

Abstract
Although dystonia is often associated with abnormal dopamine neurotransmission, dopaminergic drugs are not currently used to treat dystonia because there is a general view that dopaminergic drugs are ineffective. However, there is little conclusive evidence to support or refute this assumption. Therefore, to assess the therapeutic potential of these compounds, we analyzed results from multiple trials of dopamine receptor agonists in patients with idiopathic dystonias and also tested the efficacy of dopamine receptor agonists in a mouse model of generalized dystonia. Our results suggest that dopamine receptor agonists were effective in some, but not all, patients tested. Further, the mixed D1/D2 dopamine receptor agonist apomorphine was apparently more effective than subtype selective D2 dopamine receptor agonists. However, rigorously controlled trials are still needed. In a mouse model of dystonia, a selective D1 dopamine receptor agonist was not effective while a selective D2 dopamine receptor had modest efficacy. However, when combined, these receptor-selective agonists acted synergistically to ameliorate the dystonia. Coactivation of D1 and D2 dopamine receptors using apomorphine or by increasing extracellular concentrations of dopamine was also effective. Thus, results from both clinical trials and tests in mice suggest that coactivation of D1 and D2 dopamine receptors may be an effective therapeutic strategy in some patients. These results support a reconsideration of dopamine receptors as targets for the treatment of dystonia, particularly because recent genetic and diagnostic advances may facilitate the identification of the subtypes of dystonia patients who respond and those who do not.
Introduction
Dystonia, the third most common movement disorder after tremor and Parkinson’s disease, is characterized by involuntary sustained or intermittent muscle contractions that cause debilitating abnormal twisting movements and/or postures (Albanese et al., 2013). The pathobiology of dystonia is not understood in part because most dystonias result from neuronal dysfunction rather than overt degeneration or detectable insult. Although the cellular mechanisms that lead to the abnormal movements are unknown, a reduction in dopamine neurotransmission is observed in many forms of dystonia.
Dystonia is a prominent feature of inherited disorders that disrupt dopamine neurotransmission such as dopa-responsive dystonia, vesicular monoamine transporter 2 (VMAT2) deficiency, and dopamine transporter deficiency (Rilstone et al., 2013; Ng et al., 2014; Wijemanne and Jankovic, 2015). Dystonia may also occur in degenerative disorders that affect dopamine neurons, such as Parkinson’s disease (Jankovic, 2005; Tolosa and Compta, 2006) or in response to dopamine receptor antagonists (Mehta et al., 2015). Abnormal dopamine neurotransmission is also observed in dystonias that are otherwise not overtly associated with dopaminergic defects. For example, a reduction in D2 dopamine receptor density and abnormalities in striatal dopamine metabolites are observed in DYT1 dystonia, an inherited form of generalized dystonia caused by mutations in torsinA (Ozelius et al., 1997; Augood et al., 2002; Asanuma et al., 2005). More common idiopathic forms of dystonia such as writer’s cramp and spasmodic dysphonia are also associated with a reduction in striatal dopaminergic neurotransmission (Berman et al., 2013; Simonyan et al., 2013).
Consistent with findings in patients, striatal dopamine transmission is disrupted in mouse models of dystonia. Mouse models of DYT1 dystonia, dopa-responsive dystonia, and paroxysmal nonkinesigenic dyskinesia exhibit reduced presynaptic dopamine release and abnormal postsynaptic dopamine receptor signaling (Balcioglu et al., 2007; Page et al., 2010; Lee et al., 2012; Song et al., 2012; Rose et al., 2015; Zhang et al., 2015). A reduction in extracellular striatal dopamine concentrations is also observed in mouse models in which the dystonia originates in the cerebellum, including tottering mice and dystonia induced by an intracerebellar microinjection of kainic acid (Neychev et al., 2008; Raike et al., 2013). Thus, evidence from both patients and animal models implicates abnormal dopamine signaling even in dystonias that are not obviously caused by intrinsic dopamine neuron dysfunction.
Despite the strong association between abnormal dopamine neurotransmission and dystonia, dopaminergic agonists are not commonly prescribed to treat dystonia. Dopamine receptor agonists were tested in dystonia patients decades ago but were abandoned due to intolerable side effects and the unpredictable response across patients, which fostered the notion that dopamine receptor agonists are ineffective for the treatment of dystonia. However, the evidence supporting the presumed lack of efficacy is unclear. Therefore, to assess the validity of the claim that dopamine receptor agonists are not effective, we first analyzed the published trials of dopamine receptor agonists in patients with idiopathic dystonia. Next, we tested the efficacy of dopamine receptor agonists in a mouse model of generalized dystonia. Results from both patients and mice suggest that potentiating dopamine neurotransmission through coactivation of D1 and D2 dopamine receptors may be an effective strategy for the treatment of dystonia.
Materials and Methods
Clinical Trial Analysis.
PubMed searches were performed using keywords that included general (e.g., focal dystonia) and specific (e.g., blepharospasm) terms for dystonias, plus dopaminergic drugs, including amphetamine, apomorphine, bromocriptine, lisuride, pramipexole, ropinirole, and rotigotine. Studies of Parkinson’s disease patients and suspected dopa-responsive dystonia patients, as determined by positive responses to l-DOPA and diurnal fluctuation in the dystonia, were excluded. Studies were classified based on the level of evidence as described by the American Academy of Neurology (Gronseth et al., 2011).
Mice.
Normal C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and bred at Emory University. Male and female mice (8–12 weeks of age) were included in all experiments. Of the 354 mice tested, 175 were male, and 179 were female; no statistically significant differences between male and female responses within drug or vehicle conditions were observed. Mice were housed in 29-cm deep × 19-cm wide × 13-cm high cages (two to five mice per cage) in standard environmental conditions (72°C, 40%–50% relative humidity) with a 12-hour light/dark cycle and ad libitum access to food and water. All procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Emory University Animal Care and Use Committee.
Microinjections.
Cerebellar microinjections were performed as described elsewhere (Pizoli et al., 2002). The mice were anesthetized with isoflurane (Piramal Healthcare, Mumbai, India), and a midline incision was made over the skull. A small hole was drilled over the cerebellum (approximate coordinates: anteroposterior −4.0 mm lamba, lateral 0 mm), and a Hamilton syringe with a 2-mm needle was used to deliver 0.5 µl of 50 µg/ml kainic acid over 10 seconds to the midline cerebellum at the primary fissure. The incision was closed with wound glue (Vetbond; 3M, St. Paul, MN). The mice recovered within 10 minutes. The injections were performed without the aid of a stereotaxic apparatus to facilitate rapid surgeries and recoveries. With this freehand method, the interanimal injection sites are consistent and reproducible (Pizoli et al., 2002).
Test compounds were delivered intraperitoneally 10 minutes before cerebellar kainic acid microinjection, and the dystonia assessments commenced 10 minutes after microinjection. Striatal microinjections were bilateral (+0.6 mm anteroposterior, ±1.7 mm mediolateral, −3.5 mm dorsoventral) using a stereotaxic frame with a microinjection pump (Stoelting, Wood Dale, IL). The injection volume was 0.5 µl/side, and the injection rate was 0.1 µl/minute. The needle was kept in the injection site for 2 minutes. After completing the striatal microinjections, we performed cerebellar microinjection of kainate as described. Because the mice required more time to recover from the anesthesia that was needed for the combined striatal and cerebellar microinjections, the dystonia assessments started 30 minutes after cerebellar kainic acid injection. The microinjection sites were histologically verified.
Dystonia Assessments.
Animals were placed in an empty cage immediately after kainate challenge, and their behaviors were assessed for 1 hour. To quantify the dystonia, we rated the mice using timed behavioral sampling. The observers were blinded to the treatment. Each mouse was rated for 1 minute every 10 minutes using a well-established rating scale (Jinnah et al., 2000), where 0 = normal motor behavior; 1 = slightly slowed or abnormal movements; 2 = fleeting abnormal postures; 3 = moderate impairment, limited ambulation even when disturbed, frequent abnormal postures; and 4 = severe impairment, sustained abnormal postures. The six scores obtained during the 1-hour scoring period were summed to obtain a total score for the entire scoring period for each mouse. Therefore, the highest cumulative score was 24. Cumulative scores lower than 10, which would be achieved by ratings of 1 or 2 in each scoring interval, suggested abnormal motor behavior such as motor slowing or an unsteady gait but not overt dystonia. The mice were gently prodded at the start of each scoring bin to encourage movement because dystonia is movement induced.
Drugs.
Kainic acid [(2S,3S,4S)-carboxy-4-(1-methylethenyl)-3-pyrrolidineacetic acid], SKF-81297 [(±)-6-chloro-2,3,4,5-tetrahydro-1-phenyl-1H-3-benzazepine hydrobromide], and quinpirole were obtained from Tocris (Bristol, United Kingdom) and were dissolved in 0.9% saline or phosphate-buffered saline. Amphetamine was obtained from Sigma-Aldrich (St Louis, MI) and dissolved in 0.9% saline. Nisoxetine, citalopram, GBR-12909 (1-[2-[Bis-(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine dihydrochloride), and apomorphine were purchased from Tocris and dissolved in water. For all compounds, the behaviorally active doses were selected based on previously published studies (Yamashita et al., 2006; Fan and Hess, 2007; Laloux et al., 2007; Rose et al., 2015).
Data Analysis.
An arithmetic sum of the disability scores was used to calculate a total score for the entire 1-hour session. These data approximate a continuous variable when total scores from one animal are added together, so the data were analyzed using parametric tests, in this case one-way analysis of variance (ANOVA). GraphPad Prism (GraphPad Software, San Diego, CA) was used for statistical analyses. All results are expressed as mean ± S.E.M.
Results
We analyzed reports examining the efficacy of dopamine receptor agonist treatment of idiopathic dystonia. After eliminating the studies examining Parkinson’s disease patients, our search retrieved 25 reports. Of these, two examined dopamine receptor agonist responses in suspected dopa-responsive dystonia patients, so these reports were excluded. A total of 23 reports describing 191 unique cases of presumptive idiopathic dystonias were included in our analysis (Table 1). The types of dystonia were varied and comprised of focal, segmental, and generalized dystonias.
TABLE 1 .
Patient studies examining the efficacy of dopaminergic drugs for the treatment of dystonias
| Drug | Type of Dystonia | Patients Improved/Patients Testeda | Type of Study | Level of Evidenceb | Year | Reference |
|---|---|---|---|---|---|---|
| Amphetamine | ||||||
| Cervical, multifocal (cervical and foot) | 2/2 | Case reports | IV | 1942 | Myerson and Loman (1942) | |
| Cervical | 1/1 | Case report | IV | 1943 | Patterson and Little (1943) | |
| Apomorphine | ||||||
| Cervical | 2/7 | Double-blind, placebo crossover and open-label | IV | 1978 | Tolosa (1978) | |
| Cervical | 3/3 | Double-blind, placebo crossover | III | 1982 | Frattola et al. (1982) | |
| Craniofacialc | 5/5 | Single-blind, placebo crossover | IV | 1979 | Tolosa and Lai (1979) | |
| Craniofacial | 2/2 | Case reports | IV | 1982 | Micheli et al. (1982) | |
| Craniofacial | 12/23 | Single-blind, placebo crossover | IV | 1986 | Micheli and Fernandez Pardal (1986) | |
| Generalized | 2/2 | Case reports | IV | 1973 | Braham and Sarova-Pinhas (1973) | |
| Generalized | 1/1 | Case report | IV | 1996 | Zuddas et al. (1996) | |
| Segmental (cervical, spasmodic dysphonia, and craniofacial) | 1/1 | Case report | IV | 1993 | Vidailhet et al. (1993) | |
| Bromocriptine | ||||||
| Cervical | 0/8 | Single-blind, placebo crossover | IV | 1976 | Lees et al. (1976) | |
| Cervical | 0/14 | Randomized, double-blind, placebo crossover | III | 1979 | Juntunen et al. (1979) | |
| Craniofacial | 0/2 | Case reports | IV | 1982 | Micheli et al. (1982) | |
| Generalized, craniofacial plus cervical | 2/3 | Double-blind, placebo crossover | IV | 1981 | Stahl and Berger (1981) | |
| Generalized, cervical, craniofacial | 5/8 | Double-blind, placebo crossover | IV | 1982 | Stahl and Berger (1982) | |
| Generalized, segmental (cervical/limb), truncal, cervical | 2/13 | Blinded raters, no placebo | IV | 1982 | Girotti et al. (1982) | |
| Generalized, unspecified focal | 2/10 | Open-label followed by double-blind study in the two responders | IV | 1984 | Obeso and Luquin (1984) | |
| Generalized, hemidystonia, cervical, segmental (oromandibular and laryngeal), multifocal (cervical and foot) blepharospasm | 7/13 | Randomized, double-blind, placebo crossover | III | 1985 | Newman et al. (1985) | |
| Lisuride | ||||||
| Cervical | 3/3 | Double-blind, placebo crossover | III | 1982 | Frattola et al. (1982) | |
| Craniofacial | 2/2 | Case reports | IV | 1982 | Micheli et al. (1982) | |
| Craniofacial | 1/7 | Open label | IV | 1983 | Marsden et al. (1983) | |
| Craniofacial | 13/23 | Single-blind, placebo crossover | IV | 1986 | Micheli and Fernandez Pardal (1986) | |
| Craniofacial | 1/1 | Case report | IV | 1988 | Micheli et al. (1988) | |
| Craniofacial | 1/9 | Randomized, double-blind, placebo crossover | II | 1988 | Bassi et al. (1982) | |
| Generalized, cervical | 6/7 | Double-blind, placebo crossover | III | 1982 | Ransmayr et al. (1988) | |
| Generalized, unspecified focal | 0/10 | Open-label | IV | 1984 | Obeso and Luquin (1984) | |
| Generalized, segmental (unspecified regions), hand, cervical, craniofacial, myoclonic, Parkinsonism with dystonia, tardive, unspecified | 8/42 | Open-label followed by randomized, double-blind, placebo crossover for the eight patients who improved | IV | 1985 | Quinn et al. (1985) | |
| Generalized, cervical, craniofacial | 6/9 | Double-blind, placebo crossover | IV | 1985 | Nutt et al. (1985) |
The definition of “improved” varied across publications and is summarized here exactly as reported.
Based on the updated scheme for “Classification of Evidence for Therapeutic Studies” established by the American Academy of Neurology in 2015.
Craniofacial is used synonymously with Meige syndrome or orofacial dystonia.
The dopamine receptor agonists tested included the indirect-acting agonist amphetamine and the direct-acting agonists apomorphine, bromocriptine, and lisuride; some patients were tested with more than one drug. Our searches did not retrieve any published reports examining the efficacy of the newer dopamine receptor agonist medications pramipexole, ropinirole, or rotigotine. Indeed, most of the studies were performed before the introduction of these drugs.
Treatment with amphetamine, which increases extracellular dopamine concentrations, thus indirectly activating both D1 and D2 dopamine receptor subtypes, was reported for only three patients and was effective in all three. The mixed D1/D2 dopamine receptor agonist apomorphine was effective in 28 (63%) of 44 patients tested. Bromocriptine D2 dopamine receptor-selective agonist, was effective in 18 (25%) of 71 patients. Lisuride, which is a D2 dopamine receptor agonist that also acts at serotonergic and adrenergic receptor subtypes, was effective in 41 (36%) of 113 patients. There was no apparent relationship between the response to dopamine receptor agonist treatment and the type of dystonia.
Of the 23 reports identified, none achieved the level of evidence that is required for a class I therapeutic trial as defined by the American Academy of Neurology (Gronseth et al., 2011). Class I trials are randomized controlled clinical trials with rigorously defined criteria for study design, patient recruitment, outcomes measures, and statistical analyses. Only one study met the criteria for a class II trial, a somewhat lower level of rigor than class I. A few class III studies were performed, but the vast majority were class IV due to less stringent study designs, which is a consequence of the small samples sizes available for trials in rare disorders such as dystonia. Additionally, because many of the studies were performed decades ago, before the modern understanding of the etiologic heterogeneity of dystonia, the trials often included several different types of dystonia. These results demonstrate that rigorous trials to determine the efficacy of dopamine agonists for the treatment of dystonia have not yet been performed.
To further explore the efficacy of dopamine receptor agonists in dystonia, we used a mouse model of dystonia to dissect the underlying pharmacologic mechanisms. A mouse model in which generalized dystonia is induced in the cerebellum through local application of kainic acid (Pizoli et al., 2002) was used because 1) striatal extracellular dopamine is reduced upon induction of dystonia (Neychev et al., 2008), similar to the decrease in dopamine neurotransmission observed in many different types of dystonia, and 2) the cerebellar excitation caused by kainic acid mimics the abnormal increase in cerebellar activity observed in many forms of dystonia, including DYT1, blepharospasm, cervical dystonia, writer’s cramp, and spasmodic dysphonia (Neychev et al., 2011).
Because the dystonia is initiated in the cerebellum yet induces a reduction in striatal dopamine, we first determined the role of dopaminergic transmission in the expression of dystonia by using the indirect-acting dopamine agonist amphetamine to increase extracellular dopamine concentrations before the induction of dystonia. Mice were challenged with peripheral administration (intraperitoneal) of 1–4 mg/kg amphetamine, which are doses that reliably increase both extracellular dopamine and locomotor activity without inducing stereotypic behaviors (Fan and Hess, 2007). Amphetamine dose-dependently reduced the severity of the dystonia caused by intracerebellar microinjection of kainic acid (Fig. 1A). Indeed, at the highest dose of amphetamine, dystonia was nearly abolished.
Fig. 1.

Effects of enhanced monoaminergic transmission on dystonia induced by cerebellar microinjection of kainate. (A) Peripheral administration of amphetamine, which nonselectively increases the extracellular concentrations of the monoamines dopamine, norepinephrine, and serotonin, dose-dependently reduced the severity of dystonia induced by microinjection of kainate to the cerebellum (one-way ANOVA; F3,28 = 4.813, P < 0.01, n = 8/dose). (B) The selective norepinephrine reuptake inhibitor nisoxetine did not affect the disability score (one-way ANOVA; F3,27 = 0.468, NS, n = 7 to 8/dose). (C) Citalopram, a selective serotonin reuptake inhibitor, did not affect the disability score (one-way ANOVA; F3,26 = 0.317, NS, n = 7 to 8/dose). (D) The selective dopamine reuptake inhibitor GBR-12909 dose-dependently reduced the severity of dystonia (one-way ANOVA; F3,28 = 3.853, P < 0.05, n = 8/dose). Disability scores lower than 10 (dashed lines) suggest abnormal motor behavior, such as an unsteady gait, but not overt dystonia. Values represent the mean of the cumulative disability score in 1 hour ± S.E.M. *P < 0.05; **P < 0.01 compared with vehicle, Holm-Sidak multiple comparisons test. NS, not statistically significant.
Because amphetamine increases the extracellular concentrations of norepinephrine and serotonin in addition to dopamine (Kuczenski et al., 1995), we examined the effect of increasing the extracellular concentration of each individual monoamine on kainate-induced dystonia to identify the specific neurotransmitter(s) involved. To accomplish this, the norepinephrine reuptake inhibitor nisoxetine or the selective serotonin reuptake inhibitor citalopram was administered (intraperitoneally) before intracerebellar kainate administration. Neither nisoxetine nor citalopram affected the dystonia caused by intracerebellar microinjection of kainic acid (Fig. 1, B and C).
To determine the role of enhanced dopaminergic tone, the selective dopamine reuptake inhibitor GBR-12909 was administered (intraperitoneally) before kainate challenge. GBR-12909 dose-dependently reduced the severity of the dystonia caused by intracerebellar microinjection of kainic acid (Fig. 1D).
To determine whether dopamine receptor agonists have similar effects, we challenged mice with intracerebellar kainic acid plus peripheral administration of the direct-acting mixed D1/D2 dopamine receptor agonist apomorphine. Apomorphine dose-dependently reduced the severity of the dystonia induced by cerebellar kainic acid challenge (Fig. 2A).
Fig. 2.

Effects of dopamine receptor agonists on dystonia induced by cerebellar microinjection of kainate. (A) The nonselective dopamine receptor agonist apomorphine dose-dependently reduced the severity of dystonia (one-way ANOVA; F3,29 = 3.259, P < 0.05, n = 8–10/dose). (B) The D1 dopamine receptor selective agonist SKF-81297 did not affect the severity of cerebellar kainic acid-induced dystonia (one-way ANOVA; F3,29 = 0.325, NS, n = 7–10/dose). (C) The D2 dopamine receptor selective agonist quinpirole significantly reduced the severity of dystonia (one-way ANOVA; F3,30 = 7.233, P < 0.001, n = 8 to 9/dose). Disability scores lower than 10 (dashed lines) suggest abnormal motor behavior, such as an unsteady gait, but not overt dystonia. Values represent the mean of the cumulative disability score in 1 hour ± S.E.M. *P < 0.05; **P < 0.01 compared with vehicle, Holm-Sidak multiple comparisons test.
To dissect the signaling mechanisms, the mice were challenged with selective dopamine receptor agonists. In contrast to the mixed D1/D2 dopamine receptor agonist apomorphine, administration of the D1 dopamine receptor agonist SKF-81297 (intraperitoneally) did not affect the dystonia induced by cerebellar microinjection of kainic acid (Fig. 2B). Administration of the selective D2 dopamine receptor agonist quinpirole (intraperitoneally) resulted in a modest but significant reduction in the severity of the dystonia induced by intracerebellar administration of kainic acid (Fig. 2C).
Because the response to quinpirole alone was less robust than that observed with amphetamine and because SKF-81297 alone had no effect, we hypothesized that D1 plus D2 dopamine receptor coactivation contributes to the amelioration of dystonia. To test this hypothesis, we administered a fixed low dose of the D2 dopamine receptor agonist quinpirole (0.5 mg/kg, i.p.), which alone had no effect on cerebellar kainate-induced dystonia, along with several different doses of the D1 dopamine receptor agonist SKF-81297 (1, 2 or 5 mg/kg, i.p.). As expected, the severity of the cerebellar kainate-induced dystonia observed after 0.5 mg/kg quinpirole was comparable to that observed after saline administration. However, in the presence of this behaviorally inactive dose of quinpirole, SKF-81297 dose-dependently reduced the severity of dystonia (Fig. 3), although SKF-81297 alone did not affect the severity of dystonia. These results suggest that D1 and D2 dopamine receptor signaling acts synergistically to mediate the severity of dystonia.
Fig. 3.
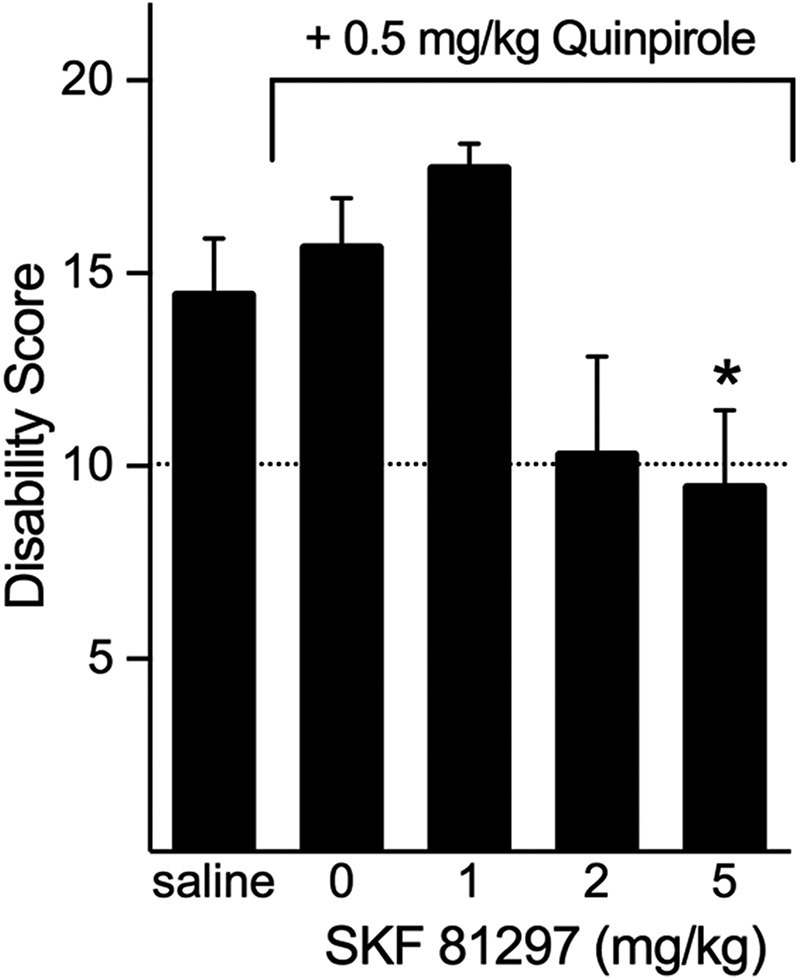
Synergistic effects of D1 plus D2 dopamine receptor activation on the severity of cerebellar mediated dystonia. Administration of a low dose of quinpirole (0.5 mg/kg) did not affect the severity of dystonia compared with vehicle administration (two-tailed Student’s t test, t = 0.629). However, administration of quinpirole plus SKF-81297 significantly reduced the severity of dystonia induced by cerebellar microinjection of kainate (one-way ANOVA; F3,30 = 4.841, P < 0.01, n = 7–11/dose). Disability scores lower than 10 (dashed lines) suggest abnormal motor behavior, such as an unsteady gait, but not overt dystonia. Values represent the mean of the cumulative disability score in 1 hour ± S.E.M.; *P < 0.05 compared with quinpirole alone, Holm-Sidak multiple comparisons test.
To identify brain region(s) associated with the ameliorating effects of dopamine agonists, we microinjected apomorphine into brain regions that are thought to play a prominent role in dystonia. Recent evidence suggests that dystonia is a network disorder involving cortico-striato-pallido-thalamo-cortical and cerebello-thalamo-cortical pathways (Carbon et al., 2010; Neychev et al., 2011; Niethammer et al., 2011; Lehericy et al., 2013). Therefore, the striatum and cerebellum were targeted for microinjection. Bilateral striatal microinjection of apomorphine significantly reduced the severity of the cerebellar-induced dystonia (P < 0.05) compared with striatal microinjection of saline. Microinjection of apomorphine into midline cerebellum had no effect on intracerebellar kainate-induced dystonia compared with saline (Fig. 4). These results suggest that dopaminergic signaling within the striatum mediates the severity of dystonia induced by abnormal cerebellar signaling.
Fig. 4.
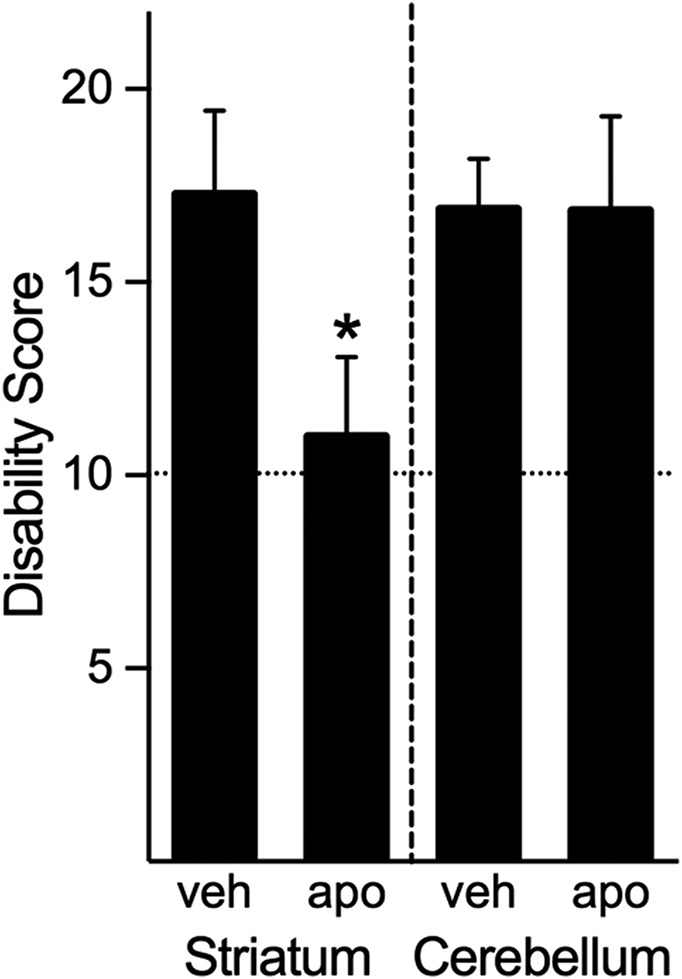
Regional analysis of the effects of apomorphine on dystonia induced by cerebellar microinjection of kainate. Microinjection of apomorphine into the striatum significantly reduced the severity of the dystonia (Student’s t test, one-tailed, P < 0.05 compared with saline), whereas cerebellar microinjection of apomorphine did not affect the severity of dystonia (Student’s t test, one-tailed, NS compared with saline). Disability scores lower than 10 (dashed lines) suggest abnormal motor behavior, such as an unsteady gait, but not overt dystonia. Values represent the mean of the cumulative disability score in 1 hour ± S.E.M.; *P < 0.05 compared with microinjection of vehicle in the same brain region.
Discussion
Understanding the role of dopamine in the pathogenesis of dystonia is critically important for therapeutic strategies, in light of the strong association between dopamine and dystonia. However, dopaminergic drugs are not currently used to treat dystonia and were abandoned decades ago because they were thought to be ineffective. Table 1 clearly illustrates that this assumption, which has become dogma, is neither supported nor refuted by the evidence. Despite this limitation, the analysis presented in Table 1 did reveal trends in patient responses. A positive response was reported for more than two-thirds of patients who received the mixed D1/D2 dopamine receptor agonist apomorphine. Amphetamine, which also results in the activation of both classes of dopamine receptors, was reportedly effective, but it is important to note that only a few patients were tested. In contrast, the more selective D2 dopamine receptor agonists bromocriptine and lisuride were not nearly as effective. This distinction between selective and nonselective dopamine receptor agonists was not obvious before our analysis. Although far from conclusive, the results of Table 1 suggest the hypothesis that coactivation of dopamine receptor subtypes is more efficacious than subtype selective dopamine receptor agonists for the treatment of dystonia.
We used a mouse model of dystonia to test this hypothesis and to dissect the pharmacology of the response. Coactivating D1 and D2 dopamine receptors by increasing extracellular concentrations of dopamine using amphetamine or the dopamine reuptake inhibitor GBR-12909 reduced the severity of dystonia, while increasing extracellular concentrations of norepinephrine or serotonin had no effect. Apomorphine, which acts at both dopamine receptor subtypes, also ameliorated the dystonia in mice.
In contrast, selective activation of D1 dopamine receptors was ineffective. In the striatum, D1 dopamine receptors are expressed on a subset of medium spiny neurons (DeLong and Wichmann, 2007; Gerfen and Surmeier, 2011); medium spiny neurons provide the only striatal efferents, suggesting that restoring dopaminergic neurotransmission to only a subset of efferents is insufficient to correct the dystonia.
Selective activation of D2 dopamine receptors produced a modest reduction in the severity of the dystonia. D2 dopamine receptors are expressed in a subset of medium spiny neurons that, for the most part, does not overlap with D1 dopamine receptor-expressing medium spiny neurons. D2 dopamine receptors are also located on both nigrostriatal and corticostriatal presynaptic terminals where they mediate afferent signals (DeLong and Wichmann, 2007; Gerfen and Surmeier, 2011). It is possible that the synergistic effects of D1 and D2 dopamine receptor activation arise through concomitant restoration of dopaminergic signaling at both afferents and efferents, which act in concert to control movement. Alternatively, the synergistic effects may be attributable to activation of D1/D2 receptor heteromers as ∼5% of medium spiny neurons coexpress D1 and D2 dopamine receptors (Bertran-Gonzalez et al., 2008; Matamales et al., 2009). These heteromeric complexes induce intracellular calcium signaling (Rashid et al., 2007) and mediate behavioral effects that are not attributable independent effects of D1 or D2 dopamine receptors (Shen et al., 2015). The development of compounds that distinguish heteromeric complexes from dopamine receptor monomers will be necessary to determine the role of heteromers in the synergistic effects observed here. Regardless of the precise mechanism, the striking parallels between the human and mouse responses suggest that potentiating dopamine transmission through both D1 and D2 dopamine receptor subtypes may be an effective therapeutic strategy.
Our results demonstrate that enhancing striatal dopamine neurotransmission ameliorates generalized dystonia that is initiated in the cerebellum. It was previously shown that a reduction in extracellular striatal dopamine concentration occurs when dystonia is caused by abnormal cerebellar signaling, including in the kainate-induced model used here (Neychev et al., 2008; Raike et al., 2013). The anatomic connectivity between the cerebellum and substantia nigra (Snider et al., 1976) and between the cerebellum and basal ganglia (Hoshi et al., 2005) is well established. Further, the dystonia in a mouse model of rapid-onset dystonia-parkinsonism is mediated by abnormal striatal signals induced by cerebellar dysfunction (Calderon et al., 2011). However, the relevance of diminished dopamine transmission to the expression of dystonia where the etiology is not overtly associated with dopaminergic defects in either patients or animal models is not known. Our results demonstrate that potentiating dopaminergic neurotransmission within the striatum, but not in the cerebellum where the pathophysiology originated, was sufficient to ameliorate the dystonia. Thus, enhancing striatal dopaminergic neurotransmission may be effective even in those dystonias that are not overtly caused by dopaminergic defects but nonetheless exhibit abnormalities in dopamine signaling such as writer’s cramp and spasmodic dysphonia (Berman et al., 2013; Simonyan et al., 2013).
Although the experiments in mice demonstrate that coactivation of D1 and D2 dopamine receptors is more effective than selective agonists and the patient studies imply similar efficacy, dopamine receptor agonists are not currently used to treat dystonia. Most currently prescribed dopamine receptor agonists, including pramipexole, ropinirole, rotigotine, bromocriptine, and lisuride are selective for D2 dopamine receptors. These selective compounds are predicted to be less effective than nonselective agonists, based on the results from the mouse model and human studies.
Apomorphine is currently the only mixed D1/D2 dopamine receptor agonist available for use in humans. While acutely-administered apomorphine appeared to be effective in the limited number of patients tested, it is not useful for the treatment of dystonia for several reasons. First, apomorphine is short-acting, with effects lasting 60–90 minutes (Neef and van Laar, 1999). Second, apomorphine has poor oral bioavailability, so it must be delivered by subcutaneous injection (Neef and van Laar, 1999). Third, the side effects include nausea, psychosis, headache, sweating, and dizziness (Kimber et al., 2017). Thus, patients would require multiple injections per day while coping with intolerable side effects, which is neither practical nor suitable for the chronic treatment of dystonia. The development of novel mixed agonists or positive allosteric modulators, which might achieve the same results without the side effects of a full mixed agonist, would provide an opportunity for more rigorous trials.
Our results suggest that it was premature to discard dopamine receptors as targets for the treatment of dystonia. Systematic trials testing dopamine receptor agonists in homogeneous patient populations are needed to provide conclusive evidence. With the modern understanding of the heterogeneous etiologies underlying the pathogenesis of dystonia, which was not apparent several decades ago when dopamine receptor agonists were tested, it may be possible to identify subtypes of dystonia patients who respond while others may not. Indeed, given the broad range of etiologies and phenotypes associated with dystonia, it is unlikely that dopamine receptor agonists would be effective in all forms of dystonia. Overall, the trends that emerged from our analysis of previous studies in humans in combination with the results in mice and the strong evidence implicating dopamine dysfunction in dystonia support a reconsideration of dopamine receptors as targets for the treatment of dystonia.
Abbreviations
- ANOVA
analysis of variance
- GBR-12909
1-[2-[Bis-(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine dihydrochloride
- SKF-81297
(±)-6-chloro-2,3,4,5-tetrahydro-1-phenyl-1H-3-benzazepine hydrobromide
Authorship Contributions
Participated in research design: Fan, Jinnah, Hess.
Conducted experiments: Fan, Donsante.
Performed data analysis: Fan, Hess.
Wrote or contributed to the writing of the manuscript: Fan, Donsante, Jinnah, Hess.
Footnotes
This work was supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke [Grant R01 NS088528].
References
- Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, et al. (2013) Phenomenology and classification of dystonia: a consensus update. Mov Disord 28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronseth GS, Woodroffe LM, Getchius TSD. (2011) Clinical Practice Guideline Process Manual. American Academy of Neurology, St. Paul, MN. [Google Scholar]
- Asanuma K, Ma Y, Okulski J, Dhawan V, Chaly T, Carbon M, Bressman SB, Eidelberg D. (2005) Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation. Neurology 64:347–349. [DOI] [PubMed] [Google Scholar]
- Augood SJ, Hollingsworth Z, Albers DS, Yang L, Leung JC, Muller B, Klein C, Breakefield XO, Standaert DG. (2002) Dopamine transmission in DYT1 dystonia: a biochemical and autoradiographical study. Neurology 59:445–448. [DOI] [PubMed] [Google Scholar]
- Balcioglu A, Kim MO, Sharma N, Cha JH, Breakefield XO, Standaert DG. (2007) Dopamine release is impaired in a mouse model of DYT1 dystonia. J Neurochem 102:783–788. [DOI] [PubMed] [Google Scholar]
- Bassi S, Ferrarese C, Frattola L, Sbacchi M, Trabucchi M. (1982) Lisuride in generalised dystonia and spasmodic torticollis. Lancet 1:514–515. [DOI] [PubMed] [Google Scholar]
- Berman BD, Hallett M, Herscovitch P, Simonyan K. (2013) Striatal dopaminergic dysfunction at rest and during task performance in writer’s cramp. Brain 136:3645–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. (2008) Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci 28:5671–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braham J, Sarova-Pinhas I. (1973) Apomorphine in dystonia musculorum deformans. Lancet 1:432–433. [DOI] [PubMed] [Google Scholar]
- Calderon DP, Fremont R, Kraenzlin F, Khodakhah K. (2011) The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci 14:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, Argyelan M, Eidelberg D. (2010) Functional imaging in hereditary dystonia. Eur J Neurol 17 (Suppl 1):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong MR, Wichmann T. (2007) Circuits and circuit disorders of the basal ganglia. Arch Neurol 64:20–24. [DOI] [PubMed] [Google Scholar]
- Fan X, Hess EJ. (2007) D2-like dopamine receptors mediate the response to amphetamine in a mouse model of ADHD. Neurobiol Dis 26:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattola L, Albizzati MG, Bassi S, Ferrarese C, Trabucchi M. (1982) “On-off” phenomena, dyskinesias and dystonias. Comparison of lisuride versus apomorphine acute treatment. Acta Neurol Scand 66:227–236. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. (2011) Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 34:441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti F, Scigliano G, Nardocci N, Angelini L, Broggi G, Giovannini P, Caraceni T. (1982) Idiopathic dystonia: neuropharmacological study. J Neurol 227:239–247. [DOI] [PubMed] [Google Scholar]
- Hoshi E, Tremblay L, Féger J, Carras PL, Strick PL. (2005) The cerebellum communicates with the basal ganglia. Nat Neurosci 8:1491–1493. [DOI] [PubMed] [Google Scholar]
- Jankovic J. (2005) Dystonia and other deformities in Parkinson’s disease. J Neurol Sci 239:1–3. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Sepkuty JP, Ho T, Yitta S, Drew T, Rothstein JD, Hess EJ. (2000) Calcium channel agonists and dystonia in the mouse. Mov Disord 15:542–551. [DOI] [PubMed] [Google Scholar]
- Juntunen J, Kaste M, Iivanainen M, Ranta T, Seppälä M. (1979) Bromocriptine treatment of spasmodic torticollis. A double-blind crossover study. Arch Neurol 36:449–450. [DOI] [PubMed] [Google Scholar]
- Kimber TE, Fang J, Huddy LJ, Thompson PD. (2017) Long-term adherence to apomorphine infusion in patients with Parkinson disease: a 10-year observational study. Intern Med J 47:570–573. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS, Cho AK, Melega W. (1995) Hippocampus norepinephrine, caudate dopamine and serotonin, and behavioral responses to the stereoisomers of amphetamine and methamphetamine. J Neurosci 15:1308–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laloux C, Derambure P, Jacquesson JM, Bordet R, Destée A, Monaca C. (2007) The effects of serotoninergic, noradrenergic, cholinergic and dopaminergic drugs on vigilance states in MPTP-treated mice. Brain Res 1161:79–87. [DOI] [PubMed] [Google Scholar]
- Lee HY, Nakayama J, Xu Y, Fan X, Karouani M, Shen Y, Pothos EN, Hess EJ, Fu YH, Edwards RH, et al. (2012) Dopamine dysregulation in a mouse model of paroxysmal nonkinesigenic dyskinesia. J Clin Invest 122:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees A, Shaw KM, Stern GM. (1976) Letter: bromocriptine and spasmodic torticollis. BMJ 1:1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehéricy S, Tijssen MA, Vidailhet M, Kaji R, Meunier S. (2013) The anatomical basis of dystonia: current view using neuroimaging. Mov Disord 28:944–957. [DOI] [PubMed] [Google Scholar]
- Marsden CD, Lang AE, Sheehy MP. (1983) Pharmacology of cranial dystonia. Neurology 33:1100–1102. [DOI] [PubMed] [Google Scholar]
- Matamales M, Bertran-Gonzalez J, Salomon L, Degos B, Deniau JM, Valjent E, Hervé D, Girault JA. (2009) Striatal medium-sized spiny neurons: identification by nuclear staining and study of neuronal subpopulations in BAC transgenic mice. PLoS One 4:e4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SH, Morgan JC, Sethi KD. (2015) Drug-induced movement disorders. Neurol Clin 33:153–174. [DOI] [PubMed] [Google Scholar]
- Micheli F, Fernandez Pardal MM. (1986) Responses to lisuride in Meige’s disease and chorea. Neurology 36:445. [DOI] [PubMed] [Google Scholar]
- Micheli F, Fernandez Pardal MM, Gatto E, Paradiso G. (1988) Continuous dopaminergic stimulation in cranial dystonia. Clin Neuropharmacol 11:241–249. [DOI] [PubMed] [Google Scholar]
- Micheli F, Fernández Pardal MM, Leiguarda RC. (1982) Beneficial effects of lisuride in Meige disease. Neurology 32:432–434. [DOI] [PubMed] [Google Scholar]
- Myerson A, Loman J. (1942) Amphetamine sulfate in treatment of spasmodic torticollis. Arch Neurol Psychiatry 48:823–828. [Google Scholar]
- Neef C, van Laar T. (1999) Pharmacokinetic-pharmacodynamic relationships of apomorphine in patients with Parkinson’s disease. Clin Pharmacokinet 37:257–271. [DOI] [PubMed] [Google Scholar]
- Newman RP, LeWitt PA, Shults C, Bruno G, Foster NL, Chase TN, Calne DB. (1985) Dystonia: treatment with bromocriptine. Clin Neuropharmacol 8:328–333. [PubMed] [Google Scholar]
- Neychev VK, Fan X, Mitev VI, Hess EJ, Jinnah HA. (2008) The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain 131:2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neychev VK, Gross RE, Lehéricy S, Hess EJ, Jinnah HA. (2011) The functional neuroanatomy of dystonia. Neurobiol Dis 42:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng J, Zhen J, Meyer E, Erreger K, Li Y, Kakar N, Ahmad J, Thiele H, Kubisch C, Rider NL, et al. (2014) Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain 137:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Carbon M, Argyelan M, Eidelberg D. (2011) Hereditary dystonia as a neurodevelopmental circuit disorder: evidence from neuroimaging. Neurobiol Dis 42:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt JG, Hammerstad JP, Carter JH, deGarmo PL. (1985) Lisuride treatment of focal dystonias. Neurology 35:1242–1243. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Luquin MR. (1984) Bromocriptine and lisuride in dystonias. Neurology 34:135–136. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, et al. (1997) The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 17:40–48. [DOI] [PubMed] [Google Scholar]
- Page ME, Bao L, Andre P, Pelta-Heller J, Sluzas E, Gonzalez-Alegre P, Bogush A, Khan LE, Iacovitti L, Rice ME, et al. (2010) Cell-autonomous alteration of dopaminergic transmission by wild type and mutant (DeltaE) TorsinA in transgenic mice. Neurobiol Dis 39:318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson RM, Little SC. (1943) Spasmodic torticollis. J Nerv Ment Dis 98:571–599. [Google Scholar]
- Pizoli CE, Jinnah HA, Billingsley ML, Hess EJ. (2002) Abnormal cerebellar signaling induces dystonia in mice. J Neurosci 22:7825–7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn NP, Lang AE, Sheehy MP, Marsden CD. (1985) Lisuride in dystonia. Neurology 35:766–769. [DOI] [PubMed] [Google Scholar]
- Raike RS, Pizoli CE, Weisz C, van den Maagdenberg AM, Jinnah HA, Hess EJ. (2013) Limited regional cerebellar dysfunction induces focal dystonia in mice. Neurobiol Dis 49:200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransmayr G, Kleedorfer B, Dierckx RA, Poewe W, Kemmler GW, Gerstenbrand F. (1988) Pharmacological study in Meige’s syndrome with predominant blepharospasm. Clin Neuropharmacol 11:68–76. [DOI] [PubMed] [Google Scholar]
- Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, O’Dowd BF, George SR. (2007) D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci USA 104:654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rilstone JJ, Alkhater RA, Minassian BA. (2013) Brain dopamine-serotonin vesicular transport disease and its treatment. N Engl J Med 368:543–550. [DOI] [PubMed] [Google Scholar]
- Rose SJ, Yu XY, Heinzer AK, Harrast P, Fan X, Raike RS, Thompson VB, Pare JF, Weinshenker D, Smith Y, et al. (2015) A new knock-in mouse model of l-DOPA-responsive dystonia. Brain 138:2987–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MY, Perreault ML, Bambico FR, Jones-Tabah J, Cheung M, Fan T, Nobrega JN, George SR. (2015) Rapid anti-depressant and anxiolytic actions following dopamine D1-D2 receptor heteromer inactivation. Eur Neuropsychopharmacol 25:2437–2448. [DOI] [PubMed] [Google Scholar]
- Simonyan K, Berman BD, Herscovitch P, Hallett M. (2013) Abnormal striatal dopaminergic neurotransmission during rest and task production in spasmodic dysphonia. J Neurosci 33:14705–14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider RS, Maiti A, Snider SR. (1976) Cerebellar pathways to ventral midbrain and nigra. Exp Neurol 53:714–728. [DOI] [PubMed] [Google Scholar]
- Song CH, Fan X, Exeter CJ, Hess EJ, Jinnah HA. (2012) Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis 48:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl SM, Berger PA. (1981) Bromocriptine in dystonia. Lancet 2:745. [DOI] [PubMed] [Google Scholar]
- Stahl SM, Berger PA. (1982) Bromocriptine, physostigmine, and neurotransmitter mechanisms in the dystonias. Neurology 32:889–892. [DOI] [PubMed] [Google Scholar]
- Tolosa E, Compta Y. (2006) Dystonia in Parkinson’s disease. J Neurol 253 (Suppl 7):VII7–VII13. [DOI] [PubMed] [Google Scholar]
- Tolosa ES. (1978) Midification of tardive dyskinesia and spasmodic torticollis by apomorphine. Possible role of dopamine autoreceptors. Arch Neurol 35:459–462. [DOI] [PubMed] [Google Scholar]
- Tolosa ES, Lai C. (1979) Meige disease: striatal dopaminergic preponderance. Neurology 29:1126–1130. [DOI] [PubMed] [Google Scholar]
- Vidailhet M, Bouchard C, Jedynak PJ, Serdaru M. (1993) Acute and long-term response to apomorphine in cranial dystonia. Mov Disord 8:237–239. [DOI] [PubMed] [Google Scholar]
- Wijemanne S, Jankovic J. (2015) Dopa-responsive dystonia--clinical and genetic heterogeneity. Nat Rev Neurol 11:414–424. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Fukushima S, Shen HW, Hall FS, Uhl GR, Numachi Y, Kobayashi H, Sora I. (2006) Norepinephrine transporter blockade can normalize the prepulse inhibition deficits found in dopamine transporter knockout mice. Neuropsychopharmacology 31:2132–2139. [DOI] [PubMed] [Google Scholar]
- Zhang L, McCarthy DM, Sharma N, Bhide PG. (2015) Dopamine receptor and Gα(olf) expression in DYT1 dystonia mouse models during postnatal development. PLoS One 10:e0123104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuddas A, Pintor M, DeMontis N, Giovanna Marrosu M, Cianchetti C. (1996) Continuous infusion of apomorphine improves torsion dystonia in a boy unresponsive to other dopaminergic drugs. J Child Neurol 11:343–345. [DOI] [PubMed] [Google Scholar]