system consisted of an inverted microscope (Eclipse TE-200; Nikon) with an epifluorescence attachment and a mercury lamp for excitation. Illumination was controlled by a Lambda 10-2 filter wheel controller and Lambda SC Smart Shutter controller (Sutter Instruments); an Andor Clara Interline CCD camera (Andor Technology) was used for high-resolution digital image acquisition. MetaFluor software (Molecular Devices) provided computer interface and controlled the whole system and was also used for image processing. A Plan Fluor objective (20 × UV, numerical aperture 0.50; Nikon) was used for both fluorescent and transmitted light imaging with phase contrast. Calcium imaging was performed using the fluorescent calcium indicator fura-2 acetoxymethyl ester (fura-2 AM) as described previously (Ferrari et al., 2016; Khomula et al., 2017). Briefly, neurons were loaded with 5 μm fura-2 AM by incubation for 20 min directly in the recording chamber. Then cells were perfused with Tyrode's solution for 10 min before the beginning of the recording to allow for complete deesterification of the fura-2 AM. Measurement of the intracellular concentration of free calcium ions ([Ca2+]i) was performed by ratiometric imaging. Fluorescence was excited at 340 and 380 nm for 2–10 ms each and the emitted light was long filtered at 520 nm using a standard Fura-2 filter set (Chroma Technology). Using MetaFluor software (Molecular Devices) corresponding pairs of digital images were acquired every 1–10 s, depending on the rate of the examined process, to minimize UV exposure and excitotoxicity; the fluorescence ratio (F340/F380) was calculated on a pixel-by-pixel basis with background correction and averaged for the region of interest defined for each neuron (Ferrari et al., 2016; Khomula et al., 2017). Fluorescence ratio was used to characterize [Ca2+]i without recalculation into concentration.
Data analysis
All data are presented as mean ± SEM of n independent observations. Statistical comparisons were made using GraphPad Prism 5.0 statistical software. A p-value <0.05 was considered statistically significant.
In the behavioral experiments, the dependent variable was change in mechanical paw-withdrawal threshold, expressed as percentage change from baseline. No significant difference in mechanical nociceptive thresholds was observed before the injection of central or peripheral fentanyl and immediately before injection of PGE2 [average mechanical nociceptive threshold before priming stimuli (fentanyl): 139.1 ± 1.8 g; average mechanical nociceptive threshold before PGE2 injection: 138.5 ± 1.6 g; n = 317 rats; paired Student's t test, t(316) = 0.7883, p = 0.4323]. In addition, 84 rats were used in Figures 1, 6, and 12. As specified in the figure legends, Student's t test or two-way repeated-measures ANOVA, followed by Bonferroni post hoc test, was performed to compare the magnitude of the hyperalgesia induced by fentanyl or PGE2 injection in the different groups, or to compare the effect produced by different treatments on the prolongation of the PGE2-induced hyperalgesia (evaluated 4 h after injection) with the control groups.
Figure 6.

Systemic fentanyl induces priming at central and peripheral terminals. A, Rats were treated with subcutaneous fentanyl (20 μg/kg, × 4, every 15 min). Forty-eight hours later, when the mechanical nociceptive threshold were not different from pre-fentanyl baseline (before fentanyl: 142.8 ± 1.9 g; 48 h after fentanyl: 143.3 ± 1.7 g), vehicle (black bars; 10 μl), cordycepin (gray bars; 4 μg/10 μl) or the combination (dotted bars) of SU 6656 (10 μg/5 μl) and U0126 (10 μg/5 μl) were injected intrathecally followed by PGE2 (400 ng/10 μl) injected at the same site. Mechanical nociceptive threshold was evaluated 30 min and 4 h after intrathecal PGE2. Two-way repeated-measures ANOVA, followed by Bonferroni post hoc test, showed that, whereas the hyperalgesia induced by PGE2 in the groups treated with vehicle or cordycepin was still present at the fourth hour, in the group pretreated with the combination of SU 6656 and U0126, it was completely blocked (F(2,30) = 304.62, ***p < 0.0001, when the hyperalgesia in the vehicle- and the combination of SU 6656 and U0126-treated groups is compared at the fourth hour after intradermal PGE2). B, Fifteen days after intrathecal treatment with vehicle, cordycepin, or the combination of SU 6656 and U0126, when the mechanical nociceptive threshold was not different from the pre-vehicle/inhibitors baseline (t(5) = 1.815; p = 0.1291, for the vehicle-treated group, t(5) = 0.8771; p = 0.4206, for the cordycepin-treated group, and t(5) = 0.8647; p = 0.4267, for the combination of SU 6656 and U0126-treated group, when the mechanical nociceptive threshold is compared before and after treatments; paired Student's t test), PGE2 (400 ng/20 μl) was again injected intrathecally. In the group previously treated with the combination of SU 6656 and U0126, PGE2-induced hyperalgesia was not present at the fourth hour (F(2,30) = 311.24, ***p < 0.0001, when the hyperalgesia in the vehicle-treated and the combination of SU 6656 and U0126-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). C, Thirty days after intrathecal treatment with vehicle, cordycepin, or the combination of SU 6656 and U0126, PGE2 (400 ng) was again injected intrathecally. In the group previously treated with the combination of inhibitors, the prolongation of PGE2-induced hyperalgesia was markedly inhibited at the fourth hour (F(2,30) = 194.44, ***p < 0.0001, when the hyperalgesia in the vehicle- and the combination of inhibitors-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas in the groups treated with vehicle or the combination of SU 6656 and U0126 the hyperalgesia was present at the fourth hour. These data support the suggestion that systemic fentanyl produces type II priming in the central terminal. D, A different group of rats were also treated with subcutaneous fentanyl (20 μg/kg, × 4, every 15 min). Forty-eight hours later, when mechanical nociceptive threshold was not different from pre-fentanyl baseline (before fentanyl: 137.9 ± 1.5 g; 48 h after fentanyl: 135.7 ± 2.1 g), vehicle (black bars; 5 μl), cordycepin (gray bars; 1 μg/5 μl), or the combination (dotted bars) of SU 6656 (1 μg/3 μl) and U0126 (1 μg/3 μl) were injected intradermally followed 10 min later by PGE2 (100 ng/5 μl) injected at the same site. Mechanical nociceptive threshold was evaluated 30 min and 4 h after intradermal PGE2. In the group treated with intradermal cordycepin, the prolongation of PGE2-induced hyperalgesia was markedly attenuated (F(2,30) = 459.43, ***p < 0.0001, when the hyperalgesia in the vehicle- and the cordycepin-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas in the groups treated with vehicle or the combination of SU 6656 and U0126, the hyperalgesia was present at the fourth hour. E, Fifteen days after intradermal treatment with vehicle, cordycepin, or the combination of SU 6656 and U0126, when the mechanical nociceptive threshold was not different from the pre-vehicle/inhibitors baseline (t(5) = 0.7133; p = 0.5076, for the vehicle-treated group, t(5) = 0.3561; p = 0.7363, for the cordycepin-treated group, and t(5) = 1.504; p = 0.1929, for the combination of SU 6656 and U0126-treated group, when the mechanical nociceptive threshold is compared before and after treatments; paired Student's t test), PGE2 (100 ng/5 μl) was again injected intradermally. The prolongation of PGE2-induced hyperalgesia was still inhibited in the group previously treated with cordycepin (F(2,30) = 389.49, ***p < 0.0001, when cordycepin-treated is compared with the vehicle-treated group at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), but not in the groups treated with vehicle or the combination of SU 6656 and U0126. F, Thirty days after intradermal vehicle, cordycepin, or the combination of SU 6656 and U0126, PGE2 (100 ng) was again injected intradermally. Prolongation of PGE2-induced hyperalgesia was markedly inhibited in the group previously treated with cordycepin (F(2,30) = 406.02, ***p < 0.0001, when the hyperalgesia in the cordycepin-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas in the groups treated with vehicle or the combination of SU 6656 and U0126, the hyperalgesia was present at the fourth hour. These data indicate that systemic fentanyl produces type I priming in the peripheral terminal of the nociceptor (n = 6 paws/6 rats per group).
Figure 12.

Systemic fentanyl induces ER Ca2+-dependent priming. A, Rats were treated intradermally with vehicle (5 μl, black bars) or dantrolene (1 μg/5 μl, gray bars) followed 10 min later by subcutaneous fentanyl (20 μg/kg, × 4, every 15 min). Forty-eight hours later, when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 0.7892; p = 0.5207, for the vehicle-treated group, and t(5) = 0.9004; p = 0.1744, for the dantrolene-treated group, when the mechanical nociceptive threshold is compared before and after subcutaneous fentanyl; paired Student's t test), PGE2 (100 ng/5 μl) was injected intradermally on the dorsum of the hindpaw and the mechanical nociceptive threshold evaluated 30 min and 4 h after injection. In the group treated with dantrolene, PGE2 did not induce prolonged hyperalgesia (F(2,20) = 280.79, ***p < 0.0001; when the hyperalgesia in the dantrolene-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), supporting the suggestion that subcutaneous fentanyl-induced priming in the peripheral terminal of the nociceptor is dependent on ER Ca2+ signaling. B, A different group of rats received intrathecal vehicle (20 μl, black bars) or dantrolene (10 μg/20 μl, gray bars) followed 10 min later by subcutaneous fentanyl (20 μg/kg, × 4, every 15 min). Forty-eight hours later, PGE2 (400 ng/20 μl) was injected intrathecally and mechanical nociceptive threshold evaluated 30 min and 4 h later. Treatment with dantrolene markedly blocked the prolongation of PGE2-induced hyperalgesia (F(2,20) = 164.24, ***p < 0.0001; when the hyperalgesia in the vehicle- and the dantrolene-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that priming induced in the central terminal, by subcutaneous fentanyl, is also dependent on ER Ca2+ release (n = 6 paws/6 rats per group).
Calcium imaging results are presented as amplitudes of the responses to drug applications. The amplitude of a response was measured as the difference between fluorescence ratios at the peak and the base of the response. Differences between means of several groups were analyzed using one-way ANOVA followed by Bonferroni post hoc analysis of differences between all pairs.
Results
Fentanyl-induced hyperalgesia
To verify that fentanyl induces hyperalgesia, the mechanical nociceptive threshold was evaluated 5, 10, 15, 20, 30, 60, 120, 180, and 240 min after intrathecal injection of vehicle (saline) or fentanyl (100 ng, diluted in 20 μl of saline) (Fig. 1A). Intrathecal fentanyl-induced hyperalgesia was detected at 30 min (20.9% of reduction in the mechanical nociceptive threshold), further increasing by 60 min (35.8% of reduction in the mechanical nociceptive threshold) (Fig. 1A). Peak hyperalgesia persisted to 180 min (29.9% of reduction; F(1,90) = 756.59, p < 0.0001, when fentanyl-treated group was compared with saline, at 30, 45, 60, 120, and 180 min after the intrathecal; two-way repeated-measures ANOVA), returning to the pre-fentanyl baseline at 240 min (Fig. 1A). Intradermal fentanyl (100 ng, diluted in 5 μl of saline) also induced mechanical hyperalgesia that was significant by 5 min (Fig. 1B; 10.8% of reduction in the mechanical nociceptive threshold; p < 0.05), peaking by 60 min (36.4% of reduction in the mechanical nociceptive threshold; F(1,80) = 118.84, p < 0.0001, when the fentanyl-treated group is compared with saline; two-way repeated-measures ANOVA) and returning to baseline by 180 min (Fig. 1B).
Fentanyl-induced priming
When PGE2 (400 ng, diluted in 20 μl of saline) was injected intrathecally 4 h after intrathecal fentanyl, the prolongation of PGE2 hyperalgesia was not observed (Fig. 2A). However, when injected 8 or 12 h after fentanyl, PGE2-induced hyperalgesia was prolonged (Fig. 2A; F(2,40) = 161.66, p < 0.0001, when saline- and fentanyl 8 h- or 12 h-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA). Therefore, intrathecal fentanyl requires between 4 and 8 h for priming to develop at the central terminal. When PGE2 (100 ng, diluted in 5 μl of saline) was injected intradermally 8, 12, or 24 h after rats received intrathecal fentanyl (Fig. 2B) at 8 and 12 h, PGE2-induced hyperalgesia was not prolonged. However, at 24 h, intradermal PGE2 produced prolonged hyperalgesia (Fig. 2B; F(2,40) = 299.73, p < 0.0001, when the saline- and fentanyl 24 h-treated groups are compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA), indicating that priming takes between 12 and 24 h, after intrathecal fentanyl, to develop in the peripheral terminal.
Figure 2.
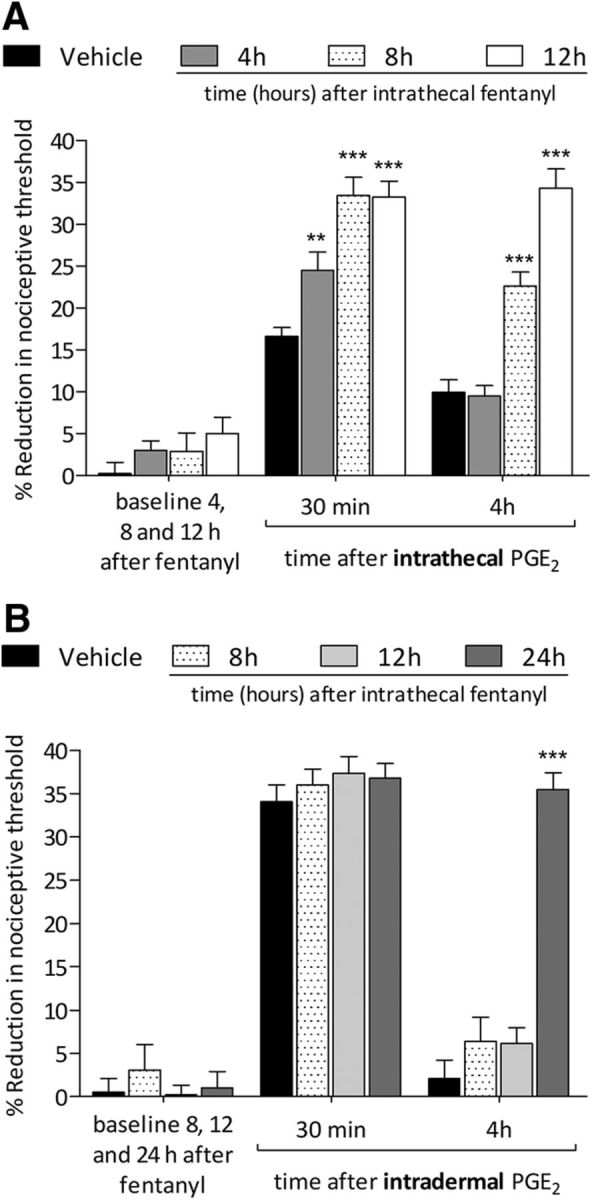
Latency to onset of priming induced by intrathecal fentanyl. A, Rats were treated intrathecally with vehicle (saline, 20 μl; black bars) or fentanyl (100 ng/20 μl). Four hours (black bars), 8 h (dotted bars), or 12 h (white bars) later, when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline [t(5) = 1.926; p = 0.1121, for the vehicle-treated group; t(5) = 1.529; p = 0.1867, for the fentanyl (4 h)-treated group; t(5) = 1.477; p = 0.1998, for the fentanyl (8 h)-treated group, and t(5) = 0.1287; p = 0.9026, for the fentanyl (12 h)-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test], PGE2 (400 ng/20 μl) was injected at the same intrathecal site and the mechanical nociceptive threshold evaluated 30 min and 4 h later. PGE2 induced hyperalgesia in all treated groups at 30 min after its injection; however, in the groups that were previously treated with intrathecal fentanyl, the hyperalgesia was more pronounced (**p < 0.01 for 4 h; ***p < 0.0001 for 8 and 12 h, when fentanyl-treated groups were compared with saline-treated group at 30 min after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). In the groups that received intrathecal fentanyl 8 and 12 h before the intrathecal injection of PGE2, the prolongation of PGE2-induced hyperalgesia was present at the fourth hour (F(2,40) = 161.66, ***p < 0.0001, when fentanyl-treated groups are compared with saline-treated group at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test) whereas the group treated with fentanyl 4 h before the injection of PGE2, did not demonstrate prolongation of PGE2 hyperalgesia. The saline-treated group received an intrathecal injection of PGE2 8 h later. Therefore, priming at the central nociceptor terminal, induced by intrathecal fentanyl, can be detected by 8 h. B, Vehicle (saline, 20 μl; black bars) or fentanyl (100 ng/20 μl) were injected intrathecally. Eight hours (dotted bars), 12 h (light gray bars), or 24 h (dark gray bars) later, when the mechanical nociceptive threshold was not different from pre-fentanyl baseline [t(5) = 0.5976; p = 0.5761, for the vehicle-treated group; t(5) = 0.3744; p = 0.7234, for the fentanyl (8 h)-treated group; t(5) = 0.1070; p = 0.9190, for the fentanyl (12 h)-treated group, and t(5) = 0.2666; p = 0.8004, for the fentanyl (24 h)-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test], PGE2 (100 ng/5 μl) was injected intradermally and the mechanical nociceptive threshold evaluated 30 min and 4 h after injection. In the groups treated with fentanyl 8 and 12 h prior, PGE2 was not able to induce prolonged hyperalgesia. However, 24 h after intrathecal fentanyl, an intradermal injection of PGE2 induced prolonged hyperalgesia (F(2,40) = 299.73, ***p < 0.0001, when the group treated 24 h prior was compared with groups treated 8 and 12 h prior; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Intradermal injection of PGE2 induced hyperalgesia in all groups when evaluated 30 min after injection. The intrathecal saline-treated group received an intradermal injection of PGE2 12 h after administration. Therefore, intrathecal fentanyl requires between 12 and 24 h to induce priming at the peripheral terminal of the nociceptor (n = 6 paws/6 rats per group).
Three hours after intradermal injection of fentanyl, when the mechanical nociceptive threshold returned to baseline, PGE2 was injected, inducing prolonged hyperalgesia at central terminal (Fig. 3A; F(2,20) = 166.61, p < 0.0001, when saline- and fentanyl-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA) and peripheral terminal (Fig. 3B; F(2,20) = 82.15, p < 0.0001, when the saline- and fentanyl-treated groups are compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA). In addition, PGE2 was injected intrathecally (Fig. 3A) or intradermally (Fig. 3B) in different groups of rats treated 1 h prior with intradermal fentanyl. PGE2 was able to induce prolonged hyperalgesia at both central (Fig. 3A, dotted box) and peripheral terminals (Fig. 3B, dotted box). These findings support a rapid onset for fentanyl-induced priming in both terminals. Because the hyperalgesia induced by intradermal fentanyl is gone by 3 h after administration, we chose this time point in the following experiments.
Figure 3.

Intradermal fentanyl: latency to priming. A, Three hours after the intradermal injection of vehicle (saline, 5 μl; black bars) or fentanyl (100 ng/5 μl; gray bars), when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 0.2666; p = 0.8004, for the vehicle-treated group and t(5) = 1.899; p = 0.1160, for the fentanyl-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test), PGE2 (400 ng/20 μl) was injected intrathecally and the mechanical nociceptive threshold evaluated 30 min and 4 h later. Mechanical hyperalgesia was present in both vehicle- and fentanyl-treated groups 30 min after intrathecal PGE2. However, at the fourth hour after PGE2, hyperalgesia was present only in the group that previously received fentanyl (F(2,20) = 166.61, ***p < 0.0001, when the fentanyl-treated group is compared with vehicle-treated group at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Intrathecal PGE2 (400 ng/20 μl) was also injected 1 h after intradermal fentanyl (100 ng/5 μl) in a different group of rats. The prolongation of PGE2-induced hyperalgesia was present at the fourth hour (dotted box), indicating that intradermal fentanyl requires ∼1 h to induce priming in the nociceptor central terminal. B, A different groups of rats received intradermal vehicle (5 μl) or fentanyl (100 ng/5 μl) and were treated 3 h later with intradermal PGE2 (100 ng/5 μl). PGE2 induced mechanical hyperalgesia 30 min after injection in both groups. However, in the fentanyl-treated group, PGE2 hyperalgesia was prolonged (F(2,20) = 82.15, ***p < 0.0001, when the fentanyl-treated group is compared with vehicle-treated group at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). In a different group of rats, intradermal PGE2 (100 ng/5 μl) was injected 1 h after intradermal fentanyl (100 ng/5 μl). At the fourth hour, prolonged hyperalgesia was present (dotted box), indicating that intradermal fentanyl requires ∼1 h to induce priming in the nociceptor peripheral terminal (n = 6 paws/6 rats per group).
Intrathecal fentanyl induces type II priming in the central and type I priming in the peripheral terminal
Maintenance of type I priming is dependent on protein translation in the nociceptor terminal, being reversed by local injection of cordycepin (Ferrari et al., 2013), whereas maintenance of type II priming is dependent on the simultaneous activation of Src and MAPK (Araldi et al., 2017a). Five days after an intrathecal injection of fentanyl, rats were treated at the same site with vehicle, cordycepin, or the combination of a Src and a MAPK inhibitor (SU 6656 and U0126, respectively), followed by an intrathecal injection of PGE2. In the group that received the combination of SU 6656 and U0126, PGE2 was not able to induce prolonged hyperalgesia (Fig. 4A; F(2,30) = 118.88, p < 0.0001, when the vehicle-treated and the combination of inhibitors-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA). When PGE2 was injected again 15 d (Fig. 4B) and 30 d (Fig. 4C) after the inhibitors, it still failed to produce prolonged hyperalgesia (15 d: F(2,30) = 118.57, p < 0.0001; 30 d: F(2,30) = 126.75, p < 0.0001, when the combination of inhibitors-treated group is compared with vehicle at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA). However, the prolongation of PGE2-induced hyperalgesia was present in the group treated with cordycepin, as well as in the vehicle-treated group. These findings indicate that intrathecal fentanyl induces type II priming in the central terminal of the nociceptor.
Figure 4.

Intrathecal fentanyl induces type II priming in the central and type I priming in the peripheral terminal of the nociceptor. A, Rats received an intrathecal injection of fentanyl (100 ng/20 μl). Five days later, vehicle (10 μl; black bars), cordycepin (4 μg/10 μl; gray bars), or the combination (dotted bars) of SU 6656 (10 μg/5 μl) and U0126 (10 μg/5 μl) were injected intrathecally followed by an injection of PGE2 (400 ng/10 μl) at the same site. Mechanical nociceptive threshold was evaluated 30 min and 4 h later. In the group treated with the combination of SU 6656 and U0126, PGE2 did not induce hyperalgesia at the fourth hour (F(2,30) = 118.88, ***p < 0.0001, when SU 6656 and U0126-treated group is compared with the vehicle- or cordycepin-treated groups at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test); the prolongation of PGE2-induced hyperalgesia was, however, present in the vehicle- and cordycepin-treated groups. B, Fifteen days after treatment with vehicle or the inhibitors, when the mechanical nociceptive threshold was not different from the pre-vehicle/inhibitor baseline (t(5) = 1.823; p = 0.1280, for the vehicle-treated group, t(5) = 1.277; p = 0.2576, for the cordycepin-treated group, and t(5) = 1.171; p = 0.2943, for the combination of SU 6656 and U0126-treated group, when the mechanical nociceptive threshold is compared before and after treatments; paired Student's t test), PGE2 (400 ng/20 μl) was again injected intrathecally and the mechanical nociceptive threshold evaluated 30 min and 4 h later. Again, PGE2 hyperalgesia was not present at the fourth hour in the group previously treated with the combination of SU 6656 and U0126 (F(2,30) = 118.57, ***p < 0.0001, when SU 6656 and U0126-treated group is compared with the vehicle- or cordycepin-treated groups at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). C, Similarly, 30 d after the injection of vehicle or the inhibitors, when PGE2 (400 ng/20 μl) was again injected intrathecally, the prolongation of PGE2-induced hyperalgesia was still absent in the SU 6656 and U0126-treated group (F(2,30) = 126.75, ***p < 0.0001, when SU 6656 and U0126-treated group is compared with the vehicle- or cordycepin-treated groups at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Therefore, intrathecal fentanyl induces type II priming in the nociceptor central terminal. (n = 6 paws/6 rats per group). D, Rats were treated with an intrathecal injection of fentanyl (100 ng/20 μl). Five days later, vehicle (5 μl, black bars), cordycepin (1 μg/5 μl; gray bars), or the combination (dotted bars) of SU 6656 (1 μg/2 μl) and U0126 (1 μg/2 μl) was injected intradermally followed 10 min later by injection of PGE2 (100 ng/5 μl) at the same site. In the group treated with cordycepin, the prolongation of PGE2-induced hyperalgesia was markedly inhibited (F(2,30) = 236.48, ***p < 0.0001, when cordycepin-treated group is compared with the vehicle-treated group at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas in the groups treated with vehicle or the combination of SU 6656 and U0126, PGE2 induced prolonged hyperalgesia. E, Fifteen days after treatment with vehicle or the inhibitors, when the mechanical nociceptive threshold was not different from the pre-vehicle/inhibitor baseline (t(5) = 1.388; p = 0.2239, for the vehicle-treated group, t(5) = 1.447; p = 0.2076, for the cordycepin-treated group, and t(5) = 0.2894; p = 0.7839, for the combination of SU 6656 and U0126-treated group, when the mechanical nociceptive threshold is compared before and after treatments; paired Student's t test), PGE2 (100 ng/5 μl) was again injected intradermally. The prolongation of PGE2-induced hyperalgesia was still inhibited in the group previously treated with cordycepin (F(2,30) = 248.53, ***p < 0.0001, when cordycepin-treated is compared with the vehicle-treated group at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), but not in the groups treated with vehicle or the combination of SU 6656 and U0126. F, PGE2 was again injected intradermally 30 d after treatment with vehicle or the inhibitors. In the group previously treated with cordycepin, the prolongation of PGE2-induced hyperalgesia was still significantly attenuated (F(2,30) = 224.73, ***p < 0.0001, when cordycepin-treated is compared with the vehicle- treated group at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas in the groups treated with vehicle or the combination of SU 6656 and U0126, the prolongation of PGE2-induced hyperalgesia was present. Therefore, intrathecal fentanyl produces type I priming in the peripheral terminal of the nociceptor (n = 6 paws/6 rats per group).
To determine whether intrathecal fentanyl induces type I or II priming in the peripheral terminal of the nociceptor, rats were treated intradermally with vehicle, cordycepin, or the combination of SU 6656 and U0126, followed by PGE2 at the same site, 5 d after intrathecal fentanyl (Fig. 4, bottom). In contrast to intrathecal PGE2, prolongation of intradermal PGE2 hyperalgesia was not present in the group treated with cordycepin (Fig. 4D; F(2,30) = 236.48, p < 0.0001, when the vehicle- and the cordycepin-treated groups are compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA), nor was it present when PGE2 was injected 15 d (Fig. 4E; F(2,30) = 248.53, p < 0.0001) and 30 d (Fig. 4F; F(2,30) = 224.73, p < 0.0001) later. However, intradermal PGE2 was able to induce prolonged hyperalgesia in the groups treated intradermally with vehicle and the combination of SU 6656 and U0126 (Fig. 4D–F). These data suggest that intrathecal fentanyl induces type I priming in the peripheral terminal of the nociceptor.
Priming induced by intradermal fentanyl
We also determined whether intradermal fentanyl induces type I and/or type II priming in the central and peripheral nociceptor terminal. Fentanyl was injected intradermally and, 5 d later, vehicle, cordycepin, or the combination of SU 6656 and U0126 were injected intrathecally, followed by PGE2 at the same site. In the group that received the combination of SU 6656 and U0126, the hyperalgesia induced by intrathecal PGE2 was not prolonged (Fig. 5A; F(2,30) = 156.03, p < 0.0001, when the combination of inhibitors-treated group is compared with vehicle at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA). In addition, when PGE2 was injected again, 15 d (Fig. 5B) or 30 d (Fig. 5C) after the treatment with inhibitors, prolongation of PGE2-induced hyperalgesia was still not present (15 d: F(2,30) = 257.50, p < 0.0001; 30 d: F(2,30) = 200.87, p < 0.0001, when the combination of inhibitors-treated group is compared with vehicle at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA). In contrast, in the vehicle- and cordycepin-treated groups, the intrathecal injection of PGE2 induced prolonged hyperalgesia (Fig. 5A–C). Therefore, the administration of fentanyl in the peripheral terminal of the nociceptor induces type II priming in the central terminal.
Figure 5.

Intradermal fentanyl induces type II priming at the central and type I priming at the peripheral terminal. A, Rats received an intradermal injection of fentanyl (100 ng/5 μl). Five days later, vehicle (10 μl, black bars), cordycepin (4 μg/10 μl; gray bars), or the combination (dotted bars) of SU 6656 (10 μg/5 μl) and U0126 (10 μg/5 μl) was injected intrathecally followed by an intrathecal injection of PGE2 (400 ng/10 μl). Thirty minutes after injection, PGE2 induced mechanical hyperalgesia in all groups. However, in the group treated with the combination of SU 6656 and U0126, PGE2-induced hyperalgesia at the fourth hour was inhibited (F(2,30) = 156.03, ***p < 0.0001, when SU 6655 and U0126 group is compared with the vehicle-treated group at the fourth hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas in the vehicle- and cordycepin-treated groups, PGE2 induced prolonged hyperalgesia. B, Fifteen days after treatment with vehicle, cordycepin, or the combination of SU 6656 and U0126, when the mechanical nociceptive threshold was not different from the pre-vehicle/inhibitors baseline (t(5) = 0.4416; p = 0.6772, for the vehicle-treated group, t(5) = 0.2162; p = 0.8374, for the cordycepin-treated group, and t(5) = 0.8402; p = 0.4391, for the combination of SU 6656 and U0126-treated group, when the mechanical nociceptive threshold is compared before and after treatments; paired Student's t test), PGE2 (400 ng/20 μl) was again injected intrathecally. Two-way repeated-measures ANOVA, followed by Bonferroni post hoc test, showed a significant inhibition of the prolonged hyperalgesia induced by PGE2 at the fourth hour, in the group that had been pretreated with the combination of SU 6656 and U0126, compared with the vehicle-treated group (F(2,30) = 257.50, ***p < 0.0001, when the hyperalgesia in the vehicle-treated and the combination of SU 6656 and U0126-treated groups is compared at the fourth hour), whereas PGE2 induced prolonged hyperalgesia in the cordycepin-treated group. C, When PGE2 was injected again, 30 d after treatment with vehicle or the inhibitors, in the group previously treated with the combination of SU 6656 and U0126, PGE2-induced hyperalgesia was not present at the fourth hour (F(2,30) = 200.87, ***p < 0.0001, when the hyperalgesia in the vehicle- and the combination of SU 6656 and U0126-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), demonstrating that intradermal fentanyl produces type II priming in the central terminal (n = 6 paws/6 rats per group). D, Rats received an intradermal injection of fentanyl (100 ng/5 μl). Five days later, vehicle (5 μl, black bars), cordycepin (1 μg/5 μl; gray bars) or the combination (dotted bars) of SU 6656 (1 μg/2 μl) and U0126 (1 μg/2 μl) was injected intradermally followed 10 min later by an injection of PGE2 (100 ng/5 μl) at the same site on the dorsum of the hindpaw. Mechanical nociceptive threshold was evaluated 30 min and 4 h after intradermal PGE2. Mechanical hyperalgesia was observed in all groups 30 min after the injection of PGE2. However, treatment with cordycepin prevented the prolongation of PGE2-induced hyperalgesia (F(2,30) = 207.62, ***p < 0.0001, when the hyperalgesia in the vehicle- and the cordycepin-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas treatment with the combination of SU 6656 and U0126 only partially attenuated hyperalgesia at the fourth hour after PGE2 (**p < 0.001, when the hyperalgesia in the vehicle- and the combination of SU 6656 and U0126-treated groups is compared at the fourth hour; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). E, Fifteen days after treatment with vehicle, cordycepin, or the combination of SU 6656 and U0126, when the mechanical nociceptive threshold was not different from the prevehicle/inhibitors baseline (t(5) = 1.585; p = 0.1737, for the vehicle-treated group, t(5) = 0.8682; p = 0.4250, for the cordycepin-treated group, and t(5) = 1.097; p = 0.3227, for the combination of SU 6656 and U0126-treated group, when the mechanical nociceptive threshold is compared before and after treatments; paired Student's t test), PGE2 (100 ng/5 μl) was again injected intradermally. Two-way repeated-measures ANOVA, followed by Bonferroni post hoc test, showed that, whereas the hyperalgesia induced by PGE2 in the vehicle and combination of SU 6656 and U0126 groups was still present at the fourth hour, in the group pretreated with cordycepin, it was significantly inhibited (F(2,30) = 245.80, ***p < 0.0001, when the hyperalgesia in the vehicle- and the cordycepin-treated groups is compared at the fourth hour after intradermal PGE2). F, Thirty days after treatment with vehicle, cordycepin, or the combination of SU 6656 and U0126, PGE2 (100 ng) was again injected intradermally. In the group previously treated with cordycepin, the prolongation of PGE2-induced hyperalgesia was markedly attenuated at the fourth hour (F(2,30) = 239.59, ***p < 0.0001, when the hyperalgesia in the vehicle- and the cordycepin-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas in the groups treated with vehicle or the combination of SU 6656 and U0126, the hyperalgesia was present at the fourth hour. These findings support the suggestion that intradermal fentanyl induces type I priming in the peripheral terminal (n = 6 paws/6 rats per group).
To evaluate priming at the peripheral terminal, vehicle, cordycepin, or the combination of SU 6656 and U0126 were injected intradermally 5 d after an intradermal injection of fentanyl, followed by intradermal PGE2. Prolongation of PGE2-induced hyperalgesia was not observed in the group treated with cordycepin (Fig. 5D; F(2,30) = 207.62, p < 0.0001, when the vehicle- and the cordycepin-treated groups are compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA). When PGE2 was again injected intradermally, 15 d (Fig. 5E) or 30 d (Fig. 5F) after the intradermal treatment with the inhibitors, the prolongation of PGE2 hyperalgesia was still not present in the cordycepin-treated group (F(2,30) = 245.80, p < 0.0001 for 15 d; F(2,30) = 239.59, p < 0.0001 for 30 d, when the cordycepin-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA). However, treatment with vehicle or the combination of SU 6656 and U0126 did not affect the prolongation of PGE2-induced hyperalgesia (Fig. 5D–F). Together, these results suggest that the administration of fentanyl in the peripheral terminal of the nociceptor-induced type I priming at its injection site.
Systemic fentanyl induces priming in the central and peripheral nociceptor terminal
Intrathecal and intradermal fentanyl induce type II priming in the central and type I in the peripheral terminal of the nociceptor. To determine the role of the nociceptor in the effects of systemic fentanyl, we determined whether systemic (subcutaneous) fentanyl is able to induce priming in the nociceptor, central, and peripheral terminals and if it is type I or type II. Rats were treated with fentanyl (20 μg/kg; 4 times, 15 min intervals). To evaluate priming induced by systemic fentanyl, vehicle, cordycepin, or the combination of SU 6656 and U0126 were injected intrathecally, at the central terminal, 48 h after systemic fentanyl, followed by intrathecal PGE2. In the group treated with vehicle, PGE2 induced prolonged hyperalgesia (Fig. 6A; reduction in the mechanical nociceptive threshold was 34.2%, when the vehicle-treated group is compared before and 4 h after intrathecal PGE2), indicating that systemic fentanyl induces prolongation of PGE2 hyperalgesia in the central terminal of the nociceptor. Treatment with the combination of SU 6656 and U0126 almost completely blocked the prolongation of intrathecal PGE2-induced hyperalgesia (Fig. 6A; F(2,30) = 304.62, p < 0.0001, when the combination of the inhibitors- and the vehicle-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA). When PGE2 was again injected intrathecally 15 d (Fig. 6B) or 30 d (Fig. 6C) after intrathecal treatment with the inhibitors, the prolongation of PGE2 hyperalgesia was still not present in the group treated with the combination of SU 6656 and U0126 inhibitors (15 d: F(2,30) = 311.24, p < 0.0001; 30 d: F(2,30) = 194.44, p < 0.0001, when the combination of the inhibitors is compared with vehicle at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA). However, treatment with vehicle or cordycepin did not affect the prolongation of PGE2-induced hyperalgesia (Fig. 6A–C). Together, these results suggest that systemic fentanyl induces type II priming at the central nociceptor terminal.
Another group of rats, also treated 48 h prior with systemic fentanyl, received intradermal vehicle, cordycepin, or the combination of SU 6656 and U0126. Ten minutes later, PGE2 was injected intradermally and the mechanical nociceptive threshold was evaluated 30 min and 4 h later. In the group treated with vehicle, intradermal PGE2 induced prolonged hyperalgesia (Fig. 6D; reduction in the mechanical nociceptive threshold was 35.3%, when the vehicle-treated group are compared before and 4 h after intradermal PGE2), indicating that systemic fentanyl induced priming in the peripheral terminal of the nociceptor. In the group treated with cordycepin, the hyperalgesia induced by intradermal PGE2 was not prolonged (Fig. 6D; F(2,30) = 459.43, p < 0.0001, when the vehicle- and the cordycepin-treated groups are compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA). In addition, when PGE2 was injected again, 15 d (Fig. 6E) or 30 d (Fig. 6F) after the treatment with inhibitors, prolongation of PGE2-induced hyperalgesia was still not present in the cordycepin-treated group (15 d: F(2,30) = 389.49, p < 0.0001; 30 d: F(2,30) = 406.02, p < 0.0001, when the cordycepin-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA). In contrast, in the vehicle-treated and the combination of SU 6656 and U0126 inhibitors-treated groups, intradermal injection of PGE2 induced prolonged hyperalgesia (Fig. 6D–F). Therefore, the systemic administration of fentanyl induces type I priming in the peripheral terminal of the nociceptor.
Priming is MOR dependent
To determine whether priming induced by fentanyl (intrathecal or intradermal) is mediated by its action at the MOR on nociceptors, we evaluated whether MOR AS would attenuate the induction of fentanyl-induced priming. MM-ODN or AS-ODN against MOR mRNA was administered intrathecally daily for 3 consecutive days. On the fourth day, fentanyl was injected intrathecally followed 12 h (Fig. 7A) or 24 h (Fig. 7B) later by an injection of PGE2 (intrathecal or intradermal, respectively). Treatment with MOR AS-ODN completely prevented the hyperalgesia induced by intrathecal fentanyl (Fig. 7A; F(1,30) = 51.44, p < 0.0001, when the MM-ODN- and the AS-ODN-treated groups are compared 1 h after intrathecal fentanyl; two-way repeated-measures ANOVA) and also the prolongation of PGE2-induced hyperalgesia when it was injected at the central terminal (Fig. 7A; F(3,30) = 137.36, p < 0.0001, when the AS-ODN-treated group is compared with MM-ODN at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA) or peripheral terminal (Fig. 7B; F(3,30) = 151.52, p < 0.0001, when the MM-ODN- and the AS-ODN-treated groups are compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA)of the nociceptor. Therefore, the hyperalgesia and priming that developed in the central and peripheral terminal of the nociceptor, by intrathecal fentanyl, is MOR dependent.
Figure 7.

Priming induced by intrathecal fentanyl is MOR dependent. A, Rats were treated intrathecally with MM-ODN (120 μg/20 μl/d; black bars) or AS-ODN (120 μg/20 μl/d; gray bars) against MOR mRNA, for 3 consecutive days. On the fourth day, ∼ 17 h after the last ODN injection, fentanyl (100 ng/20 μl) was injected intrathecally and the mechanical nociceptive threshold evaluated 1 h after injection. In the MOR AS-ODN-treated group, intrathecal fentanyl did not induce mechanical hyperalgesia at 1 h after its injection (F(1,30) = 51.44, ***p < 0.0001; when the hyperalgesia in the MM-ODN and the AS-ODN treated groups is compared at 1 h after intrathecal fentanyl; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Twelve hours after intrathecal fentanyl, when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 1.165; p = 0.2617, for the MM-ODN-treated group, and t(5) = 0.8123; p = 0.4536, for the AS-ODN-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test), PGE2 (400 ng/20 μl) was injected intrathecally and the mechanical nociceptive threshold evaluated 30 min and 4 h after injection. In the AS-ODN-treated group, intrathecal PGE2 was not able to induce hyperalgesia at the fourth hour (F(3,30) = 137.36, ***p < 0.0001, when the hyperalgesia in the MM-ODN- and the AS-ODN-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that both hyperalgesia and priming induced by intrathecal fentanyl are MOR dependent. B, A different group of rats that were also treated with MM-ODN or AS-ODN against MOR mRNA received intrathecal fentanyl (100 ng/20 μl) and, 24 h later, PGE2 (100 ng/5 μl) was injected intradermally on the dorsum of the hindpaw. The mechanical nociceptive threshold was evaluated 30 min and 4 h after the injection of PGE2. In the AS-ODN-treated group, intradermal PGE2 did not produce prolonged hyperalgesia (F(2,20) = 151.52, ***p < 0.0001, when the hyperalgesia in the MM-ODN- and the AS-ODN-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), suggesting that the priming induced by intrathecal fentanyl in the peripheral terminal of the nociceptor is also MOR dependent (n = 6 paws/6 rats per group).
Fentanyl was also injected intradermally in rats treated intrathecally with MOR MM-ODN or AS-ODN, daily for 3 d. Three hours after an intradermal injection of fentanyl, PGE2 was injected intrathecally at the same site (Fig. 8A) or at the opposite site, intrathecally (Fig. 8B). In the AS-ODN-treated groups, PGE2 was not able to induce prolonged hyperalgesia either in the peripheral terminal (Fig. 8A; F(3,30) = 139.31, p < 0.0001, when the AS-ODN-treated group is compared with MM-ODN at the fourth hour after intradermal PGE2) or central terminal (Fig. 8B; F(3,30) = 212.87, p < 0.0001, when the MM-ODN- and the AS-ODN-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA).
Figure 8.

Intradermal fentanyl induces MOR-dependent priming. A, Rats received intrathecal injections of MM-ODN (120 μg/20 μl/d; dark gray bars) or AS-ODN (120 μg/20 μl/d; light gray bars) against MOR mRNA, daily for 3 consecutive days. On the fourth day, ∼17 h after the last ODN injection, fentanyl (100 ng/5 μl) was injected intradermally and the mechanical nociceptive evaluated 1 h after its injection. Intradermal fentanyl was not able to induce hyperalgesia in the group treated with AS-ODN for MOR (F(1,30) = 74.95, ***p < 0.0001, when the hyperalgesia in the MM-ODN- and the AS-ODN-treated groups is compared at 1 h after intradermal fentanyl; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Three hours after intradermal fentanyl, when the mechanical nociceptive threshold was not different from pre-fentanyl baseline (t(5) = 0.1824; p = 0.8625, for the MM-ODN-treated group, and t(5) = 0.1966; p = 0.8519, for the AS-ODN-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test), PGE2 (100 ng/5 μl) was injected intradermally and mechanical nociceptive threshold evaluated 30 min and 4 h after injection. Intradermal PGE2 did not induce prolonged hyperalgesia in the AS-ODN-treated group (F(3,30) = 139.31, ***p < 0.0001, when the hyperalgesia in the MM-ODN- and the AS-ODN-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that intradermal fentanyl-induced hyperalgesia and priming in the peripheral terminal of the nociceptor is MOR dependent. B, A different group of rats, also treated with MM-ODN or AS-ODN against MOR mRNA for 3 consecutive days, on the fourth day received intradermal fentanyl (100 ng/5 μl). Three hours after fentanyl, PGE2 (400 ng/20 μl) was injected intrathecally and mechanical nociceptive threshold evaluated 30 min and 4 h later. The prolongation of PGE2-induced hyperalgesia was not present in the MOR AS-ODN-treated group (F(2,20) = 212.87, ***p < 0.0001, when the hyperalgesia in the MM-ODN- and the AS-ODN-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that priming in the central terminal of the nociceptor induced by intradermal fentanyl is MOR dependent (n = 6 paws/6 rats per group).
Involvement of peptidergic and nonpeptidergic nociceptors in priming induced by intrathecal fentanyl
Type I priming, induced by inflammatory mediators, occurs in IB4-positive nonpeptidergic nociceptors (Joseph and Levine, 2010), whereas type II priming induced by repeated exposure to DAMGO occurs in IB4-negative peptidergic neurons (Araldi et al., 2018). We evaluated whether priming in the central and peripheral nociceptor terminals, induced by intrathecal fentanyl, is dependent on peptidergic and/or nonpeptidergic nociceptors. IB4-saporin, which destroys IB4-positive nonpeptidergic neurons; SSP-saporin, which destroys IB4-negative peptidergic neurons; or their combination (IB4-saporin and SSP-saporin) was injected intrathecally and, 14 d later, fentanyl was injected intrathecally. Intrathecal fentanyl was not able to produce hyperalgesia, evaluated 1 h after its injection, in the group pretreated with the combination of IB4-saporin and SSP-saporin (Fig. 9A; F(3,30) = 236.16, p < 0.0001, when the combination of saporins-treated group is compared with vehicle 1 h after intrathecal fentanyl; two-way repeated-measures ANOVA), indicating that the acute hyperalgesia induced by intrathecal fentanyl is dependent on both nonpeptidergic and peptidergic nociceptors. Twenty-four hours later, PGE2 was injected intradermally and the mechanical nociceptive threshold evaluated 30 min and 4 h after its injection. Prolongation of PGE2-induced hyperalgesia was weakly attenuated in the group treated with IB4-saporin (F(3,60) = 20.42, p < 0.01, when the vehicle- and the IB4-saporin-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA), but was present in other treated groups (Fig. 9A; F(3,30) = 2.41, p = 0.1238, when all groups are compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA). Therefore, priming induced in the peripheral terminal of the nociceptor by intrathecal fentanyl is independent of nonpeptidergic and peptidergic neurons.
Figure 9.

Role of IB4-positive and negative nociceptors in hyperalgesia and priming induced by fentanyl. A, Rats received an intrathecal injection of vehicle (20 μl; black bars), IB4-saporin (3.2 μg/20 μl; dark gray bars), SSP-saporin (100 ng/20 μl; light gray bars), or the combination (dotted bars) of IB4-saporin (3.2 μg/10 μl) and SSP-saporin (100 ng/10 μl). Fourteen days later, when the mechanical nociceptive threshold was not different from pretreatment baseline (t(5) = 0.6428; p = 0.5487, for the vehicle-treated group, t(5) = 0.3085; p = 0.7701, for the IB4-saporin-treated group, t(5) = 0.7445; p = 0.9435, for the SSP-saporin-treated group, and t(5) = 0.9114; p = 0.4039, for the combination of saporins-treated group, when the mechanical nociceptive threshold is compared before and 14 d after treatments; paired Student's t test), fentanyl (100 ng/20 μl) was injected intrathecally and the mechanical nociceptive threshold evaluated 1 h after injection. Intrathecal fentanyl-induced hyperalgesia was blocked in the group previously treated with the combination of IB4-saporin and SSP-saporin (F(3,30) = 236.16, ***p < 0.0001; when the hyperalgesia in the vehicle-treated and the combination of saporins-treated groups is compared at the first hour after intrathecal fentanyl; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), whereas fentanyl was able to induce hyperalgesia in the vehicle-treated, IB4-saporin-treated, and SSP-saporin-treated groups. Twenty-four hours later, when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 0.7101; p = 0.5094, for the vehicle-treated group, t(5) = 0.7776; p = 0.4720, for the IB4-saporin-treated group, t(5) = 0.4406; p = 0.6779, for the SSP-saporin-treated group, and t(5) = 0.5368; p = 0.6145, for the combination of saporins-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test), PGE2 (100 ng/5 μl) was injected intradermally and the mechanical nociceptive receptor was evaluated 30 min and 4 h after injection. The prolongation of PGE2 hyperalgesia was present in the groups treated with vehicle, SSP-saporin, or the combination of saporins, but was weakly attenuated in the IB4-saporin-treated group (F(3,60) = 20.42, **p < 0.01; when the hyperalgesia in the vehicle- and IB4-saporin-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). These findings demonstrate that intrathecal fentanyl acts in different classes of neurons to induce hyperalgesia and priming in the peripheral terminal of the nociceptor. B, Groups of rats were treated with intrathecal vehicle (20 μl; black bars), IB4-saporin (3.2 μg/20 μl; dark gray bars) or SSP-saporin (100 ng/20 μl; light gray bars) followed 14 d later by an intrathecal injection of fentanyl (100 ng/20 μl). Twelve hours after fentanyl, PGE2 (400 ng/20 μl) was injected intrathecally and mechanical nociceptive threshold evaluated 30 min and 4 h later. The prolongation of PGE2-induced hyperalgesia was markedly attenuated in both groups treated IB4-saporin or SSP-saporin (F(2,30) = 237.34, ***p < 0.0001; when the hyperalgesia in the vehicle-, IB4-saporin- and SSP-saporin-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that both nonpeptidergic and peptidergic neurons are involved in priming induced at the central terminal by intrathecal fentanyl (n = 6 paws/6 rats per group). C, Rats received intrathecal vehicle (20 μl; black bars), IB4-saporin (3.2 μg/20 μl; dark gray bars), SSP-saporin (100 ng/20 μl; light gray bars), or the combination (dotted bars) of IB4-saporin (3.2 μg/10 μl) and SSP-saporin (100 ng/10 μl). Fourteen days later, fentanyl (100 ng/5 μl) was injected intradermally on the dorsum of the hindpaw and the mechanical nociceptive threshold evaluated 1 h later. Intradermal fentanyl did not induce hyperalgesia in the groups treated with SSP-saporin or the combination of saporins (F(3,12) = 109.04, ***p < 0.0001; when the hyperalgesia in the vehicle-, SSP-saporin-, and the combination of saporins-treated groups is compared at the first hour after intradermal fentanyl; two-way repeated-measures ANOVA followed by Bonferroni post hoc test); however, it did in the groups treated with vehicle or IB4-saporin, indicating that intradermal fentanyl-induced acute hyperalgesia is dependent on IB4-negative peptidergic nociceptors. Three hours after fentanyl, when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 0.6625; p = 0.5369, for the vehicle-treated group, t(5) = 1.062; p = 0.1942, for the IB4-saporin-treated group, t(5) = 0.3432; p = 0.7454, for the SSP-saporin-treated group, and t(5) = 0.3750; p = 0.7231, for the combination of saporins-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test), PGE2 (100 ng/5 μl) was injected intradermally and the mechanical nociceptive evaluated 30 min and 4 h later. In the group treated with the combination of saporins, PGE2 hyperalgesia was significantly inhibited at the fourth hour (F(3,24) = 245.45, ***p < 0.0001; when the hyperalgesia in the vehicle-treated and in the combination of saporins-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that intradermal fentanyl requires both nonpeptidergic and peptidergic nociceptors to develop priming in the peripheral terminal. D, Fourteen days after the intrathecal treatment with vehicle, IB4-saporin, SSP-saporin, or the combination of IB4-saporin and SSP-saporin, fentanyl (100 ng/5 μl) was injected intradermally on the dorsum of the hindpaw followed, 3 h later, by an intrathecal injection of PGE2 (400 ng/20 μl). The mechanical nociceptive threshold was evaluated 30 min and 4 h after the injection of PGE2. The prolongation of PGE2-induced hyperalgesia was prevented in groups treated with SSP-saporin and the combination of saporins (F(2,24) = 263.10, ***p < 0.0001; when the hyperalgesia in the vehicle-treated, SSP-saporin-treated, and the combination of saporins-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), but not in the groups treated with vehicle or IB4-saporin, supporting the suggestion that intradermal fentanyl-induced priming in the central terminal of the nociceptor is dependent on IB4-negative peptidergic nociceptor (n = 6 paws/6 rats per group).
Another group of rats were treated with IB4-saporin or SSP-saporin and 14 d later received intrathecal fentanyl. Twelve hours later, when PGE2 was injected intrathecally, the prolongation of PGE2-induced hyperalgesia was markedly attenuated in the groups treated with SSP-saporin and IB4-saporin (Fig. 9B; F(2,30) = 237.34, p < 0.0001, when the vehicle- and saporins-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA), indicating that priming induced in the central terminal, by intrathecal fentanyl, is dependent on both peptidergic and nonpeptidergic neurons.
Involvement of peptidergic and nonpeptidergic nociceptors in priming induced by intradermal fentanyl
In rats previously treated with SSP-saporin or the combination of IB4-saporin and SSP-saporin, intradermal fentanyl-induced hyperalgesia was not present (Fig. 9C; F(3,12) = 109.04, p < 0.0001, when the vehicle-treated, SSP-saporin-treated, and the combination of saporins-treated groups are compared 1 h after intradermal fentanyl; two-way repeated-measures ANOVA), indicating that hyperalgesia induced by intradermal fentanyl is dependent on IB4-negative peptidergic nociceptors. Three hours later, PGE2 was injected intradermally and the mechanical nociceptive threshold was measured 30 min and 4 h later. Although intradermal PGE2 induced prolonged hyperalgesia in the vehicle-treated, IB4-saporin-treated, and SSP-saporin-treated groups, in the group treated with the combination of saporins, intradermal PGE2 was not able to produce prolonged hyperalgesia (Fig. 9C; F(3,24) = 245.45, p < 0.0001, when the combination of saporins-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA). Therefore, intradermal fentanyl requires both peptidergic and nonpeptidergic neurons to develop priming in the peripheral terminal.
Fourteen days after vehicle, IB4-saporin, SSP-saporin, or their combination, a different group of rats received intradermal fentanyl followed, 3 h later, by intrathecal PGE2 (Fig. 9D). The prolongation of PGE2-induced hyperalgesia was blocked in the SSP-saporin and the combination of saporins-treated groups (Fig. 9D; F(2,24) = 263.10, p < 0.0001, when the vehicle-treated, SSP-saporin-treated, and the combination of saporins-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA), whereas in the vehicle-treated and IB4-saporin-treated groups, the prolongation of PGE2-induced hyperalgesia was present at the fourth hour (Fig. 9D). Thus, priming induced in the central terminal of the nociceptor by intradermal fentanyl is dependent on IB4-negative peptidergic neurons.
Calcium signaling in fentanyl-induced priming
The latency to onset of priming induced by intrathecal fentanyl detected at the central terminal of the nociceptor was between 4 and 8 h (Fig. 2A) and, at the peripheral terminal, it was between 12 and 24 h (Fig. 2B). The latency to onset of priming induced by intradermal fentanyl detected at both the central and peripheral terminals was even shorter, being present by 1 h (Fig. 3, dotted boxes). These latencies at the terminal remote to fentanyl administration were too short to be mediated by axonal transport. Because MOR activation can cause ER stress (Aoe, 2015), we considered that the signal between central and peripheral terminals, produced in response to fentanyl, might be mediated by Ca2+ signaling in the ER, initiated at its site of administration. To test this hypothesis, we pretreated rats with an intrathecal injection of dantrolene (a ryanodine receptor blocker that prevents calcium release from the ER) followed by fentanyl at the same site. Intrathecal fentanyl was not able to induce mechanical hyperalgesia in the group treated with dantrolene (Fig. 10A; F(1,30) = 73.20, p < 0.0001, when the dantrolene-treated group is compared with vehicle 1 h after intrathecal fentanyl; two-way repeated-measures ANOVA). Twelve hours (Fig. 10A) or 24 h (Fig. 10B) after the intrathecal injection of fentanyl, PGE2 was injected intrathecally (Fig. 10A) or intradermally (Fig. 10B) and the mechanical nociceptive threshold evaluated 30 min and 4 h after injection. In the intrathecal dantrolene-treated group, PGE2 at either the central terminal (Fig. 10A; F(3,30) = 142.71, p < 0.0001, when the vehicle- and the dantrolene-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA) or peripheral terminal (Fig. 10B; F(3,30) = 269.48, p < 0.0001, when the dantrolene-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA) was not able to induce prolonged hyperalgesia. These findings support the suggestion that the rapid onset of priming, in the central and peripheral terminal of the nociceptor, induced by intrathecal fentanyl is dependent on an ER Ca2+ signal.
Figure 10.

Ca2+ dependence of priming induced by intrathecal fentanyl. A, Rats were treated intrathecally with vehicle (10 μl; black bars) or dantrolene (10 μg/10 μl; gray bars) followed by fentanyl (100 ng/10 μl) at the same site. Mechanical nociceptive threshold was evaluated 1 h after intrathecal fentanyl. Intrathecal fentanyl did not induce mechanical hyperalgesia in the dantrolene-treated group (F(1,30) = 73.20, ***p < 0.0001 when the hyperalgesia in the vehicle- and the dantrolene-treated groups is compared at the first hour after intrathecal fentanyl; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Twelve hours after intrathecal fentanyl, when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 1.153; p = 0.3012, for the vehicle-treated group, and t(5) = 0.2162; p = 0.8374, for the dantrolene-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test), PGE2 (400 ng/20 μl) was injected intrathecally and the mechanical nociceptive threshold evaluated 30 min and 4 h after injection. In the dantrolene-treated group, the prolongation of PGE2-induced hyperalgesia was prevented (F(3,30) = 142.71, ***p < 0.0001; when the hyperalgesia in the dantrolene-treated group is compared with vehicle at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the priming induced by intrathecal fentanyl is dependent on ER Ca2+ signaling. B, A second group of rats received vehicle (10 μl; black bars) or dantrolene (10 μg/10 μl; gray bars) intrathecally followed by fentanyl (100 ng/10 μl) at the same site. Twenty-four hours later, when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 0.8098; p = 0.4549, for the vehicle-treated group, and t(5) = 2.101; p = 0.0997, for the dantrolene-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test), PGE2 (100 ng/5 μl) was injected intradermally on the dorsum of the hindpaw and the mechanical nociceptive threshold evaluated 30 min and 4 h after injection. Intradermal PGE2 did not induce prolonged hyperalgesia in the group previously treated with intrathecal dantrolene (F(2,20) = 269.48, ***p < 0.0001; when the hyperalgesia in the vehicle- and the dantrolene-treated groups is compared at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the priming developed in the peripheral terminal of the nociceptor by an intrathecal injection of fentanyl is also dependent on a Ca2+ signal arising from the central terminal (n = 6 paws/6 rats per group).
We also evaluated whether the priming induced by an intradermal injection of fentanyl is related to Ca2+ signaling. Rats were treated intradermally with dantrolene followed by an injection of fentanyl at the same site. Intradermal fentanyl failed to induce mechanical hyperalgesia in dantrolene-treated rats (Fig. 11A; F(1,30) = 81.47, p < 0.0001, when the vehicle- and the dantrolene-treated groups are compared 1 h after intradermal fentanyl; two-way repeated-measures ANOVA). Three hours after intradermal fentanyl, PGE2 was injected at the same site (Fig. 11A) or intrathecally (Fig. 11B) and the mechanical nociceptive threshold was evaluated 30 min and 4 h after injection. In the dantrolene-treated group, the prolongation of PGE2-induced hyperalgesia was not present in either the peripheral terminal (Fig. 11A; F(3,30) = 143.08, p < 0.0001, when the dantrolene-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA) or central terminal (Fig. 11B; F(2,20) = 114.78, p < 0.0001, when the vehicle- and the dantrolene-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA). Therefore, the rapid latency to onset of priming in the central and peripheral terminal of the nociceptor induced by an intradermal injection of fentanyl is also dependent on Ca2+ signaling in the ER in the nociceptor terminal.
Figure 11.

Ca2+ dependence of priming induced by intradermal fentanyl. A, Rats were treated intradermally with vehicle (5 μl; black bars) or dantrolene (1 μg/5 μl; gray bars) followed 10 min later by an injection of fentanyl (100 ng/5 μl) at the same site. Mechanical nociceptive threshold was evaluated 1 h after intradermal fentanyl. Fentanyl, was not able to produce mechanical hyperalgesia in the dantrolene-treated group (F(1,30) = 81.47, ***p < 0.0001; when the hyperalgesia in the vehicle- and the dantrolene-treated groups is compared at the first hour after intradermal fentanyl; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Three hours later, when mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 0.7869; p = 0.4670, for the vehicle-treated group, and t(5) = 1.835; p = 0.1259, for the dantrolene-treated group, when the mechanical nociceptive threshold is compared before and after fentanyl; paired Student's t test), PGE2 (100 ng/5 μl) was injected intradermally and the mechanical nociceptive threshold evaluated 30 min and 4 h after injection. PGE2-induced hyperalgesia at the fourth hour, was not present in the dantrolene-treated group (F(3,30) = 143.08, ***p < 0.0001; when the hyperalgesia in the dantrolene-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that both hyperalgesia and priming induced by an intradermal injection of fentanyl is dependent on ER Ca2+ signaling. B, A second group of rats were treated intradermally with vehicle (5 μl; black bars) or dantrolene (1 μg/5 μl; gray bars) followed 10 min later by an injection of fentanyl (100 ng/5 μl) at the same site. Three hours later, PGE2 (400 ng/20 μl) was injected intrathecally and the mechanical nociceptive threshold evaluated 30 min and 4 h after injection. The prolongation of PGE2-induced hyperalgesia at the fourth hour was prevented in the dantrolene-treated group (F(2,20) = 114.78, ***p < 0.0001; when the hyperalgesia in the vehicle- and the dantrolene-treated groups is compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), supporting the suggestion that intradermal fentanyl-induced priming, in the nociceptor central terminal, is dependent on a Ca2+ signal arising from the peripheral terminal (n = 6 paws/6 rats per group).
We also tested whether priming induced by systemic (subcutaneous) fentanyl is mediated through Ca2+ signaling in the ER. Rats were treated with intradermal (Fig. 12A) or intrathecal (Fig. 12B) dantrolene, followed by systemic fentanyl (20 μg/kg, 4 times, 15 min intervals). Forty-eight hours later, PGE2 was injected intradermally (in the intradermal dantrolene-treated group; Fig. 12A) or intrathecally (in the intrathecal dantrolene-treated group; Fig. 12B) and the mechanical nociceptive threshold was evaluated 30 min and 4 h after injection. In the dantrolene-treated groups, PGE2 was not able to induce prolonged hyperalgesia in either the peripheral terminal (Fig. 12A; F(2,20) = 280.79, p < 0.0001, when the dantrolene-treated group is compared with vehicle at the fourth hour after intradermal PGE2; two-way repeated-measures ANOVA) or central terminal (Fig. 12B; F(2,20) = 164.24, p < 0.0001, when the vehicle- and the dantrolene-treated groups are compared at the fourth hour after intrathecal PGE2; two-way repeated-measures ANOVA) of the nociceptor. These findings are in agreement with our previous results (Figs. 10, 11) in which we found that type II priming in the central and type I priming in the peripheral terminal of the nociceptor are dependent on an ER Ca2+ signal. Of note, the priming induced by fentanyl contrasts with that induced by another MOR selective agonist, DAMGO, which induces type II priming requiring repeated administration (hourly × 4) at the peripheral terminal with a longer latency to onset that was not prevented by intradermal dantrolene (data not shown).
The findings of our behavioral experiments reveal a crucial role of ryanodine receptors in the rapid onset hyperalgesia and priming induced by fentanyl. This provides strong but still indirect support of our main hypothesis about Ca2+ release from the ER in the nociceptor, as a signaling mechanism for initiation of priming. To provide further insight into the underlying mechanism, we performed in vitro calcium imaging experiments on cultured DRGs. These experiments only considered responses from small DRG neurons (with soma diameter <30 μm) because they predominantly represent the C-type nociceptive subpopulation of primary sensory neurons (Harper and Lawson, 1985; Gold et al., 1996). Using fluorescent calcium imaging with fura-2, we first tested whether direct extracellular application of fentanyl to nociceptors is able to stimulate Ca2+ release from the ER as revealed by increase in cytosolic free calcium ion concentration ([Ca2+]i; i.e., calcium transient). To address this question, we used fentanyl 500 pm, which is close to therapeutic concentrations of 1–10 nm (Verplaetse and Henion, 2016; Baselt, 2017). To isolate a Ca2+ signal produced by calcium release from the ER and to exclude possible Ca2+ entrance from extracellular space, experiments were conducted in nominally calcium-free solution achieved via chelating of Ca2+ by EGTA. Calcium-free solution was applied just 5 min before any drug applications to avoid the negative effect of prolonged exposure to a calcium-free medium, including depletion of calcium stores of the ER. Administration of fentanyl produced a Ca2+ transient (Fig. 13), confirming that fentanyl can increase [Ca2+]i in small DRG neurons (putative nociceptors).
Figure 13.

Fentanyl induces Ca2+ release from the endoplasmic reticulum. Bars left to right show pooled amplitudes of response to fentanyl (500 pm; black bar) alone (n = 35), fentanyl after dantrolene (1 μm; dotted bar) (n = 29), naloxone (10 μm; gray bar) alone (n = 23), and fentanyl after naloxone (n = 33; white bar). All drugs were administrated in calcium-free solution to isolate calcium signal from ER. Amplitude of the response to fentanyl after dantrolene, naloxone and fentanyl after naloxone were statistically not significantly different between each other, whereas they were all significantly smaller then response to fentanyl alone (F(3,116) = 28, p < 0.0001, one-way ANOVA followed by Bonferroni post hoc test for all pairs: when compared with fentanyl alone, t(62) = 6.1, οοοadjusted p < 0.0001 for fentanyl after dantrolene; t(56) = 7.3, ***adjusted p < 0.0001 for naloxone; t(66) = 7.9, ###adjusted p < 0.0001 for fentanyl after naloxone; t(60) = 1.5, adjusted p = 0.76 when fentanyl after dantrolene is compared with fentanyl after naloxone; t(50) = 1.6, adjusted p = 0.68 when fentanyl after dantrolene is compared with naloxone; t(54) = 0.20, adjusted p = 0.999 when fentanyl after naloxone is compared with naloxone).
To determine whether the increase in [Ca2+]i induced by fentanyl involves ryanodine receptors, their antagonist, dantrolene (1 μm), was administrated 10 min before fentanyl and fentanyl was applied while dantrolene continued to be in the experimental chamber (Fig. 13). This resulted in attenuation of response to fentanyl by 64% (t(62) = 6.1, adjusted p < 0.0001, Bonferroni post hoc test). Amplitude of responses to naloxone, fentanyl after naloxone, and fentanyl after dantrolene were not significantly different from each other, whereas they were significantly smaller than the response to fentanyl alone (Fig. 13; F(3,116) = 28, p < 0.0001, one-way ANOVA followed by Bonferroni post hoc test for all pairs). Together, our in vivo behavioral and in vitro calcium imaging findings support our hypothesis that Ca2+ release originated from activation of ryanodine receptors in the ER, in the fentanyl-stimulated terminal, might be the signal produced in response to activation of opioid receptors that leads to priming.
To confirm that calcium release is opioid receptor dependent, naloxone (10 μm) was administrated 10 min before fentanyl and then fentanyl applied with naloxone still present in the experimental chamber (Fig. 13). Changes in [Ca2+]i after application of naloxone alone were approximately 17% of response to fentanyl (t(56) = 7.3, adjusted p < 0.0001, Bonferroni post hoc test), whereas response to fentanyl was blocked by 80% (t(66) = 7.9, adjusted p < 0.0001, Bonferroni post hoc test) and was not significantly different from the small response to naloxone alone (t(54) = 0.20, adjusted p = 0.999, Bonferroni post hoc test). These small changes in [Ca2+]i (corresponding changes in ratio <1% of baseline) after application of naloxone alone and fentanyl after naloxone were close to noise resolution of recordings and approximately the same as “responses” to vehicle application (calcium-free solution itself in a separate experiment, data not shown). Therefore, we cannot fully exclude that these were responses to a switch of solutions. In agreement, in vivo experiments demonstrated that pretreatment with intradermal naloxone followed 30 min later by naloxone combined with fentanyl injected at the same site, completely prevented the hyperalgesia induced by fentanyl (by 96.7%; Fig. 14; t(10) = 17.74, p < 0.0001, when vehicle- and naloxone-treated groups are compared 1 h after intradermal fentanyl; unpaired Student's t test). Three hours after fentanyl, when PGE2 (100 ng) was injected intradermally, prolonged hyperalgesia was inhibited in the group treated with naloxone (by 90.5%; Fig. 14; t(10) = 11.57, p < 0.0001, when vehicle and naloxone-treated groups are compared at the fourth hour after intradermal PGE2; unpaired Student's t test), confirming that hyperalgesia and priming induced by fentanyl in the peripheral terminal of the nociceptor is opioid receptor dependent.
Figure 14.

Hyperalgesia and priming induced by fentanyl in the peripheral terminal is opioid receptor dependent. Rats received intradermal vehicle (saline, 5 μl, black bars) or naloxone (1 μg/5 μl; dotted bars). Thirty minutes later, vehicle (2 μl) combined with fentanyl (100 ng/2 μl) or naloxone (1 μg/2 μl) combined with fentanyl (100 ng/2 μl) were injected intradermally in the vehicle- or naloxone-treated groups, respectively. In the group treated with naloxone, fentanyl did not induce hyperalgesia as evaluated 1 h after its injection (t(10) = 17.74, ***p < 0.0001, when the vehicle and naloxone groups are compared 1 h after intradermal fentanyl, unpaired Student's t test). Three hours later, when the mechanical nociceptive threshold was not different from the pre-fentanyl baseline (t(5) = 1.712; p = 0.6022, for the vehicle-treated group and t(5) = 1.547; p = 0.8210, for the naloxone-treated group, when the mechanical nociceptive threshold is compared before and after intradermal fentanyl; paired Student's t test), PGE2 (100 ng/5 μl) was injected intradermally and the mechanical nociceptive threshold evaluated 30 min and 4 h later. PGE2 induced hyperalgesia in all treated groups 30 min after injection; however, the fourth hour of PGE2-induced hyperalgesia was completely blocked in the group treated with naloxone (t(10) = 11.57, ***p < 0.0001, when the vehicle and naloxone groups are compared at the fourth hour after intradermal PGE2, unpaired Student's t test). These data support hyperalgesia and priming induced by fentanyl in the peripheral terminal of the nociceptor is opioid receptor mediated (n = 6 paws/6 rats per group).
The following results are summarized in Table 1: (1) the signaling pathway involved in OIH (evaluated 1 h after fentanyl) and priming (evaluated by a central or peripheral injection of PGE2) induced by central fentanyl; (2) the signaling pathway involved in OIH and priming (evaluated by central or peripheral PGE2) induced by peripheral fentanyl; and (3) the signaling pathway involved in systemic fentanyl-induced priming in the central and peripheral terminal of the nociceptor.
Table 1.
Summary of the findings involved in the signaling pathway of systemic, central, or peripheral fentanyl-induced hyperalgesia and priming
| Opioid hyperalgesia | Central PGE2 | Peripheral PGE2 | |
|---|---|---|---|
| Central fentanyl | |||
| Protein translation | NE | − | + |
| Src and MAPK | NE | + | − |
| MOR | + | + | + |
| Peptidergic | − | + | − |
| Nonpeptidergic | − | + | − |
| Pep and nonpeptidergic | + | − | − |
| Calcium | + | + | + |
| Peripheral fentanyl | |||
| Protein translation | NE | − | + |
| Src and MAPK | NE | + | − |
| MOR | + | + | + |
| Peptidergic | + | + | − |
| Nonpeptidergic | − | − | − |
| Pep and nonpeptidergic | + | + | + |
| Calcium | + | + | + |
| Systemic fentanyl | |||
| Protein translation | NE | − | + |
| Src and MAPK | NE | + | − |
| Calcium | NE | + | + |
+, Involved in the priming mechanism; −, not involved in the priming mechanism; NE, not evaluated.
Discussion
Hyperalgesia has been reported to occur after a single administration of opioids, most frequently for members of the fentanyl class (Collett, 1998; Chia et al., 1999; Buntin-Mushock et al., 2005). Recently, we developed a model of OIH and priming (type II) in which repeated exposure to DAMGO, a MOR selective agonist, induced hyperalgesia and the subsequent prolongation of PGE2-induced hyperalgesia (Araldi et al., 2015, 2017a, 2018). Fentanyl administered at either central or peripheral terminals also induced mechanical hyperalgesia, peaking by 60 min after administration and returning to baseline by 240 and 180 min, for the central and peripheral site of injection, respectively.
Intrathecal, intradermal, and systemic fentanyl induced type I priming in peripheral and type II priming in central nociceptor terminal. Although repeated exposure to the MOR agonist DAMGO induced type II priming in the peripheral terminal (Araldi et al., 2015, 2017a, 2018), fentanyl, also a MOR agonist, induced type I priming in the peripheral terminal. The reason for this difference in type of priming induced by two different MOR agonists remains to be explained. However, because MOR antisense prevents both DAMGO- and fentanyl-induced priming, biased agonism at MOR may contribute (Al-Hasani and Bruchas, 2011). Although type I priming induced by agonists at receptors in the peripheral terminal that signal by activation of PKCε (Joseph and Levine, 2010) requires 72 h to develop, in the peripheral terminal (Aley et al., 2000; Bogen et al., 2012), intradermal fentanyl requires ∼1 h to induce type I priming at this site, as was observed in type I priming induced by intradermal ryanodine (data not shown). Because ryanodine induces type I priming by acting downstream of protein kinase ε (PKCε) (Ferrari et al., 2016; Khomula et al., 2017), one explanation for this rapid onset for type I priming is that it is mediated by ER Ca2+ signaling (Khomula et al., 2017).
Priming induced by intradermal fentanyl was also already detectable at 1 h in the central terminal. This latency to develop priming is too short to be mediated by axonal transport, as we have observed previously for PKCε-induced type I priming (Aley et al., 2000; Ferrari et al., 2014). Because MOR agonists induce the release of Ca2+ from ER through ryanodine receptors (Velazquez-Marrero et al., 2014), we considered that the message between central and peripheral terminals was mediated by Ca2+ signaling. In support of this hypothesis, pretreatment with dantrolene (a ryanodine receptor blocker that prevents calcium release from the ER) at the central or peripheral nociceptor terminal prevented the induction of priming induced by intrathecal, intradermal, and systemic fentanyl. We demonstrated recently that the activation of ryanodine receptors, which releases Ca2+ from the ER and induces Ca2+ signaling, induces type I priming in the peripheral terminal of the nociceptor (Khomula et al., 2017). Although chronic MOR activation may cause ER stress and alter signal transduction (Aoe, 2015), pretreatment with dantrolene did not prevent the induction of type II priming induced by repeated exposure to DAMGO (data not shown), supporting the suggestion that different MOR agonists activate different downstream second messengers (Bohn et al., 2000; Al-Hasani and Bruchas, 2011; Groer et al., 2011). Opioids produce changes in MOR signaling that cannot be fully explained by the classic G-protein-coupled receptor (GPCR) signaling pathway and may reflect switching in intracellular second messengers. Studies have identified how ligand-directed responses are crucial in understanding the complexity of opioid-induced changes in the signaling pathways downstream of MOR. GPCR phosphorylation-induced switching has been studied intensively in β-arrestin–mediated signaling. The work of Bohn et al. (2000) showed how β-arrestin 1 and β-arrestin 2 differentially mediate the regulation of MOR. β-arrestins are required for internalization, but only β-arrestin 2 can rescue morphine-induced MOR internalization, whereas both β-arrestin 1 and 2 can rescue DAMGO-induced MOR internalization (Groer et al., 2011). Together, these findings suggest that MOR regulation is dependent on the agonist used, which may be critical in understanding the mechanism underlying opioid-induced hyperalgesia and priming.
Our in vivo behavioral findings were complemented by in vitro experiments demonstrating that fentanyl induced Ca2+ transients that were markedly inhibited by pretreatment with dantrolene and naloxone. Recently, it was demonstrated that activation of the ryanodine receptor, which releases Ca2+ from the ER and induces Ca2+ signaling (Futagi and Kitano, 2015; Evans et al., 2016), is associated with the induction of type I priming (Khomula et al., 2017). We propose that fentanyl releases Ca2+ from the ER to affect multiple and diverse signaling events, including local protein synthesis and degradation (Saito and Cavalli, 2016). Importantly, these local signals can also have long-distance effects mediated by propagating Ca2+ waves (Saito and Cavalli, 2016).
Using MOR antisense, we demonstrated recently that type II priming, induced by DAMGO, is MOR dependent in the peripheral nociceptor terminal (Araldi et al., 2018). Similarly, our current findings show that hyperalgesia and prolongation of PGE2 hyperalgesia induced by intrathecal and intradermal fentanyl were also MOR dependent at both the central and peripheral nociceptor terminal. These results are in agreement with a recent report demonstrating that MORs, expressed by primary afferent nociceptors, initiate tolerance and opioid-induced hyperalgesia by chronic systemic opioid administration (Corder et al., 2017).
MOR is an inhibitory GPCR through which endogenous opioids regulate a variety of physiological functions, including analgesia (Kieffer and Gaveriaux-Ruff, 2002). MOR mediates the pain-relieving effects of some of the most clinically efficacious analgesics (Scherrer et al., 2009). Immunohistochemical studies demonstrated that MOR is expressed in a subpopulation of primary afferent “pain” fibers (nociceptors), the majority small-diameter, peptidergic afferents (Arvidsson et al., 1995; Scherrer et al., 2009; Usoskin et al., 2015). Recently, we demonstrated that the induction of OIH and type II priming by repeated exposure to the MOR agonist DAMGO was prevented by pretreatment with SSP-saporin, which eliminates IB4-negative peptidergic neurons (Araldi et al., 2018). Here, we evaluated whether intrathecal or intradermal fentanyl-induced hyperalgesia and priming are dependent on nonpeptidergic and/or peptidergic nociceptors (Table 1). Priming produced in the peripheral terminal of the nociceptor by intrathecal fentanyl was not prevented by pretreatment with a combination of both saporins, which is compatible with the presence of a novel class of nociceptors, in which fentanyl can also induce priming. A recent study found that ∼ 20% of TRPV1+ neurons are negative for both peptidergic and nonpeptidergic markers, indicating that the peptidergic and nonpeptidergic classes of C-fiber do not account for the entirety of unmyelinated primary afferents (Cavanaugh et al., 2011). We proposed that type I priming induced by direct activation of PKCε is dependent on nonpeptidergic neurons (Joseph and Levine, 2010) and type II induced by DAMGO is dependent on peptidergic nociceptors (Araldi et al., 2018); however, this cannot explain fentanyl-induced priming. Together, these data indicate complex effects of intrathecal and intradermal fentanyl in the peripheral and central terminals of nonpeptidergic and/or peptidergic nociceptors. In agreement with other studies (Abrahamsen et al., 2008; Cavanaugh et al., 2009), our results indicate that, at the level of the peripheral nociceptor terminal, there is behaviorally relevant specificity and selective regulation of the hyperalgesia and priming, induced by fentanyl, can be produced by subsets of nociceptors.
We conclude that fentanyl, acting at MOR on nociceptors, rapidly induces acute hyperalgesia and priming at both central and peripheral terminals, which are mediated by ER Ca2+ signaling. Priming in the central terminal is type II, whereas that in the peripheral terminal is type I. The current findings and proposed mechanisms involved in fentanyl-induced OIH and priming are summarized in Table 1. Given the complexity of the signaling pathway, downstream of MOR, activated after a single administration of fentanyl, our data support a way forward to develop therapeutics for selectively disrupting individual MOR signaling pathways to maintain adequate long-lasting pain control. Similarly, understanding the underlying mechanisms for opioid-induced hyperalgesia and priming may provide useful information for the design of drugs with improved therapeutic profiles to treat OIH and chronic pain.
Footnotes
This work was supported by the National Institutes of Health (Grant NS084545). We thank Marie Kern for technical assistance.
The authors declare no competing financial interests.
References
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN (2008) The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321:702–705. 10.1126/science.1156916 [DOI] [PubMed] [Google Scholar]
- Adasme T, Paula-Lima A, Hidalgo C (2015) Inhibitory ryanodine prevents ryanodine receptor-mediated Ca(2)(+) release without affecting endoplasmic reticulum Ca(2)(+) content in primary hippocampal neurons. Biochem Biophys Res Commun 458:57–62. 10.1016/j.bbrc.2015.01.065 [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD (2003) Hypotonicity induces TRPV4-mediated nociception in rat. Neuron 39:497–511. 10.1016/S0896-6273(03)00462-8 [DOI] [PubMed] [Google Scholar]
- Aley KO, Levine JD (1997) Dissociation of tolerance and dependence for opioid peripheral antinociception in rats. J Neurosci 17:3907–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Levine JD (1999) Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 19:2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Green PG, Levine JD (1995) Opioid and adenosine peripheral antinociception are subject to tolerance and withdrawal. J Neurosci 15:8031–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD (2000) Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci 20:4680–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, Bruchas MR (2011) Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115:1363–1381. 10.1097/ALN.0b013e318238bba6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoe T. (2015) Development of opioid tolerance and endoplasmic reticulum stress. J Pain Relief 4:174. [Google Scholar]
- Araldi D, Ferrari LF, Levine JD (2015) Repeated mu-opioid exposure induces a novel form of the hyperalgesic priming model for transition to chronic pain. J Neurosci 35:12502–12517. 10.1523/JNEUROSCI.1673-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araldi D, Ferrari LF, Levine JD (2016a) Gi-protein-coupled 5-HT1B/D receptor agonist sumatriptan induces type I hyperalgesic priming. Pain 157:1773–1782. 10.1097/j.pain.0000000000000581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araldi D, Ferrari LF, Levine JD (2016b) Adenosine-A1 receptor agonist induced hyperalgesic priming type II. Pain 157:698–709. 10.1097/j.pain.0000000000000421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araldi D, Ferrari LF, Levine JD (2017a) Hyperalgesic priming (type II) induced by repeated opioid exposure: maintenance mechanisms. Pain 158:1204–1216. 10.1097/j.pain.0000000000000898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araldi D, Ferrari LF, Green P, Levine JD (2017b) Marked sexual dimorphism in 5-HT1 receptors mediating pronociceptive effects of sumatriptan. Neuroscience 344:394–405. 10.1016/j.neuroscience.2016.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araldi D, Ferrari LF, Levine JD (2018) Role of GPCR (Mu-Opioid)-RTK (Epidermal Growth Factor) crosstalk in opioid-induced hyperalgesic priming (type II). Pain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronoff GM. (2016) What do we know about the pathophysiology of chronic pain? Implications for treatment considerations. Med Clin North Am 100:31–42. 10.1016/j.mcna.2015.08.004 [DOI] [PubMed] [Google Scholar]
- Arvidsson U, Riedl M, Chakrabarti S, Lee JH, Nakano AH, Dado RJ, Loh HH, Law PY, Wessendorf MW, Elde R (1995) Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J Neurosci 15:3328–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselt R. (2017) Disposition of toxic drugs and chemicals in man, Ed 11 Foster City, CA: Biomedical Publications. [Google Scholar]
- Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD (2012) Generation of a pain memory in the primary afferent nociceptor triggered by PKCepsilon activation of CPEB. J Neurosci 32:2018–2026. 10.1523/JNEUROSCI.5138-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG (2000) Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408:720–723. 10.1038/35047086 [DOI] [PubMed] [Google Scholar]
- Borle AB, Snowdowne KW (1982) Measurement of intracellular free calcium in monkey kidney cells with aequorin. Science 217:252–254. 10.1126/science.6806904 [DOI] [PubMed] [Google Scholar]
- Buntin-Mushock C, Phillip L, Moriyama K, Palmer PP (2005) Age-dependent opioid escalation in chronic pain patients. Anesth Analg 100:1740–1745. 10.1213/01.ANE.0000152191.29311.9B [DOI] [PubMed] [Google Scholar]
- Burch RM, Axelrod J (1987) Dissociation of bradykinin-induced prostaglandin formation from phosphatidylinositol turnover in swiss 3T3 fibroblasts: evidence for G protein regulation of phospholipase A2. Proc Natl Acad Sci U S A 84:6374–6378. 10.1073/pnas.84.18.6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ (2009) Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A 106:9075–9080. 10.1073/pnas.0901507106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh DJ, Chesler AT, Bráz JM, Shah NM, Julius D, Basbaum AI (2011) Restriction of transient receptor potential vanilloid-1 to the peptidergic subset of primary afferent neurons follows its developmental downregulation in nonpeptidergic neurons. J Neurosci 31:10119–10127. 10.1523/JNEUROSCI.1299-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Célèrier E, Rivat C, Jun Y, Laulin JP, Larcher A, Reynier P, Simonnet G (2000) Long-lasting hyperalgesia induced by fentanyl in rats: preventive effect of ketamine. Anesthesiology 92:465–472. 10.1097/00000542-200002000-00029 [DOI] [PubMed] [Google Scholar]
- Célèrier E, González JR, Maldonado R, Cabañero D, Puig MM (2006) Opioid-induced hyperalgesia in a murine model of postoperative pain: role of nitric oxide generated from the inducible nitric oxide synthase. Anesthesiology 104:546–555. 10.1097/00000542-200603000-00023 [DOI] [PubMed] [Google Scholar]
- Chen M, Van Hook MJ, Thoreson WB (2015) Ca2+ diffusion through endoplasmic reticulum supports elevated intraterminal Ca2+ levels needed to sustain synaptic release from rods in darkness. J Neurosci 35:11364–11373. 10.1523/JNEUROSCI.0754-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia YY, Liu K, Wang JJ, Kuo MC, Ho ST (1999) Intraoperative high dose fentanyl induces postoperative fentanyl tolerance. Can J Anaesth 46:872–877. 10.1007/BF03012978 [DOI] [PubMed] [Google Scholar]
- Choi JI, Koehrn FJ, Sorkin LS (2012) Carrageenan induced phosphorylation of akt is dependent on neurokinin-1 expressing neurons in the superficial dorsal horn. Mol Pain 8:4. 10.1186/1744-8069-8-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LF, Angst MS, Clark D (2008) Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain 24:479–496. [DOI] [PubMed] [Google Scholar]
- Cohen LB, Keynes RD, Hille B (1968) Light scattering and birefringence changes during nerve activity. Nature 218:438–441. 10.1038/218438a0 [DOI] [PubMed] [Google Scholar]
- Collett BJ. (1998) Opioid tolerance: the clinical perspective. Br J Anaesth 81:58–68. 10.1093/bja/81.1.58 [DOI] [PubMed] [Google Scholar]
- Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, Sotoudeh C, Clark JD, Barres BA, Bohlen CJ, Scherrer G (2017) Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med 23:164–173. 10.1038/nm.4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AM, Fameli N, Ogunbayo OA, Duan J, Navarro-Dorado J (2016) From contraction to gene expression: nanojunctions of the sarco/endoplasmic reticulum deliver site- and function-specific calcium signals. Sci China Life Sci 59:749–763. 10.1007/s11427-016-5071-0 [DOI] [PubMed] [Google Scholar]
- Ferrari LF, Levine JD (2015) Plasma membrane mechanisms in a preclinical rat model of chronic pain. J Pain 16:60–66. 10.1016/j.jpain.2014.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Chu C, Levine JD (2013) Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain 14:731–738. 10.1016/j.jpain.2013.01.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD (2014) Second messengers mediating the expression of neuroplasticity in a model of chronic pain in the rat. J Pain 15:312–320. 10.1016/j.jpain.2013.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Khomula EV, Araldi D, Levine JD (2016) Marked sexual dimorphism in the role of the ryanodine receptor in a model of pain chronification in the rat. Sci Rep 6:31221. 10.1038/srep31221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Khomula EV, Araldi D, Levine JD (2017) CD44 signaling mediates high molecular weight hyaluronan-induced anti-hyperalgesia. J Neurosci 38:308–321. 10.1523/JNEUROSCI.2695-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fill M, Copello JA (2002) Ryanodine receptor calcium release channels. Physiol Rev 82:893–922. 10.1152/physrev.00013.2002 [DOI] [PubMed] [Google Scholar]
- Futagi D, Kitano K (2015) Ryanodine-receptor-driven intracellular calcium dynamics underlying spatial association of synaptic plasticity. J Comput Neurosci 39:329–347. 10.1007/s10827-015-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S, Levine JD (1996) Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience 71:265–275. 10.1016/0306-4522(95)00433-5 [DOI] [PubMed] [Google Scholar]
- Groer CE, Schmid CL, Jaeger AM, Bohn LM (2011) Agonist-directed interactions with specific beta-arrestins determine mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J Biol Chem 286:31731–31741. 10.1074/jbc.M111.248310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AA, Lawson SN (1985) Conduction velocity is related to morphological cell type in rat dorsal root ganglion neurones. J Physiol 359:31–46. 10.1113/jphysiol.1985.sp015573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson T, Thomas S, Stabile V, Shotwell M, Han X, McQueen K (2016) A systematic review and meta-analysis of the global burden of chronic pain without clear etiology in low- and middle-income countries: trends in heterogeneous data and a proposal for new assessment methods. Anesth Analg 123:739–748. 10.1213/ANE.0000000000001389 [DOI] [PubMed] [Google Scholar]
- Johannes CB, Le TK, Zhou X, Johnston JA, Dworkin RH (2010) The prevalence of chronic pain in united states adults: results of an internet-based survey. J Pain 11:1230–1239. 10.1016/j.jpain.2010.07.002 [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD (2010) Hyperalgesic priming is restricted to isolectin B4-positive nociceptors. Neuroscience 169:431–435. 10.1016/j.neuroscience.2010.04.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Chen X, Bogen O, Levine JD (2008) Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J Pain 9:463–472. 10.1016/j.jpain.2008.01.335 [DOI] [PubMed] [Google Scholar]
- Joseph EK, Reichling DB, Levine JD (2010) Shared mechanisms for opioid tolerance and a transition to chronic pain. J Neurosci 30:4660–4666. 10.1523/JNEUROSCI.5530-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabov SG, Rogers SD, Ghilardi JR, Peters CM, Mantyh PW, Simone DA (2002) Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J Neurosci 22:9086–9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Gold MS, Dastmalchi S, Levine JD (1996) Selective attenuation of mu-opioid receptor-mediated effects in rat sensory neurons by intrathecal administration of antisense oligodeoxynucleotides. Neurosci Lett 218:17–20. [DOI] [PubMed] [Google Scholar]
- Khomula EV, Voitenko NV (2006) Dynamics of calcium release and uptake by the internal calcium stores in rat sensory neurons. Neurophysiology 38:305–307. 10.1007/s11062-006-0062-6 [DOI] [Google Scholar]
- Khomula EV, Ferrari LF, Araldi D, Levine JD (2017) Sexual dimorphism in a reciprocal interaction of ryanodine and IP3 receptors in the induction of hyperalgesic priming. J Neurosci 37:2032–2044. 10.1523/JNEUROSCI.2911-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer BL, Gaveriaux-Ruff C (2002) Exploring the opioid system by gene knockout. Prog Neurobiol 66:285–306. [DOI] [PubMed] [Google Scholar]
- Kras JV, Weisshaar CL, Pall PS, Winkelstein BA (2015) Pain from intra-articular NGF or joint injury in the rat requires contributions from peptidergic joint afferents. Neurosci Lett 604:193–198. 10.1016/j.neulet.2015.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landowne D. (1993) Measuring nerve excitation with polarized light. Jpn J Physiol 43:S7–S11. [PubMed] [Google Scholar]
- Laulin JP, Maurette P, Corcuff JB, Rivat C, Chauvin M, Simonnet G (2002) The role of ketamine in preventing fentanyl-induced hyperalgesia and subsequent acute morphine tolerance. Anesth Analg 94:1263–1269, table of contents. 10.1097/00000539-200205000-00040 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Yeomans DC (2014) Opioid induced hyperalgesia in anesthetic settings. Korean J Anesthesiol 67:299–304. 10.4097/kjae.2014.67.5.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L (2011) A comprehensive review of opioid-induced hyperalgesia. Pain Physician 14:145–161. [PubMed] [Google Scholar]
- Levine JD, Taiwo YO (1989) Involvement of the mu-opiate receptor in peripheral analgesia. Neuroscience 32:571–575. 10.1016/0306-4522(89)90279-0 [DOI] [PubMed] [Google Scholar]
- Maixner W, Fillingim RB, Williams DA, Smith SB, Slade GD (2016) Overlapping chronic pain conditions: implications for diagnosis and classification. J Pain 17:T93–T107. 10.1016/j.jpain.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauermann E, Filitz J, Dolder P, Rentsch KM, Bandschapp O, Ruppen W (2016) Does fentanyl lead to opioid-induced hyperalgesia in healthy volunteers?: a double-blind, randomized, crossover trial. Anesthesiology 124:453–463. 10.1097/ALN.0000000000000976 [DOI] [PubMed] [Google Scholar]
- Mestre C, Pélissier T, Fialip J, Wilcox G, Eschalier A (1994) A method to perform direct transcutaneous intrathecal injection in rats. J Pharmacol Toxicol Methods 32:197–200. 10.1016/1056-8719(94)90087-6 [DOI] [PubMed] [Google Scholar]
- Nishiguchi J, Sasaki K, Seki S, Chancellor MB, Erickson KA, de Groat WC, Kumon H, Yoshimura N (2004) Effects of isolectin B4-conjugated saporin, a targeting cytotoxin, on bladder overactivity induced by bladder irritation. Eur J Neurosci 20:474–482. 10.1111/j.1460-9568.2004.03508.x [DOI] [PubMed] [Google Scholar]
- Oliveira-Fusaro MCG, Zanoni CIS, Dos Santos GG, Manzo LP, Araldi D, Bonet IJM, Tambeli CH, Dias EV, Parada CA (2017) Antihyperalgesic effect of CB1 receptor activation involves the modulation of P2X3 receptor in the primary afferent neuron. Eur J Pharmacol 798:113–121. 10.1016/j.ejphar.2017.01.030 [DOI] [PubMed] [Google Scholar]
- Quanhong Z, Ying X, Moxi C, Tao X, Jing W, Xin Z, Li W, Derong C, Xiaoli Z, Wei J (2012) Intrathecal PLC(beta3) oligodeoxynucleotides antisense potentiates acute morphine efficacy and attenuates chronic morphine tolerance. Brain Res 1472:38–44. 10.1016/j.brainres.2012.06.030 [DOI] [PubMed] [Google Scholar]
- Reichling DB, Levine JD (2009) Critical role of nociceptor plasticity in chronic pain. Trends Neurosci 32:611–618. 10.1016/j.tins.2009.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfrey S, Downton C, Featherstone J (2003) The painful reality. Nat Rev Drug Discov 2:175–176. 10.1038/nrd1038 [DOI] [PubMed] [Google Scholar]
- Richebé P, Rivat C, Laulin JP, Maurette P, Simonnet G (2005) Ketamine improves the management of exaggerated postoperative pain observed in perioperative fentanyl-treated rats. Anesthesiology 102:421–428. 10.1097/00000542-200502000-00028 [DOI] [PubMed] [Google Scholar]
- Roeckel LA, Le Coz GM, Gavériaux-Ruff C, Simonin F (2016) Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience 338:160–182. 10.1016/j.neuroscience.2016.06.029 [DOI] [PubMed] [Google Scholar]
- Saito A, Cavalli V (2016) Signaling over distances. Mol Cell Proteomics 15:382–393. 10.1074/mcp.R115.052753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Blazquez P, Garcia-Espana A, Garzon J (1997) Antisense oligodeoxynucleotides to opioid mu and delta receptors reduced morphine dependence in mice: role of delta-2 opioid receptors. J Pharmacol Exp Ther 280:1423–1431. [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, Kieffer BL, Basbaum AI (2009) Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137:1148–1159. 10.1016/j.cell.2009.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutov L, Kruglikov I, Gryshchenko O, Khomula E, Viatchenko-Karpinski V, Belan P, Voitenko N (2006) The effect of nimodipine on calcium homeostasis and pain sensitivity in diabetic rats. Cell Mol Neurobiol 26:1541–1557. 10.1007/s10571-006-9107-z [DOI] [PubMed] [Google Scholar]
- Song MJ, Wang YQ, Wu GC (2009) Additive anti-hyperalgesia of electroacupuncture and intrathecal antisense oligodeoxynucleotide to interleukin-1 receptor type I on carrageenan-induced inflammatory pain in rats. Brain Res Bull 78:335–341. 10.1016/j.brainresbull.2008.10.009 [DOI] [PubMed] [Google Scholar]
- Stoicea N, Russell D, Weidner G, Durda M, Joseph NC, Yu J, Bergese SD (2015) Opioid-induced hyperalgesia in chronic pain patients and the mitigating effects of gabapentin. Front Pharmacol 6:104. 10.3389/fphar.2015.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Mattson MP (2011) Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol Rev 63:700–727. 10.1124/pr.110.003814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L, Wang C, Yu YH, Ren YY, Xie KL, Wang GL (2011) Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci 12:120. 10.1186/1471-2202-12-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JL, Xiao C, Lu B, Zhang J, Yuan XZ, Chen W, Yu LN, Zhang FJ, Chen G, Yan M (2013) CX3CL1/CX3CR1 regulates nerve injury-induced pain hypersensitivity through the ERK5 signaling pathway. J Neurosci Res 91:545–553. 10.1002/jnr.23168 [DOI] [PubMed] [Google Scholar]
- Sutko JL, Ito K, Kenyon JL (1985) Ryanodine: a modifier of sarcoplasmic reticulum calcium release in striated muscle. Fed Proc 44:2984–2988. [PubMed] [Google Scholar]
- Taiwo YO, Levine JD (1989) Prostaglandin effects after elimination of indirect hyperalgesic mechanisms in the skin of the rat. Brain Res 492:397–399. 10.1016/0006-8993(89)90928-1 [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD (1990) Direct cutaneous hyperalgesia induced by adenosine. Neuroscience 38:757–762. 10.1016/0306-4522(90)90068-F [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD (1989) Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience 32:577–580. 10.1016/0306-4522(89)90280-7 [DOI] [PubMed] [Google Scholar]
- Trang T, Al-Hasani R, Salvemini D, Salter MW, Gutstein H, Cahill CM (2015) Pain and Poppies: The Good, the Bad, and the Ugly of Opioid Analgesics. J Neurosci 35:13879–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk DC, Fillingim RB, Ohrbach R, Patel KV (2016) Assessment of psychosocial and functional impact of chronic pain. J Pain 17:T21–49. 10.1016/j.jpain.2016.02.006 [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P (2015) Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 18:145–153. 10.1038/nn.3881 [DOI] [PubMed] [Google Scholar]
- Velázquez-Marrero C, Ortiz-Miranda S, Marrero HG, Custer EE, Treistman SN, Lemos JR (2014) mu-opioid inhibition of Ca2+ currents and secretion in isolated terminals of the neurohypophysis occurs via ryanodine-sensitive Ca2+ stores. J Neurosci 34:3733–3742. 10.1523/JNEUROSCI.2505-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verplaetse R, Henion J (2016) Quantitative determination of opioids in whole blood using fully automated dried blood spot desorption coupled to on-line SPE-LC-MS/MS. Drug Test Anal 8:30–38. 10.1002/dta.1927 [DOI] [PubMed] [Google Scholar]
- Vierck CJ Jr, Kline RH, Wiley RG (2003) Intrathecal substance p-saporin attenuates operant escape from nociceptive thermal stimuli. Neuroscience 119:223–232. 10.1016/S0306-4522(03)00125-8 [DOI] [PubMed] [Google Scholar]
- Volkow ND, McLellan AT (2016) Opioid abuse in chronic pain–misconceptions and mitigation strategies. N Engl J Med 374:1253–1263. 10.1056/NEJMra1507771 [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Olson TH, Stone LS, Riedl MS, Elde R, Honda CN (2001) Cytotoxic targeting of isolectin IB4-binding sensory neurons. Neuroscience 108:143–155. 10.1016/S0306-4522(01)00377-3 [DOI] [PubMed] [Google Scholar]
- Waxman AR, Arout C, Caldwell M, Dahan A, Kest B (2009) Acute and chronic fentanyl administration causes hyperalgesia independently of opioid receptor activity in mice. Neurosci Lett 462:68–72. 10.1016/j.neulet.2009.06.061 [DOI] [PubMed] [Google Scholar]
- Weisshaar CL, Winkelstein BA (2014) Ablating spinal NK1-bearing neurons eliminates the development of pain and reduces spinal neuronal hyperexcitability and inflammation from mechanical joint injury in the rat. J Pain 15:378–386. 10.1016/j.jpain.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley RG, Kline RHt, Vierck CJ Jr (2007) Anti-nociceptive effects of selectively destroying substance P receptor-expressing dorsal horn neurons using [Sar9, Met(O2)11]-substance P-saporin: behavioral and anatomical analyses Neuroscience 146:1333–1345. 10.1016/j.neuroscience.2007.01.066 [DOI] [PubMed] [Google Scholar]