

Abstract
Nuclear receptor coactivator 3 (NCOA3) is a transcriptional coactivator that has elevated expression in multiple tumor types, including colorectal cancer (CRC). However, the molecular mechanisms that regulate the tumorigenic functions of NCOA3 in CRC remain largely unknown. In this study, we aimed to discover and identify the novel regulatory proteins of NCOA3 and explore their mechanisms of action. Immunoprecipitation (IP) coupled with mass spectrometry (IP‐MS) analysis was used to detect, identify, and verify the proteins that interacted with NCOA3 in CRC cells. The biological functions of the candidate proteins and the underlying molecular mechanism were investigated in CRC cells and mouse model in vitro and in vivo. The clinical significance of NCOA3 and its interaction partner protein in CRC patients was also studied. We identified mitotic arrest deficient 2‐like protein 2 (MAD2L2, also known as MAD2B or REV7), with two signal peptide sequences of LIPLK and EVYPVGIFQK, to be an interaction partner of NCOA3. Overexpression of MAD2L2 suppressed the proliferation, migration, and clonogenicity of CRC cells by inducing the degradation of NCOA3. The mechanism study showed that increased MAD2L2 expression in CRC cells activated p38, which was required for the phosphorylation of NCOA3 that led to its ubiquitination and degradation by the proteasome. Moreover, we found that MAD2L2 predicted favorable prognosis in CRC patients. We have discovered a novel role of MAD2L2 in the regulation of NCOA3 degradation and proposed that MAD2L2 serves as a tumor suppressor in CRC.
Keywords: colorectal cancer, degradation, MAD2L2, NCOA3, ubiquitination
Abbreviations
- ANT
adjacent normal tissues
- CHX
cycloheximide
- CIN
chromosomal instability
- CRC
colorectal cancer
- ERK
Extracellular signal‐regulated kinase
- ESCs
embryonic stem cells
- HAT
histone acetyltransferase
- HR
homologous recombination
- IHC
Immunohistochemical
- JNK
c‐Jun N‐terminal protein kinase
- LOH
loss of heterozygosity
- MAD2L2
mitotic arrest deficient 2‐like protein 2
- MMS
Methyl methanesulfonate
- MS
mass spectrometry
- MSI
microsatellite instability
- NCOA3
Nuclear receptor coactivator 3
- NHEJ
nonhomologous end joining
- TLS
translesion synthesis
1. Introduction
Colorectal cancer (CRC) is the second most common cancer in women and the third most common in men (Tariq and Ghias, 2016), with global incidence, mortality, and 5‐year prevalence of 9.7%, 8.5%, and 10.9%, respectively, according to GLOBOCAN 2012 (Ferlay et al., 2015). The molecular mechanisms in colorectal cancer development include chromosomal instability (CIN) (Grady and Carethers, 2008), microsatellite instability (MSI) (Walther et al., 2009), and CpG island methylation (CIMP) (Issa et al., 2005), and different mechanisms can individually or simultaneously exist in colorectal cancer. CIN is the major pathway in CRC pathogenesis, mainly caused by replication stress‐related chromosomal breaks, with structural chromosome abnormalities precipitating chromosome missegregation in mitosis (Burrell et al., 2013). It is associated with 65%–75% of sporadic CRC (Pino and Chung, 2010) and featured with an abnormal number of chromosomes (aneuploidy) and loss of heterozygosity (LOH) (Lin et al., 2003). Moreover, mutations of a group of specific tumor suppressor genes and oncogenes, such as KRAS, PIK3CA, APC, TP53, and so forth, are found to accumulate in CRCs with the typical karyotypic abnormalities caused by CIN (Colussi et al., 2013), though it remains unclear whether these mutations initiate CIN or vice versa.
Nuclear receptor coactivator 3 (NCOA3, also known as SRC3, AIB1, RAC3, or ACTR) is a member of the p160/SRC coactivator family and has intrinsic histone‐acetyltransferase (HAT) activity. It binds nuclear receptors in a hormone‐dependent fashion, remodels chromatin DNA to become more accessible to the transcription machinery, recruits additional transcription factors and coregulators, and thus functions as a central player in the assembly of a coactivator complex to promote gene expression (Anzick et al., 1997; Chen et al., 1997, 1999; Li et al., 1997; Xu et al., 2009). NCOA3 has been found to be elevated in breast cancer, liver cancer, prostate cancer, and colorectal cancer, correlated with poor prognosis in most cases, and a vital regulator in the process of tumorigenesis, progression, metastasis, and survival (Anzick et al., 1997; Chen et al., 2012; Shi et al., 2015; Xie et al., 2005; Xu et al., 2010; Zhou et al., 2005). The breast cancer, gastric cancer, and nonsmall cell lung cancer patients with high expression of NCOA3 have significantly shorter overall survival times, indicating that NCOA3 may be an indicator of poor prognosis (Cai et al., 2010; Sakakura et al., 2000; Zhao et al., 2003). However, some studies have reported that amplification of NCOA3 appears to be independently associated with poor prognosis in patients with hepatocellular carcinoma, and moderate or high expression of NCOA3 is associated with poor disease‐specific survival in patients with prostate disease (Gnanapragasam et al., 2001; Song et al., 2012). Nevertheless, the molecular mechanisms that regulate the tumorigenic functions of NCOA3 in CRC remain unclear so far. To identify protein regulators that interact with NCOA3 in CRC cells, we performed immunoprecipitation coupled with mass spectrometry (IP‐MS) and found mitotic arrest deficient 2‐like protein 2 (MAD2L2, also known as MAD2B or REV7) to be one of the candidates.
Mitotic arrest deficient 2‐like protein 2 is a multifunctional protein with roles in DNA damage repair, cell cycle regulation, gene expression, and carcinogenesis. MAD2L2 was originally identified as a subunit of DNA polymerase ζ critical for DNA translesion synthesis (TLS) (Murakumo et al., 2001) and a component of the mitotic spindle assembly checkpoint that inhibits the anaphase‐promoting complex (Chen and Fang, 2001). Recent studies have revealed that MAD2L2 blocks homologous recombination (HR) and promotes nonhomologous end joining (NHEJ) by inhibiting 5’ end resection downstream of 53BP1 and RIF1 and thus functions in DSB repair pathway choices (Boersma et al., 2015) (Xu et al., 2015). Besides, MAD2L2 has been reported to promote Elk‐1 phosphorylation by c‐Jun N‐terminal protein kinase (JNK) and thus lead to the up‐regulation of Elk‐1 target genes in the presence of DNA damage, which suggests that MAD2L2 might be a central player in coordinating the cellular response to DNA damage (Zhang et al., 2007). Moreover, MAD2L2 has been reported to regulate the epigenetic reprogramming of germ cells (Watanabe et al., 2013; Zhang et al., 2007) and the maintenance of pluripotency in embryonic stem cells (ESCs) (Pirouz et al., 2015), and promote the open chromatin configuration through DPPA3 in ESCs (Rahjouei et al., 2017). Accordingly, it is not surprising that dysregulation of MAD2L2 has been found in multiple cancers. For instance, MAD2L2 was overexpressed in glioma, epithelial ovarian cancer, and breast cancer (Feng et al., 2016; Niimi et al., 2014; Zhao et al., 2011), while inactivation of MAD2L2 sensitized nasopharyngeal carcinoma cells to DNA‐damaging agents (Cheung et al., 2006).
Our study identified MAD2L2 as an interaction protein of NCOA3 and revealed that MAD2L2 suppressed CRC growth both in vitro and in vivo. Our clinical data indicated that high expression of MAD2L2 was associated with good prognosis in CRC patients. Noteworthily, we showed for the first time that MAD2L2 inhibited CRC development by down‐regulating the protein level of NCOA3. We further demonstrated that increased MAD2L2 expression in CRC cells activated p38, which phosphorylated NCOA3 for its subsequent degradation by the ubiquitin–proteasome pathway. Our study has discovered a novel role of MAD2L2 in regulating the degradation of NCOA3 and suggested that MAD2L2 functions as a tumor suppressor in CRC.
2. Materials and methods
2.1. Cell culture and chemicals
The human FHC, SW620, SW480, HCT116, HT29, RKO, and DLD1 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). FHC cells were cultured in DMEM, and the other cells were cultured in RPMI‐1640 supplemented with 10% fetal bovine serum, 100 units·mL−1 penicillin, and 100 μg·mL−1 streptomycin. All cells were maintained in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37 °C.
MG132, SB203580, cisplatin, and cycloheximide (CHX) were purchased from Sigma (St. Louis, MO, USA) and dissolved in DMSO. The stock solution of MG132 and SB203580 was 10 mm, and the stock solution of CHX was 10 mg·mL−1. Methyl methanesulfonate (MMS) was dissolved in PBS, and the concentration of the stock solution was 100 mm. All the stock solutions were stored at −20 °C before use.
2.2. siRNA and stable cell lines
The MAD2L2 siRNA and flag‐tagged MAD2L2 overexpression adenovirus were purchased from GenePharma Co., Ltd (Suzhou, China). MAD2L2 knockdown adenovirus was purchased from Hanbio Biotechnology Co., Ltd (Shanghai, China). NCOA3 overexpression adenovirus was purchased from GeneChem (Shanghai, China). Cells were transfected with siRNA duplexes (100 nm) using EndoFectinTM MAX (GeneCopoeia, Inc., Rockville, MD, USA). HCT116 and SW480 cells were used to establish stable cell lines by selection with 1 μg·mL−1 puromycin for 4 weeks.
2.3. Cell proliferation
Cell viability was determined by MTS assay (Promega Biotech Co., Ltd., Madison, WI, USA). Cells were seeded in 96‐well plates (10 000 cells/well) 24 h after MAD2L2 siRNA transfection. Cell viability was detected 48 h after transfection. Cell viability of stable cell lines with MAD2L2 overexpression was detected 48 h after plating in 96‐well plates (5000 cells/well).
2.4. Scratch assay
Cells were transfected with MAD2L2 siRNA, seeded in 6‐well plates, and cultured overnight to a density of 70%–80%. Cell monolayers were scratched with a 100 μL pipette tip and washed with PBS two times to remove detached cells. The scratches were imaged using an Olympus microscope at 0 h, 36 h, and 48 h, respectively, according to their growth rate. The widths of the gap at 0 h (w1) and 36 h or 48 h (w2) were measured, and the relative migration rate was calculated as (w1‐w2)/w1 * 100%.
2.5. Tumor‐induced clonogenicity assay
Different stable cell lines were seeded in 6‐well plates (500 cells/well) and incubated. Two weeks later, cells were fixed with formalin and stained by crystal violet. The images of the clones were captured, and the numbers of the clones were counted by the software Image‐Pro Plus 6.0.
2.6. RNA extraction and quantitative RT‐PCR (qRT‐PCR)
Total RNA from cells was extracted using RaPure Total RNA Micro Kit (Magen, Guangzhou, China). Endogenous cDNA was generated using ReverTra Ace® qPCR RT Master Mix kit (ToYoBo, Shanghai, China). The primers for qRT‐PCR were purchased from GeneCopoeia, Inc. (Rockville, MD, USA): MAD2L2 (HQP000552), NCOA3 (HQP020041), and GAPDH (HQP006940). qRT‐PCR was performed with the SYBR® Green Real‐time PCR Master Mix (ToYoBo, Shanghai, China).
2.7. Antibodies and western blot analysis
Equal amounts of protein lysates were separated by SDS/PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were sequentially incubated with primary and secondary antibodies, and the protein bands were detected by enhanced chemiluminescence. Anti‐MAD2L2, anti‐GAPDH, and anti‐Flag were purchased from Proteintech (Wuhan, China); anti‐NCOA3, anti‐p‐p38, anti‐p‐ERK1/2, anti‐p‐JNK, anti‐ubiquitin, and anti‐histone H3 were from Cell Signaling Technology (Danvers, MA, USA); and antiphosphoserine/threonine was from Bioss (Woburn, MA, USA).
2.8. Co‐immunoprecipitation (co‐IP) assays
Protein extracts were prepared and incubated with the antibodies for NCOA3, flag, or IgG for 24 h at 4 °C on a rotating wheel. Then, the sepharose‐conjugated protein‐A/G beads (Santa Cruz Biotechnology, Dallas, TX, USA) were added and incubated at 4 °C for another 24 h on a rotating wheel. After extensive washings with cold PBS containing PMSF, the beads were boiled, and the precipitated proteins were separated by SDS/PAGE and transferred to PVDF membranes for further analysis.
2.9. Silver staining and mass spectroscopy (MS)
After electrophoresis, the protein gel was immersed in stationary liquid with 10% acetic acid, 50% ethanol, and 40% water at room temperature on shaker overnight, and then, the protein bands were visualized by the Fast Silver Stain Kit (Beyotime, Haimen, China) and analyzed by MS by Honortech (Beijing, China).
2.10. Animal experiments
Female BALB/c nude mice (4 weeks old) were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and quarantined for 1 week before use for tumor formation experiments. All animal experiment procedures were approved by the Animal Care and Use Committee of Sun Yat‐sen University, and every effort was made to reduce the suffering of animals. 3 × 106 cells were suspended in 100 μL of PBS and subcutaneously injected into BALB/c mice. The weight of the mice and the volume of the tumors were measured every 2 days for 3 weeks. At the end of the experiments, the mice were sacrificed, and the tumors were excised, photographed, and processed for immunohistochemical analyses.
2.11. Immunohistochemistry (IHC)
Tissue microarrays with 190 samples were purchased from Outdo Biotech Co., Ltd (Shanghai, China). The microarrays were incubated with anti‐MAD2L2 and anti‐NCOA3 primary antibodies and secondary antibodies, and after color development, scoring was done. The antibodies’ specificity against MAD2L2 and NCOA3 was detected in Fig. S1.
2.12. Statistical analysis
Statistical analyses were performed using the SPSS statistical software package (version 17.0). Chi‐square test and t‐test were applied for variance analysis, Spearman rank correlation method was for correlation analysis, and Kaplan–Meier analysis was for survival analysis. The mean ± SEM was calculated by GraphPad Prism 6.0 and presented in graphs. P < 0.05 was considered statistically significant.
3. Results
3.1. MAD2L2 was inversely correlated with NCOA3 and predicted favorable prognosis in colorectal cancer (CRC) patients
To identify protein regulators that interacted with NCOA3 in CRC cells, we performed immunoprecipitation combined with mass spectrometry (IP‐MS) in SW620, HCT116, and HT29 CRC cells with IgG control or NCOA3 antibodies. MAD2L2, with two signal peptide sequences of LIPLK and EVYPVGIFQK, was found to be a candidate interacting with NCOA3 (Fig. 1A), and the interaction between NCOA3 and MAD2L2 was further confirmed by co‐IP in HCT116 cells (Fig. 1B). Next, the expression of MAD2L2 and NCOA3 in a panel of CRC cell lines were detected by western blot, and the basic expression levels of MAD2L2 and NCOA3 in CRC cells tend to be inversely correlated (Fig. 1C).
Figure 1.
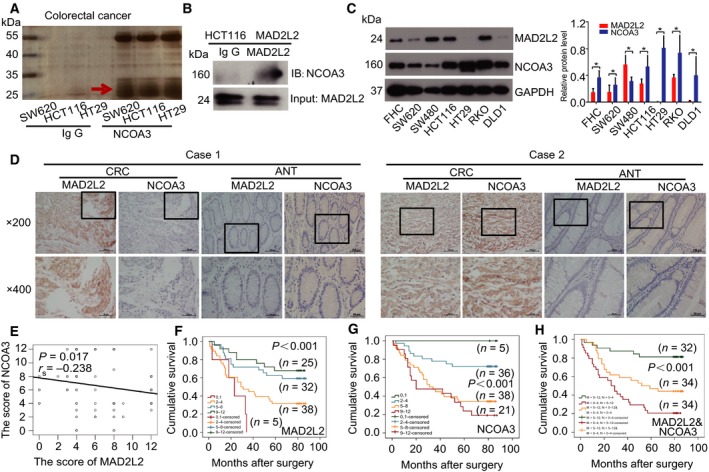
MAD2L2 expression was inversely correlated with NCOA3 expression and predicted favorable prognosis in CRC patients. (A) NCOA3 interaction partners were identified in SW620, HCT116, and HT29 cells stably overexpressing NCOA3 by IP‐MS. (B) The interaction between MAD2L2 and NCOA3 was confirmed by co‐IP in HCT116 cells with stable MAD2L2 overexpression. (C) The basic expression of MAD2L2 and NCOA3 in FHC colon epithelial cells and different CRC cells (SW620, SW480, HCT116, HT29, RKO, and DLD1) and the relative protein level in different CRC cells were shown in C right. (D) Representative images of MAD2L2 and NCOA3 expression in CRC tissues and adjacent normal tissues (ANT). (E) The expression of MAD2L2 and NCOA3 was negatively correlated with CRC tissues. (F) High expression of MAD2L2 was associated with good prognosis in CRC patients. (G) High expression of NCOA3 was related to poor prognosis in CRC patients. (H) High expression of MAD2L2 with low expression of NCOA3 (green curve) predicted favorable prognosis in CRC patients, compared with low expression of MAD2L2 and high expression of NCOA3 (red curve), along with both low and high expression of MAD2L2 and NCOA3 (yellow curve). M means MAD2L2 and N means NCOA3.
We then examined whether the expression of MAD2L2 and NCOA3 was also correlated with CRC patients. Immunohistochemical (IHC) analysis revealed that both proteins had significantly higher expression in CRC tissues than in adjacent normal tissues (ANT) (P < 0.001, Table 1), and the patients with high MAD2L2 expression tend to have a low level of NCOA3 (Fig. 1D case 1), while the patients with low MAD2L2 expression tend to have a high level of NCOA3 (Fig. 1D case 2), and the Spearman rank correlation analysis showed that MAD2L2 was in inverse correlation with NCOA3 (r s = −0.238, P = 0.017) (Fig. 1E). Moreover, the Kaplan–Meier survival analysis of 100 CRC patients showed that high levels of MAD2L2 predicted favorable prognosis (Fig. 1F), high expression of NCOA3 was associated with poor prognosis (Fig. 1G), and patients with high MAD2L2 combined with low NCOA3 had the best outcome, whereas patients with low MAD2L2 combined with high NCOA3 had the worst outcome (Fig. 1H).
Table 1.
The expression of MAD2L2 and NCOA3 in CRC is higher than ANT
| Variable | CRC | ANT | χ 2 | P |
|---|---|---|---|---|
| n (%) | n (%) | |||
| MAD2L2 | ||||
| −/1+ | 43 (43) | 58 (72.5) | 15.71 | <0.001 |
| 2 + /3+ | 57 (57) | 22 (27.5) | ||
| NCOA3 | ||||
| −/1+ | 41 (41) | 69 (86.3) | 38.29 | <0.001 |
| 2 + /3+ | 59 (59) | 11 (13.7) | ||
We further investigated the relationship between the expression of MAD2L2 and NCOA3 and the patients’ clinicopathological characteristics. As is shown in Table 2, high MAD2L2 expression was correlated with small tumor volume (P = 0.017), superficial infiltration (P = 0.023), rare metastasis (P = 0.008), and good clinical staging (P = 0.046). In contrast, high expression of NCOA3 was correlated with deep infiltration (P = 0.026).
Table 2.
Correlation between MAD2L2, NCOA3, and clinicopathological characteristics with CRC
| Variable | n | MAD2L2 | χ 2 | P | NCOA3 | χ 2 | P | ||
|---|---|---|---|---|---|---|---|---|---|
| −/1+ | 2 + /3+ | −/1+ | 2 + /3+ | ||||||
| Age | |||||||||
| < 70 | 55 | 27 (49.1) | 28 (50.9) | 1.85 | 0.174 | 27 (49.1) | 28 (50.9) | 3.308 | 0.069 |
| ≥ 70 | 45 | 16 (35.6) | 29 (64.4) | 14 (31.1) | 31 (68.9) | ||||
| Gender | |||||||||
| Male | 58 | 27 (46.6) | 31 (53.4) | 0.711 | 0.399 | 22 (37.9) | 36 (62.1) | 0.538 | 0.463 |
| Female | 42 | 16 (38.1) | 26 (61.9) | 19 (45.2) | 23 (54.8) | ||||
| Pathological type | |||||||||
| Canalicular adenoma | 83 | 34 (41.0) | 49 (59.0) | 0.826 | 0.363 | 32 (38.6) | 51 (61.4) | 1.207 | 0.272 |
| Mucinous adenocarcinoma | 17 | 9 (52.9) | 8 (47.1) | 9 (52.9) | 8 (47.1) | ||||
| Pathological grade | |||||||||
| I+II | 70 | 32 (45.7) | 38 (54.3) | 0.701 | 0.402 | 28 (40.0) | 42 (60.0) | 0.096 | 0.756 |
| III | 20 | 1 (36.7) | 19 (63.3) | 13 (43.3) | 17 (56.7) | ||||
| Tumor volume (cm3) | |||||||||
| < 30 | 51 | 16 (31.4) | 35 (68.6) | 5.741 | 0.017 | 25 (49.0) | 26 (51.0) | 2.767 | 0.096 |
| ≥ 30 | 49 | 27 (55.1) | 22 (44.9) | 16 (32.7) | 33 (67.3) | ||||
| General type | |||||||||
| Infiltrate type | 25 | 10 (40.0) | 15 (60.0) | 1.851 | 0.604 | 10 (40.0) | 15 (60.0) | 1.542 | 0.673 |
| Gel type | 8 | 4 (50.0) | 4 (50.0) | 4 (50.0) | 4 (50.0) | ||||
| Ulcerative type | 47 | 18 (38.3) | 29 (61.7) | 21 (44.7) | 26 (55.3) | ||||
| Protrude type | 20 | 11 (55.0) | 9 (45.0) | 6 (30.0) | 14 (70.0) | ||||
| Tumor location | |||||||||
| Left hemicolon | 47 | 20 (42.6) | 27 (57.4) | 0.007 | 0.932 | 15 (31.9) | 32 (68.1) | 3.026 | 0.082 |
| Right hemicolon | 53 | 23 (43.4) | 30 (56.6) | 26 (49.1) | 27 (50.9) | ||||
| Depth of invasion | |||||||||
| T1/T2/T3 | 68 | 24 (35.3) | 44 (64.7) | 5.148 | 0.023 | 33 (48.5) | 35 (51.5) | 4.98 | 0.026 |
| T4 | 32 | 19 (59.4) | 13 (40.6) | 8 (25.0) | 24 (75.0) | ||||
| Lymph node metastases | |||||||||
| N0 | 52 | 23 (44.2) | 29 (55.8) | 0.067 | 0.796 | 20 (38.5) | 32 (61.5) | 0.289 | 0.591 |
| N1/N2/N3 | 48 | 20 (41.7) | 28 (58.3) | 21 (43.7) | 27 (56.3) | ||||
| Distant metastasis | |||||||||
| M0 | 95 | 38 (40.0) | 57 (60.0) | 6.977 | 0.008 | 39 (41.1) | 56 (58.9) | 0.002 | 0.963 |
| M1 | 5 | 5 (100) | 0 (0) | 2 (40.0) | 3 (60.0) | ||||
| Clinical staging | |||||||||
| I | 4 | 1 (25.0) | 3 (75.0) | 8.001 | 0.046 | 1 (25.0) | 3 (75.0) | 0.518 | 0.915 |
| II | 47 | 21 (44.7) | 26 (55.3) | 19 (40.4) | 28 (59.6) | ||||
| III | 44 | 16 (36.4) | 28 (63.6) | 19 (43.2) | 25 (56.8) | ||||
| IV | 5 | 5 (100) | 0 (0) | 2 (40.0) | 3 (60.0) | ||||
Altogether, these results indicated that the expression of MAD2L2 was inversely related to that of NCOA3 in CRC, and MAD2L2 was associated with good prognosis in CRC patients, which suggested that MAD2L2 might inhibit the development of CRC.
3.2. MAD2L2 inhibited the proliferation, clonogenicity, and migration of CRC cells by down‐regulating NCOA3
To investigate the effect of MAD2L2 on CRC development, we knocked down MAD2L2 with its specific shRNA in HCT116 and SW480 cells and found that MAD2L2 knockdown promoted the proliferation of CRC cells and western blots showed that the level of NCOA3 was elevated when MAD2L2 was down‐regulated (Fig. 2A,C). In contrast, overexpression of MAD2L2 inhibited CRC cell proliferation, which was reversed by overexpression of NCOA3, whereas NCOA3 expression was repressed when MAD2L2 was overexpressed (Fig. 2B,D). Next, clonogenicity experiment showed that knockdown of MAD2L2 increased the clonogenicity of CRC cells (Fig. 2E,G), while overexpression of MAD2L2 decreased the cell clonogenicity and this decrease was reversed by NOCA3 overexpression (Fig. 2F,H). Finally, scratch assay showed that knockdown of MAD2L2 accelerated the migration rate of CRC cells (Fig. 3A,C,E, and G), while overexpression of MAD2L2 reduced the cell migration rate and this reduction was reversed by NCOA3 overexpression (Fig. 3B,D,F and H). To explore the downstream target genes of NCOA3 on CRC development, we knocked down MAD2L2 in HCT116 cells and found that NCOA3 activates the key targets of PI3K/AKT and Notch signaling pathway in RNA levels, which are involved in CRC progression (Fig. 4A). Collectively, these cellular experiments supported the inhibitory role of MAD2L2 in CRC development and suggested that MAD2L2 functioned as a tumor suppressor in CRC through the down‐regulation of NCOA3.
Figure 2.
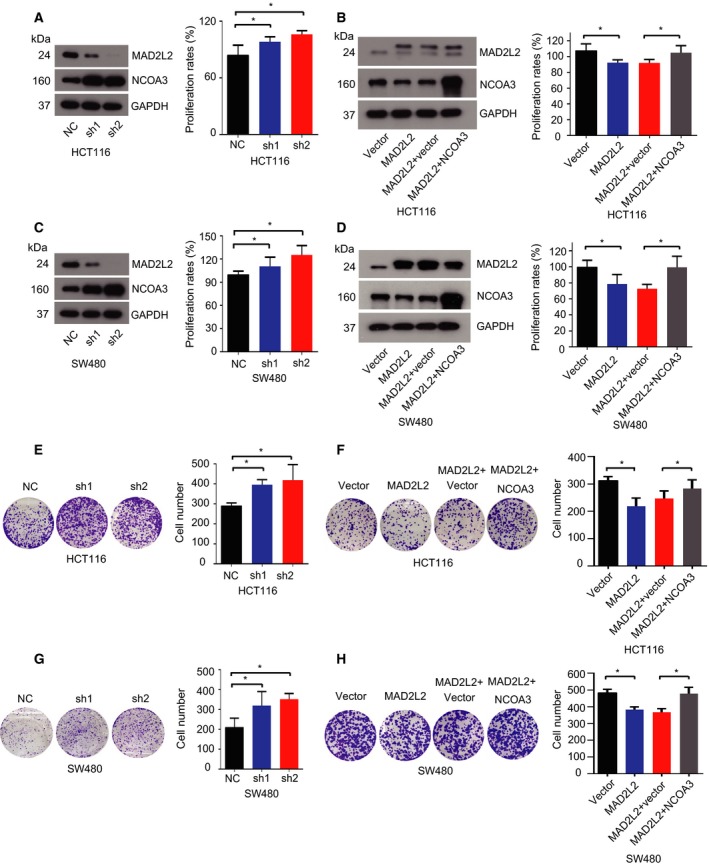
MAD2L2 suppressed CRC cell proliferation and inhibited the clonogenicity of CRC cells by down‐regulating NCOA3. (A,C) Knockdown of MAD2L2 elevated the expression of NCOA3 and promoted the proliferation of HCT116 and SW480 cells * P < 0.05 (n = 3). (B,D) Overexpression of MAD2L2 inhibited the expression of NCOA3 and suppressed CRC cell proliferation, which was reversed by NCOA3 overexpression. * P < 0.05 (n = 3). (E, G) Knockdown of MAD2L2 increased the clonogenicity of CRC cells. * P < 0.05 (n = 3). (F, H) Overexpression of MAD2L2 inhibited the clonogenicity of CRC cells, which was reversed by NCOA3 overexpression. * P < 0.05 (n = 3).
Figure 3.
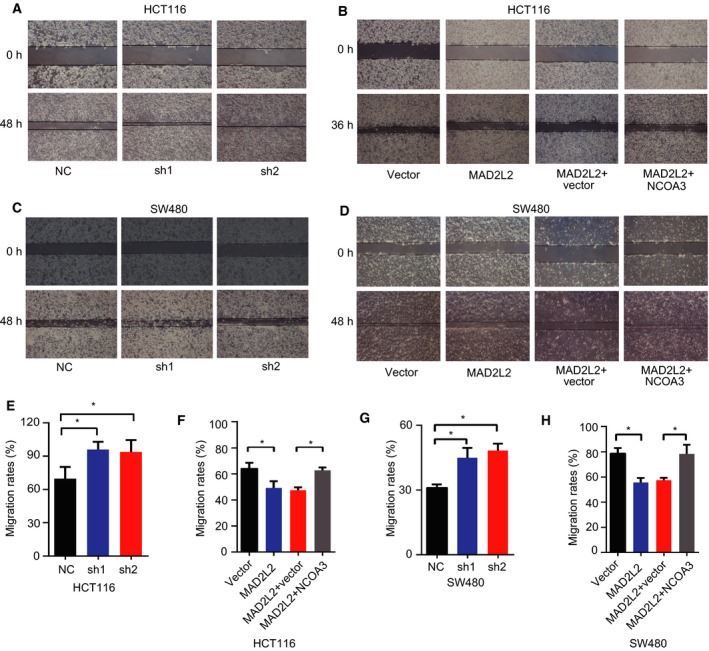
MAD2L2 inhibited CRC cell migration. (A,C,E,G) Knockdown of MAD2L2 promoted CRC cell migration. (B,D,F,H) Overexpression of MAD2L2 inhibited CRC cell migration, which was reversed by NCOA3 overexpression. * P < 0.05 (n = 3).
Figure 4.
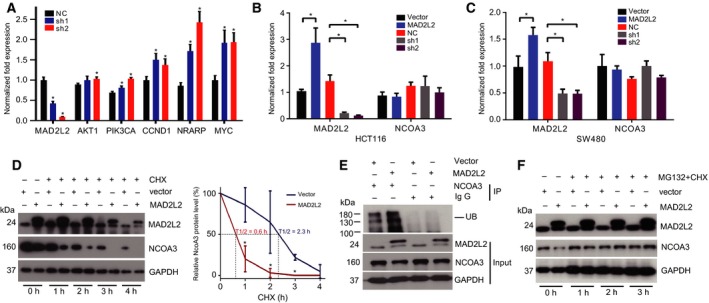
MAD2L2 promoted the degradation of NCOA3 in CRC cells. (A) The RNA levels of known downstream target genes of NCOA3 when MAD2L2 was knocked down in HCT116 cells. * P < 0.05 (n = 3). (B, C) NCOA3 mRNA remained unchanged when MAD2L2 was overexpressed (MAD2L2) or knocked down (sh1, sh2) in HCT116 and SW480 cells. * P < 0.05 (n = 3). (D) HCT116 cells without (vector) or with MAD2L2 overexpression (MAD2L2) were treated with 1 μg·mL−1 CHX for 0, 1, 2, 3, and 4 h, and the expression of NCOA3 was detected by western blot. The MAD2L2‐induced degradation of NCOA3 was enhanced in MAD2L2 overexpression cells. The half‐life curve of NCOA3 showed a considerable short half‐life of 0.6 h, compared to the vector cells with a half‐life of 2.3 h. * P < 0.05 (n = 3). (E) Extracts from HCT116 cells without or with MAD2L2 overexpression were immunoprecipitated with NCOA3 antibodies and analyzed for NCOA3 ubiquitination by western blot. (F) The MAD2L2‐induced degradation of NCOA3 was inhibited by MG132 (0.5 μm). CHX (2 μm) was used to inhibit protein synthesis.
3.3. MAD2L2 promoted NCOA3 degradation in CRC cells
To explore the molecular mechanism by which MAD2L2 down‐regulated the expression of NCOA3, we first tested whether MAD2L2 suppressed the transcription of NCOA3. As is shown in Fig. 4B,C, the mRNA level of NCOA3 remained largely unchanged when MAD2L2 was overexpressed or knocked down, which indicated that MAD2L2 did not regulate NCOA3 on the transcription level. We then tested whether MAD2L2 regulated the protein turnover of NCOA3. HCT116 cells without (vector) or with MAD2L2 overexpression (MAD2L2) were treated with 1 μg·mL−1 CHX for 0, 1, 2, 3, and 4 h to inhibit protein synthesis, and the expression of NCOA3 was detected by western blot. Excitingly, we found that NCOA3 was stilled detected in the control cells 4 h after CHX treatment (Fig. 4D), but it became barely detectable in cells overexpressing MAD2L2 only 2 h after CHX treatment (Fig. 4D). The half‐life curve of NCOA3 showed a considerable short half‐life of 0.6 h, compared to the vector cells with a half‐life of 2.3 h. These results indicated that overexpression of MAD2L2 accelerated the degradation of NCOA3.
Next, we examined whether MAD2L2 induced NCOA3 degradation by the ubiquitin–proteasome pathway. Immunoprecipitation (IP) experiments were performed with NCOA3 antibodies in HCT116 cells, and the amount of ubiquitinated NCOA3 was larger in cells overexpressing MAD2L2 (Fig. 4E). Moreover, the degradation of NCOA3 induced by MAD2L2 overexpression was inhibited by the proteasome inhibitor MG132 (Qiang et al., 2017) (Fig. 4F), confirming that NCOA3 was degraded by the proteasome.
3.4. MAD2L2 activated p38 to phosphorylate NCOA3 for its subsequent ubiquitination and degradation
Previous studies had reported that ubiquitination of NCOA3 was mediated by its phosphorylation (Ferry et al., 2011; Gianni et al., 2006; Wu et al., 2007), so we investigated whether MAD2L2 affected the phosphorylation of NCOA3. IP with NCOA3 antibodies in HCT116 cells showed that the phosphorylation of NCOA3 was enhanced in cells with MAD2L2 overexpression (Fig. 5A). Multiple kinases can differentially phosphorylate NCOA3, and it has several MAPK phosphorylation sites (Wu et al., 2004). We therefore tested whether MAD2L2 activated p38, JNK, or ERK1/2 to phosphorylate NCOA3. As is shown in Fig. 5B, the level of p‐p38 was positively correlated with the expression MAD2L2 and negatively with NCOA3. Furthermore, we detected interaction among MAD2L2, NCOA3, and p‐p38 by co‐IP in HCT116 cells with flag‐tagged MAD2L2 overexpression (Fig. 5C). In addition, the p38 kinase inhibitor SB203580 consistently inhibited the degradation of NCOA3 induced by MAD2L2 (Fig. 5D). Collectively, these findings supported our hypothesis that MAD2L2 activated p38 to phosphorylate NCOA3, which primed NCOA3 for the subsequent ubiquitination and degradation by the proteasome.
Figure 5.
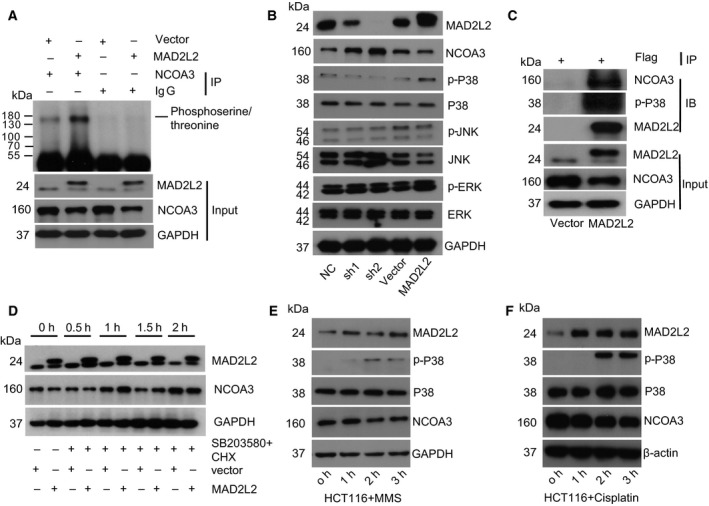
MAD2L2 activated p38 to phosphorylate NCOA3 for its subsequent ubiquitination and degradation. (A) Extracts from HCT116 cells without or with MAD2L2 overexpression were immunoprecipitated with NCOA3 antibodies and analyzed for NCOA3 phosphorylation by western blot. (B) The levels of p‐p38, p‐JNK, and p‐ERK in HCT116 cells with MAD2L2 knockdown or overexpression were detected by western blot. (C) The interaction among MAD2L2, NCOA3, and p‐p38 was detected by co‐IP in HCT116 cells overexpressing flag‐tagged MAD2L2. (D) The MAD2L2‐induced degradation of NCOA3 was inhibited by p‐p38 inhibitor SB203580 (0.5 μm). CHX (2 μm) was used to inhibit protein synthesis. (E) The DNA‐damaging agent MMS induced MAD2L2 expression and subsequent p38 activation and NCOA3 degradation. (F) The DNA‐damaging chemotherapy drug cisplatin (4 μg·mL−1) induced MAD2L2 expression and subsequent p38 activation and NCOA3 degradation.
Considering that chromosome instability is the major cause of CRC (Pino and Chung, 2010), and the expression of MAD2L2 was higher in CRC cells and tissues (Fig. 1C, Table 1), we presumed that MAD2L2 had elevated expression in response to DNA damages to serve as a protective factor in CRC. Cisplatin, a DNA‐damaging chemotherapy drug in the clinical treatment of colorectal cancer, can interact with the DNA guanine bases and prevent the replication of DNA. We treated HCT116 cells with the DNA‐damaging agent MMS and cisplatin and found that the expression of MAD2L2 was significantly increased within 1 h after MMS treatment, while noticeable p38 activation and NCOA3 degradation were detected about 2 h after MMS and cisplatin treatment (Fig. 5E,F).
3.5. MAD2L2 knockdown promoted CRC growth in a mouse xenograft model
The significant association of MAD2L2 with NCOA3 expression revealed in cellular experiments and clinical outcomes led us to further verify the roles of MAD2L2 and NCOA3 in CRC in a mouse xenograft model. HCT116 cells were subcutaneously injected into the left flank of nude mice, and tumors developed at the injection sites after 1 week. Tumor volumes were measured and recorded every 2 days, and the tumor xenografts were harvested, weighed, and processed for IHC staining 3 weeks after CRC cell injection. As is shown in Fig. 6A‐D, MAD2L2 knockdown promoted tumor growth, while MAD2L2 overexpression inhibited tumor growth, and this inhibition was rescued by elevated NCOA3 expression. IHC analysis showed that the levels of MAD2L2 and NCOA3 were inversely correlated (Fig. 6E). These in vivo results were consistent with our in vitro observations and confirmed the tumor suppressor role of MAD2L2 in CRC.
Figure 6.
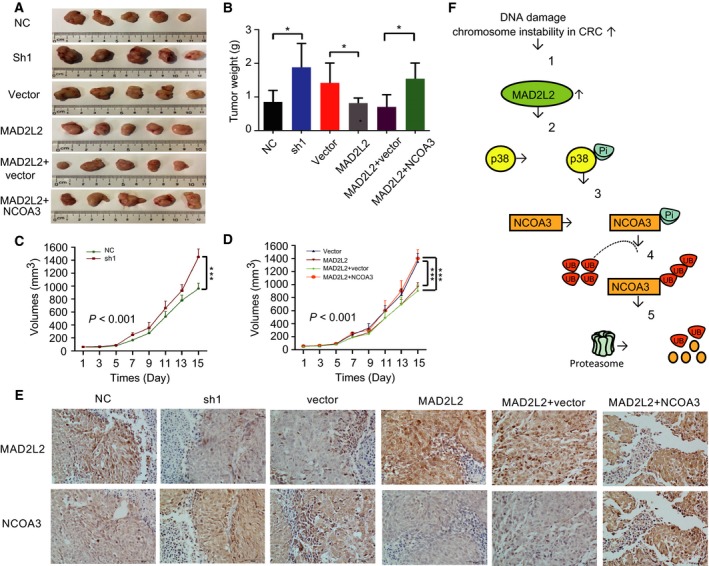
MAD2L2 knockdown promoted CRC growth in a mouse xenograft model and MAD2L2‐regulated NCOA3 phosphorylation, ubiquitination, and degradation in CRC cells. Nude mice were subcutaneously injected with HCT116 cells with nonspecific siRNA (NC), MAD2L2 knocked down by its specific shRNA (sh1), vector, MAD2L2 overexpression (MAD2L2), MAD2L2 + vector, MAD2L2 + NCOA3 overexpression (NCOA3). (A) Images of the CRC tumor xenograft from each mouse (n = 5 mice/group). (B) Tumor weights were analyzed. *P < 0.05 (n = 5) (C, D) Tumor volumes were recorded and analyzed. ***P < 0.001 (n = 5) (E) The expression of MAD2L2 and NCOA3 in tumor tissues was analyzed by IHC staining. (F) (1) MAD2L2 had elevated expression in response to the increased DNA damage and chromosome instability in CRC cells. (2) MAD2L2 activated p38. (3) p‐p38 phosphorylated NCOA3. (4) Phosphorylated NCOA3 was ubiquitinated. (5) Ubiquitinated NCOA3 was degraded by the proteasome.
4. Discussion
Accumulating evidence shows that NCOA3 is highly expressed in a various human cancers (Anzick et al., 1997; Xu et al., 2010; Zhou et al., 2005), and it can interact with nuclear receptors and other transcription factors to regulate the expression of their target genes involved in many signaling pathways, including EGFR, Akt, MAPK, E2F1, and Notch (Long et al., 2012; Louie et al., 2004; Mo et al., 2015; Yan et al., 2006). However, little is known about how NCOA3 is regulated in colorectal cancer (CRC). In this study, we discovered that MAD2L2 interacted with NCOA3 and regulated its protein level in CRC. MAD2L2 is a regulatory subunit of DNA polymerase ζ that is involved in DNA translesion synthesis (TLS) (Boersma et al., 2015), and the alterations of MAD2L2 are implicated in the pathogenesis of a wide variety of tumors (Feng et al., 2016; Niimi et al., 2014; Okina et al., 2015). Our in vivo and in vitro results showed that MAD2L2 suppressed CRC development by down‐regulating NCOA3, and our clinical data suggested that MAD2L2 predicted favorable prognosis in CRC patients. Our mechanism study showed that MAD2L2 had increased expression in the presence of DNA damage and activated p38 to phosphorylate NCOA3 for its subsequent degradation by the ubiquitin–proteasome pathway.
Colorectal cancer is one of the most common cancers and continued to be a serious public health problem in clinic. To provide valuable information for the clinical outcome prediction, we analyzed the expression of MAD2L2 and NCOA3 in CRC patients. Our results showed that there was a reverse correlation between MAD2L2 and NCOA3 expression in CRC tissues (Fig. 1D,E), which was in accordance with our findings in CRC cells (Fig. 1C). Moreover, higher expression of MAD2L2 was associated with lower tumor volume, earlier TNM stage, less invasion, and a smaller chance of distant metastasis in CRC patients (Table 2), which suggested that MAD2L2 was a suppressor of CRC growth and metastasis. Consistently, survival analysis indicated that MAD2L2 suppressed but NCOA3 promoted CRC development (Fig. 1F,G). Interestingly, the expression of both MAD2L2 and NCOA3 was higher in CRC tissues than normal tissues (Table 1). Given that CRC cells have increased DNA damage and chromosome instability (Guo et al., 2016; Ribeiro et al., 2008; Xia et al., 2016), and MAD2L2 plays a critical role in DNA repair, we proposed that the expression of MAD2L2 was elevated in CRC tissues as a stress response, and this was supported by our result that MMS and cisplatin treatment induced MAD2L2 expression (Fig. 5E,F). Collectively, our data have revealed that MAD2L2 is a protective factor in the pathogenesis of CRC.
To further study the biological relationship between MAD2 l2 and NCOA3, we knocked down or overexpressed MAD2L2 in CRC cells to determine the effects of MAD2L2 on the protein level of NCOA3, cell proliferation, colony formation, and migration capacity. Our data demonstrated that knockdown of MAD2L2 increased NCOA3 expression and enhanced the proliferation, colony formation, and migration of CRC cells, whereas overexpression of MAD2L2 had the opposite effects, which were reversed by NCOA3 overexpression (Fig. 2,3 and 6A‐D). Consistent with our findings, knockdown of NCOA3 decreased cell proliferation, colony formation, and tumorigenesis of CRC cells in vitro and in vivo (Mo et al., 2015), suggested that MAD2L2 was a novel regulator of NCOA3 in CRC progression. However, the effects of MAD2L2 on cell proliferation were not the only suppressor mechanism, and it has been reported that other mechanisms also play an important role in tumorigenesis of CRC cells (Kramer et al., 2016; Siraj et al., 2017).
To validate that the observed effects on proliferation and migration are reflected at the functional level of NCOA3, the mRNA levels of known downstream target genes of NCOA3 were detected when MAD2L2 was knocked down in HCT116 cells (Fig. 4C). Studies have shown NCOA3 activates the PI3K/AKT pathway and its downstream effectors in mammary tumor cells derived from AIB1‐tg mice (Torres‐Arzayus et al., 2004). As the key genes of PI3K/AKT pathway, the mRNA levels of AKT1, PIK3CA, and CCND1 were significantly increased, suggested that NCOA3 promotes CRC progression through regulating the PI3K/AKT pathway‐related genes. Increasing evidence has shown that Notch signaling is related to CRC progression, and NRARP represents Notch signaling activity in CRC (Kim et al., 2012; Mo et al., 2015). Moreover, Notch signaling can directly activate MYC, and a protooncogene holds a central role in regulating tumor growth (Jitschin et al., 2015; Xiao et al., 2011). Our study found that the mRNA levels of NRARP and MYC was significantly elevated, and revealed that typical target gene of Notch signaling plays an important role in CRC development. Further study showed that MAD2L2 did not regulate NCOA3 on the transcription level (Fig. 4B,C), but promoted the protein degradation of NCOA3 (Fig. 4D). Moreover, we confirmed that the degradation of NCOA3 induced by MAD2L2 happened through the ubiquitin–proteasome pathway (Fig. 4E, 5A), which controls the degradation of the majority of regulatory proteins in mammalian cells (Naujokat and Saric, 2007; Vriend and Reiter, 2015). Previously, phosphorylation of NCOA3 was found to promote its ubiquitination and degradation (Ferry et al., 2011; Wu et al., 2007). NCOA3 can be phosphorylated by kinases including MAPKs, GSK3, PKA, and CKI (Wu et al., 2004). Among them, MAPKs are key signaling molecules in cell growth, proliferation and development, and functionally important for NCOA3 phosphorylation (Ferry et al., 2011). Extracellular signal‐regulated kinase (ERK), c‐Jun N‐terminal protein kinase (JNK), and p38 kinase are the three major MAPKs (Chang and Karin, 2001), and Wu et al. found that p38 and JNK were able to phosphorylate multiple sites of NCOA3 (Wu et al., 2004). In this study, we identified that p38 was the chief mediator of MAD2L2‐induced NCOA3 ubiquitination and degradation (Fig. 5B‐D). Here, we propose a model for the MAD2L2‐regulated NCOA3 phosphorylation, ubiquitination, and degradation in CRC cells (Fig. 6F): In response to the increased DNA damage and chromosome instability in CRC cells, MAD2L2 had elevated expression and activated p38, which then phosphorylated NCOA3 for subsequent degradation through the ubiquitin–proteasome pathway.
In summary, we have discovered that MAD2L2 inhibited CRC development by promoting NCOA3 degradation. Our work has demonstrated that modulation of the MAD2L2 gene product has the potential to become a new therapy for CRC.
Author contribution
YL, LL, MC, XY, XW, and WD conceived and designed the project; YL, LL, MC, and ZG acquired the data; YL, LL, MC, ZG, HQ, GQ, QL, XF, TL, WL, DS, ML, TK, WH, and XW analyzed and interpreted the data; YL, MC, and WD wrote the paper; WD supervised the project.
Ethics approval
All animal procedures were performed following the Guide for the Care and Use of Laboratory Animals (NIH publication Nos. 80‐23, revised 1996) and the Institutional Ethical Guidelines for Animal Experiments developed by Sun Yat‐sen University.
Supporting information
Fig. S1. The specificity of the antibodies against MAD2L2 and NCOA3 were examined in CRC cells. MAD2L2 and NCOA3 proteins were detected by their antibodies by western blots of the entire gel with SW620, SW480 and HCT116 cell extracts.
Acknowledgments
This work was supported by the funds from the National Natural Science Foundation of China (81772925, 81472178), Guangzhou Science Technology and Innovation Commission (201607020038), Natural Science Foundation of Guangdong Province (2016A03031100; 2015A030313018), Guangdong Esophageal Cancer Institute (2015A09), and the State “973 Program” of China. (2014CB542005).
Contributor Information
Meihua Luo, Email: luomeihua888@163.com.
Xiaojun Wu, Email: wuxj@sysucc.org.cn.
Wuguo Deng, Email: dengwg@sysucc.org.cn.
References
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X‐Y, Sauter G, Kallioniemi O‐P, Trent JM and Meltzer PS (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277, 965–968. [DOI] [PubMed] [Google Scholar]
- Boersma V, Moatti N, Segura‐Bayona S, Peuscher MH, van der Torre J, Wevers BA, Orthwein A, Durocher D and Jacobs JJ (2015) MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature 521, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E et al (2013) Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Shames DS, Raso MG, Xie Y, Kim YH, Pollack JR, Girard L, Sullivan JP, Gao B, Peyton M et al (2010) Steroid receptor coactivator‐3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer Res 70, 6477–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LF and Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40. [DOI] [PubMed] [Google Scholar]
- Chen J and Fang GW (2001) MAD2B is an inhibitor of the anaphase‐promoting complex. Genes Dev 15, 1765–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Li W, Wan Y, Xia X, Wu Q, Chen Y, Lai Z, Yu C and Li W (2012) Amplified in breast cancer 1 enhances human cholangiocarcinoma growth and chemoresistance by simultaneous activation of Akt and Nrf2 pathways. Hepatology 55, 1820–1829. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y and Evans RM (1997) Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 90, 569–580. [DOI] [PubMed] [Google Scholar]
- Chen HW, Lin RJ, Xie W, Wilpitz D and Evans RM (1999) Regulation of hormone‐induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 98, 675–686. [DOI] [PubMed] [Google Scholar]
- Cheung HW, Chun ACS, Wang Q, Deng W, Hu L, Guan XY, Nicholls JM, Ling MT, Wong YC, Tsao SW et al (2006) Inactivation of human MAD2B in nasopharyngeal carcinoma cells leads to chemosensitization to DNA‐damaging agents. Can Res 66, 4357–4367. [DOI] [PubMed] [Google Scholar]
- Colussi D, Brandi G, Bazzoli F and Ricciardiello L (2013) Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci 14, 16365–16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Wei W, Heng Z, Yantao H and Chunbo W (2016) Knockdown of REV7 inhibits breast cancer cell migration and invasion. Oncol Res 24, 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136, E359–E386. [DOI] [PubMed] [Google Scholar]
- Ferry C, Gaouar S, Fischer B, Boeglin M, Paul N, Samarut E, Piskunov A, Pankotai‐Bodo G, Brino L and Rochette‐Egly C (2011) Cullin 3 mediates SRC‐3 ubiquitination and degradation to control the retinoic acid response. Proc Natl Acad Sci USA 108, 20603–20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni M, Parrella E, Raska I, Gaillard E, Nigro EA, Gaudon C, Garattini E and Rochette‐Egly C (2006) P38MAPK‐dependent phosphorylation and degradation of SRC‐3/AIB1 and RAR alpha‐mediated transcription. EMBO J 25, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE and Robson CN (2001) Expression of RAC 3, a steroid hormone receptor co‐activator in prostate cancer. Br J Cancer 85, 1928–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady WM and Carethers JM (2008) Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 135, 1079–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Li X, Wang R, Yu J, Ye M, Mao L, Zhang S and Zheng S (2016) Association between oxidative DNA damage and risk of colorectal cancer: sensitive determination of urinary 8‐Hydroxy‐2 ‘‐deoxyguanosine by UPLC‐MS/MS analysis. Sci Rep 6, 32581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP, Shen L and Toyota M (2005) CIMP, at last. Gastroenterology 129, 1121–1124. [DOI] [PubMed] [Google Scholar]
- Jitschin R, Braun M, Qorraj M, Saul D, Le Blanc K, Zenz T and Mougiakakos D (2015) Stromal cell‐mediated glycolytic switch in CLL cells involves Notch‐c‐Myc signaling. Blood 125, 3432–3436. [DOI] [PubMed] [Google Scholar]
- Kim HA, Koo BK, Cho JH, Kim YY, Seong J, Chang HJ, Oh YM, Stange DE, Park JG, Hwang D et al (2012) Notch1 counteracts WNT/beta‐catenin signaling through chromatin modification in colorectal cancer. J Clin Invest 122, 3248–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer HB, Lai CF, Patel H, Periyasamy M, Lin ML, Feller SM, Fuller‐Pace FV, Meek DW, Ali S and Buluwela L (2016) LRH‐1 drives colon cancer cell growth by repressing the expression of the CDKN1A gene in a p53‐dependent manner. Nucleic Acids Res 44, 582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Gomes PJ and Chen JD (1997) RAC3, a steroid/nuclear receptor‐associated coactivator that is related to SRC‐1 and TIF2. Proc Natl Acad Sci USA 94, 8479–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JK, Chang SC, Yang YC and Li AFY (2003) Loss of heterozygosity and DNA aneuploidy in colorectal adenocarcinoma. Ann Surg Oncol 10, 1086–1094. [DOI] [PubMed] [Google Scholar]
- Long W, Foulds CE, Qin J, Liu J, Ding C, Lonard DM, Solis LM, Wistuba II, Qin J, Tsai SY et al (2012) ERK3 signals through SRC‐3 coactivator to promote human lung cancer cell invasion. J Clin Invest 122, 1869–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie MC, Zou JX, Rabinovich A and Chen HW (2004) ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol 24, 5157–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo P, Zhou Q, Guan L, Wang Y, Wang W, Miao M, Tong Z, Li M, Majaz S, Liu Y et al (2015) Amplified in breast cancer 1 promotes colorectal cancer progression through enhancing notch signaling. Oncogene 34, 3935–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakumo Y, Ogura Y, Ishii H, Numata S, Ichihara M, Croce CM, Fishel R and Takahashi M (2001) Interactions in the error‐prone postreplication repair proteins hREV1, hREV3, and hREV7. J Biol Chem 276, 35644–35651. [DOI] [PubMed] [Google Scholar]
- Naujokat C and Saric T (2007) Concise review: role and function of the ubiquitin‐proteasome system in mammalian stem and progenitor cells. Stem Cells 25, 2408–2418. [DOI] [PubMed] [Google Scholar]
- Niimi K, Murakumo Y, Watanabe N, Kato T, Mii S, Enomoto A, Asai M, Asai N, Yamamoto E, Kajiyama H et al (2014) Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells. Cancer Sci 105, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okina S, Yanagisawa N, Yokoyama M, Sakurai Y, Numata Y, Umezawa A, Higashihara M and Murakumo Y (2015) High expression of REV7 is an independent prognostic indicator in patients with diffuse large B‐cell lymphoma treated with rituximab. Int J Hematol 102, 662–669. [DOI] [PubMed] [Google Scholar]
- Pino MS and Chung DC (2010) The chromosomal instability pathway in colon cancer. Gastroenterology 138, 2059–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirouz M, Rahjouei A, Shamsi F, Eckermann KN, Salinas‐Riester G, Pommerenke C and Kessel M (2015) Destabilization of pluripotency in the absence of Mad2 l2. Cell Cycle 14, 1596–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Sui F, Ma J, Li X, Ren X, Shao Y, Liu J, Guan H, Shi B and Hou P (2017) Proteasome inhibitor MG132 induces thyroid cancer cell apoptosis by modulating the activity of transcription factor FOXO3a. Endocrine 56, 98–108. [DOI] [PubMed] [Google Scholar]
- Rahjouei A, Pirouz M, Di Virgilio M, Kamin D and Kessel M (2017) MAD2L2 promotes open chromatin in embryonic stem cells and derepresses the Dppa3 Locus. Stem Cell Reports 8, 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro ML, Priolli DG, Miranda DDC, Arcari DP, Pedrazzoli J Jr and Martinez CAR (2008) Analysis of oxidative DNA damage in patients with colorectal cancer. Clin Colorectal Cancer 7, 267–272. [DOI] [PubMed] [Google Scholar]
- Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, Kimura A, Nakamura Y, Inazawa J, Abe T et al (2000) Amplification and over‐expression of the AIB1 nuclear receptor co‐activator gene in primary gastric cancers. Int J Cancer 89, 217–223. [PubMed] [Google Scholar]
- Shi J, Liu W, Sui F, Lu R, He Q, Yang Q, Lv H, Shi B and Hou P (2015) Frequent amplification of AIB1, a critical oncogene modulating major signaling pathways, is associated with poor survival in gastric cancer. Oncotarget 6, 14344–14359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siraj AK, Masoodi T, Bu R, Pratheeshkumar P, Al‐Sanea N, Ashari LH, Abduljabbar A, Alhomoud S, Al‐Dayel F, Alkuraya FS et al (2017) MED12 is recurrently mutated in Middle Eastern colorectal cancer. Gut, pii: gutjnl‐2016‐313334. [Epub ahead of print] https://doi.org/10.1136/gutjnl-2016-313334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JM, Lu M, Liu FF, Du XJ and Xing BC (2012) AIB1 as an independent prognostic marker in hepatocellular carcinoma after hepatic resection. J Gastrointest Surg 16, 356–360. [DOI] [PubMed] [Google Scholar]
- Tariq K and Ghias K (2016) Colorectal cancer carcinogenesis: a review of mechanisms. Cancer Biol Med 13, 120–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres‐Arzayus MI, Font de Mora J, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR and Brown M (2004) High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 6, 263–274. [DOI] [PubMed] [Google Scholar]
- Vriend J and Reiter RJ (2015) Breast cancer cells: modulation by melatonin and the ubiquitin‐proteasome system–a review. Mol Cell Endocrinol 417, 1–9. [DOI] [PubMed] [Google Scholar]
- Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I and Kerr D (2009) Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer 9, 489–499. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Mii S, Asai N, Asai M, Niimi K, Ushida K, Kato T, Enomoto A, Ishii H, Takahashi M et al (2013) The REV7 subunit of DNA polymerase zeta is essential for primordial germ cell maintenance in the mouse. J Biol Chem 288, 10459–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RC, Feng Q, Lonard DM and O'Malley BW (2007) SRC‐3 coactivator functional lifetime is regulated by a phospho‐dependent ubiquitin time clock. Cell 129, 1125–1140. [DOI] [PubMed] [Google Scholar]
- Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ and O'Malley BW (2004) Selective phosphorylations of the SRC‐3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell 15, 937–949. [DOI] [PubMed] [Google Scholar]
- Xia T, Konno H, Ahn J and Barber GN (2016) Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep 14, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Ye Y, Zou X, Jones S, Yearsley K, Shetuni B, Tellez J and Barsky SH (2011) The lymphovascular embolus of inflammatory breast cancer exhibits a Notch 3 addiction. Oncogene 30, 287–300. [DOI] [PubMed] [Google Scholar]
- Xie D, Sham JS, Zeng WF, Lin HL, Bi J, Che LH, Hu L, Zeng YX and Guan XY (2005) Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum Pathol 36, 777–783. [DOI] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M et al (2015) REV7 counteracts DNA double‐strand break resection and affects PARP inhibition. Nature 521, 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Chen Q, Li W, Su X, Chen T, Liu Y, Zhao Y and Yu C (2010) Overexpression of transcriptional coactivator AIB1 promotes hepatocellular carcinoma progression by enhancing cell proliferation and invasiveness. Oncogene 29, 3386–3397. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu RC and O'Malley BW (2009) Normal and cancer‐related functions of the p160 steroid receptor co‐activator (SRC) family. Nat Rev Cancer 9, 615–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Yu C‐T, Ozen M, Ittmann N, Tsai SY and Tsai M‐J (2006) Steroid receptor coactivator‐3 and activator protein‐1 coordinately regulate the transcription of components of the insulin‐like growth factor/AKT signaling pathway. Can Res 66, 11039–11046. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yang S‐H and Sharrocks AD (2007) Rev7/MAD2B links c‐Jun N‐terminal protein kinase pathway signaling to activation of the transcription factor Elk‐1. Mol Cell Biol 27, 2861–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liu S, Wang H, Zhang X, Kang T, Li Z, Deng H, Yue W and Cao S (2011) Mitotic arrest deficient protein MAD2B is overexpressed in human glioma, with depletion enhancing sensitivity to ionizing radiation. J Clin Neurosci 18, 827–833. [DOI] [PubMed] [Google Scholar]
- Zhao C, Yasui K, Lee CJ, Kurioka H, Hosokawa Y, Oka T and Inazawa J (2003) Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer 98, 18–23. [DOI] [PubMed] [Google Scholar]
- Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, Ittmann M, Tsai SY and Tsai MJ (2005) SRC‐3 is required for prostate cancer cell proliferation and survival. Can Res 65, 7976–7983. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The specificity of the antibodies against MAD2L2 and NCOA3 were examined in CRC cells. MAD2L2 and NCOA3 proteins were detected by their antibodies by western blots of the entire gel with SW620, SW480 and HCT116 cell extracts.