

Abstract
The key role of extracellular matrices in alveolar epithelial cell (AEC) biology is highlighted by the phenotypes of primary AECs cultured on a soft laminin gel contrasted with that on a stiff, fibronectin matrix. On laminin, AECs maintain an epithelial phenotype, and progenitor cells within this population proliferate. In contrast, on fibronectin, AECs rapidly lose surfactant expression and spread extensively, changes that depend on activation of latent TGF-β1 by engagement of fibronectin-binding integrins. The progenitor subpopulation responding to TGF-β1 undergoes epithelial mesenchymal transition (EMT). Although it remains uncertain to what degree EMT contributes directly to collagen 1 production, signaling pathways critical to EMT are important for repair and fibrosis, implying that EMT is part of the general program of lung repair. EMT reprogramming requires not only Smad signaling but also pY654–β-catenin. Generation of pY654–β-catenin requires assembly of complexes of the integrin α3β1, E-cadherin, and TGF-β1 receptors, and such assembly is a function of cell–cell and cell–matrix contacts. Sequestration of α3β1 or E-cadherin in such contacts prevents complex assembly, TGF-β1 induced pY654–β-catenin generation and EMT. Disruption of these contacts is a signal for the cells to initiate repair. Critical remaining questions center around better definition of direct versus indirect effects of EMT on collagen deposition and the nature of AEC progenitors differentiating during fibrogenesis. Elucidation of specific inhibitors of EMT should further test the question of whether the process is important to fibrosis in vivo and a viable therapeutic target.
Keywords: integrin, TGF-β1, β-catenin, collagen, signaling
The lung parenchyma is supported by an elaborate but delicate interstitial network of collagen and elastin. In addition, the epithelial and endothelial lining cells reside on stereotypical basement membranes comprised mainly of type IV collagen (along with many other minor collagen subtypes), laminins, and proteoglycans (1–3). Collectively, this scaffolding provides stable microenvironmental contacts for the maintenance of epithelial and endothelial differentiation and does so within the requirement of the normal lung for remarkably high compliance and distensibility. In this setting, there is very slow turnover of mature epithelial cells and of components of the extracellular matrix (ECM), such as elastin (4–6).
Lung injury to the parenchyma with leakage of plasma-like fluid or frank blood into the alveolar compartment produces drastic changes in the ECM with profound impacts on gas-exchanging surfaces (Figure 1). Perhaps because the alveolar compartment is essentially open to the environment, the lung surfaces are endowed constitutively with tissue factor and small amounts of locally produced clotting factors to rapidly initiate coagulation of plasma leakage (7, 8). If not subsequently removed by the fibrinolytic system, this process leads to the deposition in alveoli of a provisional, much less compliant ECM comprised of fibrin and fibronectin. This provisional ECM is conducive to the ingrowth of fibroblasts (9, 10), although the accumulation of a number of other chemokines/cytokines (e.g., prostaglandin E2) strongly influences the fibroblast response (Figure 1) (11, 12). In experimental systems, the pattern of fibronectin deposition early after injury correlates with subsequent regions of fibroblast and collagen accumulation during the repair phase (10). In humans, perhaps the most striking example is the intraalveolar fibronectin accumulation recognizable as Masson bodies that serve as a hallmark of the sites of granulation tissue expansion in organizing pneumonia (13).
Figure 1.
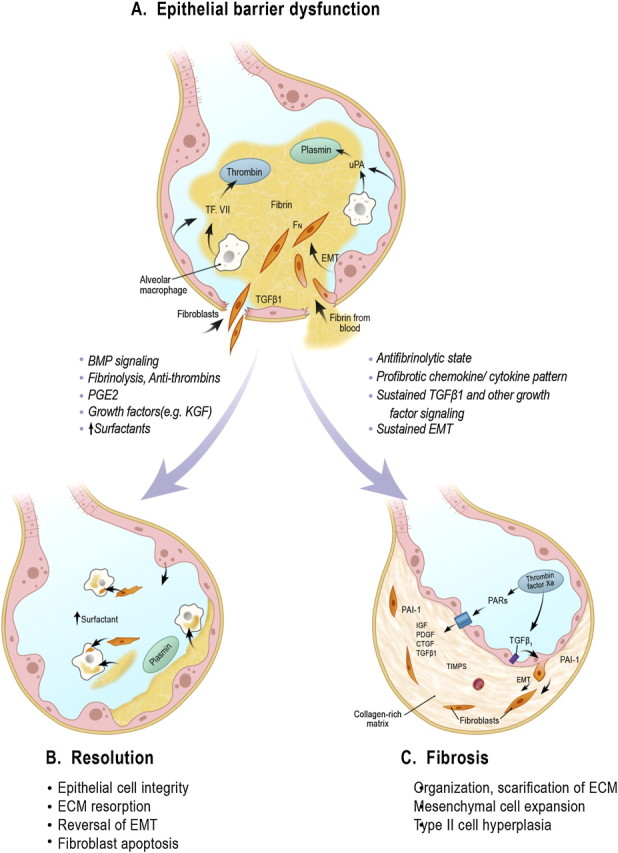
The schematics highlight early events in the development of lung injury followed by responses of the lung that favor repair and resolution or progressive fibrosis. (A) Epithelial cell loss (bottom) is depicted as an early event leading to airway barrier breakdown, deposition of alveolar fibrin and a fibronectin-rich provisional matrix, and phenotypic changes in epithelial cells promoting their transition toward a flattened squamous cell or epithelial mesenchymal transition (EMT), allowing these cells and resident fibroblasts to invade the alveolar matrix and initiate repair. (B) Limited loss of barrier integrity with resorption of the extracellular matrix (ECM) and reversal of fibroblast accumulation (by apoptosis and/or reverse EMT) favors return to a normal state, whereas (C) continuation of a profibrotic cytokine and chemokine milieu with excessive TGF-β1 and protease-activated receptor (PAR) activation promotes ongoing ECM accumulation, EMT, and hyperplasia of type II cells. CTGF = connective tissue growth factor; Fn = fibronectin; PAI-1 = plasminogen activator-1; PDGF = platelet-derived growth factor; TIMPS = tissue inhibitor of metalloproteases; TF. VII = Factor VII; uPA = urokinase. Adapted by permission from Reference 51.
It is increasingly recognized that activation of mediators of coagulation (e.g., Factor Xa and thrombin, the fibrinolytic system, various chemokines and cytokines, and the altered ECM) directly affect distal airway and alveolar repair in the setting of injury (Figure 1). Several recent reviews of important aspects of this pathophysiology are available (14–16). This article focuses on the influence of the ECM on the epithelium.
Epithelial Mesenchymal Transition and the ECM
Epithelial mesenchymal transition (EMT) is a process of reprogramming epithelial cells from a fully differentiated epithelial state toward a more mesenchymal state in which adherens junctions and other epithelial markers are lossed (or nearly lossed) and a mesenchymal profile of proteases, matrix proteins, and cytofilaments are newly expressed (17, 18). EMT is reversible and responsive to signals that promote or antagonize EMT programming. EMT is also a continuum of mesenchymal change and not an all-or-none phenomenon. EMT reprogramming is well established during embryogenesis, and there is accumulating evidence for EMT in the context of tumor invasion, especially breast tumor invasion. Partial, reversible EMT events are likely much more common than permanent mesenchymal transitions, and this may be especially so in the context of wound repair because the repair process is normally self-limited. For example, a temporary EMT that promoted stem cell expansion and lung repair was recently reported in distal airway epithelial cells after cytotoxic injury to the epithelium (19).
The influence of the ECM on the state of AEC differentiation is highlighted by the divergent outcomes of the culture of primary AECs on laminins or fibronectin. AECs cultured on a compliant, nonfibrillar type IV collagen/laminin gel (Matrigel; BD Biosciences, Sparks, MD) results in maintenance of a cuboidal epithelial phenotype and the formation of extensive E-cadherin–rich cell–cell contacts (20). In 3D cultures of matrigel, AECs form highly polarized structures (21). In contrast, AECs cultured on a stiffer fibronectin matrix spread extensively and rapidly lose surfactant expression. A similar influence of the ECM on breast epithelial cells cultured in 3D collagen gels can be seen as the stiffness of the collagen is increased: Clusters of the epithelial cells begin to spread and lose organization compared with the highly polarized epithelial phenotype observed when cells are cultured in a compliant matrix (22). The stiffness of the ECM dictates the degree of integrin and FAK kinase activation by cells engaging the matrix, leading to additional downstream signaling that influences cell growth and differentiation (23–25). The stiffness of the ECM (or the presence of fibrotic tissue in the tumor) correlates with tumor progression and poor outcome (26). Components of the ECM, including fibronectin, collagen, and proteoglycans, bind a number of growth factors that can be released by proteases to promote growth or tissue remodeling. Thus, the ECM can contribute indirectly to growth and differentiation via its reservoir of bound cytokines. One of these directly related to mesenchymal expansion is latent TGF-β1 (27, 28).
Activation of TGF-β1 is critically dependent on ECM engagement by integrins; in the case of distal airway and alveolar epithelial cells, TGF-β1 is activated predominantly by the integrin αvβ6 (29). TGF-β1 is synthesized in a latent form requiring cleavage of a pro-piece to generate the active peptide, but the cleaved pro-piece, termed the “latency binding peptide” (LBP), remains associated with and blocks the receptor-binding capacity of the active TGF-β1 peptide (30). Either further proteolytic processing of the LBP or a conformational change in the whole complex is required for TGF-β1 activity. LBP contains a canonical RGD integrin binding motif. One mechanism of TGF-β1 activation is binding of LBP to the epithelial integrin αvβ6 through the RGD motif followed by activating conformational change when the integrin is activated by engagement of epithelial cells with ECM or certain chemokines (Figure 2). Because LBP is known to bind and to be sequestered in fibronectin-rich ECMs, fibronectin is thought to be a major ECM protein whose engagement by integrins leads to TGF-β1 activation (28). Primary cells missing αvβ6 fail to activate TGF-β1 (as judged by pSmad in immunoblotting) and hence maintain a surfactant-rich epithelial state (Figure 2).
Figure 2.

Alveolar epithelial cell epithelial mesenchymal transition (EMT) requires αvβ6 and a stiff matrix for TGF-β1 activation. (A) Primary murine alveolar epithelial cells (AECs) were cultured for 2 days on Matrigel/Col 1 or fibronectin (Fn) and then lysed and analyzed by immunoblot for p-Smad2 and total Smad2/3. Epithelial cells culture on Matrigel fail to activate TGF-β1. (B) Primary AECs from wild-type (WT) and β6-null mice were cultured on Fn ± SB431542 (10 μM) for 2 days and then lysed and immunoblotted for p-Smad2 and total Smad2/3. AECs missing β6 fail to activate TGF-β1. (C and D) AECs from WT (C) and β6-null (D) mice were cultured on Fn for 4 days and stained for α-smooth muscle actin (α-SMA) and pro-surfactant protein C (SPC), illustrating the EMT response of WT but not β6-null AECs on Fn. Reprinted from Reference 20.
TGF-β1 is a key cytokine driving EMT reprogramming, and this is highlighted in Figure 2 by extensive expression of α-smooth muscle actin (α-SMA) when cells are cultured on fibronectin. TGF-β1 induces prototypical transcription factors implicated in EMT, such as Snail1 and Twist, and alters a profile of epithelial microRNAs that sustain a more mesenchymal state. However, activation of αvβ6 by Fn engagement appears to be an example of outside-in signaling in which activation of G-protein–coupled receptors or other Fn-binding integrins leads to Rho kinase activation, which alters the conformation of αvβ6 via its cytoskeletal connections, leading to activation of its bound ligand TGF-β1 (31). In principle, other stiff matrices that activate integrins and Rho kinase could have similar effects (32). This likely in part accounts for the strong positive influence of fibrillar collagens, such as collagen 1, on promoting a mesenchymal phenotype. Cells cultured on the more compliant Matrigel by contrast exhibit little or no TGF-β1 activation (Figure 2), as judged by the lack of pSmad generation, consistent with the persistence of a mature epithelial phenotype. Indeed, the addition of exogenous active TGF-β1 to primary epithelial cell cultures on Matrigel favors their apoptosis (20).
Regulation Of Emt Signaling
Although TGF-β1 is a key cytokine necessary for EMT, prior studies indicate that TGF-β1 signaling per se is insufficient to initiate an EMT program. Indeed, constitutive epithelial cell TGF-β1 signaling appears important to maintaining an antiinflammatory state within the pulmonary alveolar compartment and for suppressing epithelial cell proliferation (29). Deletion of TGF-β1 activation and signaling in a number of epithelial cells promotes tumor initiation and tissue inflammation (33). Therefore, initiation of the EMT program requires input from other signaling pathways, which indicates the need for an injury response, and this step is again regulated by the ECM. Prior studies have demonstrated that EMT ex vivo in kidney and lung AECs requires the presence of the prominent epithelial integrin α3β1 (34). α3β1 is a laminin and type IV collagen receptor but also physically accumulates in adherens junctions at least in part through interactions with E-cadherin (35). The function of the integrin in EMT centers around the colocalization of α3β1 with E-cadherin and the transcription factor β-catenin. Accumulating evidence indicates that EMT requires crosstalk between Smad and β-catenin signaling pathways, which requires physical association between Smads and β-catenin (36). The function of α3β1 is linked to this crosstalk because the integrin presence is required for tyrosine phosphorylation of β-catenin at Y654 after TGF-β1 stimulation. The accumulation of pY654–β-catenin strongly correlates with AEC EMT ex vivo and in vivo in experimental models (36). Selective deletion of epithelial α3β1 in lung epithelial cells abrogates EMT as well as myofibroblast and collagen 1 accumulation in the bleomycin injury model. PY654–β-catenin also selectively accumulates in the nuclei of lung myofibroblasts in patients with idiopathic pulmonary fibrosis, pointing to the relevance of this pathway to myofibroblast accumulation in humans.
The requirement for integrin α3β1 also implicates a key role for cell contacts in the regulation of EMT. This concept is illustrated schematically in Figure 3. TGF-β1–mediated β-catenin phosphorylation is blocked by sequestration of the integrin onto laminin 5 (Ln-5), a major component of basement membranes. This is indicated in Figure 3 by the loss of β-catenin/pSmad2 complexes when AECs are cultured on Ln-5, resulting in suppression of an EMT response. Smad phosphorylation and nuclear translocation is not affected by integrin sequestration, and thus it is likely that the antiinflammatory and antiproliferative functions of TGF-β1 remain intact. A similar suppression of β-catenin phosphorylatioin and EMT is observed when cells are cultured near cell confluence where formation of adherens junctions sequestering E-cadherin and α3β1 is extensive (34, 37). In the context of significant injury with loss of cell contacts or disruption of basement membranes, the activation of TGF-β1 could be expected to lead to pY654–β-catenin and an EMT response. Presumably, EMT wanes as new basement membranes are deposited and normal cell contacts are restored. Thus, the provisional matrices deposited in the lung as a consequence of injury promote epithelial-organized repair through a number of complementary pathways: the stiff, integrin-engaging provisional ECM promotes TGF-β1 activation, and the loss of normal epithelial contacts promotes an EMT response and not simply cell cycle arrest. It is likely that the cytoskeletal changes effected by integrin signaling on fibronectin also support EMT responses through mechanisms similar to that recently described for fibroblasts (38).
Figure 3.

Regulation of β-catenin/p-Smad2 complex formation and epithelial mesenchymal transition (EMT) by cell contacts. Upper panel: α3β1 engagement on Ln5 limits the formation of β-catenin/p-Smad2 complex formation. Lysates of TGF-β1 stimulated α3 wild-type cells from fibronectin (Fn)- or laminin 5 (Ln5)-coated plates were subject to β-catenin immunoprecipitation followed by p-Smad2 immunoblot. The β-catenin/p-Smad2 complex formation is only seen with cells plated on Fn and not with Ln5. Lower schematic: Cells cultured on Ln5 fail to activate pY654–β-catenin and form β-catenin/pSmad complexes. These cells phosphorylate Smads normally after TGF-β1 stimulation but do not under an EMT response. Cells cultured on Fn activate pY654–β-catenin, which cooperates with pSmads in translocating to the nucleus and initiating an EMT program. Similar inhibition of EMT occurs when E-cadherin/α3β1 complexes are sequestered in cell–cell junctions (not shown). Reprinted from Reference 34.
Evidence for EMT In Vivo
Although EMT is well described ex vivo and in vivo during embrogenesis and tumor progression, there is conflicting evidence for the role of EMT in vivo during fibrogenesis. A number of investigators have applied the tools of lineage tracing to the question of whether myofibroblasts in vivo derive from EMT, but the results have been ambiguous. In this approach, a differentiation marker of a specific cell lineage is used to irreversibly mark and identify the lineage's fate during embryonic development, steady states in the adult, or after injury. Lineage tracing is generally achieved by activation of a latent reporter activity (e.g., GFP or β-galactosidase) via cre recombinase expression in a time- and/or cell-specific manner. Early studies in the lung used an inducible β-galactosidase reporter activated in most, if not all, populations of distal airway and alveolar epithelial cells (20). This was achieved in a triple transgenic system composed of (1) a human surfactant protein C (SPC) promoter sequence driving a doxycycline-activatible rtTA that acts on (2) a rtTA responsive–cre transgene to drive cre transcription that in turn (3) permanently removes an inhibitory DNA sequence in the third transgene, allowing reporter (β-galactosidase) expression (20). Widespread epithelial reporter expression was achieved by exposure of mice to doxycycline throughout gestation because SPC is expressed in the progenitors of virtually all distal AECs during embryogenesis. Using this system in the context of adenoviral-mediated active TGF-β1 expression in mouse lungs, cells bearing the lineage trace could be directly recoverned from injured lungs expressing vimentin and to a lesser extent α-smooth muscle active (α-SMA), indicating EMT. More recently, using a similar system but with an e Enhanced green fluorescent protein reporter after bleomycin-induced lung injury, GFP-expressing cells also expressing vimentin, collagen 1, or α-SMA were isolated directly from lung on Day 17 after bleomycin exposure. The percentages of recovered epithelial cells expressing any of the mesenchymal proteins was low (< 6%), but the numbers are difficult to interpret because the great majority of epithelial cells are not involved in the heavily injured areas destined to become fibrotic in the bleomycin model (35). Other groups have reported similar findings in the bleomycin model (39, 40).
Although these findings provide direct evidence for EMT in vivo in the context of fibrogenesis, they do not answer the question of whether the expansion of collagen-producing cells and/or myofibroblasts during fibrosis derives appreciably from epithelial cells. The appearance of mesenchymal markers observed in a fraction of AECs after lung injury may represent a partial EMT that does not reflect the major pathway of mesenchymal cell expansion. Indeed, two studies also using lineage tracing in the last few years have concluded that EMT has little role in fibrosis. Humphries and colleagues reported pericytes and not epithelial cells as the main source of α-SMA–expressing cells in a model of renal fibrosis (41). More recently, Rock and colleagues, using a tamoxifen-inducible cre recombinase engineered within the endogenous mouse SPC gene and fluorescent reporters, found multiple sources of collagen-producing cells after bleomycin injury but little if any contribution of mature type II cells or Clara cells to the production of mesenchymal markers or collagen 1 (5). These findings call into question whether EMT has a significant role in fibrogenesis. A potentially important difference between earlier work with the SPC transgenes and recent work with the endogenous SPC “knockin” is that the SPC-creER only marks mature type II cells and not other AECs. This is relevant because of prior studies linking the capacity for EMT among epithelial cells with a stem cell–like differentiation state (42). Our recent observations suggest this principle may also apply to AECs.
Identification Of Aec Progenitor Cells: Role In Emt And Regeneration
The possibility of subpopulations of AECs in preparations of type II cells from normal adult murine lung is revealed by inspection of ex vivo EMT of primary AECs cultured on fibronectin (Figure 4). Although virtually all of the cells spread on fibronectin, only a small percentage of cells (< 10%) undergo obvious EMT, as judged by α-SMA in green. The great majority of cells maintain E-cadherin and visible cell–cell junctions. We recently observed that the population of primary cells with a high capacity for EMT is identifiable by their expression of the integrin α6β4 (44). Separation of AECs into α6β4+ and α6β4− cells indicated that the bulk of α-SMA appears in the α6β4+ fraction (Figure 4). Further characterization of these cells revealed that freshly isolated α6β4+ AECs are largely SPC and CC10 negative and possess colony-forming potential, suggesting a stem/progenitor cell population. Indeed, in an in vivo model of embryonic lung development, these adult cells formed SPC+ saccular-like or CC10+ airway-like structures, confirming their progenitor potential.
Figure 4.

The β4+ alveolar epithelial cell (AEC) subpopulation preferentially undergoes epithelial mesenchymal transition (EMT) when cultured on fibronectin (Fn). (A) Immunostaining for E-cadherin (red) or α-smooth muscle actin (α-SMA) (green) in purified AECs plated for 6 days on Fn. AECs were purified by standard methods followed by flow cytometry for E-cadherin expression. EMT develops only in a subfraction of AECs. Multiple 20× images were captured and tiled into a single mosaic image. Scale bar = 200 μm. Inset: Immunoblot confirming α-SMA preferentially develops in β4+ AECs. Adapted by permission from Reference 43.
Does the lung require AEC progenitors for repair after injury? To address this question, we recently engineered a mouse with tamoxifen-dependent cre recombinase in the endogenous SPC locus, similar to Rock and colleagues (5). Using an enhanced green fluorescent protein reporter mouse, we observed that 60 to 70% of the mature type II cells could be labeled after two injections of tamoxifen and that there was little label dilution of mature type II cells over the next 30 days (43). However, when mice were injured with bleomycin, a striking finding was that the fibrotic areas contained few labeled cells, indicating their death and removal. The type II cells that appear in these most injured areas (typical type II cell hyperplasia) were unlabeled, indicating they must arise from a progenitor cell during the repair process. Similar findings were also recently reported by Rock and colleagues (5). In nonfibrotic lung areas, there was obvious transdifferentiation of labeled cells into type I cells, and overall the total GFP signal in the whole injured lung is higher than that of uninjured control mice, implying net proliferation of type II cells after injury, at least in the bleomycin model.
Collectively, these studies led to the current model for conceptualizing the role of distal airway and alveolar progenitors in lung maintenance and repair (Figure 5). In the adult normal lung, AECs are maintained by self-replenishing type II cells and their transdifferentiation to type I cells when needed. This concept has been inferred from many prior studies of lung parenchymal epithelial cells but is now more directly demonstrable with new lineage-tracing methods (44). More severe lung injury can lead to almost total regional loss of the existing type II cells. In this case, a relatively undifferentiated stemprogenitor cell expands and differentiates to replenish type II cells (and presumably type I cells over time). The nature of these progenitor cells is unclear, although we believe it is contained within a somewhat heterogenous population identified by α6β4 expression. Other previously reported putative stem cells, such as bronchoalveolar stem cells or invariant Clara cells or basal cells in the human, could be part of, or distinct from, the α6β4+ population (45, 46). This is an area of active investigation.
Figure 5.

Schematic summarizing epithelial and fibroblast transitions in the lung after lung injury. Type II cells function to replenish the type II cell population after injury and to transdifferentiate into type I cells to maintain epithelial integrity. If the injury is more severe, an epithelial stem/progenitor cell(s) proliferates and differentiates into type II cells. The stem and progenitor cells are within a population marked by surface α6β4 expression and likely reside in distal airways and within alveoli. Bronchoalveolar stem cells and other putative stem cells, such as in variant Clara cells, may be contained in this population. The stem and progenitor cells also capable of initiating an epithelial mesenchymal transition program regulated as described in the text. Resident mesenchymal cells also expand after injury and appear to be the major source of myofibroblasts/collagen 1. Crosstalk between epithelial cells responding to canonical transcription factors promoting EMT and mesenchymal cells, as indicated in the figure, is likely important to the extent of collagen 1 accumulation.
We hypothesized that AEC progenitor cells are the major cell directly involved in differentiation along a mesenchymal pathway (EMT) in the setting of injury signals such as TGF-β1 (Figure 5). As indicated in Figure 5, the evidence favors two principal roles for EMT in repair. The first discovered was direct expression of ECM proteins via EMT (47). Although this is easily documented ex vivo and in vivo, the available data suggest that this is a small contribution to the total increase in collagen 1 during a repair or fibrotic process. No critical information on this point exists in human diseases of pulmonary fibrosis. The second pathway is promotion of fibrosis through signaling crosstalk with resident lung fibroblasts. Such crosstalk may operate not only via release of profibrotic cytokines but also by recruitment of bone-derived cells into the lung that then activate fibroblasts (48). Although the exact contribution of EMT to fibrogenesis remains uncertain, there are several lines of evidence in experimental models that support an important role for the EMT signaling program. First, epithelial cell signaling responses to TGF-β1 are critical for fibrogenesis in the bleomycin model, indicating that the functional responses of the epithelium to TGF-β1 and not simply the production of TGF-β1 by the epithelium is vital (49). Second, genetic deletion of integrin α3β1 specifically in epithelial cells blocks EMT and the expansion of myofibroblasts and collagen 1 deposition in the same model (35). Finally, using conditional deletion of Snail1 specifically in epithelial cells, it has been recently observed in kidney and liver injury models that the EMT program is critical to fibrosis (50). Collectively, these findings underscore the important role for EMT in fibrogenesis even though the direct contribution of epithelial-derived fibroblast-like cells to the myofibroblast population and collagen deposition remains unsettled.
Summary And Future Directions
Epithelial cells in the lung parenchyma are increasingly recognized as a site of disease initiation and as a key organizer of the host response to injury. A central pathway of epithelial responses to injury involves activation of the cytokine TGF-β1. TGF-β1 activation and the cellular responses to its activation are intricately connected to integrins and the ECM. Normal basement membranes and cell–cell contacts minimize TGF-β1 activation and EMT, whereas stiffer provisional matrices and accumulating collagen 1 favor ongoing activation and mesenchymal conversion. It remains uncertain to what degree EMT directly contributes to the mesenchymal pool of collagen 1–producing cells and which epithelial cells undergo EMT within the lung. The recent evidence for an important role of epithelial stem and progenitor cells in lung repair and the known links between a progenitor state and epithelial plasticity demonstrates the need for further study and lineage tracing of epithelial progenitors as important fields for future investigation.
The specific signaling pathways through which epithelial cells organize a repair process remain incompletely understood. It is appealing, though unproven, that pathways identified in studies of epithelial–fibroblast and fibroblast–epithelial crosstalk within carcinomas may shed important insight to similar biology in tissue remodeling (25). If so, it is likely that not only direct crosstalk via release of profibrotic cytokines such as PDGF-α and TGF-β1 are involved but also indirect profibrotic signaling via epithelial cell recruitment of bone marrow–derived cells, especially monocytes and macrophages, to the injured areas. Accumulation of monocytes and macrophages and other innate immune cells at sites of tissue injury are strongly linked to wound healing as well as dysfunctional fibrogenesis in tumors and fibrotic tissues. This remains an important area for future investigation.
Footnotes
Supported by National Institutes of Health grant NIH HL44712.
References
- 1.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer 2003;3:422–433. [DOI] [PubMed] [Google Scholar]
- 2.Izvolsky KI, Shoykhet D, Yang Y, Yu Q, Nugent MA, Cardoso WV. Heparan sulfate-FGF10 interactions during lung morphogenesis. Dev Biol 2003;258:185–200. [DOI] [PubMed] [Google Scholar]
- 3.Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn 2000;218:213–234. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest 1991;87:1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA 2011;108:E1475–E1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rawlins EL, Hogan BL. Ciliated epithelial cell lifespan in the mouse trachea and lung. Am J Physiol Lung Cell Mol Physiol 2008;295:L231–L234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Neut R, Cachaco AS, Thorsteinsdottir S, Janssen H, Prins D, Bulthuis J, van der Valk M, Calafat J, Sonnenberg A. Partial rescue of epithelial phenotype in integrin beta4 null mice by a keratin-5 promoter driven human integrin beta4 transgene. J Cell Sci 1999;112:3911–3922. [DOI] [PubMed] [Google Scholar]
- 8.Chapman HA, Bertozzi P, Reilly JJ. Role of enzymes mediating thrombosis and thrombolysis in lung disease. Chest 1988;93:1256–1263. [DOI] [PubMed] [Google Scholar]
- 9.Kuhn C, Boldt J, King TE, Crouch E, Vartio T, McDonald JA. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis 1989;140:1693–1703. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda Y, Ishizaki M, Masuda Y, Kimura G, Kawanami O, Masugi Y. The role of intraalveolar fibrosis in the process of pulmonary structural remodeling in patients with diffuse alveolar damage. Am J Pathol 1987;126:171–182. [PMC free article] [PubMed] [Google Scholar]
- 11.Moore BB, Peters-Golden M, Christensen PJ, Lama V, Kuziel WA, Paine R, Toews GB. Alveolar epithelial cell inhibition of fibroblast proliferation is regulated by MCP-1/CCR2 and mediated by PGE2. Am J Physiol Lung Cell Mol Physiol 2003;284:L342–L349. [DOI] [PubMed] [Google Scholar]
- 12.Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, Polosukhin V, Wain J, Karimi-Shah BA, Kim ND, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med 2008;14:45–54. [DOI] [PubMed] [Google Scholar]
- 13.Kuhn C, McDonald JA. The roles of the myofibroblast in idiopathic pulmonary fibrosis: ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol 1991;138:1257–1265. [PMC free article] [PubMed] [Google Scholar]
- 14.Strieter RM, Mehrad B. New mechanisms of pulmonary fibrosis. Chest 2009;136:1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chambers RC. Procoagulant signalling mechanisms in lung inflammation and fibrosis: novel opportunities for pharmacological intervention? Br J Pharmacol 2008;153:S367–S378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 2010;298:L715–L731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871–890. [DOI] [PubMed] [Google Scholar]
- 18.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volckaert T, Dill E, Campbell A, Tiozzo C, Majka S, Bellusci S, De Langhe SP. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J Clin Invest 2011;121:4409–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA 2006;103:13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu W, Fang X, Ewald A, Wong K, Hunt CA, Werb Z, Matthay MA, Mostov K. Formation of cysts by alveolar type II cells in three-dimensional culture reveals a novel mechanism for epithelial morphogenesis. Mol Biol Cell 2007;18:1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton 2005;60:24–34. [DOI] [PubMed] [Google Scholar]
- 23.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 2003;278:12384–12389. [DOI] [PubMed] [Google Scholar]
- 24.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009;139:891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 2010;18:884–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasebe T, Iwasaki M, Akashi-Tanaka S, Hojo T, Shibata T, Kinoshita T, Tsuda H. Important histologic outcome predictors for patients with invasive ductal carcinoma of the breast. Am J Surg Pathol 2011;35:1484–1497. [DOI] [PubMed] [Google Scholar]
- 27.Unsold C, Hyytiainen M, Bruckner-Tuderman L, Keski-Oja J. Latent TGF-beta binding protein LTBP-1 contains three potential extracellular matrix interacting domains. J Cell Sci 2001;114:187–197. [DOI] [PubMed] [Google Scholar]
- 28.Fontana L, Chen Y, Prijatelj P, Sakai T, Fassler R, Sakai LY, Rifkin DB. Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J 2005;19:1798–1808. [DOI] [PubMed] [Google Scholar]
- 29.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999;96:319–328. [DOI] [PubMed] [Google Scholar]
- 30.Nunes I, Munger JS, Harpel JG, Nagano Y, Shapiro RL, Gleizes PE, Rifkin DB. Structure and activation of the large latent transforming growth factor-beta complex. Int J Obes Relat Metab Disord 1996;20:S4–S8. [PubMed] [Google Scholar]
- 31.Xu MY, Porte J, Knox AJ, Weinreb PH, Maher TM, Violette SM, McAnulty RJ, Sheppard D, Jenkins G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am J Pathol 2009;174:1264–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huveneers S, Danen EH. Adhesion signaling: crosstalk between integrins, Src and Rho. J Cell Sci 2009;122:1059–1069. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003;113:685–700. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y, Kugler MC, Wei Y, Kim KK, Li X, Brumwell AN, Chapman HA. Integrin alpha3beta1-dependent beta-catenin phosphorylation links epithelial Smad signaling to cell contacts. J Cell Biol 2009;184:309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiPersio CM, van der Neut R, Georges-Labouesse E, Kreidberg JA, Sonnenberg A, Hynes RO. alpha3beta1 and alpha6beta4 integrin receptors for laminin-5 are not essential for epidermal morphogenesis and homeostasis during skin development. J Cell Sci 2000;113:3051–3062. [DOI] [PubMed] [Google Scholar]
- 36.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest 2009;119:213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masszi A, Fan L, Rosivall L, McCulloch CA, Rotstein OD, Mucsi I, Kapus A. Integrity of cell-cell contacts is a critical regulator of TGF-beta 1-induced epithelial-to-myofibroblast transition: role for beta-catenin. Am J Pathol 2004;165:1955–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol 2010;190:693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demaio L, Buckley ST, Krishnaveni MS, Flodby P, Dubourd M, Banfalvi A, Xing Y, Ehrhardt C, Minoo P, Zhou B, Crandall ED, Borok Z. Ligand-independent transforming growth factor-beta type I receptor signaling mediates type I collagen-induced epithelial-mesenchymal transition. J Pathol 2012;226:633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 2009;180:657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010;176:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu T. Integrin alpha6beta4 identifies an adult distal lung epihtelial population with regenerative potential in mice. J Clin Invest 2011;121:2855–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adamson IY. Drug-induced pulmonary fibrosis. Environ Health Perspect 1984;55:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snyder JC, Teisanu RM, Stripp BR. Endogenous lung stem cells and contribution to disease. J Pathol 2009;217:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005;121:823–835. [DOI] [PubMed] [Google Scholar]
- 47.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003;425:577–584. [DOI] [PubMed] [Google Scholar]
- 48.Elkabets M, Gifford AM, Scheel C, Nilsson B, Reinhardt F, Bray MA, Carpenter AE, Jirstrom K, Magnusson K, Ebert BL, et al. Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J Clin Invest 2011;121:784–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest 2011;121:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rowe RG, Lin Y, Shimizu-Hirota R, Hanada S, Neilson EG, Greenson JK, Weiss SJ. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol Cell Biol 2011;31:2392–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim K, Chapman HA. Role of the pulmonary epithelium in interstitial lung disease. : Interstitial lung disease, 5th edition , Schwarz MI, King TE, Shelton, CT: People's Medical Publishing House; 2010. pp. 335–355. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Author disclosures