

Abstract
The atypical E2Fs, E2F7 and E2F8, act as potent transcriptional repressors of DNA replication genes providing them with the ability to induce a permanent S‐phase arrest and suppress tumorigenesis. Surprisingly in human cancer, transcript levels of atypical E2Fs are frequently elevated in proliferating cancer cells, suggesting that the tumor suppressor functions of atypical E2Fs might be inhibited through unknown post‐translational mechanisms. Here, we show that atypical E2Fs can be directly phosphorylated by checkpoint kinase 1 (Chk1) to prevent a permanent cell cycle arrest. We found that 14‐3‐3 protein isoforms interact with both E2Fs in a Chk1‐dependent manner. Strikingly, Chk1 phosphorylation and 14‐3‐3‐binding did not relocate or degrade atypical E2Fs, but instead, 14‐3‐3 is recruited to E2F7/8 target gene promoters to possibly interfere with transcription. We observed that high levels of 14‐3‐3 strongly correlate with upregulated transcription of atypical E2F target genes in human cancer. Thus, we reveal that Chk1 and 14‐3‐3 proteins cooperate to inactivate the transcriptional repressor functions of atypical E2Fs. This mechanism might be of particular importance to cancer cells, since they are exposed frequently to DNA‐damaging therapeutic reagents.
Keywords: 14‐3‐3 proteins, atypical E2Fs, cell cycle, checkpoint kinase 1, DNA damage
Subject Categories: Cell Cycle; DNA Replication, Repair & Recombination
Introduction
To cope with intrinsic and extrinsic genotoxic stress, eukaryotes have developed highly conserved DNA damage signaling pathways. Intact function of DNA damage signaling is crucial to maintain genome integrity and is important in preventing the development of cancer and other diseases (Jackson & Bartek, 2009; Ciccia & Elledge, 2010). In response to DNA lesions, checkpoints can be activated to halt cell cycle progression until the damage is repaired. Within the DNA damage signaling network, checkpoint kinase 1 (Chk1) plays a central role. It is a transducer in arresting the cell cycle, repairing DNA, and regulating transcription and apoptosis induction (Dai & Grant, 2010). Chk1 directly phosphorylates multiple substrates to regulate their activity, stability, or subcellular localization. Well‐characterized substrates of Chk1 include the CDC25 phosphatases, P53, RAD51, and FANCE, which facilitate cell cycle arrest and repair in response to DNA damage (Zhang & Hunter, 2014). Also, Chk1 functions to modulate E2F‐dependent transcription, because both E2F3 and E2F6 were identified as Chk1 substrates (Bertoli et al, 2013; Gong et al, 2016).
The E2F transcription factor family is of paramount importance in coordinating cell cycle progression. Among the known E2F family members, E2F7 and E2F8 are considered as atypical E2F transcription factors, because they do not require binding with retinoblastoma protein or heterodimerization with the DNA‐binding partner (DP) proteins to repress target genes. E2F7 and E2F8 levels peak during S phase, to mediate the downswing of many oscillating target genes involved in DNA replication, metabolism, and DNA repair. We previously found that E2F7 and E2F8 are subjected to degradation via APC/CCdh1 during G1 phase, to allow a time window for upregulation of E2F target genes by activator E2Fs, and subsequent S‐phase entry (Boekhout et al, 2016). Meanwhile, ectopic expression of E2F7 and E2F8 is sufficient to stall S‐phase progression, indicating that their activity must be carefully regulated to ensure unperturbed proliferation (Westendorp et al, 2012; Boekhout et al, 2016). Interestingly, there is accumulating evidence suggesting that atypical E2Fs also play an important role in the DNA damage response. E2F7 is a transcriptional target of P53 and mediates DNA damage‐induced senescence (Aksoy et al, 2012; Carvajal et al, 2012). Moreover, E2F7 and E2F8 redundantly inhibit DNA synthesis in primary keratinocytes under etoposide treatment (Thurlings et al, 2016). Furthermore, previous work indicated that E2F7 may relocate to DNA breaks to regulate homologous recombination and that E2F7 and E2F8 can suppress E2F1‐induced apoptosis after DNA damage (Zalmas et al, 2008, 2013). We recently demonstrated that atypical E2Fs can act as tumor suppressors in two murine cancer models (Kent et al, 2016; Thurlings et al, 2016). These findings suggest that balanced activity of E2F7 and E2F8 plays a central role in cell cycle control during DNA damage and regulation of the S‐phase checkpoint. Nonetheless, the post‐translational regulation mechanisms and precise functions of E2F7 and E2F8 in the DNA damage response are still unknown.
Here, we demonstrate that Chk1 phosphorylates both E2F7 and E2F8 in response to DNA damage. Functionally, Chk1 inhibited E2F7/8 transcriptional repressor activity to prevent severe repression of E2F target genes. Blockage of this interaction caused a severe S‐phase arrest and apoptosis. Moreover, we found that reduced repressor activity of the atypical E2Fs was mediated via Chk1‐dependent 14‐3‐3 binding. In addition, a positive correlation between E2F7/8 target gene expression and 14‐3‐3 levels was found in different types of cancer, including hepatocellular carcinoma. This suggests that E2F target gene expression could be deregulated in cancer cells via Chk1/14‐3‐3‐dependent inhibition of atypical E2Fs.
Results
Chk1 phosphorylates atypical E2Fs
We asked whether atypical E2Fs undergo post‐translational modifications under DNA‐damaging conditions. To this end, we carried out label‐free mass spectrometry to detect putative phosphorylation events on E2F7 and E2F8 in response to treatment with the topoisomerase II inhibitor etoposide. Three phosphorylations on human E2F7 and E2F8 were highly induced in response to DNA damage: E2F7S410, E2F8S395, and E2F8S412 (Fig EV1A). Interestingly, E2F7S410 and E2F8S395, which locate at a similar position on each E2F, closely resemble consensus Chk1 target motifs (L/MxRxxS) and are conserved between human and mouse (Fig 1A and B). Chk1 is a serine/threonine‐specific kinase that phosphorylates protein targets resulting in the activation of the S‐phase cell cycle checkpoint to allow DNA repair and maintenance of genetic stability (Sanchez et al, 1997; Syljuasen et al, 2005; Petermann et al, 2010). To test whether Chk1 is indeed responsible for phosphorylating atypical E2Fs on the above‐described serine residues, we incubated co‐immunoprecipitated Flag‐tagged E2F7 or E2F8 with recombinant active Chk1 and performed mass spectrometry. Again, E2F7S410 and to a lesser extent E2F8S395 showed increases in phosphorylation after Chk1 incubation, and a serine at 833 was found in human E2F7 construct (Fig EV1B). Next, we performed in vitro kinase assays to confirm that E2F7/8 were indeed substrates of Chk1. In the kinase assay, E2F7 and E2F8 showed a robust phosphorylation by active Chk1 (Fig EV1C). Since Ser833 and Ser410 in human E2F7 are conserved in mouse (referred to E2F7S825 and E2F7S411 in mouse), we decided to focus on mouse constructs in our subsequent studies. To test what are the primary target amino acids of Chk1 phosphorylation, we performed site‐directed mutagenesis to generate E2F7 and E2F8 constructs in which serines are replaced by alanines (hereafter referred to E2F7S411A and E2F7S825A, E2F8S395A). In the kinase assay, we found that phosphorylation of both E2F7S411A and E2F8S395A, but not E2F7S825A, were clearly reduced compared to their wild‐type counterparts, indicating that these serine residues are indeed the main phosphorylation sites of E2F7 and E2F8 (Fig 1C).
Figure EV1. Chk1 phosphorylates atypical E2Fs.
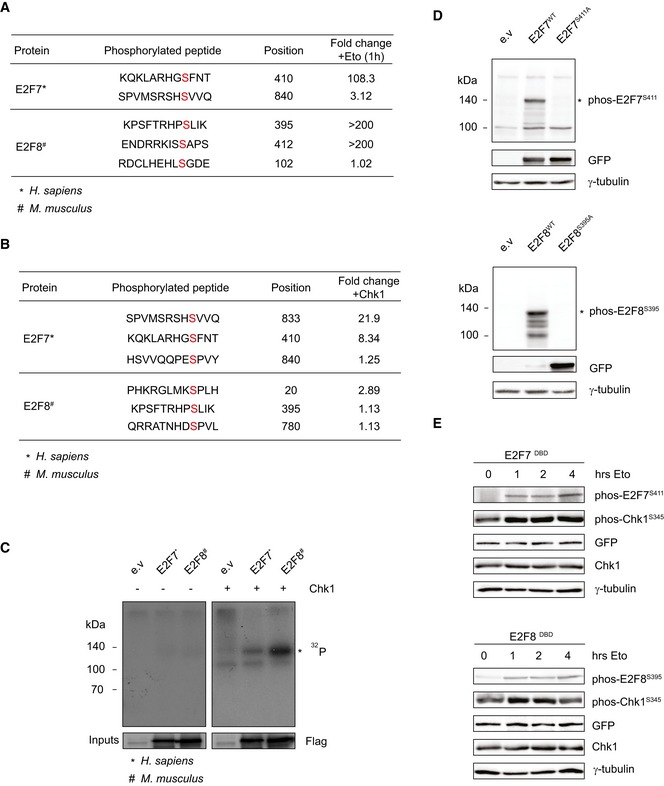
- Mass spectrometry data showing phosphorylation of E2F7 and E2F8 in response to 1‐h etoposide‐induced DNA damage. HEK cells were transfected with Flag‐tagged E2F7 or E2F8 and immunoprecipitated with anti‐Flag resins. Fold change indicates the phosphorylated peptide enrichment ratios in etoposide‐ versus DMSO‐treated condition.
- Mass spectrometry data showing phosphorylation of precipitated E2F7 and E2F8 incubated with recombinant active Chk1. HEK cells were transfected with Flag‐tagged E2F7/8, and cell lysates were precipitated with anti‐Flag resins. Fold change indicates the intensity of phosphorylation with Chk1 versus without Chk1.
- Chk1 kinase assay for human E2F7 and mouse E2F8. HEK cells were transfected with indicated plasmids, and cell lysates were precipitated with anti‐Flag resins. Precipitated E2F7 and E2F8 were incubated with or without recombinant active Chk1 and radiolabeled 32P, and then, samples were loaded on a standard SDS–PAGE gel for exposure. Asterisk indicates the approximate expected molecular weight of Flag‐tagged E2F7 and E2F8 (˜115 and ˜105 kDa, respectively).
- Immunoblots showing the detection of phosphorylated EGFP‐tagged E2F7S411 and E2F8S395 using antibodies recognizing the Chk1 phosphorylation sites in E2F7 and E2F8. HEK cells were transfected with indicated plasmids for 48 h and treated with etoposide for 8 h prior to lysis to ensure high levels of active Chk1. Asterisks indicate the full‐length fusion proteins (˜140 kDa).
- Positive correlation between Chk1 activation and E2F7/8 phosphorylation. HEK cells were transfected with DNA‐binding domain mutant versions of E2F7 and E2F8 (DBD) for 48 h. Cells were treated with etoposide and were harvested at the indicated time points after the treatment. Whole‐cell lysates were subjected to immunoblotting.
Source data are available online for this figure.
Figure 1. Chk1 phosphorylates atypical E2Fs.
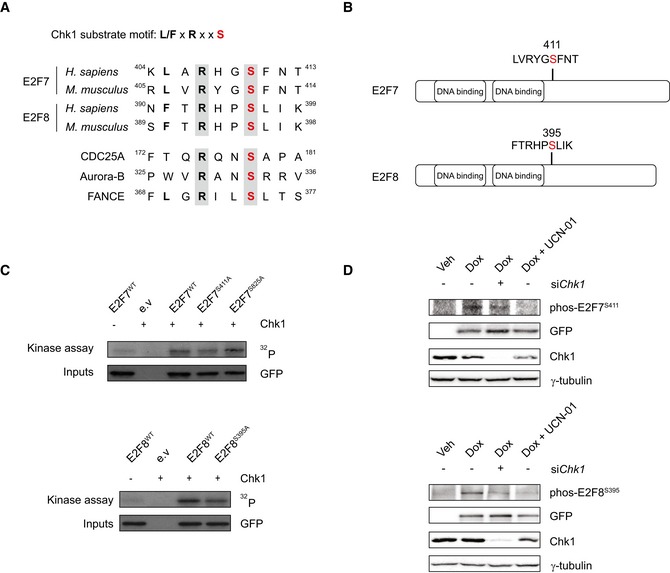
- Putative Chk1 substrate motifs in human or mouse E2F7 and E2F8 proteins, and known human Chk1 substrate motifs.
- Schematic overview of the putative Chk1 phosphorylation motifs on mouse E2F7 and E2F8.
- Kinase assay showing that Chk1 phosphorylates mouse E2F7 and E2F8 in vitro. HEK cells were transfected with EGFP‐tagged wild‐type and mutant versions of E2F7 and E2F8, lysed, and precipitated with GFP‐Trap beads. Precipitated proteins were incubated with radiolabeled 32P‐ATP, in the presence or absence of recombinant active Chk1 and loaded onto SDS–PAGE gel.
- Chk1 phosphorylates atypical E2Fs in vivo. 24 h before treatment, siRNA against Chk1 was transfected to HeLa/TO cells. Then, cells were treated either without doxycycline (Veh) or with doxycycline (Dox) for 16 h to induce the over‐expression of wild‐type E2F7 and E2F8. UCN‐01 was added to the cells simultaneously to inhibit Chk1 kinase. Whole‐cell lysates were harvested for immunoblot.
Source data are available online for this figure.
To further confirm these phosphorylation events, we devised antibodies that recognized the phosphorylation site of E2F7S411 and E2F8S395. E2F7 and E2F8 are not abundantly expressed, and therefore, we had difficulties in detecting endogenous phosphorylated E2F7 and E2F8. However, we could detect strong protein bands in cells transfected with exogenous mouse wild‐type forms of EGFP‐tagged E2F7 and E2F8, but not the alanine mutants (Fig EV1D). We then used the phospho‐specific antibodies to investigate the dynamic of the phosphorylation events on exogenous E2F7/8. To overcome the problem that exogenous E2F7/8 can perturb the cell cycle (Westendorp et al, 2012), we expressed DNA‐binding mutant versions of E2F7/8 (DBD). As shown in Fig EV1E, phosphorylation levels of E2F7S411 and E2F8S395 were low before etoposide treatment but were induced dramatically within 1 h of etoposide treatment, indicating a rapid response to the activated Chk1 kinase. To examine whether atypical E2Fs are targets of Chk1 in vivo, we measured the phosphorylation of E2F7S411 and E2F8Ser395 with siRNA against Chk1 or with a Chk1 inhibitor UCN‐01 (Chen et al, 1999; Busby et al, 2000), in HeLa cells with doxycycline‐inducible expression of EGFP‐tagged E2F7WT and E2F8WT (Tet/ON, hereafter referred to as HeLa/TO, Fig 1D). We found that inactivation of Chk1 resulted in a substantial reduction in E2F7S411 and E2F8S395 phosphorylations compared with control condition. Taken together, these data show that specific serine residues in E2F7 and E2F8 proteins are bona fide Chk1 phosphorylation sites.
Chk1‐dependent phosphorylation does not alter stabilization or subcellular relocalization of atypical E2Fs
Given that Chk1 controls the stability of many of its substrates (Mailand et al, 2000; Raleigh & O'Connell, 2000), we investigated whether Chk1‐dependent phosphorylation alters the stability of E2F7 and E2F8. We created stable HeLa cell lines with doxycycline‐inducible expression of E2F7S411A and E2F8S395A. In addition to the alanine mutants, we also generated phosphorylation‐mimicking mutants in which we replaced the respective Ser411 and Ser395 residues on E2F7 and E2F8 with aspartic acids (hereafter referred to as E2F7S411D and E2F8S395D). The EGFP tags allowed for separate detection of exogenous and endogenous E2F7/8 and the over‐expression levels were within the physiologically range and similar between wild‐type and mutant constructs (Fig EV2A). Next, HeLa/TO cells were treated with doxycycline to induce over‐expression and etoposide to cause DNA damage and Chk1 activation. Protein levels of wild‐type and mutant E2Fs were not majorly affected by etoposide treatment (Fig 2A), which implies that neither DNA damage nor Chk1 kinase has a direct impact on stabilization of E2F7 or E2F8. To evaluate whether Chk1‐dependent phosphorylation may affect stability of endogenous E2F7 and E2F8, we treated HeLa cells with etoposide to activate Chk1. Etoposide treatment increased E2F7 and E2F8 levels within 8 h (Fig 2B). However, since protein expression of E2F7 and E2F8 oscillates during the cell cycle and peaks during S phase, we hypothesized that this stabilization could merely reflect an etoposide‐induced accumulation of cells in S phase. Flow cytometry confirmed that etoposide substantially elevated the number of cells in S phase (Fig 2C). We repeated this experiment in the presence of the protein synthesis inhibitor cycloheximide. When HeLa or U2OS cells were treated with etoposide and cycloheximide, the proportion of cells in S phase and the protein expression of E2F7 and E2F8 did not change while Chk1 was strongly activated, as seen by phosphorylation of its Ser345 residue (Figs 2D and EV2B). These findings provide strong evidence that phosphorylation by Chk1 does not directly regulate the protein stability of E2F7 and E2F8.
Figure EV2. Chk1‐dependent phosphorylation does not cause stabilization or subcellular relocalization of atypical E2Fs.

- Protein expression of endogenous and exogenous atypical E2Fs in the HeLa/TO cell lines. EGFP‐tagged wild‐type, alanine and aspartic mouse constructs were incorporated in the HeLa/TO‐inducible system by single colony selection. Cells were then treated either without (Veh) or with (Dox) doxycycline for 24 h. Exogenous E2F7 was measured by anti‐GFP antibody, and endogenous E2F7 was measured by anti‐human E2F7 antibody. For E2F8, both exogenous and endogenous forms were detected by a single E2F8 antibody. Asterisks indicate specific detections from the antibodies.
- Protein expression of endogenous E2F7, E2F8, phos‐Chk1S345, and total Chk1 in U2OS cells treated with cycloheximide (CHX), in the presence or absence of etoposide.
- E2F7 and E2F8 proteins are localized in the nucleus. Inducible cell lines were treated with doxycycline and etoposide for 16 h, followed by isolation of cytosolic and nuclear protein fractions. HDAC1 expression was used as an indicator for enrichment of nuclear proteins.
- Immunofluorescence showing the localization of E2F7, E2F8, and MDC1 in response to camptothecin treatment for 8 h in U2OS cells. E2F7, E2F8, and MDC1 were labeled with GFP, and DNA damage sites are detected with an antibody against γ‐H2AX. DNA was stained with DAPI. Scale bar: 5 μm.
Source data are available online for this figure.
Figure 2. Chk1‐dependent phosphorylation does not cause stabilization or subcellular relocalization of atypical E2Fs.
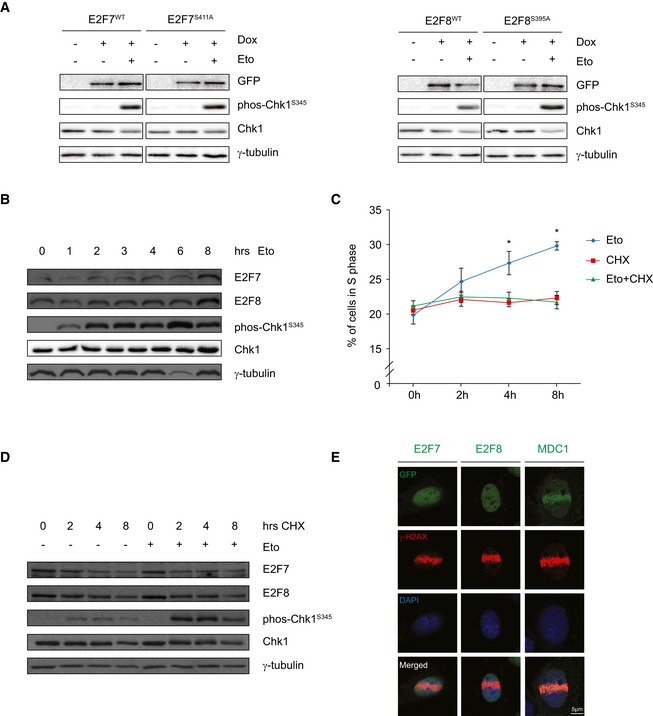
- Stability of atypical E2Fs is not affected by Chk1 phosphorylation. HeLa/TO cells were treated with doxycycline to induce over‐expression of wild‐type or alanine mutant E2F7 or E2F8, either with or without etoposide to induce DNA damage for 8 h, and protein levels were then measured by immunoblotting.
- Protein expression of endogenous E2F7, E2F8, phos‐Chk1S345, and total Chk1 in HeLa cells treated with etoposide at different time points.
- Percentage of cells in S phase was determined by flow cytometry. HeLa cells were treated with etoposide, cycloheximide (CHX), or etoposide (Eto) + CHX, harvested at the indicated time points, and subjected to flow cytometry and cell cycle analysis. Data are presented as mean ± SEM. *P < 0.05 (one‐way ANOVA test).
- Protein expression of endogenous E2F7, E2F8, phos‐Chk1S345, and total Chk1 in HeLa cells treated with cycloheximide (CHX), in the presence or absence of etoposide (Eto) at different time points.
- Immunofluorescence staining showing the localization of E2F7, E2F8, and MDC1 in response to micro‐irradiation for 1 h in HeLa cells. E2F7, E2F8, and MDC1 were labeled with GFP, and DNA damage sites are detected with an antibody against γ‐H2AX. Nuclear DNA was stained with DAPI. Scale bar: 5 μm.
Source data are available online for this figure.
As Chk1 was previously shown to mediate nuclear exclusion of some of its substrates (Lopez‐Girona et al, 1999), we then investigated whether Chk1‐dependent phosphorylation could affect the subcellular localization of E2F7 and E2F8. However, immunoblots using our phospho‐specific antibodies on fractionated protein lysates showed that both phosphorylated and non‐phosphorylated forms of E2F7 and E2F8 were almost exclusively located within the nucleus (Fig EV2C). Interestingly, previous work suggested that E2F7 may bind to DNA damage sites and contribute to the repair of double‐strand breaks (Zalmas et al, 2013). This raised the question as to whether atypical E2Fs relocalize to DNA lesions in a Chk1‐dependent manner. To visualize the E2F protein dynamics in response to DNA damage, HeLa/TO cells expressing EGFP‐tagged E2F7 and E2F8 were subjected to laser micro‐irradiation on the nucleus, followed by live microscopy imaging. Cells were stained for γ‐H2AX to confirm the presence of DNA double‐strand breaks. Surprisingly, we observed no migration of E2F7 or E2F8 to the DNA damage foci after 1 h of irradiation, while MDC1, a known mediator of DNA damage repair (Stewart et al, 2003), accumulated rapidly at DNA lesions (Fig 2E). In order to rule out that the recruitment of E2F7/8 may be delayed, or dependent on the cell line used, we tried to repeat the experiment from the previous work by transfecting U2OS cells transiently with EGFP‐E2F7/8 and treated them with camptothecin for 24 h (Zalmas et al, 2013). Again, while MDC1 localized toward γ‐H2AX foci, we did not observe recruitment of E2F7 or E2F8 by utilizing anti‐GFP antibody staining (Fig EV2D). Perhaps the severity of the damage was different from the previous work, but we did not observe that atypical E2Fs localize to DNA damage sites.
Chk1 inhibits the transcriptional repressor function of E2F7 and E2F8 to promote cell cycle progression and prevent apoptosis
The main function of atypical E2Fs is to repress the expression of a set of genes critical for cell cycle progression. Based on our findings, we hypothesized that Chk1 phosphorylation affects the repressor activity of atypical E2Fs. To test this hypothesis, we first evaluated the impact of mutating the atypical E2F‐Chk1 phosphorylation sites on cell cycle progression. Stable HeLa/TO cell lines containing inducible wild‐type and Chk1 mutant versions of atypical E2Fs were synchronized at the start of S‐phase transition by adding the ribonucleotide synthase inhibitor hydroxyurea (HU) to the medium for 16 h. Upon release from the HU block, the expression of EGFP‐tagged wild‐type and mutant versions of E2F7 and E2F8 was induced by doxycycline (Dox) and the progression of the respective cell lines until mitosis was monitored by live cell imaging (Fig 3A). We observed that approximately 60–70% of E2F7WT‐ and E2F8WT‐expressing cells completed mitosis within 24 h after HU release, similar to vehicle‐treated non‐induced cell lines. However, over‐expression of E2F7WT after HU release resulted in delayed cell cycle progression compared to vehicle‐treated cells, whereas E2F8WT over‐expression had no major impact on the speed of cell cycle progression. Importantly, cells expressing either the non‐phosphorylatable E2F7S411A or E2F8S395A showed a remarkable delay in cell cycle progression and only 10% of the cells were able to enter mitosis 24 h after HU release. In contrast, cells expressing the phospho‐mimic E2F7S411D and E2F8S395D mutants showed no changes in cell cycle progression. Flow cytometry analysis suggested that cells expressing E2F7S411A arrested in both S and G2 phase, while cells expressing E2F8S395A were mostly halted in G2 (Fig EV3A). To make sure that these effects are not only specific for hydroxyurea release, we performed synchronization experiments after a double thymidine block/release assay, which is known to induce less double‐strand DNA breaks in cells compared to HU‐treated cells (Lundin et al, 2002). After 9 h of thymidine release, HeLa/TO cells expressing wild‐type and phospho‐mimetic versions of E2F7 progressed to an ongoing S phase, while expression of alanine mutant strongly stalled the cell cycle at the onset of S phase (Fig EV3B). Moreover, E2F7WT and E2F7S411D cells had mostly completed mitosis after 14 h, while expressing E2F7S411A aggravated the S/G2‐phase arrest phenotype. These data show that cell cycle progression was severely delayed by non‐phosphorylatable forms of E2F7 and E2F8. Interestingly, during live cell imaging, we observed that a proportion of alanine mutant‐expressing cells displayed cell blebbing 24 h after HU release, which we interpreted as cell death (Fig EV3C). Flow cytometric analysis of the apoptosis marker Annexin‐V confirmed a higher percentage of apoptosis in E2F7S411A‐ and E2F8S395A‐expressing compared to wild‐type E2F7‐ and E2F8‐expressing cells (Fig 3B). Given that maintaining E2F target gene transcription ensured DNA replication and avoid replication stress‐induced DNA damage (Bertoli et al, 2016), a potential explanation for the apoptosis we observed could be that the alanine mutant versions of E2F7 and E2F8 strongly repress their target genes, resulting in DNA damage and consequently apoptosis.
Figure 3. Chk1 inhibits the transcriptional repressor function of E2F7 and E2F8 to promote cell cycle progression and prevent apoptosis.

-
ALeft panel shows a schematic view of the experimental setting. HeLa/TO cells were synchronized with hydroxyurea (HU) for 16 h, then released by washing and adding new medium containing doxycycline (Dox) to induce expression of the transgenes. Cell lines incubated with medium without doxycycline (Vehicle, Veh) were used as controls. Expression of WT and Chk1 mutant versions of E2F7/8 was monitored by EGFP fluorescence, DNA was visualized by adding fluorescent SiR‐DNA to the medium, and cell morphology was evaluated by differential interference contrast (DIC). Per condition, 100 individual cells were traced. Each cell was followed until it successfully finished mitosis and divided into two daughter cells, for a maximum of 24 h. The graphs on the right show the quantification of the mitotic events during live cell imaging. Scale bar: 10 μm.
-
BQuantification of apoptosis by flow cytometric analysis of Annexin‐V staining. Experiments were performed as outlined in (A), and cells were harvested for flow cytometry 24 h after HU release. Apoptotic cells were counted as Annexin‐positive and DAPI‐negative.
-
C, DQuantitative PCR of CDC6, RAD51, CDC25A, and CCNE1 expression in HeLa/TO cell lines expressing wild‐type and mutant versions of E2F7/8.
-
EChk1 phosphorylation does not change the promoter enrichment of E2F7 and E2F8. HEK cells were transfected with either PEI reagent alone (control) or indicated plasmids tagged with GFP. 48 h after transfection, cells were harvested for chromatin immunoprecipitation (ChIP) followed by qPCR. Histogram represents the enrichment ratio (bound/input) in E2Fs target gene promoters.
Figure EV3. Prevention of Chk1‐mediated phosphorylation of atypical E2Fs delays cell cycle progression and reduces cell survival.

- Cell cycle profile analysis by flow cytometry confirmed that over‐expression of non‐phosphorylatable forms of E2F7 or E2F8 delays cell cycle progression after HU release. HeLa/TO cells expressing wild‐type or mutant forms of E2F7/8 were harvested at 0, 9, and 14 h after HU release and doxycycline induction.
- Cell cycle profile analysis by flow cytometry confirmed that over‐expression of non‐phosphorylatable forms of E2F7 or E2F8 delays cell cycle progression after double thymidine block/release. HeLa/TO cells expressing wild‐type or mutant forms of E2F7/8 were harvested at 0, 9, and 14 h after double thymidine block/release and doxycycline induction.
- Representative montage figures from the live cell imaging show that over‐expression of alanine forms of E2F7/8 induces cell blebbing (arrows), interpreted as cell death. Scale bar: 10 μm.
- FACS sorting of cells expressing wild‐type and mutant E2F7 and E2F8. Sorting gates (rectangles) were set to isolate cells with comparable levels of expression from all E2F7/8‐EGFP‐expressing (Dox) cell lines.
Next, we determined whether alanine mutant forms of E2F7 and E2F8 possess higher transcriptional repressor activity compared to the wild‐type versions. Since HU treatment leads to replication stress and strong activation of Chk1, we treated cells for 16 h with HU and compared mRNA levels of E2F7/8 target genes after induction of wild‐type or phospho‐mutant E2F7 and E2F8. We used fluorescence‐activated cell sorting (FACS) to collect cells with similar EGFP over‐expression levels (Fig EV3D, GFP+/GFP−) and measured subsequently RNA levels of E2F target genes by quantitative PCR. Expression of E2F target genes, such as CDC6 and CCNE1, was significantly more repressed by non‐phosphorylatable alanine mutants compared to wild types and aspartic mutants (Kent et al, 2016; Westendorp et al, 2012; Fig 3C and D). This finding provides strong evidence that Chk1 phosphorylation can abrogate the repressor activity of E2F7 and E2F8.
Previous work showed that phosphorylation by Chk1 redirects E2F6 away from target gene promoters (Bertoli et al, 2013). Therefore, we tested whether Chk1 phosphorylation has a similar effect on E2F7 and E2F8. To this end, we transfected HEK 293T cells transiently with wild‐type, alanine mutant, or phospho‐mimic mutant E2F7 and E2F8, and used GFP‐trap beads to perform chromatin immunoprecipitation. A primer set designed against a distal region in the E2F1 gene served as a negative control to show that binding was specific to E2F binding motif‐containing regions (Fig 3E). However, phospho‐mutant and wild‐type versions of atypical E2Fs displayed no clear differences in enrichment on various E2F target gene promoters (Fig 3E). Thus, Chk1 phosphorylation did not alter the recruitment ability of atypical E2Fs toward E2F target promoters.
Taken together, these results indicate that Chk1 inhibits E2F7 and E2F8 function to allow resumption of the cell cycle and prevent cell death after transient replication stress and DNA damage.
Loss of Chk1 causes E2F7/8‐dependent cell cycle arrest and DNA damage
Chk1 is essential for the stabilization of replication forks, and inhibition of Chk1 therefore delays DNA replication and failure to complete S phase (Zachos et al, 2003). Since phosphorylation by Chk1 inhibits E2F7 and E2F8 repressor function, we hypothesized that Chk1 inhibition causes S‐phase arrest which depends on the repressor activity of E2F7 and E2F8. To test this, Chk1, E2F7, and E2F8 alone or in combination were knocked down with siRNAs. HeLa cells were synchronized by HU treatment for 16 h, and cell cycle profiles were analyzed at 0, 4 and 8 h after HU release by flow cytometry. We confirmed efficient knockdown of all three proteins with immunoblot analysis (Fig 4A). Moreover, we found that protein expression of Rad51 and CDC6, which are well‐described E2F target genes, were decreased by Chk1 siRNA, suggesting enhanced E2F7/8 repressor activity (Fig 4A). Flow cytometry analysis revealed that Chk1 knockdown resulted in a severe S‐phase arrest, whereas knockdown of E2F7/8 had no major impact on cell cycle progression. Importantly, no S‐phase arrest was observed when Chk1 was knocked down in combination with E2F7/8. This demonstrates that inactivation of atypical E2Fs can rescue the cell cycle phenotype induced by Chk1 inhibition (Fig 4B). In addition, we observed a similar phenotype in RPE‐hTERT cells, a non‐transformed retina pigment cell line with intact checkpoint functions (Fig EV4A). We reasoned that if knockdown of Chk1 and E2F7/8 permits the cell cycle progression, then ectopic expression of non‐phosphorylatable E2F7/8 in the same condition would arrest the S‐phase progression again, to a bigger extent than phospho‐mimetic E2F7/8. Indeed, flow cytometry showed that in the presence of siChk1 + siE2F7+8, cells overexpressing alanine mutant forms of E2F7 and E2F8 fail to continue S phase after HU release (Fig EV4B). On the contrary, a substantial amount of aspartic mutant‐expressing cells managed to progress to G2/M phase. These data confirmed our finding that cell cycle arrest by Chk1 inhibition was dependent on the deregulated repressor activity of atypical E2Fs.
Figure 4. Loss of Chk1 results in E2F7/8‐dependent cell cycle arrest and DNA damage.

-
ASchematic overview of the experimental setting and immunoblot analysis for the validation of the efficient siRNA‐mediated knockdown of Chk1, E2F7, and E2F8 at HU (0 h) or 8 h after HU release. Protein levels of atypical E2Fs targets RAD51 and CDC6 are shown. Detection of γ‐tubulin was used as loading control.
-
BFACS cell cycle profiles at 0, 4 and 8 h after HU release from propidium iodide‐stained HeLa cells incubated with siRNAs directed against Chk1 and E2F7/8. Scrambled (scr) siRNA was used as control.
-
CTranscript levels of E2F7/8 target genes CDC6, RAD51, CDC25A, and CCNE1 after HU treatment for the indicated siRNA groups measured by quantitative PCR. Fold changes were adjusted to scrambled condition.
-
D, EImmunofluorescence staining showing the intensities of Rad51 and γ‐H2AX in HeLa cells. Cells were transfected with indicated siRNA and incubated with HU for 16 h before harvest. Nuclear DNA was stained with DAPI. For quantification (right side), five fields of 40× magnification pictures were selected randomly and subjected to ImageJ software analysis. Scale bars: 20 μm.
Figure EV4. Chk1 inhibition induces an E2F7/8‐dependent cell cycle arrest and reduces cell survival.
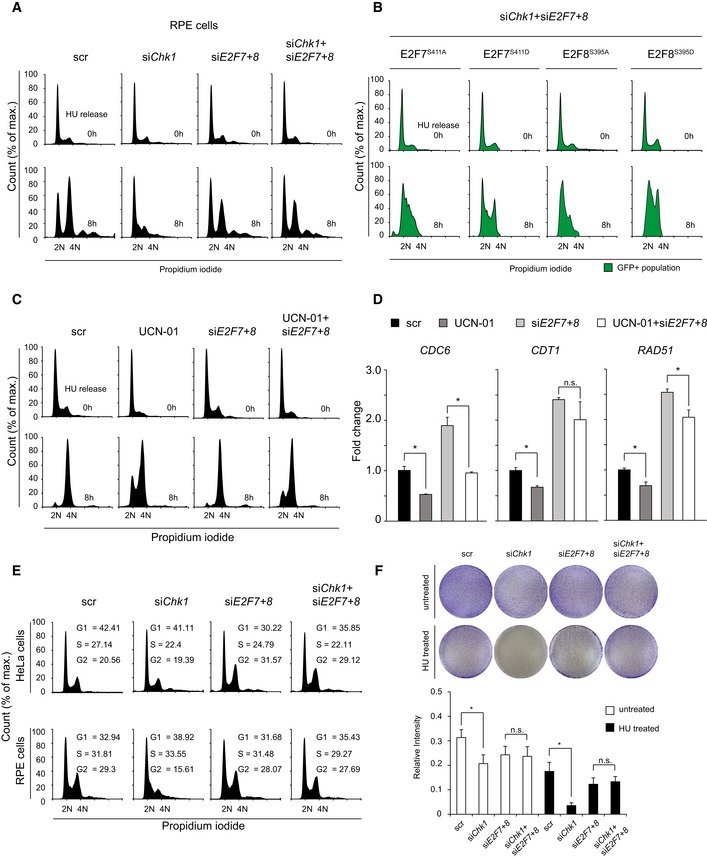
- FACS cell cycle profiles at 0 and 8 h after HU release from propidium iodide‐stained RPE cells incubated with siRNAs directed against Chk1 and E2F7/8. Scrambled (scr) siRNA was used as control.
- FACS cell cycle profiles at 0 and 8 h after HU release from propidium iodide‐stained GFP‐positive HeLa/TO cells transfected with siRNA against Chk1 and E2F7/8. Doxycycline was added to the cells 16 h before HU release.
- FACS cell cycle profiles from propidium iodide‐stained HeLa cells treated with UCN‐01. HeLa cells were first transfected with scramble or siE2F7+8, then exposed to HU and the Chk1 inhibitor UCN‐01 for 16 h, and eventually harvested at 0 and 8 h after HU release.
- Transcript levels of E2F7/8 target genes after 16 h of HU treatment, measured by qPCR. Fold changes were adjusted to the scrambled condition.
- Cell cycle distribution of HeLa and RPE cells transfected with siRNA against Chk1 and E2F7/8 under unperturbed condition. Scrambled (scr) siRNA was used as control.
- Clonogenic survival assay of HeLa cells transfection with siRNA against Chk1 and E2F7/8 under un‐treated or HU‐treated condition. Cells were transfected with indicated siRNA for 24 h and reseeded at a low density and grow for 4 days. Scrambled (scr) siRNA was used as control. Histogram shows the quantification of relative intensity from each condition (n = 2 independent experiments).
Our data suggest that Chk1 inactivation results in unrestricted activity of E2F7 and E2F8, and consequently enhanced repression of E2F target genes resulting in failure to progress through S phase after HU release. Indeed, transcription of typical E2F7/8 target genes such as CDC6, RAD51, CDC25A, and CCNE1 was all significantly repressed in HU‐treated cells with Chk1 knockdown (Fig 4C). Importantly, additional knockdown of E2F7 and E2F8 relieved this downregulation of E2F target gene expression and likely explains why cells can progress properly through the cell cycle. In addition to the usage of siRNA, we inhibited Chk1 kinase with UCN‐01. In line with Chk1 siRNA experiments, we found that UCN‐01 treatment results in enhanced E2F7/8 target gene repression and impaired recovery from a HU block, which could be rescued by E2F7 and E2F8 siRNA treatment (Fig EV4C and D).
Next, we tested whether the transcription control by the Chk1‐E2F7/8 axis affects protein levels of the target genes and its functional relevance. Rad51 is a target of E2F7 and E2F8, and it relocalizes to the DNA lesion sites to promote DNA damage repair (Shinohara et al, 1992). Consistent with the reduction in overall Rad51 protein by Chk1 knockdown (Fig 4A), immunofluorescence staining revealed a marked reduction in nuclear Rad51 intensity after knockdown of Chk1 in HU‐treated cells (Fig 4D, quantification on the right). Additionally, knockdown of E2F7 and E2F8 in addition to siChk1 rescued the expression of Rad51 demonstrating that repression of E2F target gene transcripts had a direct influence on their protein levels. Given that Rad51 promotes DNA damage repair, then repression of Rad51 can lead to accumulation of unfixed DNA lesions. To test this, γ‐H2AX intensity was measured in the siChk1 condition. Indeed, siChk1 resulted in a dramatic accumulation of γ‐H2AX compared to the control condition. Interestingly, the high‐intensity phenotype of γ‐H2AX was completely resolved in cells treated with siChk1 + siE2F7+8. These data strongly suggest that inhibition of E2F7/8 repressor activity by Chk1 is critical for preventing replication stress‐induced DNA damage.
It was documented that Chk1 is essential for the checkpoint function in an unperturbed cell cycle (Patil et al, 2013). Therefore, we investigated whether loss of Chk1 affects the progression of the cell cycle in transformed (HeLa) and non‐transformed (RPE) cell lines under unperturbed conditions. Although Chk1 decreased the percentage of RPE cells in S/G2, it had little effect on cell cycle distribution in HeLa cells (Fig EV4E). In both cell lines, the combination of Chk1 knockdown with knockdown of E2F7/8 showed cell cycle profiles highly similar to the control condition. In line with the cell cycle analysis, in a clonogenic cell survival assay we showed that siChk1 has only a mild effect on long‐term HeLa cell survival, compared to the control condition (Fig EV4F). However, we demonstrated that loss of Chk1 was detrimental to the long‐term cell survival under HU treatment, which was relieved by combinational knockdown of E2F7/8. Taken together, these data consistently show that Chk1 inhibition causes a permanent cell cycle arrest under conditions of replication stress, which is dependent on E2F7/8 repressor function.
14‐3‐3 ζ mediates Chk1‐dependent inhibition on E2F7 and E2F8
Given that Chk1 phosphorylation did not inhibit the promoter binding activity of atypical E2Fs (Figs 3E and EV3D), we evaluated whether Chk1 phosphorylation interferes with the binding of regulatory partners of atypical E2Fs. To test this, we first screened for putative regulatory E2F7‐ and E2F8‐interacting proteins. To this end, we performed mass spectrometry using SILAC (Stable Isotope Labeling with Amino acids in Cell culture). We labeled HeLa/TO cells expressing EGFP‐tagged E2F7 or E2F8 or EGFP only, and immunoprecipitated nuclear protein extracts with GFP‐trap beads. We then measured the relative abundances of proteins from EGFP‐tagged E2F7 or E2F8 over EGFP only (Fig EV5A). We selected only proteins with relative enrichment above twofold from both forward and reverse labeling experiments. With this threshold, we found 86 putative binding partners for both E2F7 and E2F8 (Table EV1). We noticed that multiple 14‐3‐3 protein isoforms were among the most highly enriched common interaction partners (Fig 5A). The 14‐3‐3 proteins are dimeric proteins, which modulate functions of their interaction partners primarily via binding in a phosphorylation‐dependent manner (Jones et al, 1995; Muslin et al, 1996). To verify these interactions, EGFP‐tagged E2F7 or E2F8 wild‐type or alanine mutants were co‐transfected with Myc‐tagged 14‐3‐3 isotypes ζ and ε in HEK 293T cells, followed by GFP immunoprecipitation and Myc immunoblotting. Strikingly, wild‐type E2F7 and E2F8 bind to 14‐3‐3 ζ and ε, and these interactions were completely lost in E2F7S411A and E2F8S395A mutants (Figs 5B and EV5B). These data show that the Chk1 phosphorylation site facilitates 14‐3‐3 binding to E2F7 and E2F8. Consistently, endogenous phosphorylated E2F7 and E2F8 were immunoprecipitated with Myc‐14‐3‐3 ζ, implying that E2F7/8 binds to 14‐3‐3 ζ in vivo (Fig EV5C). One of the known functions of 14‐3‐3 protein is to modulate the localization of its binding partner (Muslin & Xing, 2000). However, phosphorylation mutant E2F7 and E2F8 remained strictly localized in the nucleus and bind chromatin to a similar extent (Figs EV2C and EV3C, and 3E). Furthermore, co‐transfection of Myc‐tagged 14‐3‐3 ζ with GFP‐tagged E2F7 and E2F8 on U2OS cells revealed no relocalization of the atypical E2Fs to the cytosol (Fig EV5D).
Figure EV5. 14‐3‐3 proteins interact with E2F7/8 at the promoter regions.

- Experimental procedures of the SILAC assay to identify binding partners of E2F7 and E2F8.
- 14‐3‐3 ε interacts with wild‐type but not alanine mutant E2F7 and E2F8 in vitro. HEK 293T cells were transfected as indicated, and lysates were precipitated with GFP‐Trap beads and immunoblotted with antibodies directed against GFP or Myc. Inputs confirm similar expression of Myc‐14‐3‐3 ε in all samples.
- 14‐3‐3 ζ interacts with endogenous phosphorylated E2F7 and E2F8 in vivo. HEK 293T cells were transfected with Myc‐14‐3‐3 ζ and treated with or without HU. Lysates were precipitated with anti‐Myc antibodies and immunoblotted with antibodies directed against the Chk1 phosphorylation site of E2F7S410 or E2F8S396 (asterisks indicate the signal). Inputs confirm similar expression of Myc‐14‐3‐3 ζ in all samples.
- 14‐3‐3 ζ does not alter the nuclear localization of E2F7/8. U2OS cells were transfected with Myc‐14‐3‐3 ζ with either empty vector, EGFP wild‐type E2F7 or E2F8 followed by immunofluorescence staining. DAPI was used to stain nuclear DNA. Scale bar: 5 μm.
- Chromatin immunoprecipitation (ChIP) demonstrated that 14‐3‐3 proteins bind to the E2F7/8 target gene promoters. HEK cells were transfected with either PEI reagent alone (control) or indicated plasmids. 48 h after transfection, cells were harvested for ChIP assay and followed by qPCR. Histogram represents the enrichment ratio (bound/input) in E2Fs target gene promoters. A primer set designed against a distal region in the E2F1 gene served as a negative control.
- Validation of E2F7 and E2F8 knockdown efficiency of experiments shown in Fig 5F. E2F7 and E2F8 mRNA levels was determined by qPCR.
Figure 5. 14‐3‐3 ζ mediates Chk1‐dependent inhibition of E2F7/8.
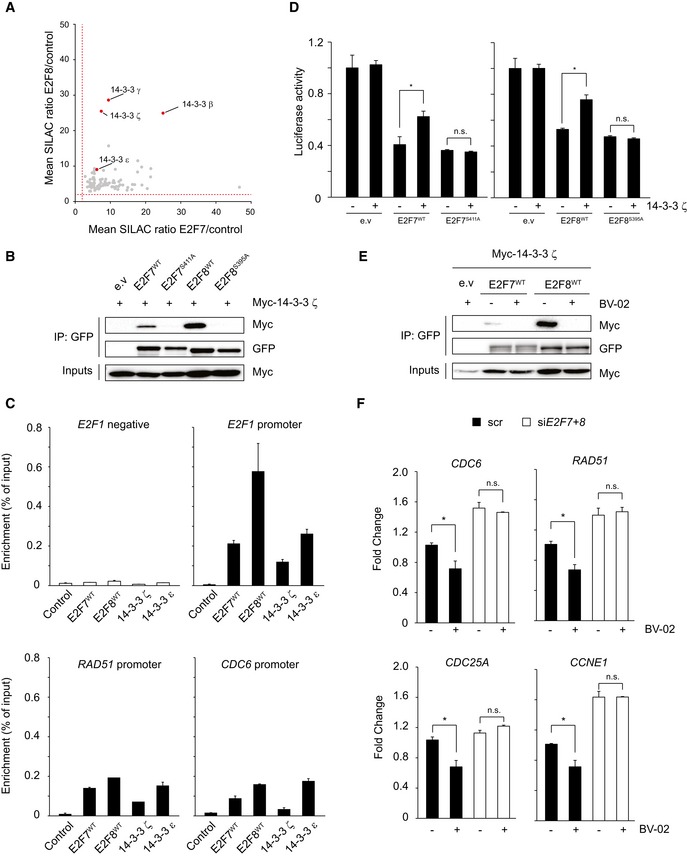
- Scatter plot shows the relative enrichment scores of common E2F7 and E2F8 binding partners. Red dashed line indicates the fold change cutoff (> 2.0). 14‐3‐3 isoforms are highlighted as red dots.
- 14‐3‐3 ζ interacts with wild‐type E2F7/8 but not with alanine mutants in vitro. HEK 293T cells were transfected with indicated constructs, and lysates were precipitated with GFP‐Trap beads and immunoblotted with antibodies directed against Myc or GFP. Inputs represent loading controls.
- Chromatin immunoprecipitation (ChIP) demonstrated that 14‐3‐3 proteins bind to the E2F7/8 target gene promoters. HEK cells were transfected with either PEI reagent alone (control) or indicated plasmids. 48 h after transfection, cells were harvested for ChIP assay and followed by qPCR. Histogram represents the enrichment ratio (bound/input) in E2Fs target gene promoters. A primer set designed against a distal region in the E2F1 gene served as a negative control.
- Luciferase reporter assay in U2OS cells using E2F1 promoter plasmid. Plasmids were co‐transfected either with wild‐type or alanine mutant, either alone or with 14‐3‐3 ζ plasmid.
- 14‐3‐3 inhibitor BV‐02 disrupts the interaction between 14‐3‐3 ζ and E2F7/8. Transfection was carried out as shown, and lysates were precipitated with GFP‐Trap beads and immunoblotted with antibodies directed against GFP or Myc. Inputs represent loading controls.
- Transcript levels of common E2F7 and E2F8 target genes after treatment with HU or HU + BV‐02 for 16 h, determined by qPCR.
Previous work demonstrated that 14‐3‐3 proteins can be found in chromatin (Milton et al, 2006). Thus, we reasoned that 14‐3‐3 proteins can bind to phosphorylated E2F7 and E2F8 at their promoter regions to modulate their functions. To test our hypothesis, we performed ChIP to investigate whether 14‐3‐3 proteins are enriched at the E2F7/8 target promoters. In the ChIP assay, wild‐type E2F7 and E2F8 were set as positive controls, and chromatin fractions were immune‐precipitated by either GFP‐trap resin or 14‐3‐3 antibody. As expected, E2F7 and E2F8 are specifically enriched at the promoter regions of their target genes such as E2F1, CHEK1, RAD51, MCM2, and CDC6 (Figs 5C and EV5E). More importantly, 14‐3‐3 proteins (ζ and ε) also showed significant enrichment toward the promoters of the same E2F7/8 target genes. Therefore, our data indicated that 14‐3‐3 proteins bind to promoter regions of E2F target genes likely via interaction with E2F7/8. Next, we investigated whether binding of 14‐3‐3 proteins to phosphorylated E2F7 and E2F8 regulates atypical E2F repressor activity by performing reporter assays. U2OS cells were co‐transfected with EGFP‐tagged E2F7 or E2F8, 14‐3‐3 ζ, and an E2F1 promoter‐luciferase reporter plasmid. 14‐3‐3 ζ constructs partially inhibited the repressor activity of wild‐type E2F7 and E2F8 on the E2F1 promoter (Fig 5D). In contrast, co‐transfection of 14‐3‐3 ζ with alanine mutant E2F7 and E2F8 could not inhibit the atypical E2F repressor activity, which suggests that 14‐3‐3 ζ binding mediates inhibition of E2F7 and E2F8 repressor function via binding to Chk1 phosphorylation sites. If 14‐3‐3 attenuates the repressor function of atypical E2Fs, then disruption of this interaction should result in an enhanced suppression of E2F target genes. To test this hypothesis, we used a 14‐3‐3 docking inhibitor BV‐02 with proven efficacy to disassociate 14‐3‐3 interactions with its binding partners (Mancini et al, 2011). Indeed, we found that BV‐02 treatment abolished the binding of 14‐3‐3 ζ to both E2F7 and E2F8 (Fig 5E). We then treated cells with HU to activate Chk1 phosphorylation and evaluated the effect of BV‐02 on E2F7/8 target gene expression. Compared to DMSO‐treated controls, BV‐02 treatment significantly downregulated transcript levels of a panel of E2F target genes, including CDC6, Rad51, CDC25A, and CCNE1, and had no impact on E2F7 and E2F8 (Figs 5F and EV5F). To evaluate whether BV‐02 downregulates E2F target genes via E2F7 and E2F8, we knocked down E2F7 and E2F8 expressions with siRNA technology (Fig EV5F). Remarkably, under siE2F7+8 conditions, BV‐02 treatment did not result in downregulation of E2F target genes anymore, indicating that BV‐02‐mediated repression is dependent on the presence of atypical E2Fs. Taken together, phosphorylation by Chk1 creates a docking site for 14‐3‐3 proteins to directly inhibit the repressor function of E2F7/8.
We next investigated whether binding of 14‐3‐3 proteins to E2F7/8 could alter the dimerization between atypical E2Fs or their interaction with E2F1. However, co‐immunoprecipitation experiments revealed that homodimerization and heterodimerization between the atypical E2Fs and their binding to E2F1 were not affected by 14‐3‐3 proteins (Appendix Fig S1A).To determine whether 14‐3‐3 binding has impact on other interaction partners of the atypical E2Fs, we performed an additional SILAC experiment. This time, we pulled down E2F7WT or E2F7S411A and investigated the differential interaction with other proteins. As expected, we could observe reduced pulldown of 14‐3‐3 proteins by the alanine mutant, confirming the validity of the assay (Appendix Table S1). However, only very few other proteins were differentially enriched, and the majority of those are suspicious contaminating proteins in mass spectrometry experiments and none of these differentially expressed proteins are known for the capability to function as co‐repressors. Taken together, this finding suggests 14‐3‐3 recruitment to E2F7/8 target promoters does not alter the dimerization or the interaction of co‐repressors of atypical E2Fs. 14‐3‐3 binding might be still able to physically hinder the intrinsic repressor function of DNA‐bound E2F7/8, which would require the design of complex studies to test the conformational status of atypical E2Fs.
Gene amplification of the 14‐3‐3 ζ is associated with upregulation of E2F7/8 target genes in human cancer
We noticed that the gene YWHAZ, which encodes for 14‐3‐3 ζ, is frequently amplified in various types of cancer and associated with oncogenic activity (Lin et al, 2009; Li et al, 2010; Chen et al, 2012). Additionally, E2F target genes are over‐expressed in many types of cancer, including hepatocellular carcinoma, despite high expression of the E2F7 and E2F8 repressors (Kent et al, 2016). Since 14‐3‐3 has the ability to inhibit the transcriptional repressor function of the atypical E2Fs, we wondered whether 14‐3‐3 expression correlates with increased expression of E2F7/8 target genes in human cancer. To this end, we first analyzed the expression data provided by the R2 platform of the AMC oncogenomics project [R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl)]. Four types of cancer, breast invasive carcinoma, prostate cancer, pancreas cancer, and liver hepatocellular carcinoma, were enrolled, based on the high frequency of YWHAZ amplifications in these types of cancer (15–30%). R value (> 0.3) and P‐value (< 0.01) were set to identify which upregulated E2F target transcripts correlate with upregulated 14‐3‐3 ζ transcripts. For this query, 79 known E2F7/8 target genes(Westendorp et al, 2012; Kent et al, 2016) were selected and are listed in Table EV2. We found a strong positive correlation between a large set of highly expressed E2F7/8 target transcripts and high transcript levels of 14‐3‐3 ζ (Fig 6A). It is noteworthy that genes encoding 14‐3‐3 proteins are not E2F target genes by themselves, neither do they oscillate throughout the cell cycle (Dougherty & Morrison, 2004). This rules out that the co‐expression of 14‐3‐3 ζ transcripts and E2F target genes could simply be explained by variation in the numbers of proliferating cells between tumor samples. In a second approach, we analyzed RNA‐sequencing data of primary hepatocellular carcinoma in The Cancer Genome Atlas (TCGA). We compared the expression of the 79 known E2F7/8 target genes in the 10% highest and lowest 14‐3‐3 ζ‐expressing tumor samples. Supporting our notion, highest 14‐3‐3 ζ RNA levels associated with amplified or copy number gains of the YWHAZ gene, supporting a gene dosage effect (Fig 6B). More importantly, the heatmap analysis demonstrated that E2F7/8 target genes were upregulated predominately in the samples with the highest 14‐3‐3 ζ expression. Similarly, high levels of 14‐3‐3 ε mRNA, the other E2F7/8 binding partner, correlated strongly with high E2F7/8 target genes (Fig 6C), indicating that 14‐3‐3 ε also negatively regulates transcription function of E2F7 and E2F8. These results indicate that upregulation of 14‐3‐3 proteins, for example, via YWHAZ copy number gains, may inactivate the atypical E2F repressor function, thereby permitting high levels of E2F‐dependent transcription in cancer.
Figure 6. High 14‐3‐3 ζ and ε expression levels are associated with high E2F7/8 target gene expression in human cancer.
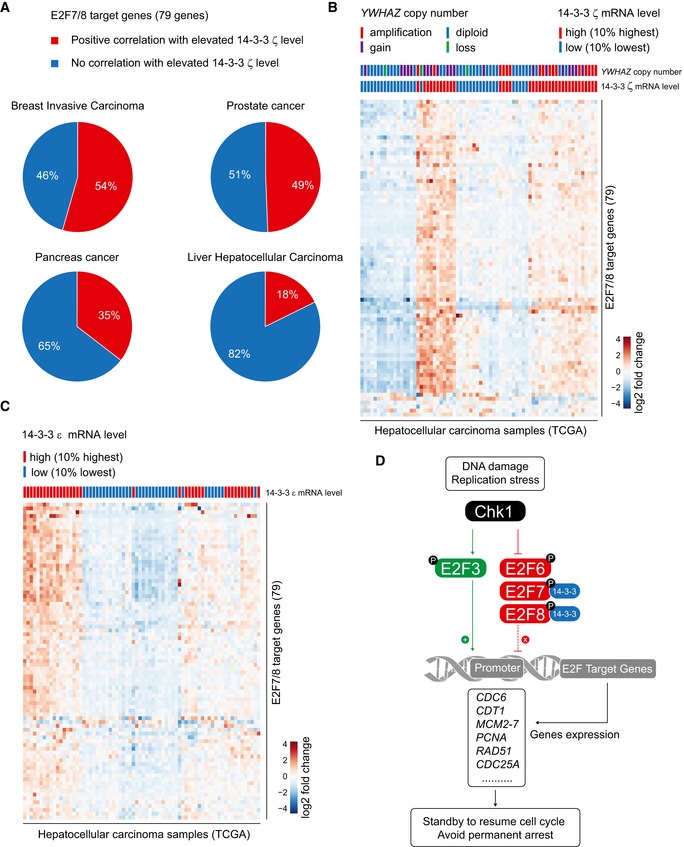
- Pie charts showing the percentage of upregulated E2F7/8 target genes that correlate with elevated 14‐3‐3 ζ expression in four types of cancer. In total, 79 known target genes were enrolled as E2F7/8 targets. In the R2 database, co‐expressed elevated 14‐3‐3 ζ and E2F7/8 target transcripts were selected based on regression analysis, then R (regression value) > 0.3 and P < 0.01 were set to distinguish the significant positive correlation.
- Heatmap showing a positive correlation between 14‐3‐3 ζ expressions with E2F7/8 target gene expression. Columns represent 82 hepatocellular carcinoma samples classified into either the 10% highest or lowest 14‐3‐3 ζ mRNA levels. Rows represent 79 E2F7/8 target genes. Amplification: multicopy gain; gain: single copy amplification; diploid: diploid chromosomes; loss: homozygous or heterozygous loss.
- Heatmap showing a positive correlation between 14‐3‐3 ε expressions with E2F7/8 target gene expression levels derived from the TCGA liver cancer RNA‐seq data. Column represents hepatocellular carcinoma samples classified into groups with either the 10% highest or lowest 14‐3‐3 ε transcript levels. Rows represent 79 E2F7/8 target genes.
- Summary model for the interaction of Chk1 and 14‐3‐3 with E2F7/8 and other Chk1‐E2Fs interplays under DNA damage and replication stress condition.
Discussion
The repressor activity of atypical E2Fs needs to be tightly regulated to avoid severe disturbances of cell cycle progression. Here, we show a novel mechanism for how the repressor activity of atypical E2Fs can be inhibited through direct phosphorylation by Chk1 which provides a docking station for 14‐3‐3 proteins to interact with E2F7 and E2F8 (Fig 6D). Previous studies have demonstrated that increased repressor activity of atypical E2Fs downregulates the expression of a large number of cell cycle genes resulting in a severe cell cycle arrest accompanied by increased DNA damage and apoptotic events (de Bruin et al, 2003; Maiti et al, 2005; Westendorp et al, 2012; Boekhout et al, 2016). Our findings provide strong evidence for a regulatory mechanism that suppresses the atypical E2F activity in order to avoid detrimental effects on cell cycle progression and cell survival under DNA‐damaging conditions.
In response to DNA damage, Chk1 is activated and whereby it inhibits the Cdc7/Dbf4 complex and facilitates the degradation of CDC25A resulting in S‐phase arrest. This sequence of molecular events transiently prevents the firing of new replication origins, contributing to the maintenance of genomic integrity (Zhao et al, 2002; Heffernan et al, 2007). Nonetheless, it is also essential to keep the DNA replication machinery ready to resume once the lesions are resolved, as long‐term replication fork stalling is catastrophic to cell survival (Dimitrova & Gilbert, 2000; Lopes et al, 2001). Recent work showed that the persistent transcription of E2F target genes during DNA repair which means that replication can readily be resumed is key to prevent further replication stress‐induced DNA damage (Bertoli et al, 2016). Chk1 plays an important role in the maintenance of E2F‐dependent transcription since it reduces the binding of the E2F6 repressor to its promoter, while the functions of the activating E2Fs (E2F1, E2F3) are stimulated by DNA damage signaling (Stevens et al, 2003; Martinez et al, 2010; Bertoli et al, 2013). Here, we identify a novel pathway by which E2F target transcription is regulated by showing that Chk1 phosphorylates and attenuates the repressor activity of E2F7 and E2F8 under DNA‐damaging conditions.
Our findings show that unlike for other E2F family members, Chk1 phosphorylation has no impact on either the stability or DNA‐binding ability of E2F7 and E2F8. Instead, Chk1 phosphorylation creates a binding site for 14‐3‐3 proteins. The 14‐3‐3 family has overwhelming functional diversity in the regulation of cellular processes including signal transduction, cell cycle progression, transcription regulation, and DNA damage signaling (Dougherty & Morrison, 2004). Previous studies showed that 14‐3‐3 stimulated E2F transcription through stabilizing E2F1 and binding to DP‐3 (Dimerization Partner 3) (Wang et al, 2004; Milton et al, 2006). Based on our data, we excluded the possibility that 14‐3‐3 proteins relocate or de‐stabilize atypical E2Fs. Since our ChIP assay demonstrated the enrichment of 14‐3‐3 at the E2F7/8 target gene promoters, 14‐3‐3 regulates the transcriptional repressor activity of the atypical E2Fs most likely in the chromatin. Previous studies have shown that 14‐3‐3 dimer binding can result in an extended conformation change of its target peptide and that 14‐3‐3 dimer can bind with very high affinity to target peptide containing two phosphoserine motifs (Yaffe et al, 1997). Although 14‐3‐3 does not alter the binding affinity between atypical E2Fs, it is possible that 14‐3‐3 can structurally alter the conformation of the E2F7/8 dimers by positioning itself in between the two individual molecules and thereby inhibit the transcriptional repressor function of the atypical E2Fs.
We also evaluated whether ATR, one of the upstream regulator of Chk1, is required for controlling cell cycle progression via E2F7/8, but inactivation of ATR had no obvious impact on cell cycle progression under HU conditions (Appendix Fig S1B). It should be noted that although Chk1 activation inhibited the repressor activity of E2F7 and E2F8, this is presumably not a complete inactivation. Firstly, the phospho‐mimetic versions of E2F7 and E2F8 (D‐mutants) could still partially repress E2F target gene expression. Secondly, we previously found that deletion of E2F7/8 abrogated the cell cycle arrest in keratinocytes treated with the topoisomerase inhibitor etoposide (Thurlings et al, 2016). Etoposide treatment causes activation of Chk1, suggesting that residual activity of E2F7/8 mediates a cell cycle arrest under these conditions. Importantly, we show here that cells expressing non‐phosphorylatable forms of E2F7/8 underwent severe apoptosis. Based on these observations, we propose that carefully balanced activity of the atypical E2Fs is critical to induce S‐phase arrest, while preventing detrimental effects on cell survival during DNA replication stress.
The novel Chk1/14‐3‐3/E2F7/8 axis that we demonstrate in the present work has substantial relevance to cancer biology. Atypical E2Fs can inhibit cell proliferation by repressing a large network of genes that are essential for S‐phase progression. In line with this function, we recently demonstrated in two different mice conditional knockout models that atypical E2Fs act as tumor suppressors (Kent et al, 2016; Thurlings et al, 2016). Paradoxically, E2F7 and E2F8 transcripts are highly expressed in multiple types of human cancer, and their high expression levels often correlated with poor prognosis (Endo‐Munoz et al, 2009; Deng et al, 2010; Park et al, 2015). It is possible that the elevated expression of E2F7 and E2F8 reflects a high proliferative status of cancer cells (Kent et al, 2016). Moreover, since atypical E2Fs can be transcriptionally upregulated by other E2Fs activators (de Bruin et al, 2003; Maiti et al, 2005), high E2F7/8 expression could result from high levels of E2F activator‐dependent transcription within a tumor sample. Crucially, our current data show that expression levels of atypical E2Fs do not necessarily represent their activity, since Chk1‐dependent binding of 14‐3‐3 proteins can attenuate their repressor function. Moreover, Chk1 protein is frequently induced in many cancers (Verlinden et al, 2007; Xu et al, 2013), and corresponding Chk1 phosphorylation of E2F7 and E2F8 was identified in proteomic screens of cancer tissues and cell lines (Zhou et al, 2013; Mertins et al, 2016). Thus, E2F7 and E2F8 could be functionally inhibited in cancer cells via Chk1 and 14‐3‐3, despite their high transcript levels.
Noteworthy is the finding that the 14‐3‐3 ζ isoform which modulates cell cycle and metabolism (Lim et al, 2015; Schoenwaelder et al, 2016) is also frequently amplified and upregulated in different types of cancer. It was found to potentiate the oncogenic capacity of cancer cells via reducing apoptosis, enhancing proliferation, and increasing chemotherapy resistance, via incompletely understood mechanisms (Matta et al, 2012). In the exploration of the TCGA transcriptome database, we identified a positive correlation between high 14‐3‐3 ζ expression and increased expression of many E2F7/8 target genes. This is consistent with our observation that 14‐3‐3 ζ inhibits E2F7/8 function. Our data suggest that 14‐3‐3 could compromise the transcriptional repressor function of E2F7 and E2F8 in cancer cells by maintaining high levels of E2F target gene expression and thereby potentially promoting uncontrolled cell proliferation.
Materials and Methods
Cell culture, cell line generation, and transfection
HeLa, U2OS, hTERT‐RPE1, and HEK 293T cell lines were obtained from the ATCC and cultured in DMEM (41966052, Thermo Fisher Scientific) containing 10% fetal bovine serum (10500064, Life Technologies). Site‐directed mutagenesis was performed by two‐step PCR amplification (PCR protocol and primers are provided in Table EV3), and mutation was confirmed with Sanger sequencing (Macrogen, Inc). Tet repressor‐expressing HeLa‐inducible cell lines (HeLa/TO) were generated as previously described (Westendorp et al, 2012). The inducible cells lines were cultured in DMEM containing 10% Tet‐approved fetal bovine serum (CL 631106, Clontech), and over‐expression was induced by adding 0.2 μg/ml doxycycline (D9891, Sigma‐Aldrich). Other drugs used in this study were as follows: etoposide (10 μM, E1383, Sigma‐Aldrich); hydroxyurea (2 mM, H8627, Sigma‐Aldrich); thymidine (2 mM, T9250, Sigma‐Aldrich); camptothecin (20 μM, C9911, Sigma‐Aldrich), cycloheximide (50 μg/ml, 01810, Sigma‐Aldrich); UCN‐01(0.3 μM, U6508, Sigma‐Aldrich); and BV‐02 (5 nM, SML0140, Sigma‐Aldrich).
Transfection of HEK cells was performed by mixing 130 μg/ml PEI (polyethylenimine, 23966, Polysciences) with desired plasmids (15 μg) at a ratio of 1:1 in DMEM. Mixtures were added to the cells for 6 h and then replaced with fresh medium. U2OS cells were transfected with Lipofectamine 2000 (11668019, Thermo Fisher Scientific) according to the protocol. ON‐Target plus SMARTpool siRNAs (2 nM) were products from GE Dharmacon; transfection was performed according to the manufacturer's protocol utilizing RNAiMax (13778075, Thermo Fisher Scientific). The following siRNA products were used: Dharmacon L‐003255‐00‐0005 (siCHEK1), Thermo Fisher HSS175354 (siE2F7), Thermo Fisher HSS128758/HSS128760 (siE2F8), Dharmacon D‐001210‐02‐05 (Scrambled).
Mass spectrometry
For SILAC experiments, HeLa/TO‐inducible cells lines were cultured with Heavy/Light Medium for 2 weeks to incorporate isotopically labeled lysine (K8, 282986440, Silantes) and arginine (R10, 282986404 Silantes). Sixteen hours after the induction of over‐expression with doxycycline, cells were lysed in RIPA buffer with 50 mM Tris–HCl, 1 mM EDTA, 150 mM NaCl, 0.25% deoxycholic acid, 1% Nonidet‐P40, 1 mM NaF and NaV3O4, and protease inhibitor cocktail (11873580001, Sigma‐Aldrich), and desired proteins were immunoprecipitated. When applicable, heavy and light samples were mixed. Precipitated proteins were denatured with 8 M urea in 1 M ammonium bicarbonate (ABC) reduced with 10 mM TCEP at RT for 30 min after which the cysteines were alkylated with chloroacetamide (40 mM end concentration) for 30 min. After fourfold dilution with ABC, proteins were on‐bead digested overnight at RT with 150 ng of Trypsin/LysC (V5071, Promega). After sample cleanup with in‐house‐made stage tips, peptides were separated on a 30‐cm pico‐tip column (50 μm ID, New Objective) and were in‐house packed with 3 μm aquapur gold C‐18 material (Dr. Maisch) using a 140‐min gradient (7–80% ACN 0.1% FA), delivered by an easy‐nLC 1000 (LC120, Thermo Scientific), and electro‐sprayed directly into an Orbitrap Fusion Tribrid Mass Spectrometer (IQLAAEGAAPFADBMBCX, Thermo Scientific). Raw files were analyzed with the MaxQuant software version 1.5.1.0. with oxidation of methionine and STY phosphorylation set as variable modifications, and carbamidomethylation of cysteine set as fixed modification. The Human protein database of UniProt, with the mouse E2F8 sequence added, was searched with both the peptide as well as the protein false discovery rate set to 1%.
Immunoblot and immunoprecipitation
Cells were washed twice with PBS and scraped from the culture dish. RIPA buffer consisting of 50 mM Tris–HCl, 1 mM EDTA, 150 mM NaCl, 0.25% deoxycholic acid, 1% Nonidet‐P40, 1 mM NaF and NaV3O4, and protease inhibitor cocktail (11873580001, Sigma‐Aldrich) was added to lyse the cells. After 12,000 g centrifugation for 10 min, supernatants were collected and proceed to a standard SDS–PAGE Immunoblot. The phospho‐specific antibodies against E2F7S411 and E2F8S395 were customized products from Davids Biotechnologie GmbH, Germany. All antibodies used in this paper are listed in Table EV3. For immunoprecipitations, cell lysates were obtained with RIPA buffer and immunoprecipitation was carried out by incubating with 4 μg of the desired antibody per IP (Table EV3) and Protein G Plus/Protein A Agarose Suspension (IP05‐1.5, Calbiochem). For Flag‐IP and GFP‐IP in this study, 20 μl of anti‐FLAG M2 Affinity Gel (A2220, Sigma‐Aldrich) and GFP‐Trap (gta‐20, ChromoTek) were used, respectively. After the pulldown, the agarose beads were washed three times with RIPA and PBS before proceeding to a standard SDS–PAGE immunoblot.
Kinase assay
The desired proteins were isolated with immunoprecipitation as described above. Proteins were washed with TE buffer (20 mM Tris pH 7.5, and 2 mM EDTA). Incubation was carried out in a reaction buffer with TE buffer plus 20 mM MgCl2, 1 mM DTT, 200 μM ATP, in addition to 1 μl ATP32 and 1 μl recombinant active Chk1 (1630‐KS‐010, R&D Systems) per reaction. Samples were incubated 30 min at 30°C with gentle shaking. Laemmli buffer was added to stop the reaction. Then samples were boiled and loaded on an SDS–PAGE gel. Radioactive signal was visualized by exposure to a film (28906836, GE Healthcare).
Flow cytometry and sorting
For the cell cycle analysis, cells were trypsinized, fixed with 70% ethanol, and stored at 4°C. Cells were washed twice with TBS and then resuspended with propidium iodide (PI, P4170, Sigma‐Aldrich) staining buffer containing 20 μg/ml PI, 250 μg/ml RNase A, and 0.1% bovine serum albumin. Samples were analyzed on a BD FACSCanto II flow cytometer. For Annexin‐V staining, cells were trypsinized and staining was performed according to manufacturer's instructions (A35110, Life Technologies). The BD Influx system was used to sort GFP‐expressing cells (Dox) and GFP‐negative cells (Veh). Cells were collected and washed twice with PBS before sorting. For each RNA isolation, 25,000 cells were harvested.
Immunofluorescence, stripe assay, and live imaging
Cells were seeded over coverslips (5 mm) and treated with desired condition as indicated. Cells were first treated with ice‐cold 0.2% Triton X‐100 for 1 min, then fixed with 4% PFA at room temperature for 20 min, followed by 0.1% Triton X‐100 at room temperature 20 min. 4% BSA or 10% goat serum were used as blocking reagents, and samples were then treated with primary and secondary antibodies, respectively, at room temperature for 2 h. Nuclei were stained with DAPI (F6057, Sigma‐Aldrich), and coverslips were mounted using Fluoroshield™. Antibodies used in IF are listed in Table EV3.
For live cell imaging, 8,000 HeLa/TO cells were subcultured into a glass‐bottom μ‐Slide 8‐well plate and treated with the desired conditions. Before live imaging, SiR‐DNA (1 μM, SC007, Spirochrome) was added to visualize DNA. For the stripe assay, micro‐irradiation of the nucleus was performed with a 405‐nm laser diode and 4 mW and 50% power, and analyzed with the Nikon A1R microscope system. Nuclear fluorescence enrichment was observed for up to 1 h. Afterward, immunofluorescence staining was performed on the cells that had undergone micro‐irradiation. Photographs were acquired from EGFP (488 nm), far‐red (652 nm), and DIC channels every 30 min.
Quantitative PCR
Isolation of RNA, cDNA synthesis, and quantitative PCR were performed as previously described (Westendorp et al, 2012). Gene transcript levels were determined using ΔΔC t method for multiple‐reference gene correction. β‐Actin and GAPDH were used as references. Primer sequences are provided in Table EV3.
Reporter assay
U2OS cells (50,000 per triplicate) were seeded to three triplicate wells in a 24‐well plate for each transfection. Each transfection mix (10 μl per well) contains 500 ng E2F1 luciferase reporter, 20 ng TK (thymidine kinase) renilla, 100 ng expression or control plasmid, and 5 μl Superfect Reagent (301305, Qiagen). After 48 h of transfection, reporter signal was measured using the Dual‐Luciferase Reporter Assay System (E1910, Promega) on a microplate luminometer (Centro LB960). TK was used for normalization of the result.
Clonogenic survival assay
HeLa cells (300,000 per duplicate) were seeded into a six‐well plate. siRNA transfection was conducted after 24 h of cell seeding. Twenty‐four hours after siRNA transfection, cells were trypsinized and reseeded to a new six‐well plate (20,000 per well). Cells were treated with or without HU for 72 h. Then, medium was removed with PBS, and cells were fixed with acetic acid/methanol 1:7 (vol/vol) for 5 min. Colonies were stained with 0.5% crystal violet for 2 h and then rinsed in tap water. Pictures were taken and loaded to Photoshop CS6 for quantification. For each condition, 10 fields (300 × 300 pixels) were randomly selected. Positive colony staining was measured with Magic Wand Tool, with tolerance value set to 32. Relative intensity was defined as the ratio of positive purple‐staining area (in pixels) over total area (300 × 300 pixels). Histograms in Fig EV4F show the quantification from two independent experiments.
R2 database and TCGA data analysis
In the R2 platform, four expression datasets were selected for the pie chart (Fig 6A): Tumor Breast Invasive Carcinoma‐TCGA‐1097‐rsem‐tcgars; Tumor Prostate Adenocarcinoma‐TCGA‐497‐rsem‐tcgars; Tumor Pancreatic adenocarcinoma‐TCGA‐178‐rsem‐tcgars; Tumor Liver Hepatocellular Carcinoma‐TCGA‐371‐rsem‐tcgars. Type of analysis: Find Correlated genes with a single gene and gene name: YWHAZ. Genes were filtered with R value (> 0.3) and P‐value (< 0.01). Analysis of TCGA data was performed as follows: All available RNA‐sequencing and copy number variation data from the liver cancer dataset (LIHC) were downloaded as level 3 data. Subsequent analysis was performed in Rstudio version 3.3.0 using the packages “rjson”, “parallel”, “GenomicRanges”, and “Hmisc” in combination with a number of custom R scripts and functions to merge all data, remove non‐tumor samples, and call segment mean values for the YWHAZ gene locus in each sample. FPKM values (Fragments Per Kilobase Million) were used to calculate log‐fold changes, and heatmaps were generated using the R package Pheatmap. All used codes are available on request.
Statistical analysis
Immunoblots (except kinase assay), immunoprecipitation, reporter assay, flow cytometry, FACS, and qPCR results were repeated three times unless otherwise stated in the figure legends. Percentages of cells in S phase were calculated using the cell cycle analysis function in FlowJo v10.0 software. Statistical analyses on FACS qPCR in Fig 3 was done using ANOVA followed by Dunnett's post hoc comparisons. Statistical analyses on Annexin‐V, qPCR and reporter assay, staining in Figs 3, 4, 5 were analyzed with the t‐test.
Author contributions
RY and BW performed experiments. JC performed the siRNA experiments in Fig 4. HRV, RMvE, and BMTB performed and designed kinase assay, mass spectrometry, and SILAC experiment. RY, BW, and AdB conceived the study design and experimental approaches, data analysis, and manuscript writing.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Ger Arkesteijn (Faculty of Veterinary Medicine, Utrecht University, NL) for providing professional assistance with the FACS sorting. Richard Wubbolts and Esther van‘t Veld in the Center for Cellular Imaging (Faculty of Veterinary Medicine, Utrecht University, NL) provided technical support with the stripe assays and live cell imaging. We thank Rachel Thomas for her contribution to manuscript revision. This work is financially supported by the China Scholarship Council (CSC) (File No. 201306380101), “Proteins at Work” program of The Netherlands Organization for Scientific Research (NWO) (project number 184.032.201), a Dutch Cancer Society funding (KWF: UU2013‐5777) to Bart Westendorp and Alain de Bruin, a ZonMW grant 91116011, and by the Netherlands Organization for Scientific Research (NWO: ALW‐IN11‐28) to Alain de Bruin.
The EMBO Journal (2018) 37: e97877
Contributor Information
Bart Westendorp, Email: b.westendorp@uu.nl.
Alain de Bruin, Email: a.debruin@uu.nl.
References
- Aksoy O, Chicas A, Zeng T, Zhao Z, McCurrach M, Wang X, Lowe SW (2012) The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev 26: 1546–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Klier S, McGowan C, Wittenberg C, de Bruin RA (2013) Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell‐cycle transcription. Curr Biol 23: 1629–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Herlihy AE, Pennycook BR, Kriston‐Vizi J, de Bruin RA (2016) Sustained E2F‐dependent transcription is a key mechanism to prevent replication‐stress‐induced DNA damage. Cell Rep 15: 1412–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhout M, Yuan R, Wondergem AP, Segeren HA, van Liere EA, Awol N, Jansen I, Wolthuis RM, de Bruin A, Westendorp B (2016) Feedback regulation between atypical E2Fs and APC/CCdh1 coordinates cell cycle progression. EMBO Rep 17: 414–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin A, Maiti B, Jakoi L, Timmers C, Buerki R, Leone G (2003) Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem 278: 42041–42049 [DOI] [PubMed] [Google Scholar]
- Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN (2000) The radiosensitizing agent 7‐hydroxystaurosporine (UCN‐01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res 60: 2108–2112 [PubMed] [Google Scholar]
- Carvajal LA, Hamard PJ, Tonnessen C, Manfredi JJ (2012) E2F7, a novel target, is up‐regulated by p53 and mediates DNA damage‐dependent transcriptional repression. Genes Dev 26: 1533–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Lowe M, Keyomarsi K (1999) UCN‐01‐mediated G1 arrest in normal but not tumor breast cells is pRb‐dependent and p53‐independent. Oncogene 18: 5691–5702 [PubMed] [Google Scholar]
- Chen CH, Chuang SM, Yang MF, Liao JW, Yu SL, Chen JJ (2012) A novel function of YWHAZ/beta‐catenin axis in promoting epithelial‐mesenchymal transition and lung cancer metastasis. Mol Cancer Res 10: 1319–1331 [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Grant S (2010) New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res 16: 376–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Wang Q, Zong WY, Zheng DL, Wen YX, Wang KS, Teng XM, Zhang X, Huang J, Han ZG (2010) E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res 70: 782–791 [DOI] [PubMed] [Google Scholar]
- Dimitrova DS, Gilbert DM (2000) Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol 2: 686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Morrison DK (2004) Unlocking the code of 14‐3‐3. J Cell Sci 117: 1875–1884 [DOI] [PubMed] [Google Scholar]
- Endo‐Munoz L, Dahler A, Teakle N, Rickwood D, Hazar‐Rethinam M, Abdul‐Jabbar I, Sommerville S, Dickinson I, Kaur P, Paquet‐Fifield S, Saunders N (2009) E2F7 can regulate proliferation, differentiation, and apoptotic responses in human keratinocytes: implications for cutaneous squamous cell carcinoma formation. Cancer Res 69: 1800–1808 [DOI] [PubMed] [Google Scholar]
- Gong C, Liu H, Song R, Zhong T, Lou M, Wang T, Qi H, Shen J, Zhu L, Shao J (2016) ATR‐CHK1‐E2F3 signaling transactivates human ribonucleotide reductase small subunit M2 for DNA repair induced by the chemical carcinogen MNNG. Biochim Biophys Acta 1859: 612–626 [DOI] [PubMed] [Google Scholar]
- Heffernan TP, Unsal‐Kacmaz K, Heinloth AN, Simpson DA, Paules RS, Sancar A, Cordeiro‐Stone M, Kaufmann WK (2007) Cdc7‐Dbf4 and the human S checkpoint response to UVC. J Biol Chem 282: 9458–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DH, Ley S, Aitken A (1995) Isoforms of 14‐3‐3 protein can form homo‐ and heterodimers in vivo and in vitro: implications for function as adapter proteins. FEBS Lett 368: 55–58 [DOI] [PubMed] [Google Scholar]
- Kent LN, Rakijas JB, Pandit SK, Westendorp B, Chen HZ, Huntington JT, Tang X, Bae S, Srivastava A, Senapati S, Koivisto C, Martin CK, Cuitino MC, Perez M, Clouse JM, Chokshi V, Shinde N, Kladney R, Sun D, Perez‐Castro A et al (2016) E2f8 mediates tumor suppression in postnatal liver development. J Clin Invest 126: 2955–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zou L, Li Q, Haibe‐Kains B, Tian R, Li Y, Desmedt C, Sotiriou C, Szallasi Z, Iglehart JD, Richardson AL, Wang ZC (2010) Amplification of LAPTM4B and YWHAZ contributes to chemotherapy resistance and recurrence of breast cancer. Nat Med 16: 214–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GE, Albrecht T, Piske M, Sarai K, Lee JT, Ramshaw HS, Sinha S, Guthridge MA, Acker‐Palmer A, Lopez AF, Clee SM, Nislow C, Johnson JD (2015) 14‐3‐3zeta coordinates adipogenesis of visceral fat. Nat Commun 6: 7671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Morrison CD, Jones S, Mohamed N, Bacher J, Plass C (2009) Copy number gain and oncogenic activity of YWHAZ/14‐3‐3zeta in head and neck squamous cell carcinoma. Int J Cancer 125: 603–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Cotta‐Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi‐Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Lopez‐Girona A, Furnari B, Mondesert O, Russell P (1999) Nuclear localization of Cdc25 is regulated by DNA damage and a 14‐3‐3 protein. Nature 397: 172–175 [DOI] [PubMed] [Google Scholar]
- Lundin C, Erixon K, Arnaudeau C, Schultz N, Jenssen D, Meuth M, Helleday T (2002) Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol 22: 5869–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J (2000) Rapid destruction of human Cdc25A in response to DNA damage. Science 288: 1425–1429 [DOI] [PubMed] [Google Scholar]
- Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R, Patil K, Tuttle J, Cleghorn W, Leone G (2005) Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem 280: 18211–18220 [DOI] [PubMed] [Google Scholar]
- Mancini M, Corradi V, Petta S, Barbieri E, Manetti F, Botta M, Santucci MA (2011) A new nonpeptidic inhibitor of 14‐3‐3 induces apoptotic cell death in chronic myeloid leukemia sensitive or resistant to imatinib. J Pharmacol Exp Ther 336: 596–604 [DOI] [PubMed] [Google Scholar]
- Martinez LA, Goluszko E, Chen HZ, Leone G, Post S, Lozano G, Chen Z, Chauchereau A (2010) E2F3 is a mediator of DNA damage‐induced apoptosis. Mol Cell Biol 30: 524–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta A, Siu KW, Ralhan R (2012) 14‐3‐3 zeta as novel molecular target for cancer therapy. Expert Opin Ther Targets 16: 515–523 [DOI] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E, Mundt F, Krug K, Tu Z, Lei JT, Gatza ML, Wilkerson M, Perou CM, Yellapantula V, Huang KL et al (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534: 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AH, Khaire N, Ingram L, O'Donnell AJ, La Thangue NB (2006) 14‐3‐3 proteins integrate E2F activity with the DNA damage response. EMBO J 25: 1046–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS (1996) Interaction of 14‐3‐3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84: 889–897 [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Xing H (2000) 14‐3‐3 proteins: regulation of subcellular localization by molecular interference. Cell Signal 12: 703–709 [DOI] [PubMed] [Google Scholar]
- Park SA, Platt J, Lee JW, Lopez‐Giraldez F, Herbst RS, Koo JS (2015) E2F8 as a novel therapeutic target for lung cancer. J Natl Cancer Inst 107: djv151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil M, Pabla N, Dong Z (2013) Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci 70: 4009–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Woodcock M, Helleday T (2010) Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci USA 107: 16090–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raleigh JM, O'Connell MJ (2000) The G(2) DNA damage checkpoint targets both Wee1 and Cdc25. J Cell Sci 113(Pt 10): 1727–1736 [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica‐Worms H, Elledge SJ (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Schoenwaelder SM, Darbousset R, Cranmer SL, Ramshaw HS, Orive SL, Sturgeon S, Yuan Y, Yao Y, Krycer JR, Woodcock J, Maclean J, Pitson S, Zheng Z, Henstridge DC, van der Wal D, Gardiner EE, Berndt MC, Andrews RK, James DE, Lopez AF et al (2016) 14‐3‐3zeta regulates the mitochondrial respiratory reserve linked to platelet phosphatidylserine exposure and procoagulant function. Nat Commun 7: 12862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara A, Ogawa H, Ogawa T (1992) Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA‐like protein. Cell 69: 457–470 [DOI] [PubMed] [Google Scholar]
- Stevens C, Smith L, La Thangue NB (2003) Chk2 activates E2F‐1 in response to DNA damage. Nat Cell Biol 5: 401–409 [DOI] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ (2003) MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421: 961–966 [DOI] [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25: 3553–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurlings I, Martinez‐Lopez LM, Westendorp B, Zijp M, Kuiper R, Tooten P, Kent LN, Leone G, Vos HJ, Burgering B, de Bruin A (2016) Synergistic functions of E2F7 and E2F8 are critical to suppress stress‐induced skin cancer. Oncogene 36: 829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlinden L, Vanden Bempt I, Eelen G, Drijkoningen M, Verlinden I, Marchal K, De Wolf‐Peeters C, Christiaens MR, Michiels L, Bouillon R, Verstuyf A (2007) The E2F‐regulated gene Chk1 is highly expressed in triple‐negative estrogen receptor/progesterone receptor/HER‐2 breast carcinomas. Cancer Res 67: 6574–6581 [DOI] [PubMed] [Google Scholar]
- Wang B, Liu K, Lin FT, Lin WC (2004) A role for 14‐3‐3 tau in E2F1 stabilization and DNA damage‐induced apoptosis. J Biol Chem 279: 54140–54152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp B, Mokry M, Groot Koerkamp MJ, Holstege FC, Cuppen E, de Bruin A (2012) E2F7 represses a network of oscillating cell cycle genes to control S‐phase progression. Nucleic Acids Res 40: 3511–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Li Y, Wang F, Wang X, Cheng B, Ye F, Xie X, Zhou C, Lu W (2013) Suppressed miR‐424 expression via upregulation of target gene Chk1 contributes to the progression of cervical cancer. Oncogene 32: 976–987 [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC (1997) The structural basis for 14‐3‐3: phosphopeptide binding specificity. Cell 91: 961–971 [DOI] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DA (2003) Chk1‐deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J 22: 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalmas LP, Zhao X, Graham AL, Fisher R, Reilly C, Coutts AS, La Thangue NB (2008) DNA‐damage response control of E2F7 and E2F8. EMBO Rep 9: 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalmas LP, Coutts AS, Helleday T, La Thangue NB (2013) E2F‐7 couples DNA damage‐dependent transcription with the DNA repair process. Cell Cycle 12: 3037–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hunter T (2014) Roles of Chk1 in cell biology and cancer therapy. Int J Cancer 134: 1013–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Watkins JL, Piwnica‐Worms H (2002) Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation‐induced S and G2 checkpoints. Proc Natl Acad Sci USA 99: 14795–14800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, Mohammed S (2013) Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res 12: 260–271 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5