

Abstract
Repeated social defeat (RSD) stress promotes the release of bone marrow-derived monocytes into circulation that are recruited to the brain, where they augment neuroinflammation and cause prolonged anxiety-like behavior. Physiological stress activates the sympathetic nervous system and hypothalamic-pituitary-adrenal gland (HPA) axis, and both of these systems play a role in the physiological, immunological, and behavioral responses to stress. The purpose of this study was to delineate the role of HPA activation and corticosterone production in the immunological responses to stress in male C57BL/6 mice. Here, surgical (adrenalectomy) and pharmacological (metyrapone) interventions were used to abrogate corticosterone signaling during stress. We report that both adrenalectomy and metyrapone attenuated the stress-induced release of monocytes into circulation. Neither intervention altered the production of monocytes during stress, but both interventions enhanced retention of these cells in the bone marrow. Consistent with this observation, adrenalectomy and metyrapone also prevented the stress-induced reduction of a key retention factor, CXCL12, in the bone marrow. Corticosterone depletion with metyrapone also abrogated the stress-induced glucocorticoid resistance of myeloid cells. In the brain, these corticosterone-associated interventions attenuated stress-induced microglial remodeling, neurovascular expression of the adhesion molecule intercellular cell adhesion molecule-1, prevented monocyte accumulation and neuroinflammatory signaling. Overall, these results indicate that HPA activation and corticosterone production during repeated social defeat stress are critical for monocyte release into circulation, glucocorticoid resistance of myeloid cells, and enhanced neurovascular cell adhesion molecule expression.
SIGNIFICANCE STATEMENT Recent studies of stress have identified the presence of monocytes that show an exaggerated inflammatory response to immune challenge and are resistant to the suppressive effects of glucocorticoids. Increased presence of these proinflammatory monocytes has been implicated in neuropsychiatric symptoms and the development of chronic cardiovascular, autoimmune, and metabolic disorders. In the current study, we show novel evidence that corticosterone produced during stress enhances the release of proinflammatory monocytes from the bone marrow into circulation, augments their recruitment to the brain and the induction of a neuroinflammatory profile. Overproduction of corticosterone during stress is also the direct cause of glucocorticoid resistance, a key phenotype in individuals exposed to chronic stress. Inhibiting excess corticosterone production attenuates these inflammatory responses to stress.
Keywords: repeated social defeat, corticosterone, HPA axis, inflammation, monocytes
Introduction
Psychological stress contributes to the development and exacerbation of anxiety-like disorders (Kendler et al., 1999; Pasquali 2012). Activation of the sympathetic nervous system (SNS) and the hypothalamic-pituitary-adrenal (HPA) axis regulates the immunological and behavioral responses to stress. For instance, HPA activation during stress corresponds with the release of glucocorticoids from the adrenal cortex. Glucocorticoids regulate glucose and energy mobilization, and immune functions in response to stress (Sapolsky et al., 2000). Chronic stress, however, leads to glucocorticoid resistance and enhanced inflammatory signaling in humans and rodents (Pace et al., 2006; Cohen et al., 2012). For instance, low socioeconomic status and prolonged caregiving stress in humans are associated with a “transcriptional fingerprint,” characterized by enhanced expression of proinflammatory signals in peripheral monocytes and resistance to the suppressive effects of glucocorticoids (Miller et al., 2008, 2014). Individuals exposed to chronic stress show high levels of circulating IL-6, which is a strong indicator of stress-induced psychiatric and cardiovascular disorders (Maes et al., 1999, 2012). Importantly, IL-6 is an acute phase protein regulated by the HPA axis (Zhou et al., 1993). Thus, overactivation of the HPA axis, along with a blunted response to corticosterone, may lead to unchecked inflammatory responses that are associated with stress-induced neuropsychiatric, metabolic, and cardiovascular diseases (Walker 2007; Sorrells et al., 2009; Marin et al., 2011).
The clinical features of chronic stress (e.g., glucocorticoid resistance, enhanced proinflammatory profile, and elevated plasma IL-6) are recapitulated in the repeated social defeat (RSD) model of stress in mice (Wohleb et al., 2014a; Reader et al., 2015). RSD increases neuronal and microglial activation, endothelial cell adhesion molecule expression, production and release of monocytes into circulation, and recruitment to tissues, including the brain (McKim et al., 2017). Increased presence of circulating monocytes has been reported in stressed individuals, and monocyte accumulation in the brain vasculature was reported in depressed suicide victims (Heidt et al., 2014; Torres-Platas et al., 2014). We have identified monocytes in the RSD brain as the pro-inflammatory (CCR2+Ly6Chi) type that propagates IL-1-receptor signaling at the brain vascular endothelium, causing prolonged anxiety-like behavior (McKim et al., 2017) and long-lasting “stress sensitization” to subsequent stressors (Wohleb et al., 2014b; McKim et al., 2016b). Other recent studies with social defeat in mice show that accumulation of pro-inflammatory monocytes in the neurovasculature triggers neuropsychiatric complications (Menard et al., 2017). Therefore, mobilization of inflammatory monocytes from the bone marrow represents an important mechanism for CNS signaling to the immune system during stress (Weber et al., 2017).
Exposure to RSD also promotes the induction of glucocorticoid resistance in myeloid cells. For instance, myeloid cells from the spleen of mice exposed to RSD are resistant to the anti-inflammatory effects of glucocorticoids (e.g., enhanced IL-6 production following LPS and sustained viability despite high corticosterone exposure) (Avitsur et al., 2001; Stark et al., 2001). This glucocorticoid-insensitive phenotype of splenocytes following RSD is significant because it is also present in peripheral monocytes from individuals suffering from chronic stress (Miller et al., 2002, 2008). Furthermore, monocytes that accumulated in the brain during RSD displayed an mRNA profile consistent with glucocorticoid resistance (i.e., reduced glucocorticoid receptor and increased IL-1β expression) (McKim et al., 2017). In addition, stress-induced glucocorticoid resistance was associated with elevated inflammatory response to subsequent innate immune challenge (Quan et al., 2001; Wohleb et al., 2012). Glucocorticoid resistance with RSD was associated with a failure of the glucocorticoid receptor in myeloid cells to translocate into the nucleus (Quan et al., 2003).
Both HPA and SNS pathways communicate with the immune system in response to stress. For instance, β-adrenergic intervention and benzodiazepines prevented the RSD-induced activation of threat appraisal and all downstream changes in the brain and the periphery (Wohleb et al., 2011; Hanke et al., 2012; Ramirez et al., 2016). Notably, inhibition of threat appraisal during RSD prevented both HPA and SNS activation. Therefore, the purpose of this study was to delineate the role of corticosterone in the peripheral immune response to stress. This is important because corticosterone generally suppresses inflammatory signaling but also has pleiotropic effects (Sorrells et al., 2009). Furthermore, the role of corticosterone on monocyte production, release, and tissue recruitment during stress is unknown. In the current study, we show, for the first time, that corticosterone production during RSD promoted mobilization of monocytes from the bone marrow into circulation. Moreover, we provide novel evidence that corticosterone caused glucocorticoid resistance in myeloid cells and enhanced cell adhesion molecule expression and inflammatory mediators in the brain during RSD.
Materials and Methods
Mice.
Male C57BL/6 mice (6- to 8-week-old) and CD-1 mice (12-month-old, retired breeders) were purchased from Charles River Breeding Laboratories. Adrenalectomized and sham C57BL/6 mice (6- to 8-week-old) were purchased from The Jackson Laboratory. All adrenalectomized mice were provided with supplemental corticosterone (25 μg/ml) in drinking water until death (Lehmann et al., 2013). Next, CXCL12-DsRed mice were generously provided by Sean J. Morrison (University of Texas Southwestern Medical Center). All experimental mice were housed in cohorts of three, whereas CD-1 mice were individually housed. Mice were kept in 11.5 × 7.5 × 6 inch polypropylene cages and were maintained at 21°C under a 12 h light/dark cycle with access to food and water ad libitum at the animal housing facility at Ohio State University. All mice were allowed to acclimate for 7–10 d before initiation of any experimental procedure. All procedures were in accordance with the National Institutes of Health Guide for the care and use of laboratory animals, and performed with approval from the Ohio State University Institutional Animal Care and Use Committee.
RSD.
The RSD model of stress involves eliciting a flight or fight in resident mice in response to an aggressive intruder. In contrast to pair fighting paradigms of stress, RSD adds a key social component that includes psychological stress caused due to disruption of the social hierarchy within an established cohort of resident mice. In the current study, we performed RSD as previously reported (Avitsur et al., 2001; Wohleb et al., 2013). In brief, a CD-1 aggressor was introduced into the cage of an established cohort (3 mice) of C57BL/6 mice for 2 h (17:00 to 19:00 h) daily for 6 consecutive nights. If the intruder mouse did not attack in the first 5 min, it was replaced by a new intruder. Different intruder mice were used on consecutive days. During each episode of stress, resident mice were monitored for submissive behaviors, including crouching, fleeing, and upright posture. At the end of the 2 h period, the intruder mice were returned to their original cages, and the resident mice were left undisturbed until the next episode of stress the following day. The health status of the mice was carefully examined throughout the experiment. In the event of an injury, mice were removed from the experiment. Consistent with our previous studies, <5% of the mice met early removal criteria. The control mice were left undisturbed in their home cages during the study.
Experimental protocols
ADX intervention and stress.
Male C57BL/6 mice were subjected to sham or adrenalectomy (ADX) surgery at The Jackson Laboratory. Mice were shipped to Ohio State University and allowed 7 d to recover. All adrenalectomized mice were provided with supplemental corticosterone (25 μg/ml) in drinking water until death (Lehmann et al., 2013). Under homeostatic conditions, corticosterone production follows a circadian oscillatory pattern (i.e., sustained increases during active phase and reduction during passive phase). This oscillatory production of corticosterone is necessary in maintaining the physiological functions (e.g., homeostatic ACTH levels) but not enough to elicit a stress response (Jacobson et al., 1988; Dhabhar et al., 2012). Because adrenalectomized mice are unable to produce corticosterone, the supplemental corticosterone in drinking water is necessary to maintain the circadian rhythmicity of corticosterone in these mice.
Next, mice were exposed to 6 cycles of RSD (Stress). Plasma for corticosterone was collected via submandibular bleeds immediately after stress (3 experiments, n = 3 or 4 per experiment). In addition, bone marrow, blood, spleen, and brain (2 experiments, n = 3 or 4 per experiment) were collected 14 h after the last cycle of stress. IL-6 protein levels were determined in plasma (2 experiments, n = 3 or 4 per experiment). Percentages of monocytes and granulocytes (2 experiments, n = 3 or 4 per experiment) were determined in the blood and bone marrow, and CXCL12 mRNA levels (2 experiments, n = 3 or 4 per experiment) were also determined in the bone marrow. For brain mRNA levels, a 1 mm coronal section from the brain (∼−0.34 mm to −1.34 mm bregma) was collected (2 experiments, n = 3 or 4 per experiment), and the rest of the brain was used to collect CD11b+ cells for flow cytometry (2 experiments, n = 3 or 4 per experiment). In a separate study, mice were treated as above and were perfused and PFA-fixed. Intercellular cell adhesion molecule-1 (ICAM-1) expression was determined by immunohistochemistry (1 experiment, n = 3 or 4).
CXCL12 reporter and stress.
Male CXCL12-DsRed mice were exposed to control or RSD (Stress). Mice were perfused, PFA-fixed, and bone marrow was collected to evaluate RFP expression (1 experiment, n = 3).
Metyrapone (MTP) intervention and stress.
Male C57BL/6 mice were injected intraperitoneally daily with either vehicle (water) or 100 mg/kg MTP (Enzo Life Sciences; catalog #BML-EI256) 30 min before control or RSD (Stress). Plasma for corticosterone was collected via submandibular bleed immediately after stress (2 experiments, n = 3 or 4 per experiment). In addition, bone marrow, blood, spleen, and brain (2 experiments, n = 3 or 4 per experiment) were collected 14 h after the last cycle of stress. IL-6 protein levels were determined in plasma (1 experiment, n = 3 or 4). Percentages of monocytes and granulocytes were determined in the bone marrow and blood (2 experiments, n = 3 or 4 per experiment). CXCL12 mRNA levels were determined in the bone marrow (1 experiment, n = 3 or 4). For mRNA analysis in the brain, a 1 mm coronal section from the brain (∼−0.34 mm to −1.34 mm bregma) was collected (2 experiments, n = 3 or 4 per experiment), and the rest of the brain was used to collect CD11b+ cells for flow cytometry. In a separate study, mice were treated as above and mice were perfused and PFA-fixed. ΔFosB, Iba-1, and ICAM-1 expression was determined by immunohistochemistry (1 experiment, n = 3 or 4). In a final study, mice were treated as above with MTP and exposed to stress. The spleen was collected 14 h after the last cycle of stress. Splenocytes were cultured ex vivo with LPS and treated with increasing doses of corticosterone. Cell survival and supernatant IL-6 levels were determined (2 experiments, n = 3 or 4 per experiment).
Isolation of CD11b+ cells from the brain.
CD11b+ cells were enriched by Percoll isolation as described previously (Wohleb et al., 2013). At 14 h after the last cycle of stress, mice were asphyxiated, perfused with ice-cold PBS, and brains were collected. Brain samples were homogenized using Glass Potter Elvehjem Tissue Grinder (OMNI International) and centrifuged at 900 × g for 6 min. Then, cells were pelleted and suspended in 70% isotonic Percoll (GE Healthcare). This suspension was layered with 50%, 35%, and 0% isotonic Percoll to create a discontinuous Percoll gradient. This gradient was centrifuged at 2070 × g for 20 min, and cells were collected from the 70%–50% Percoll interface. This interface is enriched with >90% CD11b+ cells (Wohleb et al., 2013).
Isolation of bone marrow and blood cells.
Fourteen hours after the last cycle of stress, mice were asphyxiated, and blood and bone marrow samples were collected. Bone marrow was collected from the femur and flushed out with ice-cold PBS. Samples were homogenized using a syringe plunger and filtered through a 70 μm nylon strainer. Blood samples were collected by cardiac puncture into EDTA-lined syringes, and red blood cells were lysed using lysis buffer (0.16 m NH4Cl, 10 mm KHCO3, 0.13 mm EDTA). Samples were washed and cells were counted using a Particle Count and Size Analyzer (Beckman Coulter).
Flow cytometry.
Cell surface antigens on bone marrow, blood, and Percoll-enriched brain cells were labeled as previously described (McKim et al., 2017). In brief, Fc receptors were blocked using anti-CD16/CD32 antibody (ThermoFisher Scientific, catalog #14-0161-82, RRID:AB_467133). Next, cells were incubated with fluorophore-conjugated antibodies (Ly6C, BD Biosciences, catalog #553104, RRID:AB_394628; Ly6G, BD Biosciences, catalog #551461, RRID:AB_394208; CD45, BD Biosciences, catalog #550994, RRID:AB_394003; CD11b, ThermoFisher Scientific, catalog #17–0112-83, RRID:AB_469344) for 1 h at 4°C, followed by a wash and resuspension in FACS buffer. Isotype-matched antibodies were used to determine non-specific binding. Antibody labeling was determined using FACS Calibur cytometer (BD Biosciences). Data were analyzed using FlowJo (TreeStar).
Corticosterone ELISA.
Blood samples were collected by submandibular bleeding immediately after the last cycle of stress (∼7:00 P.M.). Plasma was extracted and stored at −80°C. Corticosterone concentrations were evaluated using the Corticosterone EIA kit (Enzo; catalog #ADI-900-097) following the manufacturer's instructions.
Real-time qPCR from brain and bone marrow samples.
Fourteen hours after the last cycle of stress, mice were asphyxiated, and brain and bone marrow samples were collected. For brain mRNA analysis, a 1 mm coronal section of the brain (∼−0.34 mm to −1.34 mm bregma) was collected and flash frozen in liquid nitrogen. This brain section, collected using a mouse brain matrix (Kent Scientific, catalog #RBMS-200C), was used to evaluate the general inflammatory profile of the brain following stress and interventions. The rest of the brain was used for isolation of CD11b+ cells for cell flow cytometric analyses. RNA was extracted using a tri-reagent/Isopropranolol precipitation protocol (McKim et al., 2016a). RNA concentration and quality were determined using the NanoPhotometer (Implen). RNA was reverse-transcribed into cDNA using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time qPCR was performed using the TaqMan Gene Expression Assay-on-Demand Gene Expression protocol (Applied Biosystems). Target cDNA and reference cDNA GAPDH were amplified simultaneously using a primer/probe set consisting of an oligonucleotide probe with a 5′ fluorescent reporter dye (FAM) and a 3′ quencher dye (nonfluorescent) for each gene of interest (Invitrogen). Fluorescence was determined on an ABI PRISM 7300-sequence detection system (Applied Biosystems). Data were analyzed using the comparative threshold cycle method, and results were expressed as fold change compared with the reference gene, GAPDH.
For bone marrow mRNA analysis, femurs were flushed with ice-cold PBS and homogenized using a syringe plunger and filtered through a 70 μm strainer. Cells were centrifuged and pelleted into RNA lysis buffer (PrepEase Kit, USB), and RNA isolation was performed according to the manufacturer's instructions. RNA concentration and quality were determined using the NanoPhotometry (Implen). Reverse transcription and subsequent real-time qPCR were performed via the same procedure as for the brain (described above). Because the commonly used housekeeping gene GAPDH was altered in the bone marrow by RSD stress, Eef2 (Eukaryotic translation elongation factor 2) expression was validated and used as reference cDNA for real-time qPCR of bone marrow samples. Data were analyzed using the comparative threshold cycle method, and results were expressed as fold change compared with the reference gene, Eef2.
Immunohistochemistry and digital image analysis of ICAM-1 and ΔFosB.
Fourteen hours after the last cycle of stress, mice were asphyxiated, transcardially perfused with ice-cold PBS followed by 4% PFA. Brain samples were postfixed in formaldehyde for 24 h, followed by an additional 48 h incubation in 30% sucrose at 4°C. Fixed brain samples were frozen with isopentane (−78°C) and dry ice, and stored at −80°C until sectioning. Frozen brain samples were sectioned at 25 μm using a Microm HM550 cryostat (ThermoFisher Scientific) and free-floating sections were preserved in cryoprotectant at −20°C until labeling. Sections were washed in 1× PBS and blocked with 5% normal donkey serum (1% BSA and 0.1% Triton-X in PBS) for an hour at room temperature, followed by an overnight 4°C incubation with primary antibodies: goat anti-ICAM-1 (1:500; R&D Systems, catalog #AF796, RRID:AB_2248703), rabbit anti-ΔFosB (1:2000; Abcam, catalog #ab184938, RRID:AB_2721123), or rabbit anti-Iba1 (1:1000; Wako, catalog #019-19741, RRID:AB_839504) or rat anti-Ly6C (1:500; Abcam, catalog #ab15627, RRID:AB_302004). Sections were washed in 1× PBS and incubated with the corresponding secondary antibodies conjugated with fluorochromes (AlexaFluor-488 or AlexaFluor-594). Following 2 h of incubation at room temperature, sections were washed in DAPI (1:100), then mounted and coverslipped with Fluoromount G (Beckman Coulter), and stored at −20°C. Images were taken on a Zeiss 510 Meta confocal microscope and analyzed using ImageJ software. For the digital imaging analysis of ICAM-1 and Iba1 images, a threshold for positive labeling (full view of the labeled blood vessel, or a full view of labeled microglia with background excluded) was determined for each image. Data were processed by ImageJ using the densitometric scanning of the threshold targets, and results expressed as the average percentage area with positive labeling. For ΔFosB analysis, the number of cells positive for ΔFosB labeling was counted for each image.
Bone marrow processing and immunofluorescent labeling.
Immediately after the last cycle of stress, CXCL12-DsRed mice were asphyxiated, transcardially perfused with ice-cold PBS followed by 4% formaldehyde. Femurs were isolated and postfixed in 4% formaldehyde for 48 h at 4°C. Samples were then transferred into decalcification buffer (0.5 m EDTA in PBS) and incubated at 4°C for 48 h. Samples were then frozen with isopentane (−78°C) and dry ice and stored at −80°C until sectioning. Next, frozen bone marrow samples were sectioned at 14 μm, collected on Superfrost Plus slides (Fisher Scientific), and stored at −20°C. Slides were washed with 1× PBS, incubated with 5% normal donkey serum (1% BSA and 0.1% Triton-X in PBS) for an hour at room temperature, followed by primary antibody rabbit anti-RFP (1:500; Abcam, catalog #ab124754, RRID:AB_10971665) incubation overnight at 4°C. Slides were washed in 1× PBS and incubated with an Alexa-594 fluorochrome-conjugated antibody for 2 h at room temperature. Slides were washed, allowed to dry, and coverslipped with Fluoromount G (Beckman Coulter), and stored at −20°C. Images were taken using a Zeiss 510 Meta confocal microscope and analyzed using ImageJ software, as described above.
Glucocorticoid resistance assay.
Fourteen hours after the last cycle of stress, spleens were collected in ice-cold PBS. Spleen samples were homogenized using a syringe plunger and filtered through a 70 μm strainer. Cells were centrifuged and pelleted into ice-cold HBSS to obtain single-cell suspensions. Red blood cells were lysed using the lysis buffer (0.16 m NH4Cl, 10 mm KHCO3, 0.13 mm EDTA), and samples were washed in 10% FBS/HBSS. Samples were resuspended in media RPMI 1640 medium, GlutaMAX (ThermoFisher Scientific, catalog #61870-036) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. Cells (2 × 105 per well) were plated in triplicates on a 96-well plate (Corning, catalog #3596). LPS (Sigma-Aldrich, L-2630) was added at 1 μg/ml per well. Corticosterone (Sigma-Aldrich, catalog #27840) solutions with 0.2% ethanol were prepared in media and added to the wells at varying concentrations (0, 0.05, 0.1, 0.5, and 5 μm). Cells were cultured with LPS and corticosterone at 37°C in 5% CO2 for 18 h for IL-6 ELISA and for 48 h for cell viability analysis.
IL-6 ELISA.
To determine IL-6 production from splenocytes, supernatant samples were collected 18 h after culture and incubation (as described above) and stored at −80°C. IL-6 levels were determined using the BD OptEIA Mouse IL-6 ELISA (BD Biosciences) as previously described (Stark et al., 2001). In brief, a 96-well plate was coated with anti-mouse IL-6 capture antibodies and allowed to incubate overnight at 4°C. Standards (0–1000 pg/ml) and samples were added and incubated for 2 h at room temperature. Plates were washed and incubated with biotinylated anti-mouse IL-6 antibody. Plates were then incubated with streptavidin-HRP conjugate for 1 h. Tetramethylbenzidine substrate was added next, and reaction was stopped after a 15 min incubation. Plates were read at 450 nm using a Spectramax plate reader (Molecular Devices). For plasma IL-6 ELISA, blood samples were collected via cardiac puncture 14 h after the last cycle of stress, and plasma was stored at −80°C. IL-6 levels were determined as described above.
Cell viability assay.
Cell viability was assessed as previously described (Hanke et al., 2012). In brief, the Cell Titer 96 aqueous nonradioactive proliferation assay (Promega) was used to determine cell viability of LPS-activated splenocytes cultured ex vivo with corticosterone. At 45 h following treatment with corticosterone, tetrazolium substrate solution (20 μl) was added to each well. Samples were incubated at 37°C in 5% CO2 for 3 h, and color changes were quantified by obtaining optical density readings at 450 nm on the Spectramax plate reader (Molecular Devices). To account for differences in background activity of cells, the mean optical density of three control wells was subtracted for a given treatment from each of the corresponding LPS-stimulated values. Control wells contained untreated cells. Results were shown as the percentage of proliferation at baseline (LPS stimulation, no corticosterone treatment).
Statistical analyses.
To test for normal distribution, data were subjected to the Shapiro–Wilk test using Statistical Analysis Systems (SAS) statistical software. Observations 2 SDs above and below the mean were considered outliers and excluded from subsequent analysis. In total, 22 of 749 observations were excluded after outlier analysis. To determine significant main effects and interactions between main factors, data were analyzed using two-way (stress × intervention) ANOVA using the GLM procedures of SAS. When there was a main effect of experimental treatment or a treatment interaction effect, differences between group means were evaluated by an F-protected t test using the Least-Significant Difference procedure of SAS. Post hoc analysis results are depicted graphically in figures. All data are expressed as mean ± SEM.
Results
Stress-induced release of inflammatory monocytes from the bone marrow into circulation was prevented by ADX
RSD promotes the release of bone marrow-derived monocytes into circulation that traffic to the brain, augment neuroinflammation, and cause prolonged anxiety-like behavior (Wohleb et al., 2013; McKim et al., 2017). Physiological stress activates the SNS and the HPA axis, both of which play a role in the physiological, immunological, and behavioral responses to stress (Wohleb et al., 2011; Ramirez et al., 2016). Therefore, the aim of this study was to delineate the role of HPA activation and corticosterone production in the physiological and immunological responses to RSD.
In the first set of experiments, adrenalectomized (ADX) mice were exposed to stress (six cycles of RSD), and several physiological and immunological parameters were determined 14 h later. Stress increased plasma corticosterone levels in the sham mice (Fig. 1A; F(1,30) = 17.39; p < 0.0003). As expected, removal of the adrenal glands (ADX) ablated this increase (Fig. 1A; stress × intervention interaction, F(1,30) = 11.91; p < 0.003). In addition, stress increased plasma IL-6 levels (Fig. 1B; F(1,21) = 8.07; p < 0.02) that were prevented in ADX-stress mice compared with sham-stress mice (p < 0.05). Stress also increased spleen weight (Fig. 1C; F(1,37) = 25.29; p < 0.0001), but this increase was independent of ADX.
Figure 1.

Stress-induced release of inflammatory monocytes from the bone marrow into circulation was prevented by ADX. Male C57BL/6 mice were subjected to sham or ADX surgery and allowed to recover until exposure to RSD (Stress). Plasma for corticosterone was collected immediately after stress, and plasma for IL-6, bone marrow, and blood samples was collected 14 h later. A, Corticosterone levels (n = 9) and (B) IL-6 levels (n = 6) in the plasma, and (C) spleen weight (n = 9) were determined. D, Representative bivariate dot plots of monocytes (CD11b+ Ly6Chi) and granulocytes (CD11b+ Ly6Cint) in the bone marrow. Percentage of bone marrow (E) monocytes and (F) granulocytes (n = 6). G, Representative bivariate dot plots of CD11b and Ly6C labeling of monocytes in circulation. H, Percentage of Ly6Chi monocytes in the blood (n = 6). Error bars indicate mean ± SEM. Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other.
Stress promotes a profound increase in monocyte and granulocyte production within the bone marrow (Ramirez et al., 2016; McKim et al., 2017). Here, we examined the production of monocytes and granulocytes in adrenalectomized mice after stress exposure. Consistent with previous work (Wohleb et al., 2013), stress increased the percentage of monocytes (CD11b+/Ly6Chi) and granulocytes (CD11b+/Ly6Cint) in the bone marrow (Fig. 1D–F; p < 0.001, for each). These increases were unaffected by ADX (Fig. 1E,F). Stress also increased the percentage of circulating Ly6Chi monocytes (Fig. 1G,H; F(1,22) = 16.99; p < 0.006). Post hoc analysis revealed that ADX-stress mice had significantly fewer circulating monocytes than sham-stress mice (p < 0.005). Overall, ADX attenuated corticosterone production during stress and reduced the release of inflammatory monocytes into circulation without altering their production in the bone marrow.
Stress-associated reduction of CXCL12 in the bone marrow was attenuated by ADX
We show that ADX prevented the release of monocytes into circulation but did not alter their production in the bone marrow (Fig. 1). Therefore, we sought to determine whether the reduced release of monocytes in ADX mice was associated with altered CXCL12 expression, a key chemokine important for the retention of hematopoietic stem cells and monocytes in the bone marrow (Heidt et al., 2014). First, CXCL12-DsRed mice were exposed to stress, and CXCL12 protein levels were determined in the bone marrow. Fig. 2A shows that stress caused a marked reduction in CXCL12 protein expression in the bone marrow. This effect was paralleled in wild-type mice that had a significant reduction in CXCL12 mRNA in the bone marrow after RSD (Fig. 2B; p < 0.005). In a separate experiment, mice were adrenalectomized (ADX) before exposure to stress and CXCL12 mRNA expression was determined in the bone marrow. Again, stress reduced CXCL12 mRNA expression in the bone marrow (Fig. 2C; F(1,16) = 5.36, p < 0.05). In addition, ADX increased bone marrow CXCL12 mRNA (Fig. 2C; F(1,16) = 10.70; p < 0.01). Post hoc analysis confirmed that sham-stress mice had lower CXCL12 mRNA expression compared with sham-control mice (p < 0.05), and this reduction was not evident in ADX-stress mice compared with ADX-control. Together, stress reduced CXCL12 expression in the bone marrow, and this reduction was prevented by ADX.
Figure 2.

Stress-associated reduction of CXCL12 in the bone marrow was attenuated by ADX. Male CXCL12-DsRed mice were exposed to control or RSD (Stress). A, Representative images of RFP expression in the femur immediately after stress (n = 3). Scale bar, 125 μm. Next, male C57BL/6 mice were exposed to control or RSD (Stress). B, CXCL12 mRNA expression in the bone marrow was determined (n = 3). In a separate experiment, male C57BL/6 mice were subjected to sham or ADX surgery and allowed to recover until exposure to RSD (Stress). C, CXCL12 mRNA expression in the bone marrow 14 h later (n = 6). Error bars indicate mean ± SEM. Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other.
Stress-induced monocyte accumulation in the brain and the neurovascular induction of ICAM-1 was prevented by ADX
Next, we assessed the effects of ADX and stress on monocyte accumulation in the brain and endothelial ICAM-1 induction. Corresponding with our previous reports (McKim et al., 2016a, 2017), stress increased the presence of monocytes (CD11b+/CD45hi) in the brain (Fig. 3A,B; F(1,23) = 34.28, p < 0.0002). This monocyte accumulation in the brain with stress tended to be attenuated by ADX (Fig. 3A,B; stress × intervention, F(1,23) = 4.07; p = 0.06). Post hoc analysis confirmed that ADX-stress mice had significantly fewer monocytes in the brain compared with sham-stress mice (p < 0.02).
Figure 3.
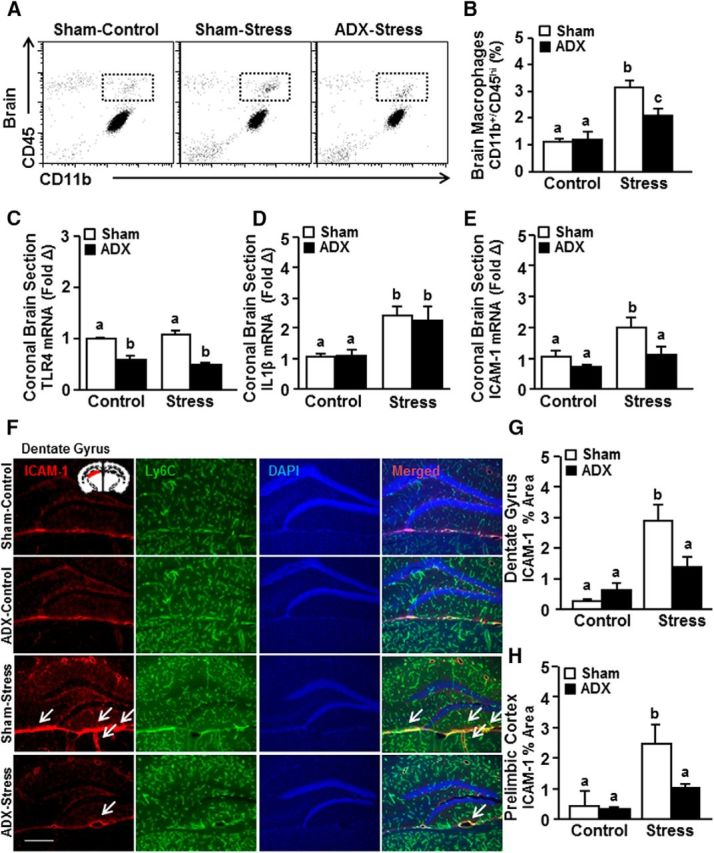
Stress-induced monocyte accumulation in the brain and the neurovascular induction of ICAM-1 were prevented by ADX. Male C57BL/6 mice were subjected to sham or ADX surgery and allowed to recover until exposure to RSD (Stress). Brain samples were collected 14 h later for flow cytometry and mRNA analyses. A, Representative bivariate dot plots. B, Percentage of CD45 and CD11b labeling of Percoll-enriched myeloid cells isolated from the brain (n = 6). mRNA expression of (C) TLR4, (D) IL-1β, and (E) ICAM-1 in a coronal brain section (n = 3–6). In a separate experiment, mice were treated as above. At 14 h after stress, brains were perfused, fixed, and labeled for ICAM-1 expression (n = 6). F, Representative images of ICAM-1 and Ly6C expression on blood vessels counterstained with DAPI in the dentate gyrus. Scale bar, 275 μm. Inset, Region used for analysis. Percentage area of ICAM-1 labeling in the (G) dentate gyrus and (H) PrL. Error bars indicate mean ± SEM. Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other. Inset, Region from which images were acquired.
We next determined mRNA expression of several key inflammatory mediators, IL-1β, TLR4, and ICAM-1, in a coronal brain section collected from the same mice used in the flow cytometric analysis above. The coronal section was used to evaluate the general inflammatory profile of the brain following stress and interventions. While ADX reduced TLR4 mRNA levels in the brain (Fig. 3C; F(1,12) = 74.89; p < 0.0001), there was no main effect of stress on TLR4 mRNA expression. Stress also increased IL-1β mRNA expression in the brain (Fig. 3D; F(1,35) = 20.47; p < 0.0001), but this increase was independent of ADX (Fig. 3D). Stress increased ICAM-1 levels in the brain (Fig. 3E; F(1,14) = 10.51; p < 0.01), and this induction was attenuated by ADX. Post hoc analysis revealed that ADX-stress mice tended to have lower mRNA expression of ICAM-1 compared with sham-stress mice (p = 0.06).
Last, ICAM-1 protein expression was determined with ADX and stress. The recruitment/accumulation of monocytes was associated with increased ICAM-1 expression in the brain endothelial cells. The selective expression of ICAM-1 labeling on endothelial cells was confirmed by colabeling with Ly6C. Ly6C is strongly expressed on endothelial cells and remains unaltered during inflammatory events (Jutila et al., 1988; Wohleb et al., 2013; Zhang et al., 2014; Liu et al., 2015). In the current study, Ly6C expression on the brain endothelial cells was unaffected by stress or ADX (data not shown). Here, ICAM-1 expression was increased with stress in the dentate gyrus (Fig. 3F,G; F(1,11) = 7.29; p < 0.05) and prelimbic cortex (PrL) (Fig. 3H; F(1,15) = 8.26; p < 0.05). Post hoc analysis confirmed that ICAM-1 expression was significantly higher in the sham-stress mice compared with ADX-stress in the dentate gyrus (p < 0.02) and tended to be higher in the PrL (p = 0.06). Collectively, increased monocyte recruitment/accumulation and ICAM-1 induction in the brain endothelial cells during stress were attenuated by ADX.
Stress-induced release of inflammatory monocytes from the bone marrow into circulation was attenuated by MTP
We show that ADX prevented the stress-induced release of inflammatory monocytes into circulation. ADX, however, may exert broad effects on homeostatic endocrine functioning (Cruz-Topete et al., 2016). Therefore, metyrapone (MTP) intervention was used during stress to ablate corticosterone production. MTP prevents corticosterone synthesis by inhibiting 11β-hydroxylase, the enzyme that converts inert corticosterone into active corticosterone (García-García et al., 2017). Here, mice were treated with vehicle or MTP 30 min before each cycle of social defeat, and several physiological and immune parameters were determined 14 h after stress. First, stress-induced neuronal activation (i.e., ΔFosB expression) within threat appraisal centers was determined with or without MTP intervention. Notably, ΔFosB (an isoform of FosB) is an immediate early gene detected in neurons (Perrotti et al., 2004) and has a longer half-life compared with other immediate early genes (e.g., c-Fos) (McClung et al., 2004). Therefore, it serves as a marker of cumulative neuronal activation over the 6 d of RSD stress (McKim et al., 2017). Here, we show that stress increased neuronal activation (i.e., ΔFosB expression) in the PrL (Fig. 4A,B; F(1,11) = 17.69, p < 0.01), and this increase was maintained independent of MTP intervention.
Figure 4.
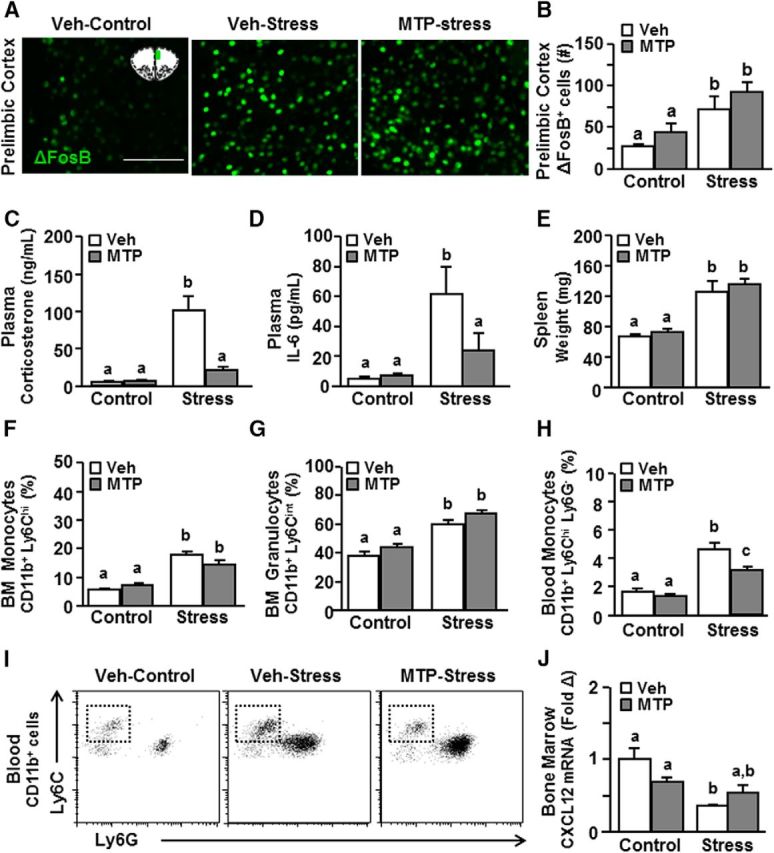
Stress-induced release of inflammatory monocytes from the bone marrow into circulation was attenuated by MTP. Male C57BL/6 mice were injected daily with either vehicle or MTP (100 mg/kg) 30 min before control or RSD (Stress). At 14 h after stress, brains were perfused, fixed, and labeled for ΔFosB. A, Representative images of ΔFosB expression in the prelimbic cortex (PrL). Scale bar, 125 μm. Inset, Region used for analysis. B, Average number of ΔFosB+ cells in the PrL (n = 3). C, Plasma corticosterone (n = 6). D, IL-6 levels (n = 3 or 4). E, Spleen weight (n = 6) was determined. Percentage of (F) CD11b+ Ly6Chi monocytes and (G) CD11b+ Ly6Cint granulocytes in the bone marrow. H, Percentage and (I) representative bivariate dot plots of CD11b+ and Ly6Chi labeling of monocytes in circulation. J, mRNA expression of CXCL12 in the bone marrow (n = 3 or 4). Error bars indicate mean ± SEM. Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other.
Next, plasma corticosterone and spleen weight were assessed after stress and MTP intervention. Similar to the ADX experiment, stress increased plasma corticosterone levels (Fig. 4C; F(1,22) = 22.21; p < 0.0002), and this increase was attenuated by MTP (Fig. 4C; stress × intervention, F(1,22) = 13.31, p < 0.002). Parallel to this, stress increased plasma IL-6 (Fig. 4D; F(1,14) = 7.38; p < 0.05), which was also attenuated by MTP. For example, post hoc analysis revealed that MTP-stress mice had significantly lower IL-6 levels in the plasma compared with vehicle-stress mice (p < 0.05). Stress also increased spleen weight (Fig. 4E; F(1,26) = 48.31; p < 0.0001), but this increase was independent of MTP. Together, MTP attenuated activation of the HPA axis during stress without affecting threat appraisal (neuronal activation) of the stressor.
Next, we assessed the effects of MTP intervention on monocyte production and release in response to stress. Consistent with the ADX results, stress increased the production of monocytes (CD11b+/Ly6Chi) and granulocytes (CD11b+/Ly6Cint) in the bone marrow (Fig. 4F,G; p < 0.001, for each), but these increases were unaffected by MTP intervention. Nonetheless, the stress-induced release of Ly6Chi monocytes in circulation (Fig. 4H,I; F(1,23) = 86.41; p < 0.0001) was attenuated by MTP intervention (Fig. 4H,I; stress × intervention, F(1,23) = 5.30; p < 0.05). More specifically, the percentage of circulating Ly6Chi monocytes was lower in the MTP-treated stress mice compared with the vehicle-treated stress mice (p < 0.0005), but these levels were still higher than in control mice (p < 0.0001). Last, we determined the effects of stress and MTP intervention on CXCL12 expression, a key retention factor for bone marrow stem cells and monocytes. Stress reduced CXCL12 mRNA levels in the bone marrow (Fig. 4J; F(1,9) = 11.99; p < 0.05) in an MTP-dependent manner (Fig. 4J; stress × intervention, F(1,9) = 5.62; p < 0.05). Post hoc analysis confirmed that CXCL12 mRNA levels in the bone marrow were significantly reduced in the vehicle-stress mice compared with vehicle-controls (p < 0.05). Moreover, CXCL12 mRNA levels in the bone marrow of MTP-stress mice were not different compared with MTP-controls (Fig. 4J). Overall, MTP intervention during stress reduced the release of inflammatory monocytes into circulation by increasing their retention in the bone marrow.
MTP attenuated stress-induced glucocorticoid resistance of splenocytes
We have previously reported that myeloid cells from the spleen of mice exposed to RSD have an exaggerated inflammatory response to LPS stimulation and a resistance to the suppressive effects of corticosterone (i.e., glucocorticoid resistance) (Stark et al., 2001; Hanke et al., 2012). Here, we examined the effects of MTP on stress-induced glucocorticoid resistance of splenocytes. In this experiment, splenocytes were collected 14 h after stress, cultured ex vivo, activated with LPS, and incubated with increasing concentrations of corticosterone. Fig. 5A shows that an increasing concentration of corticosterone from 0 μm (baseline) to 5 μm reduced the survival of LPS-activated splenocytes from vehicle-control and MTP-control groups. The LPS-activated splenocytes from vehicle-stress group had a higher rate of survival (i.e., glucocorticoid resistance) under all concentrations of corticosterone compared with vehicle-controls (p < 0.01 for each dose). Moreover, the ability of corticosterone to reduce survival of splenocytes was dependent on MTP intervention (Fig. 5A; stress × intervention, F(1,18) = 6.28; p < 0.05). For example, cell survival of the splenocytes from the MTP-stress group was reduced by corticosterone at 0.5 μm (p < 0.05) and 5 μm (p < 0.01) compared with the vehicle-stress group. These findings indicate that the increased glucocorticoid resistance of splenocytes following exposure to stress was attenuated by MTP intervention.
Figure 5.

MTP attenuated stress-induced glucocorticoid resistance of splenocytes. Male C57BL/6 mice were injected daily with either vehicle or MTP (100 mg/kg) 30 min before control or RSD (Stress). Splenocytes were collected 14 h later, and cultured ex vivo in presence of LPS (1 μg/ml) and increasing concentrations of corticosterone (0, 0.05, 0.1, 0.5, and 5 μm). A, Cell survival was determined 48 h after treatment, and results are expressed as percentage baseline of cell survival at 0 μm corticosterone (as indicated by the horizontal dashed line) (n = 6). B, Supernatant samples were collected from a duplicate preparation 18 h after the beginning of culture, and IL-6 levels were determined (n = 3). Error bars indicate mean ± SEM. Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other.
Next, to assess the effect of stress and MTP on the inflammatory capacity of splenocytes, IL-6 levels were determined in supernatants from the duplicate preparation of the same ex vivo cultures as described above. LPS-activated splenocytes from vehicle-stress mice produced significantly higher IL-6 in the presence of corticosterone compared with vehicle-control mice (p < 0.0001, for all concentrations). Furthermore, this IL-6 response was significantly reduced with MTP intervention (Fig. 5B; stress × intervention, F(1,11) = 6.72; p < 0.05). Compared with MTP-stress, LPS-activated splenocytes from vehicle-stress mice produced the highest IL-6 levels in the presence of corticosterone (p < 0.007 at 0 μm; p < 0.02 at 0.05 μm; p < 0.0003 at 0.1 μm; p < 0.0001 at 0.5 μm; and p < 0.001 at 5 μm). Overall, stress augmented the proinflammatory response of splenocytes to LPS, and these splenocytes were resistant to the anti-inflammatory actions of corticosterone. Furthermore, the stress-induced exaggerated inflammatory response to LPS was attenuated by MTP intervention.
MTP attenuated monocyte recruitment to the brain and prevented neuroinflammatory signaling
Next, the effects of stress and MTP intervention on microglial morphology, monocyte accumulation in the brain, and inflammation were assessed. As expected, stress increased the morphological restructuring of microglia (i.e., increased percentage area of Iba-1 labeling) in the dentate gyrus (Fig. 6A,B; F(1,11) = 9.17; p < 0.02). There was no difference in the morphological restructuring of microglia between MTP-treated control and MTP-treated stress mice (p = 0.29). Stress also increased microglial restructuring in the prelimbic cortex (PrL) (Fig. 6C; F(1,11) = 6.82; p < 0.05), and this increase was prevented by MTP intervention (Fig. 6C; stress × intervention, F(1,11) = 6.82; p < 0.05). For example, vehicle-treated stress mice showed higher Iba-1 expression of microglia compared with MTP-treated stress mice (p < 0.05). Therefore, MTP intervention during stress may lead to region-dependent reductions in the morphological alterations of microglia.
Figure 6.
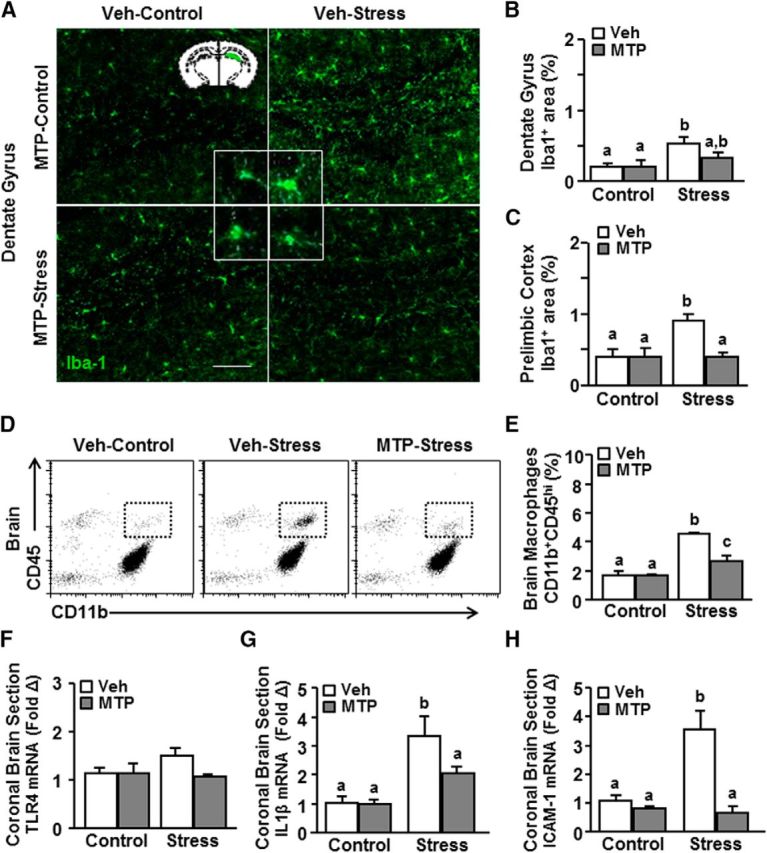
MTP attenuated monocyte recruitment to the brain and prevented neuroinflammatory signaling. Male C57BL/6 mice were injected daily with either vehicle or MTP (100 mg/kg) 30 min before control or RSD (Stress). At 14 h after stress, brains were perfused, fixed, and labeled for Iba-1. A, Representative images of Iba-1 expression in the dentate gyrus. Scale bar, 275 μm. Inset, Region used for analysis. Percentage area of Iba-1 labeling in the (B) dentate gyrus and (C) PrL (n = 3 or 4). In a separate experiment, mice were treated as above. D, Representative bivariate dot plots of CD45 and CD11b labeling of Percoll-enriched myeloid cells isolated from the brain. E, The percentage of CD45hi monocytes/macrophages in the enriched CD11b+ from the brain (n = 3). mRNA expression of (F) TLR4, (G) IL-1β, and (H) ICAM-1 in a coronal brain section (n = 3–6). Error bars indicate mean ± SEM. Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other.
Next, we examined monocyte recruitment to the brain with MTP intervention during stress. MTP attenuated the stress-induced accumulation of monocyte/macrophages (CD11b+/CD45hi) in the brain (Fig. 6D,E; stress × intervention, F(1,11) = 8.75; p < 0.001). For example, vehicle-stress mice had significantly more monocytes in the brain compared with MTP-stress mice (Fig. 6B; p < 0.005). MTP-stress mice, however, still had more monocytes in the brain than control mice (p < 0.05). In the same experiments, mRNA expression of inflammatory mediators, TLR4, IL-1β, and ICAM-1, were also determined in a coronal section of the brain. TLR4 mRNA levels were unaffected by either stress or MTP treatment (Fig. 6F). IL-1β mRNA levels were increased in the brain after stress (Fig. 6G; F(1,24) = 11.43; p < 0.0028), and this increase was attenuated by MTP (Fig. 6G; stress × intervention, F(1,24) = 4.19; p = 0.05). For example, vehicle-stress mice had significantly higher IL-1β mRNA levels in the brain (p < 0.01) compared with MTP-stress mice. ICAM-1 mRNA in the brain was also increased by stress (Fig. 6H; F(1,22) = 7.29; p < 0.01), and this increase was prevented by MTP (Fig. 6H; stress × intervention, F(1,22) = 20.96; p < 0.001). For instance, vehicle-stress mice had significantly higher ICAM-1 mRNA (p < 0.0001) compared with MTP-stress mice. Together, MTP prevented stress-induced morphological changes in microglia, recruitment of monocytes, and ICAM-1 and IL-1β mRNA induction in the brain.
MTP prevented stress-induced induction of endothelial ICAM-1 in the brain
Last, the effects of stress and MTP intervention on ICAM-1 induction were assessed. As indicated by ICAM-1 expression on Ly6C+ brain endothelial cells, there was a neurovascular induction of ICAM-1 protein by stress in the dentate gyrus (Fig. 7A,B; F(1,11) = 15.41; p < 0.006) and PrL (Fig. 7C; F(1,11) = 6.21; p < 0.05). Furthermore, this increase was attenuated by MTP intervention (Fig. 7A,B; stress × intervention, F(1,11) = 24.81, p < 0.002 for DG and Fig. 7C; F(1,11) = 7.09, p < 0.03 for PrL). Vehicle-stress mice had a higher ICAM-1 protein expression compared with MTP-stress mice in the dentate gyrus (p < 0.01) and in the PrL (p < 0.05). Thus, MTP intervention was effective in preventing stress-induced ICAM induction within threat appraisal centers.
Figure 7.

MTP prevented stress-induced induction of endothelial ICAM-1 in the brain. Male C57BL/6 mice were injected daily with either vehicle or MTP (100 mg/kg) 30 min before control or RSD (Stress). At 14 h after stress, brains were perfused, fixed, and labeled for ICAM-1 expression. A, Representative images of ICAM-1 and Ly6C expression on blood vessels counterstained with DAPI in the dentate gyrus. Scale bar, 275 μm. Inset, Region used for analysis. Percentage area of ICAM-1 labeling in the (B) dentate gyrus and (C) prelimbic cortex (PrL) (n = 3 or 4). Error bars indicate mean ± SEM. Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other.
Discussion
We and others have reported that RSD stress enhanced the production and release of inflammatory monocytes that accumulate in the brain (Wohleb et al., 2013; Menard et al., 2017). RSD also induced endothelial cell adhesion molecule expression and microglial activation, marked by the production of chemokines and proinflammatory mediators. This microglial activation was associated with the recruitment of monocytes that induced neuroinflammatory signaling and anxiety-like behavior (McKim et al., 2017). Activation of the peripheral immune compartments during RSD occurred through the SNS and the HPA axis (Hanke et al., 2012; Ramirez et al., 2016). Our objective here was to delineate the effects of HPA activation and corticosterone on immune dysregulation during RSD. We report that corticosterone produced during stress did not alter monocyte production in the bone marrow but promoted their release into circulation in a CXCL12-dependent manner. Corticosterone also enhanced neuroendothelial ICAM-1 expression that was associated with increased accumulation of monocytes and enhanced IL-1β production in the brain. Last, corticosterone production during stress was responsible for glucocorticoid resistance in the splenic myeloid cells.
An important confirmation from this study was that stress-induced HPA activation resided downstream of the threat appraisal circuitry. RSD increases the number of ΔFosB and cFos-positive cells in the threat appraisal regions, including PrL, hippocampus, and amygdala (Wohleb et al., 2011; McKim et al., 2017). Noradrenergic and GABAergic interventions (Wohleb et al., 2011; Ramirez et al., 2016) prevented this stress-induced threat appraisal activation, which also prevented monocyte release and accumulation in the brain. Because stress activates both the SNS and HPA axis, our goal was to intervene in the stress-response pathway at the level of corticosterone production, a physiological hallmark of stress response (Sapolsky et al., 2000). Corticosterone depletion prevented the increase in plasma corticosterone and IL-6 during stress but did not alter neuronal activation in the threat appraisal regions. These findings indicate that threat appraisal activation during stress preceded HPA activation and corticosterone production.
A key finding of this study was that corticosterone depletion prevented the stress-induced release of bone marrow-derived monocytes into circulation. It is important to note that enhanced production of monocytes with stress was maintained despite corticosterone depletion. These findings indicate that corticosterone depletion increased monocyte retention within the bone marrow during stress. In support of this conclusion, we show novel data that stress induced a profound reduction in CXCL12 mRNA and protein in the bone marrow. CXCL12 is a chemokine key in the retention of bone marrow cells. Disruption in the CXCL12/CXCR4 pathways caused significant impairments in cell mobilization from the bone marrow (Lévesque et al., 2003; Greenbaum et al., 2013). CXCL12 levels in the brain were not determined in the current study, but we have previously shown that brain CXCL12 levels remain unaltered by stress (Sawicki et al., 2015). In the current study, ADX and MTP intervention attenuated the stress-induced reduction of CXCL12 in the bone marrow, and this effect was associated with reduced monocyte release into circulation.
Our findings on the role of corticosterone in monocyte mobilization are novel because existing literature attributes mobilization of bone marrow cells to macrophages or noradrenergic nerve terminals at the bone marrow (Katayama et al., 2006; Chow et al., 2011). For instance, local sympathetic activity in the bone marrow was associated with increased production and release of hematopoietic stem cells and monocytes during steady state and chronic variable stress (Mendez-Ferrer et al., 2010; Heidt et al., 2014). Nonetheless, the mechanisms underlying mobilization of monocytes during psychological stress are unclear. For instance, acute psychological stress, but not adrenergic receptor activation, increased progenitor cell release into circulation (Riddell et al., 2015). Moreover, transgenic mice with deficits in hematopoietic stem cell mobilization recover normal functions when parabiotically paired with wild-type mice (Pierce et al., 2017). These studies showed that a blood-borne factor (i.e., corticosterone) mobilizes bone marrow stem cells into circulation (Pierce et al., 2017). Our findings here show that corticosterone plays an important role in mobilization of bone marrow monocytes during stress.
Related to the points above, monocyte production during stress has been attributed to enhanced sympathetic signaling in the bone marrow (Heidt et al., 2014). We have reported that interrupting the sympathetic pathway prevents production of monocytes and accumulation in tissues (Wohleb et al., 2011; Powell et al., 2013). In the current study, corticosterone depletion did not completely ablate monocyte release. Thus, the SNS likely remains at play. We propose that corticosterone does not contribute to enhanced monocyte production during stress but acts synergistically with the SNS to promote monocyte release into circulation.
Another noteworthy finding of this study was that corticosterone depletion attenuated ICAM-1 expression on the neurovascular endothelial cells in response to stress. We and others have previously reported that stress causes rapid induction of ICAM-1 on the vascular endothelium (Joachim et al., 2008; Sawicki et al., 2015). Blocking threat appraisal activation during stress prevented ICAM-1 induction (McKim et al., 2017). Nonetheless, preventing microglial activation or eliminating microglia did not alter the stress-induced increase in ICAM-1 (McKim et al., 2017). Therefore, we hypothesized that corticosterone, which is produced rapidly during stress, promotes ICAM-1 induction in the neurovascular endothelium. In support of this hypothesis, both ADX and MTP intervention prevented the stress-induced increase in ICAM-1. Corresponding with reduced ICAM-1 expression, the percentage of CD45hi monocytes in the brain was also reduced with corticosterone depletion. Collectively, corticosterone depletion during stress prevents monocyte accumulation in the brain by limiting their release into circulation and by reducing their adherence to ICAM-1 on the brain endothelial cells.
It is also relevant to discuss that reduced monocyte accumulation in the brain with MTP was associated with reduced IL-1β mRNA expression. This is consistent with our previous studies showing that monocytes accumulate in the brain during stress and propagate IL-1 signaling through the endothelial IL-1-receptor1 on the brain vasculature (McKim et al., 2017).
In addition, monocyte accumulation and increased IL-1β expression may further augment microglial activation with stress. This was apparent in the PrL where MTP attenuated monocyte accumulation in the brain and prevented morphological alterations of microglia. In the adrenalectomized mice, microglial activation was not assessed and stress-induced IL-1β mRNA was maintained despite reduction of monocyte accumulation. This discrepancy in IL-1β expression between ADX and MTP treatment may be attributed to broader effects of ADX (Cruz-Topete et al., 2016). For instance, adrenalectomized, but not MTP-treated, mice had reduced CX3CR1, nr4a1, and P2X7 mRNA expression in the brain at baseline (data not shown). Thus, ADX may introduce confounds that influence CNS homeostasis. Nonetheless, our overall results support the conclusion that corticosterone induction with RSD is critical in the release of pro-inflammatory and glucocorticoid resistant monocytes that subsequently accumulate in the brain and augment neuroinflammatory signaling.
It is also important to note that MTP blocks corticosterone production by preventing the conversion of its precursor, 11β-deoxycorticosterone, levels of which are increased following MTP treatment. Although 11β-deoxycorticosterone may exert aldosterone-like effects, its potency is low (2% potency as aldosterone) and is not expected to cause significant effects (Gomez-Sanchez et al., 2014). Furthermore, 11β-deoxycorticosterone levels would not be increased with ADX. Indeed, our findings from the ADX and MTP experiments are consistent with each other. Thus, the results of this study were not confounded by increased 11β-deoxycorticosterone levels with MTP treatment.
Another important finding here was that corticosterone depletion prevented stress-induced glucocorticoid resistance in the splenocytes. Glucocorticoids suppress inflammatory signaling by preventing transcription and post-translational modification of inflammatory genes and by triggering apoptosis of immune cells (Smoak et al., 2004). Chronic stress in humans blunts the ability of glucocorticoids to suppress inflammatory signaling, and increases risk for viral infections, cardiovascular, and other chronic inflammatory conditions (Cohen et al., 2012; Fagundes et al., 2013; Heidt et al., 2014; Miller et al., 2014). Glucocorticoid resistance in RSD was shown in splenocytes that were resistant to the apoptotic effects of corticosterone and produced exaggerated levels of IL-6 in response to LPS (Stark et al., 2001; Quan et al., 2003). This glucocorticoid resistance is caused by a failure of the glucocorticoid receptor in myeloid cells to translocate into the nucleus (Quan et al., 2003). Nonetheless, the role of corticosterone on glucocorticoid resistance in these myeloid cells was unknown. Here, we provide novel data that glucocorticoid resistance of myeloid cells was caused by overproduction of corticosterone during stress. Indeed, stress-induced glucocorticoid resistance of myeloid cells was prevented by corticosterone depletion with MTP. Thus, HPA activation and corticosterone production during stress induce glucocorticoid resistance in myeloid cells.
In conclusion, we shed light on the specific role of corticosterone in the immunomodulatory effects of RSD. We show, for the first time, that corticosterone production during stress contributes to monocyte mobilization from the bone marrow by reducing CXCL12. Furthermore, corticosterone also increases neurovascular ICAM-1 expression during stress that facilitates monocyte adherence to vasculature. Finally, our findings indicate that corticosterone production during RSD is the cause of glucocorticoid resistance in myeloid cells. These findings have implications not only in stress-induced neuropsychiatric conditions, but also in cardiovascular and inflammatory disorders associated with stress.
Footnotes
This work was supported by National Institute of Health Grants R01-MH-093473 and R01-MH097243 to J.F.S. and National Institute of Aging Grant R01-AG033028 to J.P.G. We thank Rahul Gupta for his technical assistance.
The authors declare no competing financial interests.
References
- Avitsur R, Stark JL, Sheridan JF (2001) Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav 39:247–257. 10.1006/hbeh.2001.1653 [DOI] [PubMed] [Google Scholar]
- Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N, Tanaka M, Merad M, Frenette PS (2011) Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 208:261–271. 10.1084/jem.20101688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, Turner RB (2012) Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U S A 109:5995–5999. 10.1073/pnas.1118355109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Topete D, Myers PH, Foley JF, Willis MS, Cidlowski JA (2016) Corticosteroids are essential for maintaining cardiovascular function in male mice. Endocrinology 157:2759–2771. 10.1210/en.2015-1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS, Malarkey WB, Neri E, McEwen BS (2012) Stress-induced redistribution of immune cells—from barracks to boulevards to battlefields: a tale of three hormones—Curt Richter Award winner. Psychoneuroendocrinology 37:1345–1368. 10.1016/j.psyneuen.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagundes CP, Glaser R, Kiecolt-Glaser JK (2013) Stressful early life experiences and immune dysregulation across the lifespan. Brain Behav Immun 27:8–12. 10.1016/j.bbi.2012.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-García L, Shiha AA, Fernández de la Rosa R, Delgado M, Silván Á, Bascuñana P, Bankstahl JP, Gomez F, Pozo MA (2017) Metyrapone prevents brain damage induced by status epilepticus in the rat lithium-pilocarpine model. Neuropharmacology 123:261–273. 10.1016/j.neuropharm.2017.05.007 [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez E, Gomez-Sanchez CE (2014) The multifaceted mineralocorticoid receptor. Compr Physiol 4:965–994. 10.1002/cphy.c130044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC (2013) CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495:227–230. 10.1038/nature11926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF (2012) Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun 26:1150–1159. 10.1016/j.bbi.2012.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur Muhlen C, Bode C, Fricchione GL, Denninger J, Lin CP, Vinegoni C, Libby P, Swirski FK, Weissleder R, Nahrendorf M (2014) Chronic variable stress activates hematopoietic stem cells. Nat Med 20:754–758. 10.1038/nm.3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson L, Akana SF, Cascio CS, Shinsako J, Dallman MF (1988) Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology 122:1343–1348. 10.1210/endo-122-4-1343 [DOI] [PubMed] [Google Scholar]
- Joachim RA, Handjiski B, Blois SM, Hagen E, Paus R, Arck PC (2008) Stress-induced neurogenic inflammation in murine skin skews dendritic cells towards maturation and migration: key role of intercellular adhesion molecule-1/leukocyte function-associated antigen interactions. Am J Pathol 173:1379–1388. 10.2353/ajpath.2008.080105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutila MA, Kroese FG, Jutila KL, Stall AM, Fiering S, Herzenberg LA, Berg EL, Butcher EC (1988) Ly-6C is a monocyte/macrophage and endothelial cell differentiation antigen regulated by interferon-gamma. Eur J Immunol 18:1819–1826. 10.1002/eji.1830181125 [DOI] [PubMed] [Google Scholar]
- Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS (2006) Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124:407–421. 10.1016/j.cell.2005.10.041 [DOI] [PubMed] [Google Scholar]
- Kendler KS, Karkowski LM, Prescott CA (1999) Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry 156:837–841. 10.1176/ajp.156.6.837 [DOI] [PubMed] [Google Scholar]
- Lehmann ML, Brachman RA, Martinowich K, Schloesser RJ, Herkenham M (2013) Glucocorticoids orchestrate divergent effects on mood through adult neurogenesis. J Neurosci 33:2961–2972. 10.1523/JNEUROSCI.3878-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ (2003) Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest 111:187–196. 10.1172/JCI15994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yamashita T, Chen Q, Belevych N, McKim DB, Tarr AJ, Coppola V, Nath N, Nemeth DP, Syed ZW, Sheridan JF, Godbout JP, Zuo J, Quan N (2015) Interleukin 1 type 1 receptor restore: a genetic mouse model for studying interleukin 1 receptor-mediated effects in specific cell types. J Neurosci 35:2860–2870. 10.1523/JNEUROSCI.3199-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M, Lin AH, Delmeire L, Van Gastel A, Kenis G, De Jongh R, Bosmans E (1999) Elevated serum interleukin-6 (IL-6) and IL-6 receptor concentrations in posttraumatic stress disorder following accidental man-made traumatic events. Biol Psychiatry 45:833–839. 10.1016/S0006-3223(98)00131-0 [DOI] [PubMed] [Google Scholar]
- Maes M, Song C, Yirmiya R (2012) Targeting IL-1 in depression. Expert Opin Ther Targets 16:1097–1112. 10.1517/14728222.2012.718331 [DOI] [PubMed] [Google Scholar]
- Marin MF, Lord C, Andrews J, Juster RP, Sindi S, Arsenault-Lapierre G, Fiocco AJ, Lupien SJ (2011) Chronic stress, cognitive functioning and mental health. Neurobiol Learn Mem 96:583–595. 10.1016/j.nlm.2011.02.016 [DOI] [PubMed] [Google Scholar]
- McClung CA, Ulery PG, Perrotti LI, Zachariou V, Berton O, Nestler EJ (2004) DeltaFosB: a molecular switch for long-term adaptation in the brain. Brain Res Mol Brain Res 132:146–154. 10.1016/j.molbrainres.2004.05.014 [DOI] [PubMed] [Google Scholar]
- McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP (2016a) Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J Neurosci 36:2590–2604. 10.1523/JNEUROSCI.2394-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim DB, Patterson JM, Wohleb ES, Jarrett BL, Reader BF, Godbout JP, Sheridan JF (2016b) Sympathetic release of splenic monocytes promotes recurring anxiety following repeated social defeat. Biol Psychiatry 79:803–813. 10.1016/j.biopsych.2015.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, Ramirez-Chan K, Wang Y, Roeth RM, Sucaldito AD, Sobol CG, Quan N, Sheridan JF, Godbout JP (2017) Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry. Advance online publication. Retrieved Apr. 4, 2017. doi: 10.1038/mp.2017.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, Takahashi A, Flanigan ME, Aleyasin H, LeClair KB, Janssen WG, Labonté B, Parise EM, Lorsch ZS, Golden SA, Heshmati M, Tamminga C, Turecki G, Campbell M, Fayad ZA, et al. (2017) Social stress induces neurovascular pathology promoting depression. Nat Neurosci 20:1752–1760. 10.1038/s41593-017-0010-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Battista M, Frenette PS (2010) Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci 1192:139–144. 10.1111/j.1749-6632.2010.05390.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Cohen S, Ritchey AK (2002) Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol 21:531–541. 10.1037/0278-6133.21.6.531 [DOI] [PubMed] [Google Scholar]
- Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, Ma R, Cole SW (2008) A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry 64:266–272. 10.1016/j.biopsych.2008.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Murphy ML, Cashman R, Ma R, Ma J, Arevalo JM, Kobor MS, Cole SW (2014) Greater inflammatory activity and blunted glucocorticoid signaling in monocytes of chronically stressed caregivers. Brain Behav Immun 41:191–199. 10.1016/j.bbi.2014.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM (2006) Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163:1630–1633. 10.1176/ajp.2006.163.9.1630 [DOI] [PubMed] [Google Scholar]
- Pasquali R. (2012) The hypothalamic-pituitary-adrenal axis and sex hormones in chronic stress and obesity: pathophysiological and clinical aspects. Ann N Y Acad Sci 1264:20–35. 10.1111/j.1749-6632.2012.06569.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti LI, Hadeishi Y, Ulery PG, Barrot M, Monteggia L, Duman RS, Nestler EJ (2004) Induction of δFosB in reward-related brain structures after chronic stress. J Neurosci 24:10594–10602. 10.1523/JNEUROSCI.2542-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce H, Zhang D, Magnon C, Lucas D, Christin JR, Huggins M, Schwartz GJ, Frenette PS (2017) Cholinergic signals from the CNS regulate G-CSF-mediated HSC mobilization from bone marrow via a glucocorticoid signaling relay. Cell Stem Cell 20:648–658.e4. 10.1016/j.stem.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, Kobor MS, Reader BF, Sheridan JF, Cole SW (2013) Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A 110:16574–16579. 10.1073/pnas.1310655110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan N, Avitsur R, Stark JL, He L, Shah M, Caligiuri M, Padgett DA, Marucha PT, Sheridan JF (2001) Social stress increases the susceptibility to endotoxic shock. J Neuroimmunol 115:36–45. 10.1016/S0165-5728(01)00273-9 [DOI] [PubMed] [Google Scholar]
- Quan N, Avitsur R, Stark JL, He L, Lai W, Dhabhar F, Sheridan JF (2003) Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. J Neuroimmunol 137:51–58. 10.1016/S0165-5728(03)00042-0 [DOI] [PubMed] [Google Scholar]
- Ramirez K, Niraula A, Sheridan JF (2016) GABAergic modulation with classical benzodiazepines prevent stress-induced neuro-immune dysregulation and behavioral alterations. Brain Behav Immun 51:154–168. 10.1016/j.bbi.2015.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reader BF, Jarrett BL, McKim DB, Wohleb ES, Godbout JP, Sheridan JF (2015) Peripheral and central effects of repeated social defeat stress: monocyte trafficking, microglial activation, and anxiety. Neuroscience 289:429–442. 10.1016/j.neuroscience.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell NE, Burns VE, Wallace GR, Edwards KM, Drayson M, Redwine LS, Hong S, Bui JC, Fischer JC, Mills PJ, Bosch JA (2015) Progenitor cells are mobilized by acute psychological stress but not beta-adrenergic receptor agonist infusion. Brain Behav Immun 49:49–53. 10.1016/j.bbi.2015.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU (2000) How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 21:55–89. 10.1210/edrv.21.1.0389 [DOI] [PubMed] [Google Scholar]
- Sawicki CM, McKim DB, Wohleb ES, Jarrett BL, Reader BF, Norden DM, Godbout JP, Sheridan JF (2015) Social defeat promotes a reactive endothelium in a brain region-dependent manner with increased expression of key adhesion molecules, selectins and chemokines associated with the recruitment of myeloid cells to the brain. Neuroscience 302:151–164. 10.1016/j.neuroscience.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoak KA, Cidlowski JA (2004) Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 125:697–706. 10.1016/j.mad.2004.06.010 [DOI] [PubMed] [Google Scholar]
- Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM (2009) The stressed CNS: when glucocorticoids aggravate inflammation. Neuron 64:33–39. 10.1016/j.neuron.2009.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF (2001) Social stress induces glucocorticoid resistance in macrophages. Am J Physiol Regul Integr Comp Physiol 280:R1799–R1805. 10.1152/ajpregu.2001.280.6.R1799 [DOI] [PubMed] [Google Scholar]
- Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N (2014) Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun 42:50–59. 10.1016/j.bbi.2014.05.007 [DOI] [PubMed] [Google Scholar]
- Walker BR. (2007) Glucocorticoids and cardiovascular disease. Eur J Endocrinol 157:545–559. 10.1530/EJE-07-0455 [DOI] [PubMed] [Google Scholar]
- Weber MD, Godbout JP, Sheridan JF (2017) Repeated social defeat, neuroinflammation, and behavior: monocytes carry the signal. Neuropsychopharmacology 42:46–61. 10.1038/npp.2016.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, Nelson RJ, Godbout JP, Sheridan JF (2011) β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci 31:6277–6288. 10.1523/JNEUROSCI.0450-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP (2012) Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 37:1491–1505. 10.1016/j.psyneuen.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF (2013) Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 33:13820–13833. 10.1523/JNEUROSCI.1671-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, McKim DB, Sheridan JF, Godbout JP (2014a) Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci 8:447. 10.3389/fnins.2014.00447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, Godbout JP (2014b) Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry 75:970–981. 10.1016/j.biopsych.2013.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Kusnecov AW, Shurin MR, DePaoli M, Rabin BS (1993) Exposure to physical and psychological stressors elevates plasma interleukin 6: relationship to the activation of hypothalamic-pituitary-adrenal axis. Endocrinology 133:2523–2530. 10.1210/endo.133.6.8243274 [DOI] [PubMed] [Google Scholar]