

Abstract
Receptor theory predicts that fixed-proportion mixtures of a competitive, reversible agonist (e.g., fentanyl) and antagonist (e.g., naltrexone) at a common receptor [e.g., mu-opioid receptors (MORs)] will result in antagonist proportion-dependent decreases in apparent efficacy of the agonist/antagonist mixtures and downward shifts in mixture dose-effect functions. The present study tested this hypothesis by evaluating behavioral effects of fixed-proportion fentanyl/naltrexone mixtures in a warm-water tail-withdrawal procedure in rhesus monkeys (n = 4). Fentanyl (0.001–0.056 mg/kg) alone, naltrexone (0.032–1.0 mg/kg, i.m.) alone, and fixed-proportion mixtures of fentanyl/naltrexone (1:0.025, 1:0.074, and 1:0.22) were administered in a cumulative-dosing procedure, and the proportions were based on published fentanyl and naltrexone Kd values at MOR in monkey brain. Fentanyl alone produced dose-dependent antinociception at both 50 and 54°C thermal intensities. Up to the largest dose tested, naltrexone alone did not alter nociception. Consistent with receptor theory predictions, naltrexone produced a proportion-dependent decrease in the effectiveness of fentanyl/naltrexone mixtures to produce antinociception. The maximum effects of fentanyl, naltrexone, and each mixture were also used to generate an efficacy-effect scale for antinociception at each temperature, and this scale was evaluated for its utility in quantifying 1) efficacy requirements for antinociception at 50 and 54°C and 2) relative efficacy of six MOR agonists that vary in their efficacies to produce agonist-stimuated GTPγS binding in vitro (from lowest to highest efficacy: 17-cyclopropylmethyl-3,14β-dihyroxy-4,5α-epoxy-6α-[(3′-isoquinolyl)acetamindo]morphine, nalbuphine, buprenorphine, oxycodone, morphine, and methadone). These results suggest that fixed-proportion agonist/antagonist mixtures may offer a useful strategy to manipulate apparent drug efficacy for basic research or therapeutic purposes.
Introduction
Pharmacodynamics is concerned with the affinity and efficacy of drugs at their receptor targets. Drug affinity can be precisely measured with ligand binding techniques, but drug efficacy to activate receptor signaling and produce downstream effects is a relative measure dependent in part on the signaling pathway(s) and downstream effects under consideration, and drug efficacies are typically described in relation to some standard high-efficacy ligand (Ruffolo, 1982; Kenakin, 2012). Although efficacy is challenging to measure, it is clearly relevant in drug development. For example, mu-opioid receptor (MOR) ligands differ in their efficacy to activate MOR-coupled signal transduction processes and produce MOR-mediated effects such as analgesia and respiratory depression. Fentanyl has high MOR efficacy, and increasing fentanyl doses can produce both antinociception and lethal respiratory depression (Gerak et al., 1994; Banks et al., 2010a; Ding et al., 2016). At the other extreme of the efficacy continuum, naltrexone has little or no MOR efficacy, produces no agonist effects, and functions as a competitive reversible antagonist (Walker et al., 1994; Ko et al., 1998; Bowen et al., 2002). Between these extremes are intermediate-efficacy MOR ligands such as nalbuphine and buprenorphine, which produce submaximal stimulation of MOR signaling and a subset of agonist effects that includes analgesia but only weak respiratory depression (Gerak et al., 1994; Pitts et al., 1998; Kishioka et al., 2000). Experiments to investigate the expression and consequences of ligand efficacy at MORs or other receptor targets can be useful both 1) to determine the efficacy required to produce different effects of interest, and 2) to evaluate relative efficacy of new ligands as they are developed.
One common approach to efficacy evaluations relies on the use of irreversible antagonists to evaluate the impact of reducing receptor number on expression of drug effects (Furchgott, 1966; Kenakin, 1993; Bergman et al., 2000). Efficacy requirements for different effects can be estimated, because an irreversible antagonist will produce greater antagonism of effects with high- versus low-efficacy requirements (Zernig et al., 1997). Relative efficacies of different drugs can be estimated, because an irreversible antagonist will produce greater antagonism of a low- versus high-efficacy agonist (Zimmerman et al., 1987; Walker et al., 1998). However, studies with irreversible antagonists can be logistically challenging (e.g., due to the long duration of antagonist effects), and irreversible antagonists are not available for many receptors of interest. Decreases in receptor number can also be accomplished with genetic mutations, as in wild-type, heterozygous, and homozygous receptor knockout animals (Grim et al., 2016), but the degree of control over the magnitude of that decrease is limited. Receptor theory suggests an alternative, more precise, and more flexible strategy to investigate efficacy using mixtures of competitive agonists and antagonists. Figure 1 (left panel) shows a theoretical dose-effect function for a high-efficacy agonist administered alone or in the presence of increasing fixed doses of an antagonist. The familiar result is an antagonist dose-dependent rightward shift in the agonist dose-effect curve (Ko et al., 1998; Negus et al., 2003). Figure 1 (right panel) shows theoretical effects using a different experimental design, in which the agonist is administered in combination with fixed-proportional doses of the antagonist, such that increasing agonist doses are administered in combination with increasing antagonist doses. In this design, the antagonist is expected to produce proportion-dependent downward shifts in the agonist dose-effect curve, and mixtures with decreasing agonist-to-antagonist proportions have decreasing apparent efficacies to activate the receptor. This approach has two potential advantages relative to existing strategies. First, agonist-to-antagonist proportion can be precisely manipulated to yield precise increments in efficacy. Second, this approach could be applied to any receptor system for which a competitive agonist and antagonist are available.
Fig. 1.

Theoretical curves simulated from the Furchgott equation for receptor theory (Ruffolo, 1982). Left panel shows rightward shifts in a competitive reversible agonist dose-effect function after pretreatment with increasing fixed doses of a competitive reversible antagonist. Right panel shows downward shifts in a competitive reversible agonist dose-effect function when agonist and antagonist are coadministered in fixed-proportion mixtures. Equations and definition of terms are shown below the panels. For this simulation, agonist dose A and antagonist dose B vary in KD units (i.e., at a dose of 1, dose = KD); Rt was set arbitrarily at 100, and all other variables were set arbitrarily at 1. Note that in the left panel, antagonist dose is a fixed dose B that remains constant across a range of agonist doses. For the right panel, antagonist dose is a fixed proportion p of the agonist dose A, such that B = pA and increases in agonist dose are accompanied by increases in antagonist dose.
The goal of the present study was to test the utility of this approach using the competitive MOR agonist fentanyl and antagonist naltrexone (Negus et al., 1993; Emmerson et al., 1994, 1996; Walker et al., 1994). Effects of these drugs administered alone and in fixed-proportion mixtures were determined in an assay of thermal nociception using two thermal stimulus intensities (50 and 54°C warm water) and compared with effects produced by six other MOR ligands shown previously to vary in their relative MOR efficacies in in vitro assays of agonist-stimulated GTPγS binding (Emmerson et al., 1996; Selley et al., 1998; Alt et al., 2001; Thompson et al., 2004; Yuan et al., 2015). We predicted that the effects of fentanyl, naltrexone, and the mixtures would match the predicted results in Fig. 1 (right panel). Additionally, we predicted that the maximal effects of fentanyl, naltrexone, and the mixtures could be used to generate efficacy-effect scales for quantification of both 1) MOR efficacy requirements for antinociception at 50 and 54°C, and 2) relative efficacies of the six MOR test ligands.
Methods
Subjects.
Four adult male rhesus macaques (Macaca mulatta) of Indian or Chinese origin and weighing between 10 and 14 kg served as subjects. All subjects had previous experimental histories that included exposure to opioid ligands, monoaminergic transporter ligands, and N-methyl D-aspartate antagonists. Monkeys were fed a diet of laboratory biscuits (Purina, Framingham, MA) supplemented with fresh fruits, vegetables, and nuts to maintain healthy, stable body weights. Monkeys were individually housed in a temperature and humidity controlled room that was maintained on a 12-hour light/12-hour dark cycle (lights on from 6:00 AM until 6:00 PM). Water was available ad libitum in the housing chamber. The facility was licensed by the United States Department of Agriculture and accredited by AAALAC International. Both research and enrichment protocols were approved by the Institutional Animal Care and Use Committee and in accordance with the 2011 Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). Environmental enrichment included: music, movies, puzzle feeders, and chew toys. Furthermore, monkeys were afforded opportunities to interact socially using olfactory and auditory cues; mirrors provided additional opportunities for visual interaction.
Assay of Thermal Nociception.
Monkeys were trained to sit comfortably in an acrylic restraint chair using the pole-and-collar technique such that their tails hung freely. The subject’s tail was shaved 10–12 cm from the distal end weekly and immersed in a thermal container of warm water. If the subject did not remove its tail by 20 seconds, the tail was removed by the experimenter, and a latency of 20 seconds was assigned. A stopwatch was used to record tail-withdrawal latencies. During each 15-minute cycle, tail-withdrawal latencies were recorded from water warmed to 38, 50, and 54°C and the order of warmed water presentations varied between successive cycles. Baseline tail-withdrawal latencies at all three thermal intensities were determined in each daily test session before drug administration. Test sessions continued only if tail-withdrawal latencies from 38°C water did not occur before the 20-second cutoff. This criterion was met in every monkey during every test session. Cumulative dose test sessions consisted of four to six 15-minute cycles composed of a 10-minute drug pretreatment phase and a 5-minute testing phase. Drugs were administered intramuscularly at the start of each 15-minute cycle, and each drug dose increased the total cumulative dose by one-fourth or one-half log units. Tail-withdrawal latencies were redetermined during the 5-minute testing phase as described previously.
Initially, dose-effect functions were determined for fentanyl (0.001–0.056 mg/kg, i.m.) and naltrexone (0.032–1 mg/kg, i.m.) alone and each dose-effect function was determined twice. Subsequently, three fixed-proportion fentanyl and naltrexone mixtures were examined and each cumulative dose-effect function was determined once. The proportions of each drug in the three test mixtures were based on the published affinities (Kd) of fentanyl (1.48 nM) and naltrexone (0.11 nM) at the MOR in rhesus monkey brain (Emmerson et al., 1994). Specifically, the fixed-proportion of fentanyl to naltrexone for one mixture, denoted as the 1:1 mixture, was set to the proportion of their Kd values (1.48:0.11 = 1:0.074). Relative to the 1:1 mixture, the 3:1 fentanyl/naltrexone mixture had a 3-fold higher proportion of fentanyl to naltrexone (1:0.025), and the 1:3 fentanyl/naltrexone mixture had a 3-fold lower proportion of fentanyl to naltrexone (1:0.22). Mixtures were tested up to doses that produced maximal antinociception, undesirable physiologic effects such as respiratory depression, or antagonized fentanyl effects in other studies. Experiments were generally conducted twice per week, usually on Tuesdays and Fridays, with at least 3 days between test days.
Following these initial fentanyl/naltrexone fixed-proportion experiments, three additional studies were conducted. First, for comparison with effects of the fentanyl/naltrexone mixtures, cumulative dose-effect functions were determined for a series of six other MOR ligands that varied from low to high in their efficacy at mu receptors as determined by in vitro assays of agonist-stimuluated GTPγS binding (Emmerson et al., 1996; Selley et al., 1998; Alt et al., 2001; Thompson et al., 2004; Yuan et al., 2015): 17-cyclopropylmethyl-3,14β-dihyroxy-4,5α-epoxy-6α-[(3′-isoquinolyl)acetamindo]morphinan (NAQ) (0.1–10 mg/kg, i.m.), buprenorphine (0.032–3.2 mg/kg, i.m.), nalbuphine (0.032–3.2 mg/kg, i.m.), morphine (0.1–10 mg/kg, i.m.), oxycodone (0.01–1 mg/kg, i.m.), and methadone (0.1–5.6 mg/kg, i.m.). Each dose-effect function was determined once. Drugs were tested up to doses that produced maximal antinociception, undesirable physiologic effects such as respiratory depression, or antagonized fentanyl effects in other studies. These experiments were generally conducted twice per week, except for studies with buprenorphine, nalbuphine, and morphine, which were separated by at least 7 days to allow dissipation of long-acting drug effects and/or to minimize potential effects of antinociceptive tolerance. Second, receptor theory predicts that pretreatment with a low-efficacy agonist should attenuate the potency, but not efficacy, of a higher efficacy agonist and thus shift the higher efficacy agonist dose-effect function to the right. To test this hypothesis, fixed-dose pretreatment experiments were conducted with naltrexone (0.0032–0.032 mg/kg, i.m.), NAQ (10 mg/kg, i.m.), or 1:0.22 fentanyl/naltrexone mixture (0.032 mg/kg fentanyl + 0.007 mg/kg naltrexone, i.m.) to cumulative fentanyl (0.001–1 mg/kg, i.m.), and each experiment was singly determined. Naltrexone, NAQ, and the fentanyl/naltrexone mixture were administered 15 minutes before the first fentanyl dose. Finally, drug interactions can be influenced not only by the relative drug doses in a mixture but also by their relative time courses. Accordingly, the time course of 0.056 mg/kg fentanyl was determined when combined with naltrexone as a 1:0.074 fixed-proportion mixture for simultaneous administration of both drugs, and when the equivalent naltrexone dose (0.0041 mg/kg) in the 1:0.074 fentanyl/naltrexone mixture was administered 3 minutes before or 3 minutes after 0.056 mg/kg fentanyl alone. Tail-withdrawal latencies were redetermined 10, 30, and 100 minutes after fentanyl administration unless emergence of respiratory depression required rescue with additional naltrexone treatments. These experiments were generally conducted twice per week.
Data Analysis.
Drug effects were expressed as the percent maximum possible effect (%MPE) using the following equation:
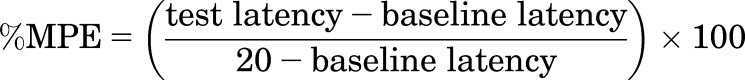 |
where test latency was the tail-withdrawal latency from either 50 or 54°C water obtained after drug administration, and baseline latency was the latency from either 50 or 54°C water obtained before drug administration. Maximum antinociceptive effects were also determined for each drug or mixture at the group mean and individual level for thermal stimulus intensity. Maximum effect was defined as the highest effect produced by any dose. Group mean maximum effects were compared using a one-way repeated-measures analysis of variance, and a Tukey post-hoc test was conducted following a significant main effect. In addition, maximum effect values were used in the analysis described in the next paragraph.
Theoretically, fentanyl/naltrexone mixtures should be useful to generate precise increments in efficacy that can be used 1) to generate mixtures with efficacies not available in existing single molecules, 2) to calibrate efficacy requirements for drug effects in different procedures, and 3) to infer efficacies of other drugs tested in those procedures. For example, if the relative efficacies of naltrexone and fentanyl are set arbitrarily at 0 and 1, respectively, then mixtures of 1:3, 1:1, and 3:1 fentanyl/naltrexone (after correcting for ligand affinity) will have relative efficacies along this continuum of 0.25, 0.5, and 0.75, respectively (i.e., relative efficacy = fractional contribution of fentanyl to the total drug in the mixture). The efficacy requirement of a given procedure can then be quantified by 1) testing effects of fentanyl and naltrexone alone and of all three mixtures, 2) generating efficacy-effect functions to relate maximum effects of each drug and mixture to the fentanyl proportion and associated relative efficacy, and 3) using nonlinear regression to determine the effective proportion of fentanyl to produce a maximum effect value equal to 50%MPE (EP50) in that procedure. The EP50 values can then be compared across procedures. Additionally, once the efficacy-effect relationships are established, efficacy of a test drug can then be estimated as the fentanyl proportion that produces maximum effects equivalent to that of the test drug. To evaluate the utility of this approach, efficacy-effect curves were generated using nonlinear regression (GraphPad Prism, La Jolla, CA) to fit maximum effects data for fentanyl alone, naltrexone alone, and each mixture at each temperature using the following equation:
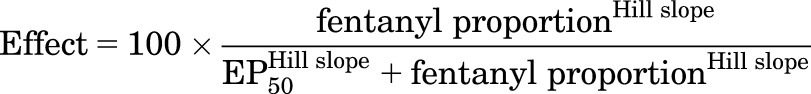 |
where fentanyl proportion was the fractional contribution of fentanyl to the total drug in the mixture, and EP50 was the fentanyl proportion that produced a maximum effect equivalent to 50%MPE. Relative efficacies of test compounds were then estimated for each individual monkey by comparing maximum effects of each drug at each temperature with the group mean efficacy-effect curves. Specifically, relative efficacy was defined as the fentanyl proportion at which maximum effects of the test drug deviated least from the efficacy-effect functions. Deviation was quantified as the sum of the differences between test drug maximum effect and efficacy-effect curve at both 50 and 54°C, and the fentanyl proportion was identified at which deviation was smallest. Individual test drug values were then averaged to yield group mean values and these data were analyzed using one-way repeated measures analysis of variance. In the presence of a significant main effect, comparisons between test drug maximum effects were made using Tukey’s test.
For pretreatment and time course studies, two-way repeated-measures analysis of variance was performed with experimental manipulation (e.g., pretreatment) and fentanyl dose or time after fentanyl administration as the main independent variables. Following a significant interaction, a Holm-Sidak post-hoc test was performed, and the criterion for significance was P < 0.05. Naltrexone pA2 values were determined as described previously (Bowen et al., 2002).
Drugs.
Fentanyl HCl, (−)-naltrexone HCl, morphine sulfate, and (−)-oxycodone HCl were supplied by the National Institute on Drug Abuse Drug Supply Program (Bethesda, MD). (−)-Nalbuphine HCl was provided by Dr. Kenner Rice (Drug Design and Synthesis Section, National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD). (±)-Methadone HCl and (±)-buprenorphine HCl were purchased from Spectrum Chemicals (Gardena, CA). NAQ HCl was synthesized and provided by Dr. Yan Zhang (Li et al., 2009). Fentanyl, naltrexone, buprenorphine, nalbuphine, oxycodone, morphine, methadone, and all mixtures were dissolved in sterile water. NAQ was dissolved in 50% dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO) and 50% sterile water. All drug doses were expressed as the salt forms listed previously, and administered intramuscularly in the thigh.
Results
Fentanyl-Naltrexone Fixed-Proportion Mixtures.
Across all baseline sessions before drug administration, monkeys always left their tail in 38°C water for 20 seconds, and the mean tail-withdrawal latencies at 50 and 54°C were 1.1 ± 0.5 and 0.7 ± 0.1 seconds, respectively. Figure 2 (left panels) shows the antinociceptive effects of fentanyl alone and following fixed naltrexone dose (0.0032–0.032 mg/kg, i.m.) pretreatments at 50 (top left panel) and 54°C (bottom left panel). Fentanyl alone produced dose-dependent and full (≥90%MPE) antinociception at both temperatures in all monkeys. Increasing naltrexone dose pretreatments produced parallel rightward shifts in the fentanyl dose-effect function at both temperatures. Mean fentanyl ED50 values are shown in Table 3, and the naltrexone pA2 values (95% confidence limits) were 8.58 (8.35, 8.82) and 8.50 (7.96, 8.52) for 50 and 54°C, respectively. Figure 2 (right panels) shows the antinociceptive effects of fentanyl alone, naltrexone alone, and the three fentanyl/naltrexone mixtures at 50 (top right panel) and 54°C (bottom right panel). Maximum effect values from mean dose-effect curves are shown in Table 1, and maximum effect values in individual monkeys are shown in Table 2. As in the left panels (Fig. 2), fentanyl alone produced dose-dependent antinociception. In contrast, naltrexone alone was ineffective at both temperatures (<5%MPE) (right panels Fig. 2). Fentanyl/naltrexone mixtures produced a naltrexone proportion-dependent decrease in maximum effects. Fentanyl alone and the 1:0.025 fentanyl/naltrexone mixture produced maximum effects that were significantly different from both naltrexone alone and the 1:0.22 fentanyl/naltrexone mixture at both 50 and 54°C (50°C: F1.5,4.4 = 16.0, P = 0.0111; 54°C: F1.9,5.7 = 31.3, P = 0.0009).
Fig. 2.

Effects of fixed-dose naltrexone pretreatments to fentanyl and fixed-proportion fentanyl/naltrexone mixtures in an assay of thermal nociception in male rhesus monkeys. Left panels show effects of fentanyl alone and after increasing naltrexone doses administered as a 15-minute pretreatment to fentanyl at 50°C (top) and 54°C (bottom) thermal intensities. Right panels show effects of fentanyl alone, naltrexone alone, and three fentanyl/naltrexone mixtures at 50°C (top) and 54°C (bottom). Abscissae: cumulative intramuscular fentanyl dose (left panels) or cumulative drug dose (right panels) in milligrams per kilogram. Note that for data with fentanyl/naltrexone mixtures in the right panels, the abscissa shows the fentanyl dose in the mixture, and the naltrexone dose = fentanyl dose × naltrexone proportion. Ordinates: %MPE. All points represent mean ± S.E.M. value of four monkeys.
TABLE 3.
Fentanyl ED50 values and (95% confidence limits) administered alone or following a 15-minute pretreatment with naltrexone (0.0032–0.032 mg/kg), 10 mg/kg NAQ, or 0.032 mg/kg fentanyl/naltrexone (1:0.22) mixture in an assay of thermal nociception at 50 and 54°C
Data are presented as the mean value of three monkeys for the naltrexone pretreatment studies and mean value of four monkeys for the NAQ and fentanyl/naltrexone mixture pretreatment studies.
| Treatment | ED50 in mg/kg (95% CL) |
|
|---|---|---|
| 50°C |
54°C |
|
| mg/kg | mg/kg | |
| Fentanyl alone | 0.006 (0.006, 0.006) | 0.018 (0.018, 0.018) |
| Fentanyl + 0.0032 mg/kg naltrexone | 0.021 (0.016, 0.028)a | 0.035 (0.018, 0.069) |
| Fentanyl + 0.01 mg/kg naltrexone | 0.018 (0.013, 0.05)a | 0.169 (0.057, 0.228)a |
| Fentanyl + 0.032 mg/kg naltrexone | 0.057 (0.043, 0.128)a | 0.257 (0.109, 0.608)a |
| Fentanyl alone | 0.006 (0.005, 0.006) | 0.014 (0.012, 0.017) |
| Fentanyl + 10 mg/kg NAQ pretreatment | 0.041 (0.014, 0.118)a | 0.155 (0.122, 0.197)a |
| Fentanyl alone | 0.006 (0.005, 0.006) | 0.014 (0.012, 0.017) |
| Fentanyl + 0.032 mg/kg fentanyl/naltrexone (1:0.22) pretreatment | <0.017 (0.002, 0.18)b | 0.035 (0.015, 0.08) |
CL, confidence limit.
Denotes nonoverlapping 95% CL.
The ED50 value could only be determined in two out of four monkeys because no fentanyl dose produced <50%MPE.
TABLE 1.
Group mean %MPEmax values and (±S.E.M.) for each fentanyl/naltrexone combination or test drug administered in an assay of thermal nociception at 50 and 54°C in rhesus monkeys (n = 4)
| Drug or Drug Mixture | %MPEmax (S.E.M.) |
|
|---|---|---|
| 50°C | 54°C | |
| Fentanyl | 100 (0)a,b | 96.4 (2.8)a,b |
| 1:0.025 Fentanyl/naltrexone | 100 (0)a,b | 95.3 (4.7)a,b |
| 1:0.074 Fentanyl/naltrexone | 70.8 (24.2) | 40.4 (14.9) |
| 1:0.22 Fentanyl/naltrexone | 14.5 (9.5) | 12.9 (10.7) |
| (−)-Naltrexone | −1.8 (5.6) | 0.9 (0.4) |
| (±)-Methadone | 100 (0) | 100 (0) |
| (−)-Oxycodone | 100 (0) | 89.5 (6.7) |
| (−)-Morphine | 100 (0) | 78.1 (9.9) |
| (−)-Nalbuphine | 100 (0) | 64.1 (14.5) |
| (±)-Buprenorphine | 93.3 (6.7) | 14.0 (6.0) |
| NAQ | 8.9 (6.9) | 5.1 (3.7) |
Significantly different from naltrexone (P < 0.05).
Significantly different from the 1:0.22 fentanyl/naltrexone mixture (P < 0.05).
TABLE 2.
Individual %MPEmax values for each fentanyl/naltrexone combination or test drug administered in an assay of thermal nociception at 50 and 54°C in rhesus monkeys
| Drug or Drug Mixture | %MPEmax |
|||||||
|---|---|---|---|---|---|---|---|---|
| 50°C |
54°C |
|||||||
| M1414 | M1473 | M1478 | M1503 | M1414 | M1473 | M1478 | M1503 | |
| Fentanyl | 100 | 100 | 100 | 100 | 87 | 100 | 100 | 100 |
| 1:0.025 Fentanyl/naltrexone | 100 | 100 | 100 | 100 | 81.1 | 100 | 100 | 100 |
| 1:0.074 Fentanyl/naltrexone | 84.2 | 100 | 100 | 3.3 | 37.6 | 62.7 | 62.2 | 4.7 |
| 1:0.22 Fentanyl/naltrexone | 9 | 6.6 | 42.2 | 10.4 | 18.1 | 10 | 44.2 | 1.1 |
| (−)-Naltrexone | 9.5 | 2.1 | −12.6 | −1.6 | 3.2 | 1.8 | −0.3 | 0.6 |
| (±)-Methadone | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| (−)-Oxycodone | 100 | 100 | 100 | 100 | 71.4 | 100 | 86.1 | 100 |
| (−)-Morphine | 100 | 100 | 100 | 100 | 74.9 | 52.8 | 84.7 | 100 |
| (−)-Nalbuphine | 100 | 100 | 100 | 100 | 52.8 | 100 | 71.9 | 31.7 |
| (±)-Buprenorphine | 73.2 | 100 | 100 | 100 | 32.1 | 18.7 | 29.3 | 16.5 |
| NAQ | 4.6 | 5.9 | 28.8 | 8 | 6.4 | 1.1 | 16.1 | 2.8 |
MOR Ligands.
Figure 3 shows the antinociceptive effects of the MOR ligands NAQ, nalbuphine, buprenorphine, oxycodone, morphine, and methadone at both 50 (top panel) and 54°C (bottom panel). Maximum effects values from mean dose-effect curves are given in Table 1, and maximum effect values in individual monkeys are given in Table 2. All drugs except NAQ produced maximum or near maximum antinociceptive effects at 50°C, and the rank of order of maximum effects at 54°C (from lowest to highest) was NAQ, buprenorphine, nalbuphine, morphine, oxycodone, and methadone. In general, the sensitivity of individual monkeys to declining efficacy of fentanyl/naltrexone mixtures paralleled sensitivity to declining efficacy of test compounds. For example, the 1:0.22 fentanyl/naltrexone mixture produced the greatest antinociceptive effect at 54°C (44.2%MPE) in M1478, and this monkey also displayed the greatest or close to the greatest maximum individual antinociceptive effect at 54°C following nalbuphine, buprenorphine, or NAQ administration. In contrast, the 1:0.074 fentanyl/naltrexone mixture produced the least antinociceptive effect at 54°C in M1503, and this monkey also showed the weakest or close to the weakest individual antinociceptive effects at 54°C following nalbuphine, buprenorphine, or NAQ administration.
Fig. 3.

Effects of six different MOR ligands in an assay of thermal nociception in male rhesus monkeys. Top panel shows effects of NAQ, nalbuphine, buprenorphine, morphine oxycodone, and methadone at 50°C; bottom panel shows effects at 54°C. Abscissae: cumulative intramuscular drug dose (milligrams per kilogram). Ordinates: %MPE. All points represent mean ± S.E.M. value of four monkeys.
Efficacy Estimates of MOR Ligands Relative to Fentanyl and Naltrexone.
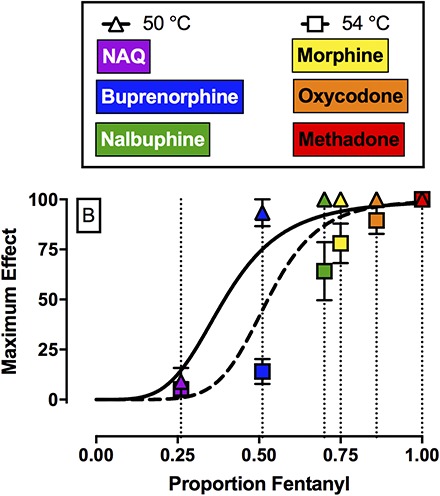
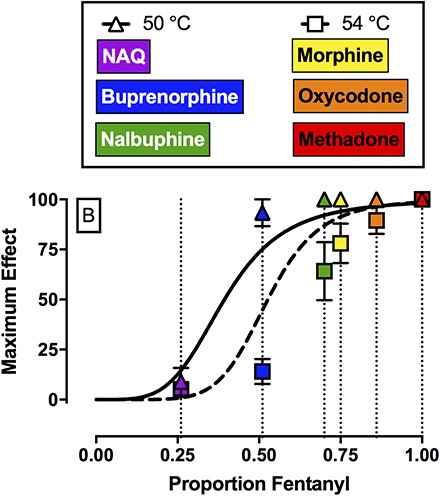
Figure 4A shows efficacy-effect curves that relate %MPEmax effects of fentanyl, naltrexone, and each mixture at 50 and 54°C to the proportion of fentanyl in the mixture from 0 (naltrexone alone) to 1 (fentanyl alone). Comparison of the nonlinear fits for the two different temperatures using the extra sum-of-squares F-test demonstrated that each temperature data set was best fit by different nonlinear functions (F2,6 = 7.3, P = 0.0249). For 50°C, the Hill slope was 4.26, the EP50 value (95% confidence limits) was 0.39 (0.34, 0.46), and the R2 value was 0.99. For 54°C, the Hill slope was 6.66, the EP50 value was 0.53 (0.41, 0.61), and the R2 value was 0.98. The 95% confidence limits for the EP50 values at 50 and 54°C overlapped. Figure 4B shows the best fit for the maximum effect of each test drug to the efficacy-effect curves defined by the naltrexone-to-fentanyl continuum. Using this analysis, the efficacy of each compound relative to the naltrexone-to-fentanyl continuum was determined and the results are reported in Table 4. Comparison of maximum effects demonstrated that fentanyl and methadone both produced significantly higher maximum effects compared with buprenorphine, NAQ, and naltrexone (F2.3, 6.8 = 23.3, P = 0.0008). In addition, buprenorphine also produced significantly higher maximum effects compared with NAQ.
Fig. 4.

Top panel (A) shows maximum antinociceptive effect at 50°C (triangles) and 54°C (squares) as a function of the fentanyl proportion in the fentanyl/naltrexone mixture in male rhesus monkeys. Bottom panel (B) shows empirically determined maximum antinociceptive effects of NAQ, buprenorphine, nalbuphine, morphine, oxycodone, and methadone. Results were fit to the model generated from the top panel, and relative efficacy of each ligand was estimated as the fentanyl proportion to produce maximum effects at 50 and 54°C that are most like the test ligand. Abscissae: Efficacy expressed as proportion fentanyl. “0” denotes naltrexone alone, “1” denotes fentanyl alone, and the efficacy of each mixture (Emix) was calculated as the fractional contribution of fentanyl to the mixture as described in Methods. Ordinates: maximum effect. All points represent the mean ± S.E.M. value of four monkeys.
TABLE 4.
Estimated efficacy of each compound relative to the naltrexone-to-fentanyl continuum in proportion fentanyl units (95% confidence limits) for each of the eight MOR ligands tested in rhesus monkeys (n = 4)
Individual %MPEmax values were fitted to the nonlinear function generated from the group mean results shown in Fig. 4A.
| Test Drug | Proportion Fentanyl (95% CL) |
|---|---|
| Fentanyl | 0.94 (0.77, 1.12)a,b,c |
| (±)-Methadone | 1 (1, 1)a,b,c |
| (−)-Oxycodone | 0.86 (0.58, 1.13) |
| (−)-Morphine | 0.75 (0.47, 1.03) |
| (−)-Nalbuphine | 0.7 (0.37, 1.03) |
| (±)-Buprenorphine | 0.51 (0.47, 0.54)b |
| NAQ | 0.26 (0.16, 0.36) |
| (−)-Naltrexone | 0.12 (−0.1, 0.33) |
CL, confidence limit.
Significantly different from naltrexone (P < 0.05).
Significantly different from NAQ (P < 0.05).
Significantly different from buprenorphine (P < 0.05).
Effects of NAQ or Fentanyl/Naltrexone (1:0.22) Pretreatment.
Figure 5 shows cumulative fentanyl dose-effect functions alone or following a 15-minute pretreatment with the low-efficacy MOR ligand NAQ (10 mg/kg; left panels) or the low-efficacy 1:0.22 fentanyl/naltrexone mixture (0.032 mg/kg; right panels) at both 50 (top panels) and 54°C (bottom panels) thermal intensities, and the fentanyl ED50 values are given in Table 3. Consistent with the results described in Fig. 2, fentanyl alone produced dose-dependent antinociception at both thermal intensities. NAQ pretreatment produced a significant (∼9-fold) increase in the fentanyl ED50 value at 54°C (Table 3) and significantly attenuated the antinociceptive effects of cumulative 0.032 mg/kg fentanyl (fentanyl dose: F2,6 = 566.3, P < 0.0001; NAQ: F1,3 = 176, P = 0.0009; interaction: F2,6 = 367.1, P < 0.0001). Conversely, pretreatment with 0.032 mg/kg 1:0.22 fentanyl/naltrexone did not significantly increase in the fentanyl ED50 value at 54°C (Table 3), although it did significantly decrease the antinociceptive effects of cumulative 0.032 mg/kg fentanyl at 54°C (fentanyl dose: F2,6 = 24.4, P = 0.0013; interaction: F2,6 = 16.1, P = 0.0038).
Fig. 5.

Effects of cumulative fentanyl (0.001–0.32 mg/kg, i.m.) administered either alone or following a 15-minute pretreatment with either 10 mg/kg NAQ (left panels) or 0.032 mg/kg fentanyl/naltrexone (1:0.22) (right panels) in rhesus monkeys. Abscissae: cumulative intramuscular fentanyl dose (milligrams per kilogram). Ordinates: %MPE. All points represent the mean ± S.E.M. value of four monkeys. Solid points denote statistical significance (P < 0.05) compared with fentanyl alone.
Time Course As a Factor in Drug-Interaction Studies.
Figure 6 shows the time course of antinociception produced at 50 and 54°C by 0.056 mg/kg fentanyl administered in combination with 0.0041 mg/kg naltrexone. When these two doses were administered simultaneously (i.e., 0.056 mg/kg of the 1:0.074 fentanyl/naltrexone mixture), submaximal antinociceptive effects were observed at both 50 and 54°C, and these effects dissipated after 30–100 minutes. The effects of this bolus mixture after 10 minutes were similar to the effects observed when the same dose of this mixture was tested as part of the cumulative dose-effect curve (from Fig. 2). Additionally, the effects of this bolus mixture dose were similar to effects observed when the fentanyl dose was administered 3 minutes after the naltrexone dose. However, when fentanyl was administered 3 minutes before naltrexone, the experiment had to be terminated because of severe sedation and respiratory depression in two monkeys that required additional naltrexone administration.
Fig. 6.
Time course of antinociceptive effects of 0.056 mg/kg fentanyl in combination with 0.0041 mg/kg naltrexone administered simultaneously as a bolus dose of the 1:0.074 fentanyl/naltrexone mixture or with the naltrexone dose administered as a 3-minute pretreatment to the fentanyl dose in rhesus monkeys. Antinociceptive effects of cumulative 0.056 mg/kg 1:0.074 fentanyl/naltrexone from Fig. 2 are also plotted for comparison. Abscissae: fentanyl dose (milligrams per kilogram). Ordinate: %MPE. Each point represents the mean ± S.E.M. value of four monkeys.
Discussion
The primary aim of the present study was to evaluate the degree to which fixed-proportion mixtures of fentanyl and naltrexone would produce effects predicted by receptor theory for mixtures of a competitive reversible agonist and antagonist targeting a common receptor. A secondary aim was to evaluate the utility of results with fentanyl/naltrexone mixtures for establishing an efficacy-effect scale that could be used to quantify 1) efficacy requirements for different drug effects, and 2) relative efficacies of different MOR ligands. There were three main findings. First, as predicted by receptor theory, the addition of naltrexone to fentanyl produced a naltrexone proportion-dependent decrease in the maximal antinociceptive effects of fentanyl/naltrexone mixtures. Second, the proportion of fentanyl in the mixtures served as a metric for efficacy of the mixtures, and this scale provided a strategy for quantifying efficacy requirements for different drug effects (i.e., antinociception at 50 vs. 54°C) and relative in vivo efficacies of different MOR ligands. Finally, the results reported here also provide insight into factors that can limit utility of this approach. Overall, these results support the potential use of agonist/antgonist mixtures as tools in basic research, while also suggesting factors that may influence the usefulness of this approach.
Fentanyl alone produced dose-dependent and thermal intensity–dependent antinociception in rhesus monkeys, whereas naltrexone alone produced <10%MPE up to the largest doses tested. These results were consistent with a large body of literature demonstrating the antinociceptive effects of fentanyl in humans (Finch and DeKornfeld, 1967), nonhuman primates (Nussmeier et al., 1991; Gatch et al., 1995; Maguire and France, 2014), and rodents (Millan, 1989; Walker et al., 1994; Minami et al., 2009). Because naltrexone failed to produce significant antinociception, one method to determine whether a behaviorally active dose range was administered would be to give naltrexone as a pretreatment to cumulative fentanyl. In this experiment, receptor theory would predict that increasing naltrexone fixed-dose pretreatments would produce parallel rightward shifts in the fentanyl dose-effect function. The present results were consistent with this hypothesis, and the naltrexone pA2 values reported in this study were consistent with previous naltrexone studies in monkeys (Rowlett et al., 2000; Bowen et al., 2002; Gerak and France, 2007). Overall, these results provide an empirical foundation to interpret the antinociceptive effects of fixed-proportion fentanyl and naltrexone mixtures.
Receptor theory predicts that fixed-proportion mixtures of a competitive reversible agonist and antagonist should produce maximal effects that decline as the proportion of agonist in the mixture declines. Results support this prediction. Specifically, the MOR agonist fentanyl produced dose-dependent antinociception at both 50 and 54°C, and mixtures of fentanyl with the MOR antagonist naltrexone produced decreasing maximal antinociceptive effects as the proportion of fentanyl in the mixture decreased. The declining maximal effects of fentanyl/naltrexone mixtures with declining fentanyl proportions resembles the declining maximal effects of mu agonists produced by pretreatments with irreversible antagonists (Zernig et al., 1994; Walker and Young, 2002). As such, fixed-proportion mixtures with competitive antagonists may serve as an alternative to use of irreversible antagonists for research on the role of efficacy as a determinant of drug effects. This approach may be especially useful in research on systems for which competitive antagonists are available, but irreversible antagonists are not.
Because the agonist/antagonist proportion determined the apparent efficacy of a mixture, this proportion could be used as a quantitative measure of in vivo efficacy. In the present study, this metric was applied in two ways. First, we evaluated the efficacy requirements for antinociception at 50 and 54°C by comparing the fentanyl proportion required to produce a maximal effect of 50%MPE at each temperature. Although the 95% confidence limits for these values overlapped, the higher mean value at 54°C agrees with other data to suggest that efficacy requirements for antinociception are higher at 54 than 50°C (Walker et al., 1993; Banks et al., 2010b; Maguire and France, 2014). Additionally, although this study compared efficacy requirements of similar endpoints (i.e., antinociception at two different stimulus intensities in rhesus monkeys), it is theoretically possible to apply this approach across multiple endpoints that could include not only other behavioral and physiologic endpoints in rhesus monkeys, but also endpoints in other species or in in vitro assays. For example, two undesirable effects of MOR agonists that limit their clinical utility are respiratory depression and abuse liability, and fentanyl/naltrexone mixtures could be used to quantify the efficacy requirement for each of these or any other MOR agonist effect of interest. These experiments would also provide empirical data on the utility of agonist/antagonist mixtures to assess the efficacy requirements of different experimental endpoints.
A second implication of the present study was that fentanyl/naltrexone mixtures could be used to stratify MOR ligands based on their in vivo antinociceptive efficacy in rhesus monkeys. In the present study, NAQ produced <10%MPE and these results are consistent with and extend previous findings in mice (Zhang et al., 2014; Yuan et al., 2015) and rats (Siemian et al., 2016). Buprenorphine (Walker et al., 1995; Maguire and France, 2014), nalbuphine (Walker et al., 1993; France and Gerak, 1994; Banks et al., 2010b), morphine (Bowen et al., 2002), oxycodone, and methadone (Stevenson et al., 2003; Banks et al., 2010b) produced dose-dependent and thermal intensity–dependent antinociception in the present study, and these results were generally consistent with the extant literature examining MOR agonists in a warm-water tail-withdrawal procedure in monkeys. With one major exception (see the results regarding nalbuphine in the next paragraph), the order of MOR efficacies for these drugs as ranked here agrees with the order of efficacies as determined by in vitro approaches such as agonist-stimulated GTPγS binding (Selley et al., 1998; Alt et al., 2001; Yuan et al., 2015). Specifically, both approaches yield a rank order of lowest-to-highest efficacy of naltrexone < NAQ < buprenorphine < morphine < oxycodone < fentanyl < methadone. By comparing effects of these mu agonists to effects of fentanyl/naltrexone mixtures, it was possible not only to rank order drug efficacies, but also to provide a quantitative measure of those relative efficacies, expressed as fentanyl proportion.
The results with nalbuphine in the present study did not agree with previous in vitro results using agonist-stimulated GTPγS binding with either mouse MOR (Selley et al., 1998) or rat MOR (Alt et al., 2001). The basis for this difference between published GTPγS results and antinociceptive efficacy in rhesus monkeys remains to be empirically determined. Although there are no published GTPγS results with any MOR ligand using monkey MOR, two lines of evidence support the conclusion that nalbuphine functions as a higher efficacy MOR ligand than buprenorphine in rhesus monkeys. First, in human embryonic kidney cells expressing MOR and examining inhibition of forskolin-stimulated cAMP accumulation, nalbuphine produced similar efficacy to morphine (Gharagozlou et al., 2003). Second, the present nalbuphine results demonstrating greater antinociceptive effects of nalbuphine compared with buprenorphine are generally consistent with previously published studies in nonhuman primates (Walker et al., 1993, 1995; Maguire and France, 2014). In addition to these antinociceptive studies, nalbuphine also shows higher efficacy than buprenorphine in an assay of schedule-controlled responding. For example, nalbuphine produced dose-dependent and near complete suppression of operant responding (Stevenson et al., 2003; Banks et al., 2010b), whereas buprenorphine decreased operant responding to approximately 65% of control (Negus et al., 2002). Overall, the present results highlight potential species differences in MOR ligand efficacy and support the utility of nonhuman primates in preclinical pharmacology research.
Although the present results support the concept that agonist/antagonist mixtures can be used to manipulate apparent in vivo efficacy, these results also revealed factors that can influence the precision of this approach. Two particular limitations will be mentioned here. First, the efficacies of the constituent drugs in a mixture define the upper and lower boundaries of efficacy that can be assessed. For example, in the present study, fentanyl served as the agonist, and studies of in vitro agonist-stimulated GTPγS binding suggest that some MOR ligands (e.g., methadone) may have higher efficacy than fentanyl (Selley et al., 1998; Alt et al., 2001). Because fentanyl defines the upper boundary of efficacy that can be achieved with fentanyl/naltrexone mixtures, these mixtures would not be useful for scaling effects of drugs such as methadone that may have higher efficacy than fentanyl. Similarly, these mixtures would not be useful for scaling effects of drugs that have lower efficacy than naltrexone.
Second, although agonist/antagonist proportions can be precisely controlled in a mixture, the pharmacokinetics and associated time courses of the constituent drugs play a key role in determining the proportional drug concentrations at receptor targets after in vivo drug administration. For example, in the present study, cumulative administration of the 1:0.074 fentanyl/naltrexone mixture could be safely studied at doses up to 0.32 mg/kg. However, bolus administration of this mixture at a dose of 0.1 mg/kg fentanyl + 0.0074 mg/kg naltrexone could not be studied due to the onset of severe sedation and respiratory depression in at least one monkey. This suggests that, after bolus administration, fentanyl distributes more quickly than naltrexone to receptors that mediate sedation and respiratory depression. This difference may be mitigated during cumulative dosing by sustained effects of naltrexone doses administered early in the dosing regimen. Additionally, the impact of these pharmacokinetic issues may be influenced by both the agonist/antagonist proportion and overall mixture dose. For example, in the present study, both cumulative and bolus administration of 0.056 mg/kg 1:0.074 fentanyl/naltrexone produced similar effects. However, administration of fentanyl just 3 minutes before naltrexone resulted in severe sedation and respiratory depression. Overall, these results highlight the time course of drug effects as a key consideration in the deployment of competitive agonist/antagonist mixtures for both basic research or clinical studies.
As a final note, the present results with fentanyl/naltrexone mixtures can be compared with development of opioid formulations that include a MOR agonist in combination with the competitive reversible antagonist naloxone (e.g., fixed-proportion formulations of oxycodone + naloxone or buprenorphine + naloxone) (Mendelson and Jones, 2003; Chen et al., 2014; Fanelli and Fanelli, 2015; O’Brien, 2015). Consumption of these products by intended enteral routes of administration results in naloxone distribution to the gastrointenstinal tract (which may reduce constipating effects of the agonist) but limited distribution to the central nervous system due to extensive first-pass metabolism by the liver (resulting in limited interference with centrally mediated agonist effects). However, parenteral administration bypasses first-pass metabolism, resulting in greater naloxone distribution to the central nervous system and potential blockade of centrally mediated agonist effects and/or precipitation of withdrawal in opioid-dependent subjects. As a result of these characteristics, naloxone combination products are thought to have fewer gastrointestinal side effects and lower abuse liability than the agonists alone. The experimental design deployed in the present study could be used to test this hypothesis, with the caveat that naloxone’s relatively short duration of action may hamper naloxone’s utility for this type of research. For example, naloxone should be more potent to produce proportion-dependent downward shifts in agonist dose-effect curves for gastrointestinal side effects than centrally mediated effects after enteral but not parenteral administration. The present study also suggests how the general concept of agonist + antagonist mixtures can be expanded beyond naloxone-containing combination products to include other antagonists such as naltrexone, or agonist + antagonist mixtures targeting other receptors, yielding mixtures with other pharmacological profiles.
Abbreviations
- EP50
effective proportion of fentanyl to produce a maximum effect equal to 50% maximum possible effect in that procedure
- MOR
mu-opioid receptor
- %MPE
percent maximum possible effect
- NAQ
17-cyclopropylmethyl-3,14β-dihyroxy-4,5α-epoxy-6α-[(3′-isoquinolyl)acetamindo]morphinan
Authorship Contributions
Participated in research design: Cornelissen, Negus, Banks.
Conducted experiments: Cornelissen.
Contributed new reagents or analytic tools: Obeng, Zhang, Rice.
Performed data analysis: Cornelissen, Negus, Banks.
Wrote or contributed to the writing of the manuscript: Cornelissen, Negus, Banks.
Footnotes
This research was supported by the National Institutes of Health [Grants R01DA037287, R01DA026946, DA024022, and DA044855]. In addition, this research was supported (in part) by the Intramural Research Program of the National Institutes of Health National Institute on Drug Abuse and the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism.
The National Institute on Drug Abuse had no role in the study design, collection, analysis, and interpretation of the data; in the writing; or in the decision to submit the manuscript for publication. The manuscript content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
References
- Alt A, McFadyen IJ, Fan CD, Woods JH, Traynor JR. (2001) Stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding in digitonin-permeabilized C6 rat glioma cells: evidence for an organized association of μ-opioid receptors and G protein. J Pharmacol Exp Ther 298:116–121. [PubMed] [Google Scholar]
- Banks ML, Folk JE, Rice KC, Negus SS. (2010a) Selective enhancement of fentanyl-induced antinociception by the delta agonist SNC162 but not by ketamine in rhesus monkeys: further evidence supportive of delta agonists as candidate adjuncts to mu opioid analgesics. Pharmacol Biochem Behav 97:205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks ML, Rice KC, Negus SS. (2010b) Antinociceptive interactions between mu-opioid receptor agonists and the serotonin uptake inhibitor clomipramine in rhesus monkeys: role of mu agonist efficacy. J Pharmacol Exp Ther 335:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman J, France CP, Holtzman SG, Katz JL, Koek W, Stephens DN. (2000) Agonist efficacy, drug dependence, and medications development: preclinical evaluation of opioid, dopaminergic, and GABAA-ergic ligands. Psychopharmacology (Berl) 153:67–84. [DOI] [PubMed] [Google Scholar]
- Bowen CA, Fischer BD, Mello NK, Negus SS. (2002) Antagonism of the antinociceptive and discriminative stimulus effects of heroin and morphine by 3-methoxynaltrexone and naltrexone in rhesus monkeys. J Pharmacol Exp Ther 302:264–273. [DOI] [PubMed] [Google Scholar]
- Chen KY, Chen L, Mao J. (2014) Buprenorphine-naloxone therapy in pain management. Anesthesiology 120:1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Czoty PW, Kiguchi N, Cami-Kobeci G, Sukhtankar DD, Nader MA, Husbands SM, Ko MC. (2016) A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proc Natl Acad Sci USA 113:E5511–E5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson PJ, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. (1996) Characterization of opioid agonist efficacy in a C6 glioma cell line expressing the mu opioid receptor. J Pharmacol Exp Ther 278:1121–1127. [PubMed] [Google Scholar]
- Emmerson PJ, Liu MR, Woods JH, Medzihradsky F. (1994) Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther 271:1630–1637. [PubMed] [Google Scholar]
- Fanelli G, Fanelli A. (2015) Developments in managing severe chronic pain: role of oxycodone-naloxone extended release. Drug Des Devel Ther 9:3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch JS, DeKornfeld TJ. (1967) Clinical investigation of the analgesic potency and respiratory depressant activity of fentanyl, a new narcotic analgesic. J Clin Pharmacol J New Drugs 7:46–51. [DOI] [PubMed] [Google Scholar]
- France CP, Gerak LR. (1994) Behavioral effects of 6-methylene naltrexone (nalmefene) in rhesus monkeys. J Pharmacol Exp Ther 270:992–999. [PubMed] [Google Scholar]
- Furchgott R. (1966) The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes, in Advances in Drug Research (Harper N, Simmonds A. eds) pp 21–55, Academic Press, New York. [Google Scholar]
- Gatch MB, Negus SS, Butelman ER, Mello NK. (1995) Antinociceptive effects of cocaine/opioid combinations in rhesus monkeys. J Pharmacol Exp Ther 275:1346–1354. [PubMed] [Google Scholar]
- Gerak LR, Butelman ER, Woods JH, France CP. (1994) Antinociceptive and respiratory effects of nalbuphine in rhesus monkeys. J Pharmacol Exp Ther 271:993–999. [PubMed] [Google Scholar]
- Gerak LR, France CP. (2007) Time-dependent decreases in apparent pA2 values for naltrexone studied in combination with morphine in rhesus monkeys. Psychopharmacology (Berl) 193:315–321. [DOI] [PubMed] [Google Scholar]
- Gharagozlou P, Demirci H, David Clark J, Lameh J. (2003) Activity of opioid ligands in cells expressing cloned mu opioid receptors. BMC Pharmacol 3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grim TW, Morales AJ, Gonek MM, Wiley JL, Thomas BF, Endres GW, Sim-Selley LJ, Selley DE, Negus SS, Lichtman AH. (2016) Stratification of cannabinoid 1 receptor (CB1R) agonist efficacy: manipulation of CB1R density through use of transgenic mice reveals congruence between in vivo and in vitro assays. J Pharmacol Exp Ther 359:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. (1993) Pharmacologic Analysis of Drug Receptor Interaction, Raven Press, New York. [Google Scholar]
- Kenakin T. (2012) Pharmacology in Drug Discovery, Elsevier, Boston. [Google Scholar]
- Kishioka S, Paronis CA, Lewis JW, Woods JH. (2000) Buprenorphine and methoclocinnamox: agonist and antagonist effects on respiratory function in rhesus monkeys. Eur J Pharmacol 391:289–297. [DOI] [PubMed] [Google Scholar]
- Ko MC, Butelman ER, Traynor JR, Woods JH. (1998) Differentiation of kappa opioid agonist-induced antinociception by naltrexone apparent pA2 analysis in rhesus monkeys. J Pharmacol Exp Ther 285:518–526. [PMC free article] [PubMed] [Google Scholar]
- Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y. (2009) Design, synthesis, and biological evaluation of 6α- and 6β-N-heterocyclic substituted naltrexamine derivatives as μ opioid receptor selective antagonists. J Med Chem 52:1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire DR, France CP. (2014) Impact of efficacy at the μ-opioid receptor on antinociceptive effects of combinations of μ-opioid receptor agonists and cannabinoid receptor agonists. J Pharmacol Exp Ther 351:383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson J, Jones RT. (2003) Clinical and pharmacological evaluation of buprenorphine and naloxone combinations: why the 4:1 ratio for treatment? Drug Alcohol Depend 70 (Suppl 2):S29–S37. [DOI] [PubMed] [Google Scholar]
- Millan MJ. (1989) Kappa-opioid receptor-mediated antinociception in the rat. I. Comparative actions of mu- and kappa-opioids against noxious thermal, pressure and electrical stimuli. J Pharmacol Exp Ther 251:334–341. [PubMed] [Google Scholar]
- Minami K, Hasegawa M, Ito H, Nakamura A, Tomii T, Matsumoto M, Orita S, Matsushima S, Miyoshi T, Masuno K, et al. (2009) Morphine, oxycodone, and fentanyl exhibit different analgesic profiles in mouse pain models. J Pharmacol Sci 111:60–72. [DOI] [PubMed] [Google Scholar]
- National Research Council (2011) Guide for the care and use of laboratory animals (8th ed.). Washington, DC: National Academies Press. [Google Scholar]
- Negus SS, Bidlack JM, Mello NK, Furness MS, Rice KC, Brandt MR. (2002) Delta opioid antagonist effects of buprenorphine in rhesus monkeys. Behav Pharmacol 13:557–570. [DOI] [PubMed] [Google Scholar]
- Negus SS, Brandt MR, Gatch MB, Mello NK. (2003) Effects of heroin and its metabolites on schedule-controlled responding and thermal nociception in rhesus monkeys: sensitivity to antagonism by quadazocine, naltrindole and β-funaltrexamine. Drug Alcohol Depend 70:17–27. [DOI] [PubMed] [Google Scholar]
- Negus SS, Burke TF, Medzihradsky F, Woods JH. (1993) Effects of opioid agonists selective for mu, kappa and delta opioid receptors on schedule-controlled responding in rhesus monkeys: antagonism by quadazocine. J Pharmacol Exp Ther 267:896–903. [PubMed] [Google Scholar]
- Nussmeier NA, Benthuysen JL, Steffey EP, Anderson JH, Carstens EE, Eisele JH, Jr, Stanley TH. (1991) Cardiovascular, respiratory, and analgesic effects of fentanyl in unanesthetized rhesus monkeys. Anesth Analg 72:221–226. [DOI] [PubMed] [Google Scholar]
- O’Brien T. (2015) The attractiveness of opposites: agonists and antagonists. J Pain Palliat Care Pharmacother 29:67–69. [DOI] [PubMed] [Google Scholar]
- Pitts RC, Allen RM, Walker EA, Dykstra LA. (1998) Clocinnamox antagonism of the antinociceptive effects of mu opioids in squirrel monkeys. J Pharmacol Exp Ther 285:1197–1206. [PubMed] [Google Scholar]
- Rowlett JK, Spealman RD, Platt DM. (2000) Cocaine-like discriminative stimulus effects of heroin in squirrel monkeys: role of active metabolites and opioid receptor mechanisms. Psychopharmacology (Berl) 150:191–199. [DOI] [PubMed] [Google Scholar]
- Ruffolo RR., Jr (1982) Review important concepts of receptor theory. J Auton Pharmacol 2:277–295. [DOI] [PubMed] [Google Scholar]
- Selley DE, Liu Q, Childers SR. (1998) Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTPγS binding in mMOR-CHO cells and rat thalamus. J Pharmacol Exp Ther 285:496–505. [PubMed] [Google Scholar]
- Siemian JN, Obeng S, Zhang Y, Zhang Y, Li JX. (2016) Antinociceptive interactions between the imidazoline I2 receptor agonist 2-BFI and opioids in rats: role of efficacy at the μ-opioid receptor. J Pharmacol Exp Ther 357:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Linsenmayer DC, Rice KC, Negus SS. (2003) Opioid interactions in rhesus monkeys: effects of δ + μ and δ + κ agonists on schedule-controlled responding and thermal nociception. J Pharmacol Exp Ther 307:1054–1064. [DOI] [PubMed] [Google Scholar]
- Thompson CM, Wojno H, Greiner E, May EL, Rice KC, Selley DE. (2004) Activation of G-proteins by morphine and codeine congeners: insights to the relevance of O- and N-demethylated metabolites at μ- and δ-opioid receptors. J Pharmacol Exp Ther 308:547–554. [DOI] [PubMed] [Google Scholar]
- Walker EA, Butelman ER, DeCosta BR, Woods JH. (1993) Opioid thermal antinociception in rhesus monkeys: receptor mechanisms and temperature dependency. J Pharmacol Exp Ther 267:280–286. [PubMed] [Google Scholar]
- Walker EA, Makhay MM, House JD, Young AM. (1994) In vivo apparent pA2 analysis for naltrexone antagonism of discriminative stimulus and analgesic effects of opiate agonists in rats. J Pharmacol Exp Ther 271:959–968. [PubMed] [Google Scholar]
- Walker EA, Young AM. (2002) Clocinnamox distinguishes opioid agonists according to relative efficacy in normal and morphine-treated rats trained to discriminate morphine. J Pharmacol Exp Ther 302:101–110. [DOI] [PubMed] [Google Scholar]
- Walker EA, Zernig G, Woods JH. (1995) Buprenorphine antagonism of mu opioids in the rhesus monkey tail-withdrawal procedure. J Pharmacol Exp Ther 273:1345–1352. [PubMed] [Google Scholar]
- Walker EA, Zernig G, Young AM. (1998) In vivo apparent affinity and efficacy estimates for mu opiates in a rat tail-withdrawal assay. Psychopharmacology (Berl) 136:15–23. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Zaidi SA, Stevens DL, Scoggins KL, Mosier PD, Kellogg GE, Dewey WL, Selley DE, Zhang Y. (2015) Design, syntheses, and pharmacological characterization of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3′-carboxamido)morphinan analogues as opioid receptor ligands. Bioorg Med Chem 23:1701–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernig G, Butelman ER, Lewis JW, Walker EA, Woods JH. (1994) In vivo determination of mu opioid receptor turnover in rhesus monkeys after irreversible blockade with clocinnamox. J Pharmacol Exp Ther 269:57–65. [PubMed] [Google Scholar]
- Zernig G, Lewis JW, Woods JH. (1997) Clocinnamox inhibits the intravenous self-administration of opioid agonists in rhesus monkeys: comparison with effects on opioid agonist-mediated antinociception. Psychopharmacology (Berl) 129:233–242. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Braithwaite A, Yuan Y, Streicher JM, Bilsky EJ. (2014) Behavioral and cellular pharmacology characterization of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3′-carboxamido)morphinan (NAQ) as a mu opioid receptor selective ligand. Eur J Pharmacol 736:124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman DM, Leander JD, Reel JK, Hynes MD. (1987) Use of beta-funaltrexamine to determine mu opioid receptor involvement in the analgesic activity of various opioid ligands. J Pharmacol Exp Ther 241:374–378. [PubMed] [Google Scholar]