

Abstract
Adenosine is known as an endogenous purine nucleoside and it modulates a wide variety of physiological responses by interacting with adenosine receptors. Among the four adenosine receptor subtypes, the A3 receptor is of major interest in this study as it is overexpressed in some cancer cell lines. Herein, we have highlighted the strategy of designing the hA3 receptor targeted novel benzothiazolylquinoline scaffolds. The radioligand binding data of the reported compounds are rationalized with the molecular docking results. Compound 6a showed best potency and selectivity at hA3 among other adenosine receptors.
Keywords: benzothiazolylquinoline, human A3 receptor antagonist, molecular docking, radioligand binding assay, glide score
1. Introduction
Adenosine is an endogenous purine nucleoside which regulates many physiological functions through the activation of four specific receptor subtypes, classified as A1, A2A, A2B and A3 adenosine receptors (AR), belonging to the family of G-protein-coupled receptors [1]. These receptors are widely distributed in mammalian tissues. The A3AR subtype is the most recently characterized member of the family which was first cloned from a rat testis cDNA library [2] and is still undergoing intensive pharmacological characterization. The A3AR subtype is implicated in various pathological conditions such as cardiac and cerebral ischaemia, neurodegenerative diseases as well as inflammatory pathologies including rheumatoid arthritis and asthma [3]. Furthermore, A3AR is overexpressed in various neoplastic cells including HL-60 leukaemia, human Jurkat T-cell lymphoma, astrocytoma, A378 melanoma, B16-F10 and solid tumour (e.g. a two–threefold increase in colon carcinomas), while low or almost no receptor expression was found in normal cells [4,5].
Similar results were found in studies of the receptor expression levels in tumour tissues derived from patients with colon, breast, small cell lung, pancreatic and hepatocellular carcinomas and melanoma in direct comparison with adjacent body normal tissues [6–15]. Higher A3AR expression in the tumour versus adjacent non-neoplastic tissue was further confirmed by reverse transcription-PCR analysis of colon and breast carcinoma. Protein analysis of A3AR expression in fresh tumours derived from colon (n = 40) or breast (n = 17) revealed 61% and 78% higher expression in the tumour than adjacent normal tissue, respectively [16]. Thus the A3 receptor could be a prospective therapeutic target and biological predictive marker in cancer therapy. The high A3AR expression level in the tumour tissues was associated with elevated nuclear factor κB and cyclin D1 levels [16]. High A3AR mRNA expression was also exhibited in other solid tumour types. Mechanistic studies demonstrated that A3AR activation by synthetic agonists or antagonists induces down-regulation of key cell growth-regulatory proteins including cyclin D1 and nuclear factor κB [10,17,18]. Hence, discovery of selective A3AR targeting ligands has been a great challenge in last two decades. Moreover, A3AR antagonists research not only aids in the development of therapeutic agents but also in the development of diagnostic agents [2,19]. Nowadays, the diagnostic approaches have been significantly developed by using fluorescently labelled pharmacophores.
In the past few years, there have been strenuous efforts to develop different heterocyclic scaffolds as hA3antagonists including pyridine and dihydropyridine analogues, flavonoid, isoquinoline, triazoloquinazolines, pyrazolo-[3,4-c]or-[4,3-c]quinolones, triazoloquinoxaline, pyrazolo-[4,3-e]1,2,4-triazolo-[1,5-c]pyrimidines, ruthenium-pyrazolopyrimidines [20–29]. However, none of the pharmacophores has been tested in A3 overexpressed cancer cell lines. The cellular imaging study and cell surface receptor localization study was also not performed because these scaffolds are nonfluorescent. Therefore, to identify the hA3 targeting novel fluorescent ligands, a molecular simplification followed by molecular docking approach was employed.
Benzothiazole possesses several biological activities such as anti-inflammatory, antimicrobial, anti-HIV, anticancer and amyloid marker [30–36]. These scaffolds are also able to arrest metal promoted amyloid fibril build-up [37]. Likewise, 8-hydroxy quinoline has also been developed as potent bioactive scaffold [38]. Herein, we have highlighted the strategy of designing the novel 2-(2′-hydroxyphenyl)benzothiazole (HBT) scaffolds having dual properties (pharmacophore and fluorophore) via molecular simplification followed by a molecular docking approach (figure 1). The designed molecules have already shown potency and selectivity in hA3AR overexpressed cancer cell lines than normal cell lines [38]. In our earlier report, the cellular localization was also observed using those scaffolds. To justify the molecular pathway of these drugs, we have initiated the molecular docking approach as well as radioligand binding study at hA3AR.
Figure 1.

Dual role: pharmacophore as well as a fluorophore. G-PCR, G-protein coupled receptor.
2. Results and discussion
2.1. Chemistry
2.1.1. Molecular simplification
Following a molecular simplification approach, we have identified the benzothiazolylquinoline ring system as an appropriate core skeleton for the design of novel fluorescent hA3AR antagonists. Here, we have simplified the structure of the tricyclic A3 antagonist (triazoloquinoxaline) into bicyclic motif (2-benzothiazolyl phenol). In the course of structure design, we have attached the bicyclic fluorescent pharmacophore (2-benzothiazolyl phenol) with another bioactive 8-hydroxy quinolone unit through a linker to enhance the A3 binding ability (figure 2).
Figure 2.

Molecular simplification approach to design the hA3 targeting ligand.
2.2. Synthesis
In our earlier report, a series of 2-(2′-hydroxyphenyl)benzothiazolylquinoline scaffolds were prepared in conventional way and also under microwave condition in one pot sequence (scheme 1) [38]. We followed the earlier reported green method to synthesize the following scaffolds and then characterized by 1H NMR, 13C NMR and LCMS study (electronic supplementary material, supporting information). Structure of compound 6c was further confirmed by single crystal X-ray study (figure 3) [38].
Scheme 1.

Preparation of 8-[2-(2-benzothiazol-2-yl-phenoxy)-alkoxy]-quinoline derivatives (6a-l) [38].
Figure 3.
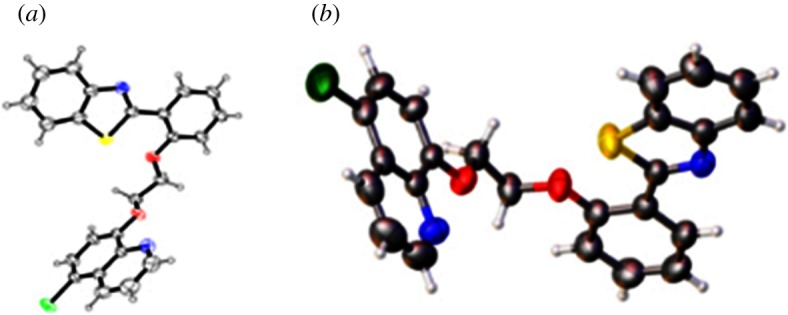
ORTEP diagrams of compound 6c drawn at the 50% probability level [38].
2.3. Molecular docking studies
2.3.1. Homology modelling
The crystallographic structure of hA2AAR complexed with ZM-241385 as a high affinity antagonist (PDB code: 3EML) [39]. It was already being used to build up a homology model of the hA3AR by our group [40]. Considering the high resolution (2.6 Å) and accuracy of the structure of hA2AAR, 3EML was selected as a template by various research groups [41,42]. Modeller 9.11 was used to perform the homology modelling [43–46] and the quality of the model was evaluated using the Ramachandran plot. Subsequently, the prediction ability of the constructed hA3AR homology model was evaluated in the molecular docking experiments using the GLIDE tool from Schrödinger maestro.
2.3.2. Molecular docking
Our approach was to select a new class of hA3AR targeting compounds from a group of hypothetically designed 2-phenylbenzothiazole (HBT)-based scaffolds. We performed molecular docking of 12 different HBT-based ligands using the GLIDE tool from Schrödinger maestro to identify the hypothetical binding mode at the hA3AR. Finally, we have identified the best inhibitor for targeted hA3AR from GLIDE scores (table 1).
Table 1.
Binding energy of benzothiazolylquinoline analogues (6a-l) with hA3.
| entry | compound | GLIDE score (Kcal mol−1) |
|---|---|---|
| 1 | 6a | −10.31 |
| 2 | 6b | −9.06 |
| 3 | 6c | −6.50 |
| 4 | 6d | −8.90 |
| 5 | 6e | −9.05 |
| 6 | 6f | −8.81 |
| 7 | 6g | −9.04 |
| 8 | 6h | −8.78 |
| 9 | 6i | −7.48 |
| 10 | 6j | −8.37 |
| 11 | 6k | −9.05 |
| 12 | 6l | −9.36 |
All the newly synthesized benzothiazolylquinoline scaffolds were docked into the orthosteric transmembrane-binding cavities of hA3AR. From the ligand docking, we have inferred that out of 12 synthesized scaffolds, compound 6a displayed the best GLIDE score with the lowest binding energy. In figure 4, the hypothetical binding pose of compound 6a is clearly observed at the hA3AR. In particular, the most prominent aromatic π–π stacking interactions are established between Phe 168 and ligands and it was anchored properly within the binding cleft. Moreover, a strong hydrogen bond with Phe 168 was also appeared within the binding pocket. Likewise, a strong π–π stacking interaction was observed between Tyr 265 and compound 6f. Compound 6g also formed a hydrogen bond with Val 169 (electronic supplementary material, figure S1)
Figure 4.

Binding interaction of most potent benzothiazolylquinoline analogue (6a) with hA3 receptor.
2.3.3. Binding affinity at hA1AR, hA2AAR, hA2BAR and hA3AR
The receptor binding affinities of the synthesized benzothiazolylquinoline derivatives (6a-l) are recapitulated in table 2. The binding affinity of the antagonist was estimated by measuring the displacement of selective radioligands which were formerly bound to the receptor expressed (Chinese hamster ovary cells (CHO) for hA1AR, hA2AAR and hA3AR) at the surface of the cell. In this particular assay, the displacement of: (i) specific [3H] CCPA (2-chloro-N(6)-cyclopentyladenosine) binding at hA1AR, (ii) specific [3H] NECA (5'-N-ethylcarboxamidoadenosine) binding at the hA2AAR, and (iii) [3H] HEMADO (2-(1-hexynyl)-N6-methyladenosine) at the hA3AR were evaluated. There is no suitable radioligand for hA2BAR found and hence the antagonists activity was determined in adenylyl cyclase experiments in CHO cells expressing the hA2BAR [47,48]. Ki (dissociation constant) value of the data was calculated using the Cheng and Prusoff equation [49], with geometric means of at least three experiments including 95% confidence intervals. From table 2, it was observed that most of the compounds exhibited a Ki value at hA3 in the range of 2–6 µM. Compound 6c and 6i showed least binding efficacy with hA3 which is clearly rationalized with their binding energy profile (GLIDE score). Compound 6a exhibited the most binding potency at hA3 in the 2.6 µM range. This compound has also shown 38-fold and 11-fold more selective potency at hA3 than hA1 and hA2A, respectively. While increasing the length of the –CH2– linker from 2 to 3, binding potency of the compound 6e at hA3 is reduced to 3.8 µM. Selectivity of this compound at hA3 with respect to hA1 and hA2 has also been reduced to some extent. Consequently, compound 6i showed much less potency and selectivity in hA3. Compounds (6f-h and 6j-l) having electronegative groups and lengthy linkers (n = 3, 4) showed good potency and selectivity at hA3. It was also observed that compound 6b having two electronegative bromine groups with a small linker (n = 2) exhibited good potency (3.2 µM) and selectivity at hA3.
Table 2.
Binding affinity (Ki) of synthesized compounds at hA1AR, hA2AAR and hA3AR and selectivity against hA1AR and hA2AAR.
| Ki, µM (95% CI)d | selectivity | |||||
|---|---|---|---|---|---|---|
| compound | hA1b | hA2Ac | hA2Ba | hA3e | hA1/A3hA2A/A3 | |
| 6a | >100 | 23.8 (21.6–26.4) | >30 | 2.6 (1.8–4.5) | >38.46 | >11.53 |
| 6b | >100 | 17.2 (15.3–19.4) | >20 | 3.2 (2.4–4.1) | >31.25 | >6.25 |
| 6c | 30.8 (26.5–32.7) | 32.8 (28.7–34.1) | >20 | 30.4 (27.6–32.1) | 1.01 | >19.80 |
| 6d | >100 | >20 | >20 | 5.6 (3.8–7.1) | >17.85 | >3.57 |
| 6e | >100 | >20 | >30 | 3.8 (2.6–4.7) | >26.31 | >7.89 |
| 6f | >100 | >20 | >30 | 6.2 (4.8–7.1) | >16.12 | >4.83 |
| 6g | >100 | >20 | >20 | 4.2 (3.1–5.4) | >23.80 | >4.76 |
| 6h | >100 | >20 | >20 | 6.1 (4.9–7.2) | >16.39 | >3.27 |
| 6i | 29.7 (27.8–32.6) | 34.9 (32.7–37.1) | >20 | 25.4 (23.6–27.1) | 1.16 | >0.78 |
| 6j | >100 | >20 | >20 | 6.4 (4.9–8.2) | >15.62 | >3.15 |
| 6k | >100 | >20 | >10 | 3.7 (2.7–4.8) | >27.02 | >2.70 |
| 6l | >100 | >20 | >30 | 3.6 (2.1–4.8) | >27.77 | >8.33 |
aAdenylyl cyclase activity of synthesized compounds at the hA2BAR.
bDisplacement of specific [3H]-CCPA binding at hA1AR expressed in Chinese hamster ovary (CHO) cells (n = 3–6).
cDisplacement of specific [3H]-5′-N-ethylcarboxamido adenosine (NECA) binding at hA2AAR expressed in CHO cells (n = 3–6).
dKi values for inhibition of NECA-stimulated adenylyl cyclase activity in CHO cells (n = 3–6).
eDisplacement of specific [3H]-2-(1-hexynyl)-N6-methyl adenosine (HEMADO) binding at hA3AR expressed in CHO cells (n = 3–6).
3. Experimental
3.1. Biology
3.1.1. Chinese hamster ovary membranes preparation
The human A1, A2A, A2B, and A3ARs were transfected in CHO cells based on the previously reported method [47–50]. The cells were grown adherently and maintained in Dulbecco's modified Eagle's medium with nutrient mixture F12 (DMEM/F12) without nucleosides, containing 10% foetal calf serum, streptomycin (100 µg ml−1), penicillin (100 µg ml−1), l-glutamine (2 mM) and Geneticin (G418, 0.2 mg ml−1) at 37°C in 5% CO2, 95% air. The membrane preparation was initiated by removal of culture medium followed by washing of the cells with phosphate buffered saline and scraped off T75 flasks in ice-cold hypotonic buffer (5 mM Tris–HCl, 2 mM EDTA, pH7.4). Then the cell suspension was homogenized with a polytron, and the homogenate was centrifuged for 10 min at 1000 g. The supernatant was then centrifuged for 30 min at 100 000 g. The membrane pellet was suspended in (i) 50 mM Tris–HCl buffer, pH 7.4, for A1ARs; (ii) 50 mM Tris–HCl, 10 mM MgCl2 buffer, pH 7.4, for A2AARs; and (iii) 50 mM Tris–HCl, 10 mM MgCl2, 1 mM EDTA buffer, pH 7.4, for A3ARs. The cell suspension was incubated with adenosine deaminase for 30 min at 37°C. Then the membrane preparation was used for binding experiments.
3.1.2. Binding at human A1, A2A and A3 adenosine receptors
For radioligand binding at A1 ARs 1 nM [3H] CCPA was used, whereas 30 nM of [3H] NECA were used for A2A and 10 nM of [3H]- HEMADO were used for A3 receptors, respectively [47–50]. Nonspecific binding of [3H]CCPA was determined in the presence of 1 mM theophylline, when [3H]NECA 100 µM R-PIA was used.
3.1.3. Adenylyl cyclase activity
The potency of antagonists at the A2BAR was determined in adenylyl cyclase experiments [47–50]. For the measurement of adenylyl cyclase activity, only one high speed centrifugation of the homogenate was used. The resulting crude membrane pellet was resuspended in 50 mM Tris–HCl, pH 7.4 and immediately used for the cyclase assay.
3.1.4. Data analysis
Inhibitory binding constants, Ki, were calculated from the IC50 values according to the Cheng and Prusoff equation Ki = IC50/(1 + [C*]/KD*), where [C*] is the concentration of the radioligand and KD* its dissociation constant [49,50]. A weighted nonlinear least-squares curve fitting program LIGAND was also used for computer analysis of inhibition experiments. Potency values (IC50) obtained in cyclic AMP assays were calculated by nonlinear regression analysis using the equation for a sigmoid concentration–response curve (Graph Pad Prism, San Diego, CA, USA). All experimental data are expressed as geometric mean with 95% confidence limits in parentheses of three or four independent experiments performed in duplicate.
4. Conclusion
In summary, we have designed a class of benzothiazolylquinoline scaffolds for the hA3 target. Molecular simplification and molecular docking approach using the GLIDE tool from Schrödinger maestro has been employed for the design of these drugs. The effective binding modes of the scaffolds with the receptor binding sites were clearly explained. In addition, we have also performed radioligand binding assay of these scaffolds at hA1AR, hA2AAR, hA2BAR and hA3AR. We observed that compound 6a exhibited maximum potency and selectivity in hA3AR with respect to hA1AR, hA2AAR and hA2BAR which is rationalized with a docking study. Finally, it was concluded that these cytotoxic molecules are selectively targeting to the hA3AR.
Supplementary Material
Acknowledgements
The authors thank VIT University for providing us with laboratory facilities.
Ethics
The department was ethically approved by UGC.
Data accessibility
Molecular docking images of selected compounds can be found in the electronic supplementary material.
Authors' contributions
B.S. and S.M. synthesized and characterized the compounds. B.S. has performed the molecular docking study. B.S. and S.M. contributed equally in this paper. G.R.J. did the radioligand binding assay for this paper. B.S., S.M. and G.R.J. drafted the manuscript. All the authors analysed and discussed the results and revised the manuscript.
Competing interests
We have no competing interests.
Funding
The authors are grateful to VIT University for seed funding. We acknowledge DST, New Delhi, India for the DST-FIST project. We also acknowledge DST-SERB, India for a young scientist grant and the DST-SERB EMR project for providing us with funding.
References
- 1.Haskó G, Linden J, Cronstein B, Pacher P. 2008. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 7, 759–770. (doi:10.1038/nrd2638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baraldi PG, Preti D, Borea PA, Varani K. 2012. Medicinal chemistry of A3 adenosine receptor modulators: pharmacological activities and therapeutic implications. J. Med. Chem. 55, 5676–5703. (doi:10.1021/jm300087j) [DOI] [PubMed] [Google Scholar]
- 3.Moro S, Gao ZG, Jacobson KA, Spalluto G. 2006. Progress in the pursuit of therapeutic adenosine receptor antagonists. Med. Res. Rev. 26, 131–159. (doi:10.1002/med.20048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gessi S, Varani K, Merighi S, Cattabriga E, Iannotta V, Leung E, Baraldi PG, Borea PA. 2002. A3 adenosine receptors in human neutrophils and promyelocytic HL60 cells: a pharmacological and biochemical study. Mol. Pharmacol. 61, 415–424. (doi:10.1124/mol.61.2.415) [DOI] [PubMed] [Google Scholar]
- 5.Gessi S, et al. 2004. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin. Cancer Res. 10, 5895–5901. (doi:10.1158/1078-0432.CCR-1134-03) [DOI] [PubMed] [Google Scholar]
- 6.Fishmana P, Bar-Yehudaa S, Liangb BT, Jacobsonc KA. 2012. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 17, 359–366. (doi:10.1016/j.drudis.2011.10.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohana G, Bar-Yehuda S, Arich A, Madi L, Dreznick Z, Rath-Wolfson L, Silberman D, Slosman G, Fishman P. 2003. Inhibition of primary colon carcinoma growth and liver metastasis by the A3 adenosine receptor agonist CF101. Br. J. Cancer 89, 1552–1558. (doi:10.1038/sj.bjc.6601315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamiya H, Kanno T, Fujita Y, Gotoh A, Nakano T, Nishizaki T. 2012. Apoptosis-related gene transcription in human A549 lung cancer cells via A3 adenosine receptor. Cell Physiol. Biochem. 29, 687–696. (doi:10.1159/000312589) [DOI] [PubMed] [Google Scholar]
- 9.Fishman P, Bar-Yehuda S, Rath-Wolfson L, Barrer F, Rath-Ochaion A, Madi L. 2003. Targeting the A3 adenosine receptor for cancer therapy: inhibition of prostate carcinoma cell growth by A3AR agonist. Anticancer Res. 23, 2077–2083. [PubMed] [Google Scholar]
- 10.Madi L, Bar-Yehuda S, Barer F, Ardon E, Ochaion A, Fishman P. 2003. A3 adenosine receptor activation in melanoma cells. J. Biol. Chem. 278, 42 121–42 130. (doi:10.1074/jbc.M301243200) [DOI] [PubMed] [Google Scholar]
- 11.Gessi S, Varani K, Merighi S, Morelli A, Ferrari D, Leung E, Baraldi PG, Spalluto G, Borea PA. 2001. Pharmacological and biochemical characterization of A3 adenosine receptors in Jurkat T cells. Br. J. Pharmacol. 134, 116–126. (doi:10.1038/sj.bjp.0704254) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merighi S, Varani K, Gessi S, Cattabriga E, Iannotta V, Ulouglu C, Leung E, Borea PA. 2001. Pharmacological and biochemical characterization of adenosine receptors in the human malignant melanoma A375 cell line. Br. J. Pharmacol. 134, 1215–1226. (doi:10.1038/sj.bjp.0704352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suh BC, Kim TD, Lee JU, Seong JK, Kim KT. 2001. Pharmacological characterization of adenosine receptors in PGT-β mouse pineal gland tumour cells. Br. J. Pharmacol. 134, 132–142. (doi:10.1038/sj.bjp.0704218) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trincavelli ML, Tuscano D, Marroni M. 2002. A3 adenosine receptors in human astrocytoma cells: agonist-mediated desensitization, internalization, and down-regulation. Mol. Pharmacol. 62, 1373–1384. (doi:10.1124/mol.62.6.1373) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auchampach JA, Xiaowei J, Tina CW, George H, Caughey GH, Linden J. 1997. Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol. Pharmacol. 52, 846–860. (doi:10.1124/mol.52.5.846) [DOI] [PubMed] [Google Scholar]
- 16.Madi L, et al. 2004. The A3 adenosine receptor is highly expressed in tumor versus normal cells: potential target for tumor growth inhibition. Clin. Cancer Res. 10, 4472–4479. (doi:10.1158/1078-0432.CCR-03-0651) [DOI] [PubMed] [Google Scholar]
- 17.Fishman P, Madi L, Bar-Yehuda S, Barer F, Del Valle L, Khalili K. 2002. Evidence for involvement of Wnt signaling pathway in IB-MECA mediated suppression of melanoma cells. Oncogene 21, 4060–4064. (doi:10.1038/sj.onc.1205531) [DOI] [PubMed] [Google Scholar]
- 18.Fishman P, Bar-Yehuda S, Barer F, Madi L, Multani AF, Pathak S. 2001. The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp. Cell Res. 269, 230–236. (doi:10.1006/excr.2001.5327) [DOI] [PubMed] [Google Scholar]
- 19.Catarzi D, et al. 2015. 1,2,4-Triazolo[1,5-a]quinoxaline derivatives and their simplified analogues as adenosine A3 receptor antagonists. Synthesis, structure–affinity relationships and molecular modeling studies. Bioorg. Med. Chem. 23, 9–21. (doi:10.1016/j.bmc.2014.11.033) [DOI] [PubMed] [Google Scholar]
- 20.Tuccinardi T, et al. 2008. Substituted Pyrazolo[3,4-b]pyridines as potent A1 adenosine antagonists: synthesis, biological evaluation, and development of an A1 bovine receptor model. Chem. Med. Chem. 3, 898–913. (doi:10.1002/cmdc.200700355) [DOI] [PubMed] [Google Scholar]
- 21.Press NJ, et al. 2004. New highly potent and selective adenosine A3 receptor antagonists. Curr. Top. Med. Chem. 4, 863–870. (doi:10.2174/1568026043451023) [DOI] [PubMed] [Google Scholar]
- 22.Poli D, Catarzi D, Colotta V, Varano F, Filacchioni G, Daniele S, Moro S. 2011. The identification of the 2-phenylphthalazin-1(2H)-one scaffold as a new decorable core skeleton for the design of potent and selective human A3 adenosine receptor antagonists. J. Med. Chem. 54, 2102–2113. (doi:10.1021/jm101328n) [DOI] [PubMed] [Google Scholar]
- 23.Colotta V, Lenzi O, Catarzi D, Varano F, Filacchioni G, Martini C, Moro S. 2009. Pyrido[2,3- e]-1,2,4-triazolo[4,3-a]pyrazin-1-one as a new scaffold to develop potent and selective human A3 adenosine receptor antagonists. Synthesis, pharmacological evaluation, and ligand–receptor modeling studies. J. Med. Chem. 52, 2407–2419. (doi:10.1021/jm8014876) [DOI] [PubMed] [Google Scholar]
- 24.Vazquez-Rodriguez S, Matos MJ, Santana L, Uriarte E, Borges F, Kachler S, Klotz K-N. 2013. Chalcone-based derivatives as new scaffolds for h A3 adenosine receptor antagonists. J. Pharm. Pharmacol. 65, 697–703. (doi:10.1111/jphp.12028) [DOI] [PubMed] [Google Scholar]
- 25.Tafi A, et al. 2006. Pharmacophore based receptor modeling: the case of adenosine A3 receptor antagonists. An approach to the optimization of protein models. J. Med. Chem. 49, 4085–4097. (doi:10.1021/jm051112+) [DOI] [PubMed] [Google Scholar]
- 26.Settimo FD, et al. 2007. 5-Amino-2-phenyl[1,2,3]triazolo[1,2- a][1,2,4]benzotriazin-1-one: a versatile scaffold to obtain potent and selective A3 adenosine receptor antagonists. J. Med. Chem. 50, 5676–5684. (doi:10.1021/jm0708376) [DOI] [PubMed] [Google Scholar]
- 27.Morizzo E, et al. 2007. Scouting human A3 adenosine receptor antagonist binding mode using a molecular simplification approach: from triazoloquinoxaline to a pyrimidine skeleton as a key study. J. Med. Chem. 50, 6596–6606. (doi:10.1021/jm070852a) [DOI] [PubMed] [Google Scholar]
- 28.Kenneth AJ, Athena MK, Dilip KT, Andei AI, Delia P, Pier GB. 2009. Handbook of experimental pharmacology. Handbook Exp. Pharmacol. 193, 123–159. (doi:10.1007/978-3-540-89615-9_5) [Google Scholar]
- 29.Paira P, Chow MJ, Venkatesan G, Kosaraju VK, Cheong SL, Klotz K-N, Ang WH, Pastorin G. 2013. Organoruthenium antagonists of human A3 adenosine receptors. Chem. Eur. J. 19, 8321–8330. (doi:10.1002/chem.201203291) [DOI] [PubMed] [Google Scholar]
- 30.Bandyopadhyay P, Sathe M, Ponmariappan S, Sharma A, Sharma P, Srivastava AK, Kaushik MP. 2011. Exploration of in vitro time point quantitative evaluation of newly synthesized benzimidazole and benzothiazole derivatives as potential antibacterial agents. Bioorg. Med. Chem. Lett. 21, 7306–7309. (doi:10.1016/j.bmcl.2011.10.034) [DOI] [PubMed] [Google Scholar]
- 31.Solomon VR, Hua C, Lee H. 2009. Hybrid pharmacophore design and synthesis of isatin–benzothiazole analogs for their anti-breast cancer activity. Bioorg. Med. Chem. 17, 7585–7592. (doi:10.1016/j.bmc.2009.08.068) [DOI] [PubMed] [Google Scholar]
- 32.Singh MK, Tilak R, Nath G, Awasthi SK, Agarwal A. 2013. Design, synthesis and antimicrobial activity of novel benzothiazole analogs. Eur. J. Med. Chem. 63, 635–644. (doi:10.1016/j.ejmech.2013.02.027) [DOI] [PubMed] [Google Scholar]
- 33.Hutchinson I, Bradshaw TD, Matthews CS, Stevens MFG, Westwell AD. 2003. Antitumour benzothiazoles. Part 20: 3′-cyano and 3′-alkynyl-substituted 2-(4′-aminophenyl)benzothiazoles as new potent and selective analogues. Bioorg. Med. Chem. Lett. 13, 471–474. (doi:10.1016/S0960-894X(02)00930-7) [DOI] [PubMed] [Google Scholar]
- 34.Chhabra M, Sinha S, Banerjee S, Paira P. 2016. An efficient green synthesis of 2-arylbenzothiazole analogues as potent antibacterial and anticancer agents. Bioorganic Med. Chem. Lett. 26, 213–217. (doi:10.1016/j.bmcl.2015.10.087) [DOI] [PubMed] [Google Scholar]
- 35.Christina CC, Caitlin B, Joanna SO, Stephen D, Jerry Y. 2012. Oligovalent amyloid-binding agents reduce sevi-mediated enhancement of HIV-1 infection. J. Am. Chem. Soc. 134, 905–908. (doi:10.1021/ja210931b) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Rodriguez C, et al. 2009. Design, selection, and characterization of thioflavin-based intercalation compounds with metal chelating properties for application in Alzheimer's disease. J. Am. Chem. Soc. 131, 1436–1451. (doi:10.1021/ja806062g) [DOI] [PubMed] [Google Scholar]
- 37.Hickey JL, et al. 2013. Diagnostic imaging agents for Alzheimer's disease: copper radiopharmaceuticals that target Aβ plaques. J. Am. Chem. Soc. 135, 16 120–16 132. (doi:10.1021/ja4057807) [DOI] [PubMed] [Google Scholar]
- 38.Chhabra M, Babu LT, Mondal A, Sun H, Paira P. 2017. Amberlite IRA 402(OH) mediated green synthesis of novel benzothiazole-quinoline conjugates as cancer theranostics. Chemistry Select 2, 2480–2486. (doi:10.1002/slct.201700066) [Google Scholar]
- 39.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien TEY, Lane JR, Ijzerman AP, Stevens RC. 2008. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217. (doi:10.1126/science.1164772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venkatesan G, Paira P, Cheong SL, Vamsikrishna K, Federico S, Klotz K-N, Spalluto G, Pastorin G. 2014. Discovery of simplified N2-substituted pyrazolo[3,4-d]pyrimidine derivatives as novel adenosine receptor antagonists: efficient synthetic approaches, biological evaluations and molecular docking studies. Bioorg. Med. Chem. 22, 1751–1765. (doi:10.1016/j.bmc.2014.01.018) [DOI] [PubMed] [Google Scholar]
- 41.Lenzi O, et al. 2009. 2-Phenylpyrazolo[4,3-d]pyrimidin-7-one as a new scaffold to obtain potent and selective human A3 adenosine receptor antagonists: new insights into the receptor–antagonist recognition. J. Med. Chem. 52, 7640–7652. (doi:10.1021/jm900718w) [DOI] [PubMed] [Google Scholar]
- 42.Cheong SL, et al. 2010. The significance of 2-furyl ring substitution with a 2-(para-substituted) aryl group in a new series of pyrazolo-triazolo-pyrimidines as potent and highly selective hA3 adenosine receptors antagonists: new insights into structure–affinity relationship and receptor–antagonist recognition. J. Med. Chem. 53, 3361–3375. (doi:10.1021/jm100049f) [DOI] [PubMed] [Google Scholar]
- 43.Eswar N, Marti-Renom MA, Webb B, Madhusudhan MS, Eramian D, Shen M, Pieper U, Sali A. 2007. Comparative protein structure modeling using Modeller. Curr. Protoc. Protein Sci. Wiley 50, 2.9.1–2.9.31. (doi:10.1002/0471140864.ps0209s50) [DOI] [PubMed] [Google Scholar]
- 44.Marti-Renom MA, Stuart A, Fiser A, Sánchez R, Melo F, Sali A. 2000. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325. (doi:10.1146/annurev.biophys.29.1.291) [DOI] [PubMed] [Google Scholar]
- 45.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. (doi:10.1006/jmbi.1993.1626) [DOI] [PubMed] [Google Scholar]
- 46.Fiser A, Do RK, Sali A. 2000. Modeling of loops in protein structures. Protein Sci. 9, 1753–1773. (doi:10.1110/ps.9.9.1753) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. 1998. Comparative pharmacology of human adenosine receptor subtypes—characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 357, 1–9. (doi:10.1007/PL00005131) [DOI] [PubMed] [Google Scholar]
- 48.Klotz KN, Cristalli G, Grifantini M, Vittori S, Lohse MJ. 1985. Photoaffinity labeling of A1-adenosine receptors. J. Biol. Chem. 260, 14659–14664. [PubMed] [Google Scholar]
- 49.Cheng YC, Prusoff WH. 1973. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. (doi:10.1016/0006-2952(73)90196-2) [DOI] [PubMed] [Google Scholar]
- 50.De Lean A, Hancock AA, Lefkowitz RJ. 1982. Validation and statistical analysis of a computer modeling method for quantitative analysis of radioligand binding data for mixtures of pharmacological receptor subtypes. Mol. Pharmacol. 21, 5–16. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Molecular docking images of selected compounds can be found in the electronic supplementary material.