

Abstract
Triphenylphosphine oxide (TPPO) and oxalyl chloride ((COCl)2) are used as novel and high-efficiency coupling reagents for the esterification of alcohols with carboxylic acids via the TPPO/(COCl)2 system at room temperature for 1 h. The reaction represents the first TPPO-promoted esterification under mild and neutral conditions with excellent yields. Furthermore, we proposed a plausible mechanism with the help of 31P NMR spectroscopy.
Keywords: triphenylphosphine oxide, coupling reagent, oxalyl chloride, esterification
1. Introduction
As is well known, carboxylic esters are fundamental organic compounds in organic synthesis and have been widely used in chemical and pharmaceutical industries, such as spices, daily chemical industries, foods, medicines, rubbers, coating materials and so on [1]. Owing to the importance of esters, numerous chemical methods have been reported to accomplish this basic transformation [2,3]. Esters are primarily prepared from the condensation of carboxylic acids with alcohols; generally, the most common methods for the preparation of ester proceed via carboxyl group activation and subsequent reaction with a suitable alcohol [4]. Among them, acid halides were recognized as powerful esterifying agents because of their complete conversion and high yields; however, to the best of our knowledge, acid halides always generate highly acidic by-products such as hydrochloric acid, which could result in decomposition of the initial materials; this method has almost no application in the synthesis of a natural product because of greater possibility of reaction with some acid-sensitive functional groups [2–4]. Moreover, acid chlorides are prone to hydrolysis under basic conditions through the standard ketene intermediate (scheme 1) [4]. Therefore, it is crucial to find a mild coupling system for the further development of chemistry.
Scheme 1.

Racemization through the ketene intermediate is a common problem associated with the use of acyl chlorides as reagents in ester (and amide) coupling reactions.
Phosphonium, phosphinic salts and phosphine oxides as frequently used coupling reagents have been reported especially for some famous reactions such as the Wittig reaction [5–10], Appel reaction [11–14], Staudinger reaction [15–20] and Mitsunobu reaction [2,21–32]. The Mitsunobu reaction, first reported by Mitsunobu et al. in 1967 [21], converts an alcohol into a variety of other functional groups including esters, and this method could generate esters in excellent yield (90%) via the condensation of a carboxylic acid and alcohol with a mixture of triphenylphosphine (Ph3P) and diethyl azodicarboxylate (DEAD). More specifically, the Mitsunobu reaction is highly stereospecific and selective; therefore, it is appropriate for preparing some products or derivatives with sensitive groups.
In the Mitsunobu reaction, the alcohol was usually activated towards nucleophilic attack from the carboxylic acid, and this activation was achieved by the reaction with a phosphine, typically Ph3P, and a dialkyl azodicarboxylate. In recent years, a number of reports have focused on generating other azo dicarboxylates such as diisopropyl azodicarboxylate [2,22], di-2-methoxyethyl azodicarboxylate [23], azodicarbonyl dimorpholide [24], di-p-nitrobenzyl azodicarboxylate [25], 5,5′-dimethyl-3,3′-azoisoxazole [26], 4-dimethylaminopyridine [27,28], di-p-chlorobenzyl azodicarboxylate [29], DEAD [30], N-chlorobenzotriazole [31] and Fe(Pc) [32].
It is worth noting that the production of carboxylic esters using the Mitsunobu reaction will generate triphenylphosphene oxide (TPPO). The difficulty of removing it from the reaction mixture not only makes this method hard to operate, but also limits large-scale applicability of some reactions. Although many reports have been directed towards modifying Ph3P to polymer phosphorus compounds [33–37], the preparation of these polymers was not simple and the treatment of TPPO by-products gave rise to a waste of resources. To overcome these drawbacks, most studies focus on the reduction of TPPO to Ph3P [38–40]. Mecinovic and co-workers reported an amide reaction mediated by the PPh3/CCl4 system, and TPPO was reduced to PPh3 via this reaction [41]. Consequently, these phosphorus oxides have been used for several catalytic reactions, such as the catalytic Wittig reaction [5–10], catalytic Appel reaction [11–14] and catalytic Staudinger reaction [15–20]. It is easy to find the operating cost so high that it is not suitable for industrial production. Therefore, it is meaningful to recycle this waste product (TPPO) and reassemble a novel system to substitute the PPh3/assistant reagents.
By searching a large number of works of the relevant literature, it is worth mentioning that the reaction of oxalyl chloride ((COCl)2) with TPPO discovered by Fukui & Masaki [42] could form an intermediate (a kind of acyl phosphosphonium salt), and then the potential usefulness of this reaction system for catalytic transformation was exploited by Denton and co-workers in the Appel reaction and other reactions under the Appel conditions [13,14,43–45]. Through the mechanisms of these reactions, we have successfully synthesized amides by using the TPPO/(COCl)2 system [46], and hypothesized that this intermediate may be applied for esterification. To our surprise, we found that this acyl phosphonium salt could promote the activation of carboxylic acids first during the experiment and react with alcohols to generate corresponding esters (scheme 2). In this system, TPPO can act as a Lewis base to promote the reaction between acid and alcohol during condensation.
Scheme 2.

TPPO/(COCl)2 applied to esters by using recyclable TPPO.
Compared with traditional esterification via acid chloride, our system has the advantages of short reaction time (just 1 h), mild condition (room temperature), excellent yields and high atom efficiency (TPPO can be recycled). Although TPPO increased the complexity of purification, this problem could be ignored with reference to the listed advantages compared with the classical Mitsunobu reaction. This neutral method is applicable for natural product synthesis. Moreover, there is great potential in the chemical industry to resolve the waste due to the by-products of TPPO. Fortunately, there is no report describing the utilization of TPPO with (COCl)2 for the esterification system thus far.
2. Material and methods
2.1. Reagents
CH3CN (greater than or equal to 99.0%), CH2Cl2 (greater than or equal to 99.5%), C2H4Cl2 (greater than or equal to 99.0%), PhMe (greater than or equal to 99.5%), Et3N (greater than or equal to 99.0%), TPPO (99%), (COCl)2 (98%), PhCOOH (greater than 99.0%), NO2-PhCOOH (greater than 99.0%), CH3O-PhCOOH (greater than 99.0%), CH3-PhCOOH (greater than or equal to 98.0%), Cl-PhCOOH (99.0%), trans-cinnamic acid (greater than or equal to 98.0%), picolinic acid (99%), 1-naphthoic acid (greater than or equal to 99.0%), diphenylacetic acid (greater than or equal to 99.0%), PhCH2OH (greater than or equal to 99.0%), CH3(CH2)3OH (greater than or equal to 99.0%), CH3OH (99.9%), Ph(CH2)2OH (greater than or equal to 99.0%), 1-adamantanol (99%), CH3(CH2)11OH (greater than or equal to 99.5%) and Ph2CH2OH (99%).
2.2. Methods
2.2.1. Preparation of esters by using TPPO/(COCl)2
To a cold solution of TPPO (1.4 g, 5 mmol) in acetonitrile (CH3CN, 5 ml), (COCl)2 (0.55 ml, 6.5 mmol) was added slowly in drops under magnetic stirring. After 10 min, carboxylic acid (1 equiv, 5 mmol) was added and stirred for 10 min. Then, alcohol (1.3 equiv, 6.5 mmol) and Et3N (0.67 ml, 5 mmol) were added in sequence. The reaction was carried out under the protection of nitrogen gas, and the reaction temperature was room temperature. Stirring was continued for 1 h. The progress of reaction was followed by thin layer chromatography (TLC). After the reaction, mixture was evaporated in vacuo and the final product was purified by column chromatography with petroleum ether/ethyl acetate (8 : 1) as the eluent. All esters presented in table 3 are previously known and reported compounds.
Table 3.
Condensation of carboxylic acids with different alcohols in the presence of the TPPO/(COCl)2/Et3N system.
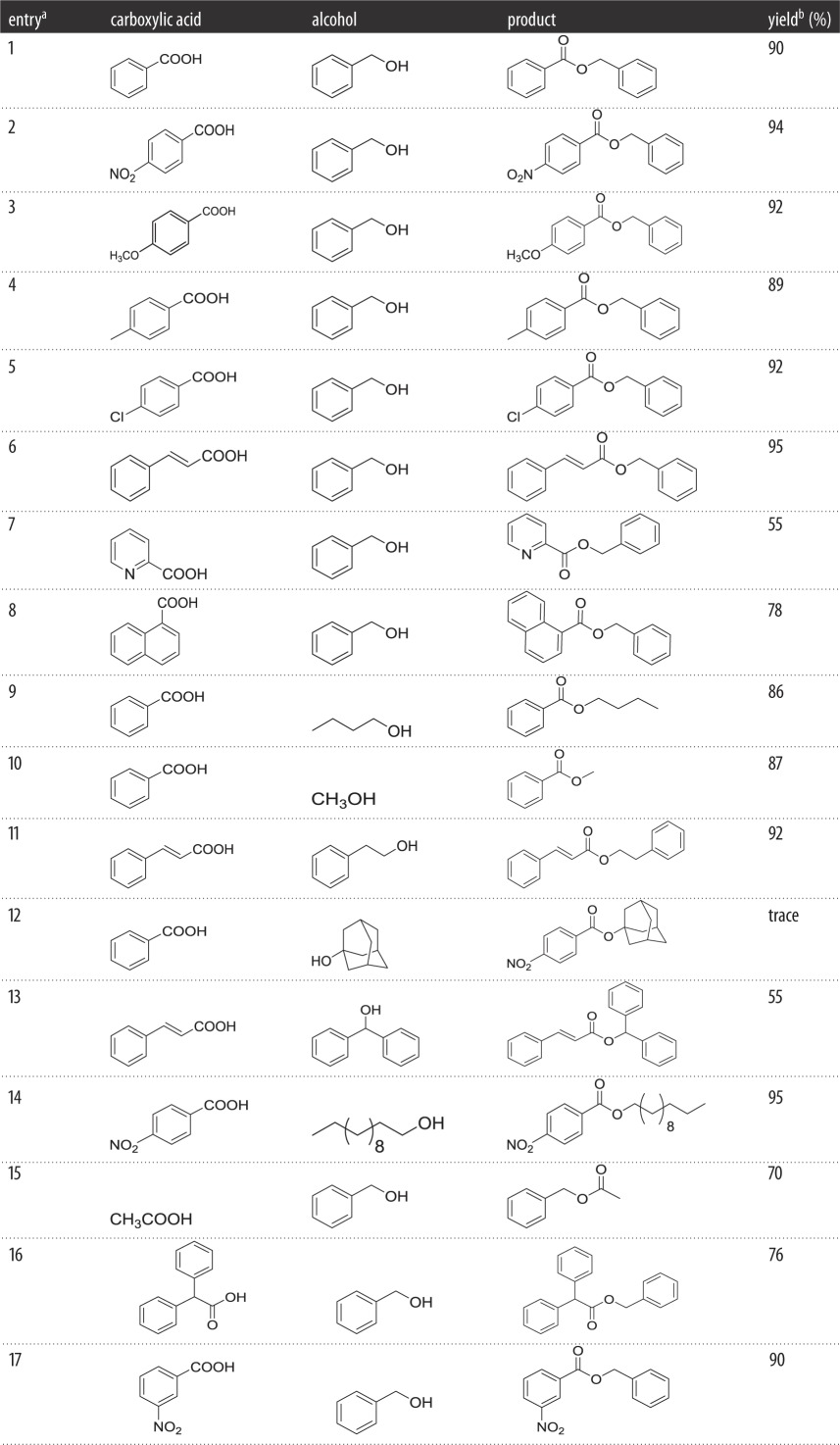 |
aReactions were carried out with RCOOH (5 mmol, 1 equiv), R1OH (6.5 mmol, 1.3 equiv), TPPO (5 mmol, 1 equiv), (COCl)2 (6.5 mmol, 1.3 equiv) and Et3N (5 mmol, 1 equiv) in CH3CN (5 mL) at room temperature for 1 h.
bIsolated yield.
2.2.2. Procedure for esterification (table 1)
Table 1.
Esterification of benzoic acid and benzyl alcohol by using different organic solvents under different temperatures and reaction times.
| entrya | organic solvent | temperature (°C) | time (h) | isolated yield (%) |
|---|---|---|---|---|
| 1 | CH3CN | 28 | 1 | 90 |
| 2 | CH3CN | 28 | 2.5 | 89 |
| 3 | CH3CN | 30 | 4 | 86 |
| 4 | PhMe | 30 | 1 | 86 |
| 5 | CH2Cl2 | 30 | 1 | 89 |
| 6 | C2H4Cl2 | 30 | 1 | 88 |
| 7 | CH3CN | 50 | 1 | 90 |
| 8 | CH3CN | 70 | 1 | 90 |
aReaction conditions: benzoic acid (5 mmol), benzyl alcohol (1.3 × 5 mmol), solvent (5 ml), Et3N (5 mmol) under nitrogen.
TPPO (1.4 g, 5 mmol), CH3CN (5 ml), (COCl)2 (0.55 ml, 6.5 mmol), PhCOOH (0.67 g, 5 mmol), PhCH2OH (0.67 ml, 6.5 mmol) and Et3N (0.67 ml, 5 mmol) were added as in the previous procedure. The reaction was carried out under different reaction temperatures and times. In the end, the yield was measured by TLC (table 1).
2.2.3. Procedure for recycling TPPO
At the end of the reaction, the mixture was evaporated in vacuo and corresponding esters with TPPO were separated by column chromatography. The residue was purified with an eluent of 8 : 1 petroleum ether/ethyl acetate and then the polarity of the eluent was changed to 2 : 1, and TPPO was obtained. The crude product was evaporated and dried in vacuo. The white solid obtained was the reusable TPPO.
3. Results and discussion
As mentioned above, in order to optimize the reaction conditions, we firstly chose benzoic acid (1 equiv) and benzyl alcohol (1.3 equiv) as reaction substrates, which were stirred in CH3CN for 1 h at room temperature under the protection of nitrogen gas and then was added TPPO (1 equiv)/(COCl)2 (1.3 equiv); the reaction mixture was neutralized by triethylamine (Et3N), and the desired ester, benzyl benzoate, was obtained in a 90% yield (table 1, entry 1). Initially, we tested the influence of reaction time from 1 to 4 h and different organic solvents including CH3CN, PhMe, CH2Cl2 and C2H4Cl2 at different temperatures to find the most suitable conditions for the reaction; the best results are summarized in table 1.
According to the results obtained in different organic solvents (table 1, entries 4–6), the esterification yields had no obvious difference. As is well known, TPPO has a better solubility in CH2Cl2 or C2H4Cl2 than CH3CN and PhMe, and during the experimental phenomena, we found that the solid in CH3CN was dissolved rapidly after the addition of (COCl)2, and there were no obvious changes in the phenomena in other solvents; hence we chose CH3CN as the reaction solvent.
Moreover, by monitoring the reaction using TLC, we found that the reactant was consumed within 1 h and that extended reaction time cannot improve product yields appreciably. By contrast, as the reaction progressed, some generated esters were decomposed because of the reversibility of the reaction (table 1, entries 1–3). Generally, temperature is an important factor for various reactions. However, it had no obvious influence in this system as seen in table 1, entries 6–8.
We also examined the effect of different ratios of TPPO/(COCl)2/PhCOOH/PhCH2OH/Et3N in CH3CN at room temperature for the conversion to benzyl benzoate (scheme 3); the results are summarized in table 2.
Scheme 3.

Reaction giving benzyl benzoate in the presence of TPPO with (COCl)2.
Table 2.
Esterification with different ratios of TPPO/(COCl)2/PhCOOH/PhCH2OH/Et3N in CH3CN at room temperature.
| entrya | molar ratio TPPO/(COCl)2/R1COOH/ROH/Et3N | isolated yield / % |
|---|---|---|
| 1 | 1/1/1/1.3/1 | 79 |
| 2 | 1/1.3/1/1.3/1 | 90 |
| 3 | 1/2/1/1.3/1 | 88 |
| 4 | 1/0.75/1/1.3/1 | 70 |
| 5 | 0.75/1.3/1/1.3/1 | 50 |
| 6 | 2/1.3/1/1.3/1 | 89 |
| 7 | 1/1.3/1/1/1 | 68 |
| 8 | 1/1.3/1/2/1 | 90 |
| 9 | 1/1.3/1/1.3/1.3 | 90 |
| 10 | 1/1.3/1/1.3/2 | 90 |
| 11 | 1/1.3/1/1.3/0.75 | 85 |
| 12 | 1/0/1/1.3/1 | nr |
| 13 | 0/1.3/1/1.3/1 | nr |
aReaction conditions: solvent (5 ml), room temperature, 1 h, under nitrogen.
As the esterification reaction of benzoic acid to benzyl benzoate gave a 90% yield (table 3, entry 1), we first applied our reaction conditions to the carboxylic acids carrying both electron-donating groups (--OMe and --Me) (table 3, entries 3 and 4) and electron-withdrawing groups (--NO2 and --Cl) (table 3, entries 2, 5 and 17), and these carboxylic acids gave their corresponding esters in good yields. However, there was a little difference between electron-donating groups and electrophilic groups. The position of substituent groups could affect the esterification yields (table 3, entries 2 and 17); it was easy to find that m-nitrobenzoic acid has lower conversion (90%) compared to p-nitrobenzoic acid (94%). By now, we clearly realized that the double bond had a crucial role in the esterification (table 3, entry 6); it improved the yield (95%). Of course, steric effects via acids were examined. It was evident that the steric hindrance of acids is an influencing factor for the esterification yield (table 3, entries 7, 8 and 16), which reduces the conversion appreciably.
In comparison, aliphatic alcohols had lower reactivity than aromatic alcohols (table 3, entries 1 and 9, 1 and 10). However, as is to be expected, this impact could be ignored when employing electron-deficient carboxylic acids (table 3, entries 6 and 11, entries 2 and 14). Moreover, the aromatic acid was more reactive than the aliphatic acid (table 3, entries 1 and 15) just as we expected. Tertiary alcohols because of their steric hindrance were tested to react with benzoic acid; as we expected, there was little corresponding ester generated (table 3, entry 12); besides, secondary alcohols gave lower yields for the same reason (table 3, entries 6 and 13).
To explore the effect between substrates and corresponding yields accurately, we have provided a summary about the times of reaction for different substrates in table 4. The progress of the reaction was followed by TLC, and the final product was purified by column chromatography.
Table 4.
Optimization of triphenylphosphine oxide promoted esterification.
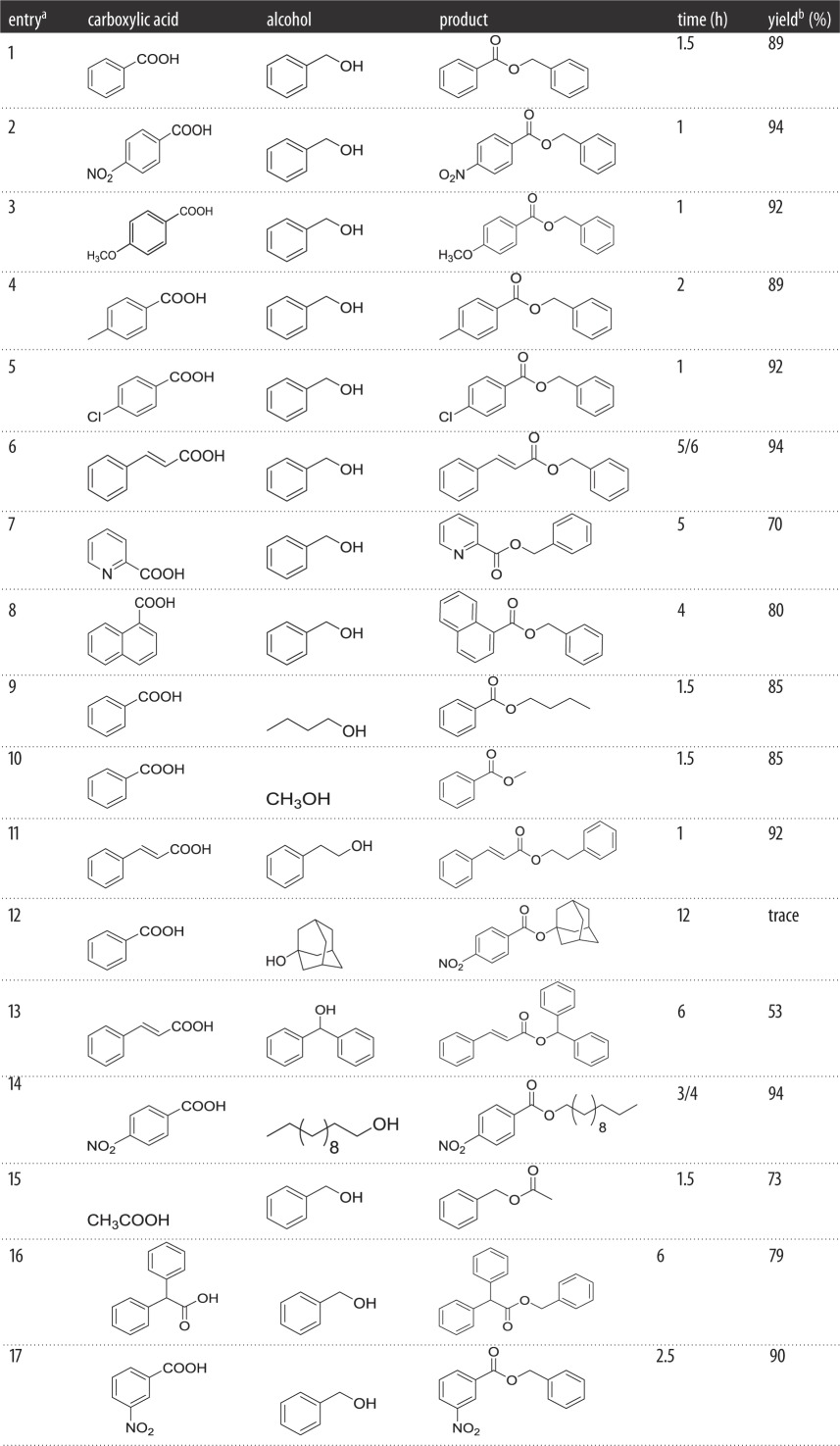 |
aReactions were carried out with RCOOH (5 mmol, 1 equiv), R1OH (6.5 mmol, 1.3 equiv), TPPO (5 mmol, 1 equiv), (COCl)2 (6.5 mmol, 1.3 equiv) and Et3N (5 mmol) in CH3CN (5 ml) at room temperature.
bIsolated yield.
As shown in table 4, some cases indicated that the reactants were consumed at 1 h, and most substrates proved that increased reaction time will not change yields greatly, with the exception of table 4, entry 7 by TLC. In conclusion, esterification was difficult to be carried out for substrates with bulky chemical constitution.
3.1. Benzyl benzoate (table 3, entry 1)
1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 7.8 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.53–7.36 (m, 7H), 5.41 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 165.57, 135.86, 131.14, 128.77, 128.69, 128.64, 128.42, 128.29, 66.98; HRMS (FT ICR-MS) m/z: [M + Na]+ calcd for C14H12O2 235.07295; found 235.07294.
3.2. Benzyl 4-nitrobenzoate (table 3, entry 2)
1H NMR (500 MHz, CDCl3) δ 8.28 (dd, J = 20.8, 8.9 Hz, 4H), 7.56–7.35 (m, 5H), 5.46 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 164.54, 150.62, 135.53, 135.29, 130.84, 128.77, 128.67, 128.45, 123.56, 67.66; MS m/z: [M] calcd for C14H11O4N 257.24; found 257.
3.3. Benzyl 4-methoxybenzoate (table 3, entry 3)
1H NMR (500 MHz, CDCl3) δ 8.09 (t, J = 13.7 Hz, 2H), 7.42 (ddd, J = 25.3, 20.3, 7.2 Hz, 5H), 6.96 (t, J = 14.7 Hz, 2H), 5.38 (s, 2H), 3.88 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.20, 163.48, 136.36, 131.77, 128.59, 128.17, 128.12, 122.59, 113.66, 66.41, 55.43; MS m/z: [M + Na]+ calcd for C15H14O3 242.27; found 242.
3.4. Benzyl 4-methylbenzoate (table 3, entry 4)
1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 8.0 Hz, 2H), 7.48–7.30 (m, 5H), 7.26 (d, J = 7.9 Hz, 2H), 5.39 (s, 2H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.52, 143.73, 136.33, 129.81, 129.15, 128.63, 128.22, 128.16, 127.54, 66.53, 21.66; HRMS (FT ICR-MS) m/z: [M + Na]+ calcd for C15H14O2 249.08860; found 249.088703.
3.5. Benzyl 4-chlorobenzoate (table 3, entry 5)
1H NMR (500 MHz, CDCl3) δ 8.05 (t, J = 11.4 Hz, 2H), 7.47 (d, J = 6.4 Hz, 2H), 7.45–7.37 (m, 5H), 5.41 (d, J = 20.2 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 165.60, 139.53, 135.81, 131.13, 128.76, 128.67, 128.60, 128.41, 128.28, 66.97; MS m/z: [M + Na]+ calcd for C14H11O2Cl 246.69; found 246.
3.6. Benzyl cinnamate (table 3, entry 6)
1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 16.0 Hz, 1H), 7.56 (dd, J = 6.1, 3.0 Hz, 2H), 7.49–7.34 (m, 8H), 6.54 (d, J = 16.0 Hz, 1H), 5.30 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 166.80, 145.20, 136.13, 134.41, 130.37, 128.93, 128.63, 128.31, 128.14, 117.94, 66.38; MS m/z: [M + Mn]+ calcd for C16H14O2 293.28; found 293.2.
3.7. Benzyl 1-H-pyrrole-2-carboxylate (table 3, entry 7)
1H NMR (500 MHz, CDCl3) δ 8.78 (d, J = 4.3 Hz, 1H), 8.16 (d, J = 7.8 Hz, 1H), 7.86 (td, J = 7.7, 1.4 Hz, 1H), 7.43–7.28 (m, 6H), 5.48 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 164.89, 149.83, 147.85, 137.21, 135.60, 128.63, 128.54, 127.03, 125.34, 67.60; MS m/z: [M + H]+ calcd for C13H11O2N 214.23; found 214.2.
3.8. Benzyl 1-naphthoate (table 3, entry 8)
1H NMR (500 MHz, CDCl3) δ 9.3–9.20 (m, 1H), 8.47–8.33 (m, 1H), 8.07 (d, J = 8.2 Hz, 1H), 8.01–7.92 (m, 1H), 7.90–7.27 (m, 9H), 5.62 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 167.34, 136.40, 134.05, 133.72, 131.71, 130.63, 128.85, 128.78, 128.45, 128.04, 127.05, 126.42, 126.07, 124.67, 66.92; HRMS (FT ICR-MS) m/z: [M + H]+ calcd for C18H14O2 263.10666; found 263.10599.
3.9. Butyl benzoate (table 3, entry 9)
1H NMR (500 MHz, CDCl3) δ 8.07 (d, J = 7.5 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 4.34 (t, J = 6.6 Hz, 2H), 1.80–1.73 (m, 2H), 1.54–1.46 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.64, 132.76, 130.56, 129.53, 128.30, 64.79, 30.79, 19.28, 13.74; MS m/z: [M] calcd for C11H14O2 178.23; found 178.
3.10. Ethyl benzoate (table 3, entry 10)
1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 7.4 Hz, 2H), 7.55 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.7 Hz, 2H), 3.91 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.07, 132.89, 131.40, 130.18, 129.56, 128.35, 52.03; MS m/z: [M + Na]+ calcd for C8H8O2 136.15; found 136.
3.11. Phenethyl cinnamate (table 3, entry 11)
1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 16.0 Hz, 1H), 7.63–7.49 (m, 2H), 7.46–7.34 (m, 5H), 7.33–7.27 (m, 3H), 6.47 (d, J = 16.0 Hz, 1H), 4.47 (t, J = 7.1 Hz, 2H), 3.13–3.02 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 166.92, 144.89, 137.93, 134.44, 130.33, 128.98, 128.92, 128.57, 128.13, 126.62, 118.10, 65.05, 35.25; MS m/z: [M + Na]+ calcd for C17H16O2 275.31; found 275.3.
3.12. Benzhydryl cinnamate (table 3, entry 13)
1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 16.0 Hz, 1H), 7.61–7.55 (m, 2H), 7.42–7.33 (m, 11H), 7.32–7.27 (m, 2H), 7.06 (s, 1H), 6.61 (d, J = 16.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 166.00, 145.46, 142.23, 140.29, 134.37, 130.43, 128.93, 128.55, 128.40, 128.18, 127.95, 127.44, 127.28, 127.21, 118.04, 80.01; MS m/z: [M + Na]+ calcd for C22H18O2 337.38; found 337.3.
3.13. Dodecyl benzoate (table 3, entry 14)
1H NMR (500 MHz, CDCl3) δ 8.31 (d, J = 8.8 Hz, 2H), 8.23 (d, J = 8.8 Hz, 2H), 4.76–4.21 (m, 2H), 1.81 (m, 2H), 1.46 (m, 2H), 1.40–1.25 (m, 16H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 164.76, 150.48, 135.90, 130.66, 123.52, 66.14, 31.92, 29.64, 29.57, 29.51, 29.35, 29.25, 28.61, 25.99, 22.69, 14.11; MS m/z: [M] calcd for C19H29O4N 335.44; found 335.
3.14. Phenylmethyl acetate (table 3, entry 15)
1H NMR (500 MHz, CDCl3) δ 7.43–7.38 (m, 4H), 7.38 (s, 1H), 5.15 (s, 2H), 2.11 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.84, 136.02, 128.60, 128.29, 66.30, 20.99; MS m/z: [M] calcd for C9H10O2 150.17; found 150.
3.15. Benzyl 2,2-diphenylacetate (table 3, entry 16)
1H NMR (500 MHz, CDCl3) δ 7.37–7.27 (m, 15H), 5.22 (s, 2H), 5.11 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 172.32, 138.57, 128.64, 128.59, 128.51, 128.22, 128.17, 127.28, 66.91, 57.06; MS m/z: [M + Mn]+ calcd for C21H18O2 357.37; found 357.3.
3.16. Benzyl 3-nitrobenzoate (table 3, entry 17)
1H NMR (500 MHz, CDCl3) δ 8.91 (s, 1H), 8.50–8.37 (m, 2H), 7.68 (t, J = 8.0 Hz, 1H), 7.44 (ddd, J = 18.2, 17.5, 6.4 Hz, 5H), 5.44 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 164.35, 148.33, 135.40, 135.28, 131.95, 129.66, 128.77, 128.67, 128.51, 127.50, 124.70, 67.65; MS m/z: [M] calcd for C14H11O4N 257.24; found 257.
Based on previous reports [32,47–50], the intermediates of the reaction of TPPO and (COCl)2 had been identified as chlorotriphenylphosphonium salt (scheme 4, intermediate 1), which could also be generated in the reaction of phosphine (from the reduction of TPPO) with CCl4; we offer a plausible esterification mechanism with the help of 31P NMR spectroscopy (figure 1) in scheme 4 for the sake of finding out the role of TPPO with (COCl)2 to promote esterification.
Scheme 4.

Proposed mechanism of ester synthesis mediated by TPPO and (COCl)2.
Figure 1.

31P NMR spectra for the synthesis of RCOOR1 (benzyl benzoate). I: TPPO (1 equiv), (COCl)2 (1.3 equiv), CH3CN (5 ml). II: after addition of RCOOH (benzoic acid, 1 equiv). III: R1OH (benzyl alcohol, 1.3 equiv) with Et3N (1 equiv) were added to solution II.
Firstly, the solution after adding TPPO (δ = 29.26 ppm) [46] with (COCl)2 showed a strong singlet at 65.11 ppm (figure 1, I) (δ = 65.5 ppm [51]), indicating the formation of intermediate 1. Secondly, after adding benzoic acid, a new singlet was formed at δ = 44.43 ppm (figure 1, II) [46], which we hypothesized was due to the formation of an acyl phosphonium salt 2. To exclude the effect of a base, we tested the mixture of TPPO, CH3CN with benzoic acid and did not find the analogous singlet; furthermore, we could only see a singlet at 29 ppm. Finally, the salt 2 reacts with alcohol (R1OH) to produce corresponding esters and results in a shift of resonance (δ = 42.29 ppm) (figure 1, III). To exclude the effect of solvent, we chose equivalent CH3CN to replace alcohol; what surprised us was that there was hardly any change in the singlet. Through the post-processing, a sharp singlet of TPPO (δ = 29.26 ppm) appeared again. Therefore this particular mechanism needs further study.
4. Conclusion
In conclusion, we developed a new and efficient method for the synthesis of esters with excellent yields by the TPPO/(COCl)2 system. In comparison with the previous methods for the esterification between carboxylic acids and alcohols, this system offered several advantages. Firstly, this system reduced the side reactions that occurred during the classical Mitsunobu reaction, improved the atom efficiency and reduced the reaction cost, because the raw material TPPO is an industrial by-product of the production of various chemicals and has the characteristic of being widely available, and at the end of this reaction, TPPO could be recycled and only CO, CO2 and HCl are wasted. Secondly, the corresponding esters could be generated with excellent yields in mild and neutral conditions, and can be applied to some substrates bearing sensitive groups in contrast to esterification via the formation of acid chloride. Finally, this system has simple experimental operation at the end of the reaction and the reaction liquid was purified by column chromatography directly. Moreover, we also proposed a plausible mechanism according to 31P NMR spectroscopy. In our laboratory, we will conduct further investigation of the modified reaction system to extend the application of TPPO.
Supplementary Material
Acknowledgements
The authors are grateful to the Shanghai Institute of Technology for providing the laboratory.
Data accessibility
All data used in this research are included in figures, tables and the electronic supplementary material.
Authors' contributions
M.J. performed all the experiments, and the characterization and analysis of the data. M.J., L.J., F.N. and X.S. were involved in the conception and design of the experiments. Y.Z. performed parts of the experiments. All the authors were involved the drafting, revision and approval of the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the School of Chemical and Environment Engineering, Shanghai Institute of Technology. The work was supported by the Shanghai Alliance Program (no. LM 201666), the Shanghai Students’ Science and Technology Innovation Activities Key Projects (SH 2016001) and the Capacity-building Projects in Shanghai Local University (no. 15120503700).
References
- 1.Otera J. 2003. Esterification: methods, reactions and applications. Weinheim, Germany: Wiley-VCH. [Google Scholar]
- 2.Tsakos M, Schaffert ES, Clement LL, Villadsen NL, Poulsen TB. 2015. Ester coupling reactions---an enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 32, 605–632. (doi:10.1039/c4np00106k) [DOI] [PubMed] [Google Scholar]
- 3.Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. 2009. Mitsunobu and related reactions: advances and applications. Chem. Rev. 109, 2551–2651. (doi:10.1021/cr800278z) [DOI] [PubMed] [Google Scholar]
- 4.Siengalewicz P, Mulzer J, Rinner U. 2014. Synthesis of esters and lactones. Compr. Org. Synth. II. 6, 355–410. (doi:10.1016/B978-0-08-097742-3.00612-1) [Google Scholar]
- 5.O’ Brien CJ, Tellez JL, Nixon ZS, Kang LJ, Carter AL, Kunkel SR, Przeworski KC, Chass GA. 2009. Recycling the waste: the development of a catalytic Wittig reaction. Angew. Chem. Int. Ed. 48, 6836–6839. (doi:10.1002/anie.200902525) [DOI] [PubMed] [Google Scholar]
- 6.O'Brien CJ, Lavigne F, Covle EE, Holohan AJ, Doonan BJ. 2013. Breaking the ring through a room temperature catalytic Wittig reaction. Chem. Eur. J. 19, 5854–5858. (doi:10.1002/chem.201300546) [DOI] [PubMed] [Google Scholar]
- 7.O'Brien CJ, et al. 2013. Part I: the development of the catalytic Wittig reaction. Chem. Eur. J. 19, 15 281–15 298. (doi:10.1002/chem.201301444) [Google Scholar]
- 8.Covle EE, Doonan BJ, Holohan AJ, Walsh KA, Lavigne F, Krenske EH, O'Brien CJ. 2014. Catalytic Wittig reactions of semi- and nonstabilized ylides enabled by ylide tuning. Angew. Chem. 126, 13 121–13 125. (doi:10.1002/ange.201406103) [DOI] [PubMed] [Google Scholar]
- 9.Schirmer ML, Adomeit S, Werner T.. 2015. First base-free catalytic Wittig reaction. Org. Lett. 17, 3078–3081. (doi:10.1021/acs.orglett.5b01352) [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann M, Deshmukh S, Werner T. 2015. Scope and limitation of the microwave-assisted catalytic Wittig reaction. Eur. J. Org. Chem. 46, 4532–4543. (doi:10.1002/ejoc.201500310) [Google Scholar]
- 11.Ramage R. 1979. Organophosphorus reagents in the synthesis of peptides. In Organophosphorus reagents in organic synthesis (ed. JIG Cadogan), pp. 387–424. New York, NY: Academic; Press. [Google Scholar]
- 12.van Kalkeren HA, Bruins J, Rutjes FPJT, van Delft FL. 2012. Organophosphorus-catalysed Staudinger reduction. Adv. Synth. Catal. 354, 1417–1421. (doi:10.1002/adsc.201100967) [Google Scholar]
- 13.Denton RM, Tang X, Przeslak A. 2010. Catalysis of phosphorus(V)-mediated transformations: dichlorination reactions of epoxides under Appel conditions. Org. Lett. 12, 4678–4681. (doi:10.1021/ol102010h) [DOI] [PubMed] [Google Scholar]
- 14.Denton RM, An J, Adeniran B. 2010. Phosphine oxide-catalysed chlorination reactions of alcohols under Appel conditions. Chem. Commun. 46, 3025–3027. (doi:10.1039/c002825h) [DOI] [PubMed] [Google Scholar]
- 15.van Kalkeren HA, Leenders SHAM, Hommersom CRA, Rutjes FPJT, van Delft FL. 2011. In situ phosphine oxide reduction: a catalytic Appel reaction. Chem. Eur. J. 17, 11 290–11 295. (doi:10.1002/chem.201101563) [DOI] [PubMed] [Google Scholar]
- 16.van Kalkeren HA, Bruins JJ, Rutjes FPJT, van Delft FL. 2012. ChemInform abstract: organophosphorus-catalyzed Staudinger reduction. Adv. Synth. Catal. 354, 1417–1421. (doi:10.1002/chin.201240074) [Google Scholar]
- 17.Kosal AD, Wilson EE, Ashfeld BL. 2012. Phosphine-based redox catalysis in the direct traceless Staudinger ligation of carboxylic acids and azides. Angew. Chem. Int. Ed. 51, 12 036–12 040. (doi:10.1002/anie.201206533) [DOI] [PubMed] [Google Scholar]
- 18.van Kalkeren HA, Grotenhuis CT, Haasjes FS, Hommersom CA, Rutjes FPJT, van Delft FL. 2013. Catalytic Staudinger/Aza-Wittig sequence by in situ phosphane oxide reduction. Eur. J. Org. Chem. 45, 7059–7066. (doi:10.1002/ejoc.201300585) [Google Scholar]
- 19.Wang L, Wang Y, Chen M, Ding MW. 2014. ChemInform abstract: reversible P(III)/P(V) redox: catalytic Aza-Wittig reaction for the synthesis of 4(3H)-quinazolinones and the natural product vasicinone. Adv. Synth. Catal. 356, 1098–1104. (doi:10.1002/adsc.201300950) [Google Scholar]
- 20.Wang L, Xie YB, Huang NY, Yan JY, Hu WM, Liu MG, Ding MW. 2016. Catalytic Aza-Wittig reaction of acid anhydride for the synthesis of 4H-benzo[d] [1,3] oxazin-4-ones and 4-benzylidene-2-aryloxazol-5(4H)-ones. ACS Catal. 6, 4010–4016. (doi:10.1021/acscatal.6b00165) [Google Scholar]
- 21.Mitsunobu O, Yamada M, Mukaiyama T. 1967. Preparation of esters of phosphoric acid by the reaction of trivalent phosphorus compounds with diethyl azodicarboxylate in the presence of alcohols. Bull. Chem. Soc. Jpn. 40, 935–939. (doi:10.1246/bcsj.40.935) [Google Scholar]
- 22.Fitzjarrald VP, Pongdee R. 2007. A convenient procedure for the esterification of benzoic acids with phenols: a new application for the Mitsunobu reaction. Tetrahedron Lett. 48, 3553–3557. (doi:10.1016/j.tetlet.2007.03.095) [Google Scholar]
- 23.Hagiya K, Muramoto N, Misaki T, Sugimura T. 2009. DMEAD: a new dialkyl azodicarboxylate for the Mitsunobu reaction. Tetrahedron 65, 6109–6114. (doi:10.1016/j.tet.2009.05.048) [Google Scholar]
- 24.Lanning ME, Fletcher S. 2013. Azodicarbonyl dimorpholide (ADDM): an effective, versatile, and water-soluble Mitsunobu reagent. Tetrahedron Lett. 54, 4624–4628. (doi:10.1016/j.tetlet.2013.06.049) [Google Scholar]
- 25.Yang J, Dai L, Wang X, Chen Y. 2011. Di-p- nitrobenzyl azodicarboxylate (DNAD): an alternative azo-reagent for the Mitsunobu reaction. Tetrahedron 67, 1456–1462. (doi:10.1016/j.tet.2010.12.036) [Google Scholar]
- 26.Iranpoor N, Firouzabadi H, Khalili D. 2010. 5,5′- Dimethyl-3,3′-azoisoxazole as a new heterogeneous azo reagent for esterification of phenols and selective esterification of benzylic alcohols under Mitsunobu condition. Org. Biomol. Chem. 8, 4436–4443. (doi:10.1039/c004357e) [DOI] [PubMed] [Google Scholar]
- 27.Tian J, Gao W, Zhou D, Zhang C. 2012. Recyclable hypervalent iodine(III) reagent iodosodilactone as an efficient coupling reagent for direct esterification, amidation, and peptide coupling. Org. Lett. 14, 3020–3023. (doi:10.1021/ol301085v) [DOI] [PubMed] [Google Scholar]
- 28.Carle MS, Shimokura GK, Murphy GK. 2016. Iodobenzene dichloride in the esterification and amidation of carboxylic acids: in-situ synthesis of Ph3PCl2. Eur. J. Org. Chem. 3, 3930–3933. (doi:10.1002/ejoc.201600714) [Google Scholar]
- 29.Lipshutz BH, Chung DW, Rich B, Corral R. 2006. Simplification of the Mitsunobu reaction. Di-p-chlorobenzyl azodicarboxylate: a new azodicarboxylate. Org. Lett. 8, 5069–5072. (doi:10.1021/ol0618757) [DOI] [PubMed] [Google Scholar]
- 30.Iranpoor N, Firouzabadi H, Khalili D, Motevalli S. 2008. Easily prepared azopyridines as potent and recyclable reagents for facile esterification reactions. An efficient modified Mitsunobu reaction. J. Org. Chem. 73, 4882–4887. (doi:10.1021/jo8000782) [DOI] [PubMed] [Google Scholar]
- 31.Rouhisaadabad H, Akhlaghinia B. 2014. Direct, rapid and convenient synthesis of esters and thioesters using PPh3/N-chlorobenzotriazole system. J. Braz. Chem. Soc. 25, 253–263. (doi:10.5935/0103-5053.20130291) [Google Scholar]
- 32.Taniguchi T, Hirose D, Ishibashi H. 2011. Esterification via iron-catalyzed activation of triphenylphosphine with air. ACS Catal. 1, 1469–1474. (doi:10.1021/cs2003824) [Google Scholar]
- 33.Pathak G, Rokhum L. 2015. Selective monoesterification of symmetrical diols using resin-bound triphenylphosphine. ACS Comb. Sci. 17, 483–487. (doi:10.1021/acscombsci.5b00086) [DOI] [PubMed] [Google Scholar]
- 34.Roller S, Zhou H, Haag R. 2005. High-loading polyglycerol supported reagents for Mitsunobu- and acylation-reactions and other useful polyglycerol derivatives. Mol. Divers. 9, 305–316. (doi:10.1007/s11030-005-8117-y) [DOI] [PubMed] [Google Scholar]
- 35.Lizarzaburu ME, Shuttleworth SJ. 2002. Synthesis of aryl ethers from aminoalcohols using polymer-supported triphenylphosphine. Tetrahedron Lett. 43, 2157–2159. (doi:10.1016/S0040-4039(02)00222-8) [Google Scholar]
- 36.Blumel J. 2008. Linkers and catalysts immobilized on oxide supports: new insights by solid-state NMR spectroscopy. Coord. Chem. Rev. 252, 2410–2423. (doi:10.1016/j.ccr.2008.06.013) [Google Scholar]
- 37.Friesen CM, Montgomery CD, Temple SAJU. 2012. The first fluorous biphase hydrogenation catalyst incorporating a perfluoropolyalkylether: [RhCl(PPh2(C6H4C(O)OCH2CF(CF3) (OCF2CF(CF3))nF))3] with n = 4–9. J. Fluorine Chem. 144, 24–32. (doi:10.1016/j.jfluchem.2012.09.001) [Google Scholar]
- 38.Hérault D, Nguyen DH, Nuel D, Buono G. 2015. Reduction of secondary and tertiary phosphine oxides to phosphines. Chem. Soc. Rev. 44, 2508–2528. (doi:10.1039/c4cs00311j) [DOI] [PubMed] [Google Scholar]
- 39.van Kalkeren HA, van Delft FL, Rutjes FPJT. 2013. Catalytic Appel reactions. Pure Appl. Chem. 85, 817–828. (doi:10.1351/PAC-CON-12-06-13) [Google Scholar]
- 40.Schirmer ML, Jopp S, Holz J, Spannenberg A, Werner T. 2016. Organocatalyzed reduction of tertiary phosphine oxides. Adv. Synth. Catal. 358, 26–29. (doi:10.1002/adsc.201500762) [Google Scholar]
- 41.Lenstra DC, Rutjes FPJT, Mecinovic J. 2014. Triphenylphosphine-catalysed amide bond formation between carboxylic acids and amines. Chem. Commun. 50, 5763–5766. (doi:10.1039/C4CC01861C) [DOI] [PubMed] [Google Scholar]
- 42.Masaki M, Fukui K. 1977. Reaction of tertiary phosphine dichlorides with thiols in the presence of triethylamine. A convenient method for the reduction of phosphine oxides to phosphines. Chem. Lett. 6, 151 (doi:10.1246/cl.1977.151) [Google Scholar]
- 43.Tang X, An J, Denton RM. 2014. A procedure for Appel halogenations and dehydrations using a polystyrene supported phosphine oxide. Tetrahedron Lett. 55, 799–802. (doi:org/10.1016/j.tetlet.2013.11.098) [Google Scholar]
- 44.Denton RM, An J, Lindovska P, Lewis W. 2012. Phosphonium salt-catalysed synthesis of nitriles from in situ activated oximes. Tetrahedron 68, 2899–2905. (doi:org/10.1016/j.tet.2012.01.067) [Google Scholar]
- 45.Denton RM, An J, Adeniran B, Blake AJ, Lewis W, Poulton AM. 2011. Catalytic phosphorus(V)-mediated nucleophilic substitution reactions: development of a catalytic Appel reaction. J. Org. Chem. 76, 6749–6767. (doi:10.1021/jo201085r) [DOI] [PubMed] [Google Scholar]
- 46.Jiang LX, Niu FF, Zhang DRD, Sun XL. 2017. A high-efficient method for the amidation of carboxylic acids promoted by triphenylphosphine oxide and oxalyl chloride. Heteroatom Chem. 28, e21364 (doi:10.1002/hc.21364) [Google Scholar]
- 47.Byrne PA, Rajendran KV, Muldoon J, Gilheany DG. 2012. A convenient and mild chromatography-free method for the purification of the products of Wittig and Appel reactions. Org. Biomol. Chem. 10, 3531–3537. (doi:10.1039/c2ob07074j) [DOI] [PubMed] [Google Scholar]
- 48.Lee JB. 1966. Preparation of acyl halides under very mild conditions. J. Am. Chem. Soc. 88, 3440–3441. (doi:10.1021/ja00966a052) [Google Scholar]
- 49.Kumar A, Akula HK, Lakshman MK. 2010. Simple synthesis of amides and Weinreb amides via use of PPh3 or polymer-supported PPh3 and iodine. Eur. J. Org. Chem. 14, 2709–2715. (doi:10.1002/ejoc.200901420) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duangkamol C, Jaita S, Wangngae S, Phakhodee W, Pattarawarapan M. 2015. Catalytic role of PPh3 and its polymer bound analog in the amidation of carboxylic acids mediated by 2,4,6-trichloro-1,3,5-triazine. Tetrahedron Lett. 56, 4997–5001. (doi:10.1016/j.tetlet.2015.07.012) [Google Scholar]
- 51.Godfrey SM, McAuliffe CA, Pritchard RG, Sheffield JM. 1998. Structural dependence of the reagent Ph3PCl2 on the nature of the solvent, both in the solid state and in solution; X-ray crystal structure of trigonal bipyramidal Ph3PCl2, the first structurally characterised five-coordinate R3PCl2 compound. Chem. Commun. 921–922. (doi:10.1039/A800820E) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this research are included in figures, tables and the electronic supplementary material.