

Miyai et al. used multipotent progenitors harboring a tamoxifen-inducible form of Id3—where virtually all cells became B cells within 6 d by withdrawing 4-OHT—to identify a three-step transcription factor network model during specification of multipotent progenitors toward the B-cell lineage.
Keywords: transcriptional network, epigenetics, single-cell RNA-seq, B-cell differentiation, lineage commitment, transcription factor
Abstract
Stem cell fate is orchestrated by core transcription factors (TFs) and epigenetic modifications. Although regulatory genes that control cell type specification are identified, the transcriptional circuit and the cross-talk among regulatory factors during cell fate decisions remain poorly understood. To identify the “time-lapse” TF networks during B-lineage commitment, we used multipotent progenitors harboring a tamoxifen-inducible form of Id3, an in vitro system in which virtually all cells became B cells within 6 d by simply withdrawing 4-hydroxytamoxifen (4-OHT). Transcriptome and epigenome analysis at multiple time points revealed that ∼10%–30% of differentially expressed genes were virtually controlled by the core TFs, including E2A, EBF1, and PAX5. Strikingly, we found unexpected transcriptional priming before the onset of the key TF program. Inhibition of the immediate early genes such as Nr4a2, Klf4, and Egr1 severely impaired the generation of B cells. Integration of multiple data sets, including transcriptome, protein interactome, and epigenome profiles, identified three representative transcriptional circuits. Single-cell RNA sequencing (RNA-seq) analysis of lymphoid progenitors in bone marrow strongly supported the three-step TF network model during specification of multipotent progenitors toward B-cell lineage in vivo. Thus, our findings will provide a blueprint for studying the normal and neoplastic development of B lymphocytes.
Cell fate is controlled by lineage-associated transcription factors (TFs) and epigenetic modification. TFs contribute mainly to the lineage specification process, which is followed by epigenetic maintenance that stabilizes the cell fate. Once cells are committed to a certain lineage, they never become other types of cells under physiological conditions. Therefore, both TFs and epigenetic factors collaborate to induce lineage commitment and promote cellular differentiation.
B cells are generated from pluripotent hematopoietic stem cells (HSCs) through a successive series of lineage restriction steps. HSCs gradually lose their self-renewal capacity and multipotency as cells progress through various lineages and then differentiate into more lymphoid-biased progenitors followed by a final commitment to the B-cell lineage. Many essential TFs, such as IKAROS, PU.1, E2A, EBF1, and PAX5, have been implicated in regulating cell fate choice in the B-cell lineage (Boller and Grosschedl 2014). IKAROS and PU.1 are important at the stage when HSCs become lymphoid-primed multipotent progenitors (LMPPs) (Rothenberg 2014). E2A proteins (E12 and E47) are members of the E-protein family that also includes HEB and E2-2 (Engel and Murre 2001). E2A proteins are essential for the development of LMPPs by regulating the TF FOXO1 (Welinder et al. 2011). The transcriptional activity of E2A is regulated by its natural inhibitors, the Id proteins (Id1–Id4), which heterodimerize with E2A through a helix–loop–helix (HLH) domain and suppress its DNA-binding activity (Engel and Murre 2001). Interaction of E2A–Id proteins controls the differentiation and maintenance of HSCs (Semerad et al. 2009; Mercer et al. 2011), the development of innate lymphoid cells (Constantinides et al. 2014), and the generation of T-cell subsets (Miyazaki et al. 2015). E2A regulates expression of many genes essential for early B-cell development, including Ebf1, Bcl11a, Irf4, and Irf8 (Lin et al. 2010; Mercer et al. 2011). E2A and EBF1 then act in concert to induce the expression of Pax5 (Rothenberg 2014). Thus, EBF1 and PAX5 are downstream from E2A and are essential for early B-cell development, as a similar block of B-cell differentiation is observed in their deficient mice. Once EBF1 and PAX5 are activated, they collaborate to initiate B-cell-specific gene programs, including the expression of the pre-BCR components Cd79a, Cd79b, Igll1, and Vpreb1 and genes involved in signal transduction; receptors; and cellular metabolism (Cobaleda et al. 2007; Boller and Grosschedl 2014). Notably, E2A, EBF1, and PAX5 are proposed to suppress differentiation of alternative cell fates (Boller and Grosschedl 2014). The inactivation of any of these genes in B-cell progenitors led to the disruption of their genetic program and loss of B-cell identity. Moreover, committed progenitors deficient for these genes acquire multipotency and self-renewal activity (Nutt et al. 1997; Ikawa et al. 2004; Pongubala et al. 2008), indicating the essential function of these master regulators in the generation and maintenance of B-cell progenitors. However, transcriptional networks underlying the generation of these B-lineage programs during cell fate determination remain unexplored because of the lack of suitable experimental systems.
We recently established a system that can construct and validate gene regulatory networks during lymphoid lineage specification from HSCs (Ikawa et al. 2015). We overexpressed an Id3-ERT2 (estrogen receptor) fusion protein whose nuclear translocation is induced by 4-hydroxytamoxifen (4-OHT) in hematopoietic progenitors and cultured them under B-cell differentiation conditions. In the presence of 4-OHT, B-cell development of Id3 transduced cells was blocked at an early stage, and the cells grew enormously while still maintaining multipotency, just like E2A- or EBF1-deficient hematopoietic progenitors. We named these multipotent progenitors “induced leukocyte stem” (iLS) cells, as they have the potential to give rise to T, B, and myeloid cells both in vivo and in vitro. The cells can be cultured with stromal cells in the presence of SCF, IL-7, Flt-3L, and 4-OHT for at least several months without losing their differentiation potential. Notably, almost all cells became CD19+ B cells within 6 d by simply withdrawing 4-OHT.
Here, we used this system to characterize global gene expression patterns and histone modifications at multiple time points. The expression of most of B-lineage-associated TFs, such as Ebf1, Pax5, and Foxo1, was dramatically up-regulated after 48 h of induction. The transcript levels of genes involved in the maintenance of HSCs and differentiation of other lineage cells were reciprocally reduced. Strikingly, we observed a rapid and sequential up-regulation of TFs before the onset of the key TF program. The earliest responding TFs, such as Egr1 and Klf4, had a peak at 0.5–2 h following a continuous stream of genes (Tead1, Cebpb, and Gata2) within 24 h. Knockdown of the early responding genes and a subset of epigenetic regulators impaired the differentiation of HSCs into B cells. Based on the analysis, we constructed global genetic networks in which these regulators were separated into three waves according to their expression patterns, protein interactions, and chromatin modifications. Digital transcriptional profiling of single cells at the multipotent and committed state in bone marrow (BM) strongly supported the three-step TF network model. Taken together, our findings provide a genome-wide view of the dynamic association of TFs and epigenetic programs in B-cell fate determination.
Results
Establishment of an inducible culture system to monitor B-lineage commitment
To develop an inducible system for B-cell commitment, BM Lin−c-kit+Sca-1+ (LSK) cells from B6CD45.1 mice were transduced with an Id3-ERT2 retrovirus and cultured on TSt-4 stromal cells that support the generation of B cells in medium supplemented with SCF, IL-7, and Flt3-L in the presence of 4-OHT (Supplemental Fig. S1A). Consistent with our previous report (Ikawa et al. 2015), the Id3-ERT2 transduced cells expanded over time under these conditions. After 1 mo of culture, a homogeneous cell population was generated. The cells expressed cell surface CD34, Sca-1, c-Kit, IL-7Rα, and Flt-3 but did not express any lineage markers, a phenotype indicating that they were LMPP-like cells (Supplemental Fig. S1B–D). To determine their lymphoid and myeloid differentiation potential, the Id3-ERT2 transduced cells were cultured on TSt-4 or TSt-4/DLL1 stromal cells in the absence of 4-OHT. The cells gave rise to B and myeloid cells on TSt-4 cells and to T and NK cells on TSt-4/DLL1 cells, respectively (Supplemental Fig. S1E,F). The Id3-ERT2 transduced cells generated T-, B-, and myeloid lineage cells upon transfer into irradiated immunodeficient (NOG) mice, confirming their myeloid and lymphoid reconstitution potential (Supplemental Fig. S1G–K). The Id3-ERT2 transduced cells were stable for at least several months; the phenotype and the developmental potential were unchanged after 10 mo of culture, although they ceased proliferation and reduced in size after commitment to B-lineage cells, just like normal pre-B cells (Supplemental Fig. S1L,M). Thus, the Id3-ERT2 transduced cells were considered as iLS cells that retain self-renewal activity and multipotency, as reported previously (Ikawa et al. 2015).
Dynamic gene expression profiles during B-cell fate determination
To examine the exact mechanisms of B-cell fate specification, 4-OHT was removed to induce the differentiation of iLS cells into B-lineage cells in vitro (Fig. 1A). The cells began differentiating into B cells within 24 h, and virtually all cells became CD19+ cells within 6 d (Fig. 1B). Id3-ERT2 protein quickly disappeared from the nucleus within 24 h of induction (Fig. 1C). Quantitative RT–PCR (qRT–PCR) analysis demonstrated the up-regulation of B-cell-specific genes, including core TFs (Ebf1 and Pax5) and B-cell-associated genes (Cd19, Rag1, Vpreb1, and Igll1). Conversely, genes involved in the proliferation and differentiation of multipotent progenitors, such as Flt3 and Ikzf1, were gradually down-regulated (Supplemental Fig. S2A). Chromatin immunoprecipitation (ChIP)-PCR analysis showed increased E2A occupancy on the regulatory regions of B-lineage-specific genes (Supplemental Fig. S2B). Differentiation into B-lineage cells was confirmed by the gradual increase in V(D)J and Vκ–Jκ rearrangements of immunoglobulin heavy chain (IgH) genes and κ light chain (IgL) genes, respectively (Supplemental Fig. S2C).
Figure 1.

Genome-wide transcriptome analysis using time-course samples of iLS cells differentiating into B cells. (A) Scheme of B-cell induction from iLS cells. Time-course samples were prepared at the indicated time points. (B) Flow cytometric profiles of B220 and CD19 expression. Cultured iLS cells were analyzed at the indicated time point during B-cell commitment. (C) Western blot analysis for localization of hId3-ERT2 fusion protein. α-Tubulin and Lamin A/C are shown as loading controls for cytosolic and nuclear fractions. (D) Reproducibility matrix between every pair of 16 time points from three independent experiments. (E) Principal component analysis (PCA) of the gene expression pattern of differentially expressed genes (n = 4290) in each time point. Genes were selected for their variance (more than twofold difference with q-values <0.01) using ANOVA. (F) k-means clustering of differentially expressed genes (n = 4290) among time points. Respective gene expression is shown in gray, and representative expression pattern is highlighted in red. Heat maps of each cluster are shown at the right. See also Supplemental Figure S3 for gene ontology analysis and representative genes of each cluster. A complete list of genes in each cluster is in Supplemental Table 3.
To evaluate the changes in genome-wide gene expression during the B-cell commitment process, we performed RNA sequencing (RNA-seq) analysis using samples at 16 time points (Fig. 1A). To assess the early events in transcriptional regulation of B-cell commitment, samples were taken frequently at the early time points (0.5–12 h) and then daily at later time points. The gene expression profiles among three biological replicates confirmed a high correlation between every pair of time-course samples after B-cell induction (Fig. 1D). We were able to identify 4290 genes that were differentially expressed specifically during B-cell fate commitment by ANOVA. Using these data sets, we performed principal component analysis (PCA) to clarify the characteristics of the gene expression patterns. Of note, the sequential changes in gene expression were aligned in an arc (Fig. 1E). Moreover, there were three differentiation phases: early (0.5–4 h), intermediate (∼6–48 h), and late (72–168 h). These three stages were clearly separable, and their transitions were consecutive.
To investigate the global gene expression profiles, we performed k-means clustering among the 4290 genes. The results showed that they could be divided into 10 clusters based on their expression patterns (Fig. 1F; Supplemental Fig. S3; Supplemental Table S3). Representative genes in respective clusters exhibited the wave-like structure of expression patterns during specification into the B lineage (Fig. 1F). Cluster I contained genes whose expression was down-regulated during the commitment process and included Cd34, Flt3, and Dnmt3a, which are already known to be involved in the maintenance of hematopoietic stem/progenitor cells (HSPCs). Cluster II represented the “first wave” and was enriched for the earliest responding genes (Jun, Egr1, and Klf4), whose transcripts quickly increased but then decreased within a few hours after induction. Cluster III to cluster VII represented the second to fourth waves, which appear to be in a transition state prior to the onset of the master regulators in clusters VIII and IX. Most of the B-lineage-associated genes, including Ebf1, Bach2, and Foxo1, were enriched in clusters VIII and IX. The expression of this cluster of genes may represent the final stage of B-cell commitment. There was also a cluster (Hhex, Myb, and Sox4) that exhibited a U-shaped expression pattern curve (cluster X). Together, the transcriptome data sets from multiple time points identified dynamic and continuous transition of gene expression status during B-cell commitment with high reproducibility.
E2A, EBF1, and PAX5 play major roles in B-cell commitment
To understand the roles of E2A, EBF1, and PAX5, we examined the expression of these master TFs in this system. The mRNA levels of Tcf3, Ebf1, and Pax5 gradually increased during the culture (Fig. 2A). To determine how E2A, EBF1, and PAX5 contribute to B-lineage commitment, the frequency of the target genes of each TF among each cluster was examined. To pick up the target genes of each TF, the published ChIP-seq (ChIP combined with high-throughput sequencing) data were used (Lin et al. 2010; Treiber et al. 2010; Revilla-i-Domingo et al. 2012). About 10%–30% of the differentially expressed genes were regulated by E2A, EBF1, and PAX5 or a combination of these TFs. Cotarget genes of these TFs were also identified among clusters. Of note, ∼30% of the genes in cluster IX were the targets of the E2A, EBF1, and PAX5 (Fig. 2B). Since cluster IX contains most of B-cell-associated genes whose expression increased at the late stage of B-cell induction (Fig. 1F), the expression of Ebf1, Pax5, and B-lineage-associated genes increased synchronously. The up-regulation of target genes of E2A, EBF1, and PAX5 at the late stage of commitment was confirmed by the time-course analysis (Fig. 2C). These results indicate that E2A, EBF1, and PAX5 play key roles in inducing a subset of the B-cell-associated genes during B-cell commitment.
Figure 2.

Transition of core TF targets during B-cell commitment in the iLS system. (A) mRNA expression profiles of Tcf3 (E2A), Ebf1, and Pax5. (B) Composition of TF (E2A, EBF1, and PAX5) targets among clusters defined in Figure 1F. The Y-axis represents the percentage of target genes of each TF, and the actual numbers of targets are also shown in bars. (C) Box plot representation for expression of TF target and non-TF target genes. The notches in the box plot indicate 95% confidence interval.
Histone modification changes in association with gene expression during B-cell fate determination
Gene expression regulated by lineage-specific TFs during development is normally well correlated with histone modifications on the gene regulatory regions. To assess histone modifications of the lineage-associated loci at each time point, we performed ChIP-seq analysis to enrich for DNA associated with three histone 3 (H3) modifications: H3K4me3, H3K9me3, and H3K27me3. H3K4me3 is functionally linked to activation at transcriptional start sites (TSSs), H3K9me3 is a marker for heterochromatin, and H3K27me3 is associated with transcriptional silencing (Bannister and Kouzarides 2005). Transcriptional regulatory regions of B-cell-associated genes (Ebf1, Pax5, or Vpreb1) were initially occupied by suppressive histone mark H3K27me3 or H3K9me3. During B-cell differentiation, these modifications were gradually replaced by the transcriptional activation mark H3K4me3, consistent with increased transcript levels for these genes (Supplemental Fig. S4A). In contrast, the H3K4me3 mark disappeared, and H3K27me3 was increased in stem cell-associated gene loci (Flt3 and Cd34) (Supplemental Fig. S4B). Lineage-inappropriate gene loci such as Csf2ra (GM-CSF [granulocyte–macrophage colony-stimulating factor] receptor) and Epor (erythropoietin receptor) were strongly suppressed, based on high occupancy with H3K9me3 or H3K27me3 (Supplemental Fig. S4C). The coordination between gene expression and histone modifications (H3K4me3 and H3K27me3) was confirmed in genome-wide comparisons of down-regulated and up-regulated gene loci (Supplemental Fig. S4D).
Three-step transcriptional networks that establish B-cell identity
We next constructed transcriptional networks by combining gene expression profiles at multiple time points, our database of genome-wide TF occupancy, and the protein–protein interaction data (Fig. 3A). The differentially expressed genes that displayed wave behavior and the genes that were constantly expressed were isolated from transcriptome data sets based on their expression level and variance (Fig. 3B; Supplemental Table S4). The regulatory relationships between factors and their targets were inferred from the database integrated by the multiple ChIP-seq records deposited in the Gene Expression Omnibus (GEO) and the ChEA2 database (Kawakami et al. 2016). The protein–protein interactions between TFs were also taken into account by interrogating the STRING database. These could be divided into three time phases: early (up to 4 h), mid (6–48 h), and late (72–168 h) based on the expression variance of regulatory factors (Fig. 3B). Each time phase shaped its own network: At an early stage, the activity of Junb, Klf4, and Egr1 plays a central role to promote B-cell fate specification, as described before (Fig. 3C, “early”). Activation by Tead family genes (Tead1, Tead2, and Tead4) and the suppression by Gata2 or Smad1 were shown at the mid stage (Fig. 3C, “mid”). At the late stage of B-cell commitment, the factors operative at early and mid phases were replaced by the major B-cell TFs Ebf1, Pax5, and Foxo1 (Fig. 3C, “late”). Of note, the epigenetic modifiers Ezh2 and Smarca4 were found to collaborate to suppress the activities of lineage-inappropriate genes. The genes that are constantly expressed at high levels throughout B-cell commitment were closely associated with the clusters at each time phase (Fig. 3D; Supplemental Fig. S5; Supplemental Table S5). Many transcriptional and epigenetic factors were among this group. At any of the developmental stages, the TFs and epigenetic factors among the gene regulatory circuits were directly connected to each other (Supplemental Fig. S5B). Strikingly, the sequential changes in transcriptional activities were also revealed by this analysis. For example, the Polycomb-repressive complex 2 (PRC2) components Ezh2 and Suz12 were suggested to be strong negative regulators at the later stages of differentiation, although they initially seemed to be activators at an early phase (Fig. 3C,D). Of note, the activities of continuously expressed TFs such as Myc and Ikaros (Ikzf1) were dramatically changed from “repressor” to “activator” during differentiation and affected the regulatory networks at all three phases (Fig. 3D). Collectively, these results suggested that orchestrated regulation of TFs and epigenetic factors are essential for initiation and promotion of the B-cell-specific gene program.
Figure 3.

Proposed gene regulatory network in B-cell fate determination. (A) Strategy for the construction of a network. (B) Gene expression profiles of highly variable TFs. Three major wave factors (early, mid, and late) are indicated as green, red, and black bars at the left. See also Supplemental Table 4 for the details of the expression profile (log fold change). (C,D) Transcriptional regulatory network during B-cell commitment. A network of wave factors (C) and constant factors (D) is shown in three time phases (early, mid, and late). The color of each node represents the transcriptional activity (activation or suppression of expression of their postulated downstream target genes) at each time frame. The thickness of the edge indicates the probability of protein–protein interaction (experimental score in the STRING database).
Digital transcriptional profiling of single cells during B-cell fate determination
To further evaluate transcriptional changes in individual progenitors during B-lineage commitment, single-cell RNA-seq analysis was performed using the iLS system. The iLS cells maintained at an undifferentiated state were used as multipotent progenitor cells. The iLS cells differentiated into B cells by culturing them in the absence of 4-OHT for 8 d were used as committed B-cell progenitors (Supplemental Fig. S6A). The mRNA expression in individual cells was analyzed by a droplet-based system that enables 3′ mRNA digital counting of thousands of single cells per sample (Zheng et al. 2017). Data from multiple sequencing runs were merged using the Cell Ranger pipelines. At ∼30,000 reads per cell, the median numbers of genes and unique molecular identifier (UMI) counts detected per cell were ∼1000 and ∼2500, respectively (Supplemental Fig. S6B).
We performed clustering analysis to examine cellular heterogeneity among the cells. k-means clustering identified two clusters in multipotent progenitors and one cluster in committed B-cell progenitors, which were visualized in two-dimensional projection of t-distributed stochastic neighbor embedding (t-SNE) (Supplemental Fig. S6C). The heat map of the top 239 variable genes ranked by normalized dispersion demonstrated that the expression of most of the B-lineage-specific genes, such as Ebf1, Cd79b, and Igll1, was prominent in committed B-cell progenitors but not in multipotent progenitors (Supplemental Fig. S6D). This is consistent with data of the bulk RNA-seq analysis. The t-SNE projection with each cell was colored based on their normalized expression of the B-lineage-associated genes and indicated the high enrichment in committed but not in multipotent progenitors (Supplemental Fig. S6E). In contrast, the genes involved in the maintenance of hematopoietic stem/progenitors were expressed predominantly in multipotent progenitors at the single-cell level (Supplemental Fig. S6F). These results indicated the synchronous and correlated expression of cell type-specific genes in individual cells, which play critical roles in regulating B-cell fate specification.
To further characterize the transcriptional profiling of B-cell commitment in vivo, single-cell RNA-seq analysis was performed using the progenitors isolated from BM. LMPP, common lymphoid progenitor (CLP), and pro-B cells were sorted from BM, and single-cell RNA-seq analysis was performed (Fig. 4A; Supplemental Fig. S7A). At ∼300,000 reads per cell, the median numbers of genes and UMI counts detected per cell were ∼2000 and ∼6000, respectively (Supplemental Fig. S7B). k-means clustering identified two clusters in CLP and pro-B cells but one cluster in LMPP cells, which were visualized in two-dimensional projection of t-SNE (Fig. 4B; Supplemental Fig. S7C; Supplemental Table S6). The heat map on the differentially expressed genes demonstrated the ordered and synchronous expression of lineage-associated genes, including TFs (Fig. 4C; Supplemental Table S7). Consistent with the data of the iLS differentiation system, the genes involved in multipotency expressed exclusively in LMPP or CLP cells. In contrast, the transcripts of B-lineage-associated genes were induced in pro-B cells (Fig. 4C,D; Supplemental Table S8). Interestingly, the clustering analysis revealed the heterogeneity of CLP and pro-B cells. The CLP cells were separated into two clusters. The genes expressed in each population were quite similar, whereas the expression levels, such as Spi1 and Ikzf1, in individual cells were higher in CLP1 and lower in CLP2. The pro-B cells were also separated into two clusters. The cells of both clusters expressed B-lineage-specific genes, whereas pro-B1 cells expressed Tcf3, Ebf1, and Pax5 at relatively higher levels than pro-B2 cells. Moreover, pro-B1 cells specifically expressed the cell cycle-related genes Cdk1, Cdc20, and E2f2. Consistent with previous findings, gradual and cascading expression of key TFs Spi1, Ikzf1, Foxo1, Tcf3, Ebf1, and Pax5 was also disclosed. We also examined the mRNA expression of each component among the three-step transcriptional networks shown in Figure 3. Consistent with the bulk data, the transcript levels of Fos, Junb, and Egr1 of early wave components were high in LMPP cells and gradually reduced in CLP and pro-B cells. The transcripts of Cebpb and Fosl2 of mid-wave components increased at the CLP stage but decreased at the pro-B-cell stage. The mRNA expression of Ezh2, Smarca4, and Spib of late wave components was dramatically induced at pro-B cells, whereas Suz12 and Chd4 were constantly expressed from LMPP to pro-B cells (Fig. 5). These results strongly supported the transcriptional profiling revealed by the iLS system at the single-cell level.
Figure 4.

Digital single-cell RNA-seq analysis of B-cell precursors in BM. (A) Cell populations used in this analysis. The detailed sorting strategy is shown in Supplemental Figure S7A. (B) t-SNE projection of LMPP, CLP, and pro-B cells. Cells were categorized by their expression characteristics using k-means clustering (k = 5). t-SNE projection without clustering is shown in Supplemental Figure S7C. Lists of differentially expressed genes of each cluster are in Supplemental Table S6. (C) Gene expression profiles of respective cell populations. Genes were clustered with the Ward method. The representative genes in each cluster are displayed at the right of the heat map. The expression level (UMI count) of B-cell-associated TFs in individual cells is at the bottom. Complete gene lists of each cluster are in Supplemental Table S7. (D) t-SNE projection with each cell colored based on their normalized expression of B-cell-associated TFs: Spi1 (PU.1), Ikzf1 (IKAROS), Tcf3 (E2A), Foxo1, Ebf1, and Pax5. The numbers indicate the frequency of expressing cells in each cluster. See Supplemental Table S8 for detailed frequency data of all other genes.
Figure 5.
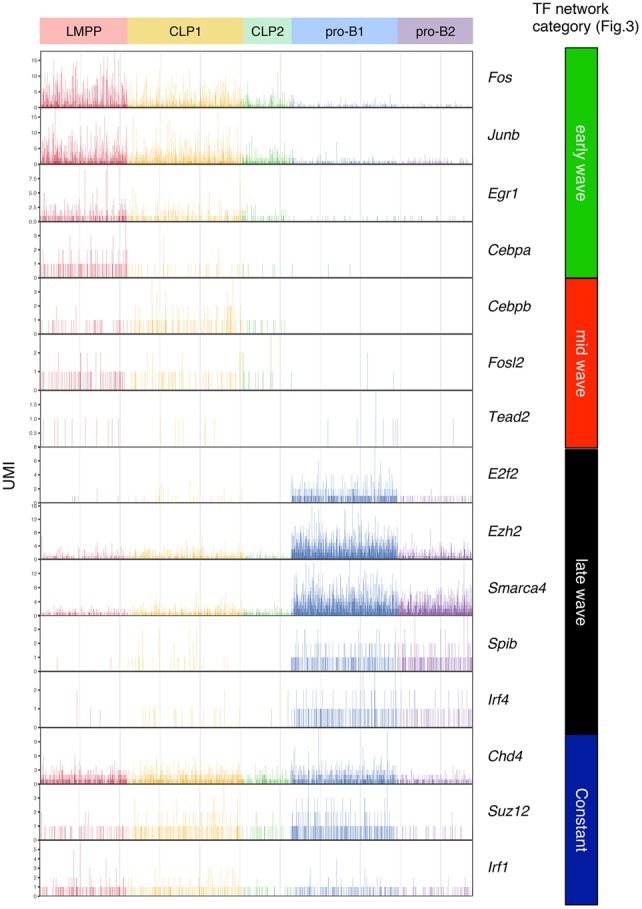
Sequential priming of multiple TFs in single B-cell precursors in BM. The expression level (UMI count) of each gene among TF networks (shown in Fig. 3) in the indicated populations of BM is shown.
Early responding factors are essential for proper B-cell commitment
Next, we focused on the TFs in cluster II, which contained early responding factors (Fig. 1F; Supplemental Fig. S3). Among 104 genes in this cluster, 22 were TFs. Immediate early genes (IEGs) such as Junb, Nr4a1/2, Egr1/2, and Krüppel-like factors (Klf2, Klf4, Klf6, and Klf7) were found in this cluster (Supplemental Fig. S8A). We suspected that these factors might trigger B-cell lineage determination even though they are not B-cell-specific factors. To test this hypothesis, we investigated the function of these factors in regulating the B-lineage program in more detail. qRT–PCR analysis of Id3 transduced cells (not inducible) cultured with or without 4-OHT demonstrated that the mRNA levels of Junb, Nfil3, and Klf4 were not changed upon 4-OHT withdrawal (Supplemental Fig. S8B), excluding the possibility that the gene expression was induced simply by 4-OHT. Single-cell RNA-seq analysis demonstrated that Junb, Nr4a1, Egr1, and Klf4 were highly expressed in LMPP and CLP cells (Supplemental Fig. S8C). Knockdown of Nr4a2, Klf4, and Egr1 significantly inhibited B-cell generation from iLS cells (Supplemental Fig. S8D–F). Moreover, overexpression of shRNA targeting Egr1 and Klf4 impaired the differentiation of BM progenitors into B cells (Supplemental Fig. S8G,H), suggesting the functional importance of these TFs at the first step in B-cell commitment.
Among the early responding TFs, we also found Nfil3 (E4BP4) (Supplemental Fig. S8A), which was reported previously to be important in the development and function of NK cells, CD4+ T cells, innate lymphoid cells, and CD8α+ conventional dendritic cells (Kashiwada et al. 2011; Motomura et al. 2011; Geiger et al. 2014; Seillet et al. 2014). The Nfil3 mRNA levels were high in the LMPP stage but decreased thereafter along with B-cell differentiation (Fig. 6A,B; Supplemental Figs. S9, S10A). To determine the role of Nfil3 in B-cell differentiation, we examined the Eμ-Nfil3 transgenic (Tg) mice, in which the Nfil3 expression is controlled by the Eμ promoter (Fig. 6C,D; Supplemental Fig. S10B). A significant reduction of fraction C′, pre-B, and immature B cells was observed in Eμ-Nfil3 Tg mice (Fig. 6E,F). Gene expression profiles of pro-B as well as pre-B cells of wild-type and Eμ-Nfil3 Tg mice showed that the expression of several B-cell-specific TFs such as Bach2, Irf4, Spib, and Ikzf3 was severely reduced in pro-B cells of Eμ-Nfil3 Tg mice, whereas Jun and Id3 expression was up-regulated in both pro-B and pre-B cells (Fig. 6G,H; Supplemental Table S9). We also found a mild increase of progenitor B cells in BM of Nfil3−/− mice or in shRNA-mediated Nfil3 knockdown HSCs differentiating into B cells (Supplemental Fig. S10C–F). These results suggested that suppression of Nfil3 expression at the appropriate time point is required for proper B-cell fate determination.
Figure 6.

Prolonged Nfil3 expression perturbs normal B-cell commitment. (A) qRT–PCR analysis of Nfil3 in HSPCs and early B-cell progenitors in BM. Values represent mean ± SD in a representative of two independent experiments. The gating strategy for each fraction is shown in Supplemental Figure S8. (B) Expression profile of Nfil3 in BM progenitors at the single-cell level. The t-SNE projection and UMI count of Nfil3 in LMPP, CLP, and pro-B cells in BM are shown. The number in the t-SNE projection indicates the percentage of expressing cells in each cluster. (C) Western blotting of Flag tag in B220+ cells in the spleens of Eμ-Nfil3 Tg and littermate control (wild-type [WT]) mice. See also Supplemental Figure S10B for a schematic of the construct. (D) Intracellular staining of Flag tag in B220+ cells in the spleens of Eμ-Nfil3 Tg and wild-type mice. (E,F) FACS profiles (E) and cell number (F) of B-cell progenitor population in wild-type and Eμ-Nfil3 Tg mice. Values represent mean ± SD in a representative of three independent experiments. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (G) Volcano plot for a comparison of gene expression status in pro-B (top) or pre-B (bottom) cells in BM of wild-type and Eμ-Nfil3 Tg mice. The X-axis indicates the expression ratio of Eμ-Nfil3 Tg versus wild-type cells, and the Y-axis indicates the statistical significance. The expression differences and significance of selected genes (difference between Eμ-Nfil3 and wild-type is greater than twofold; P-value is <0.01) are listed in Supplemental Table S9. (H) qRT–PCR analysis of differentially expressed genes in pro-B and pre-B cells in BM of wild-type and Eμ-Nfil3 Tg mice. Values represent mean ± SD in a representative of two independent experiments. (***) P < 0.001.
Transcriptional and epigenetic regulation that determines B-cell fate
We next investigated the transcriptional and epigenetic control of B-cell fate determination. We focused on clusters VIII and IX, which contain genes up-regulated at the late stage of B-cell commitment (e.g., Ebf1, Bach2, and Foxo1) (Fig. 1F; Supplemental Fig. S3). Not only B-cell-related TFs but also various epigenetic factors—such as the PRC1 component Cbx2, DNA methyltransferase Dnmt1 and the related gene Uhrf1, and histone H2A deubiquitinase Mysm1—were found in this group (Fig. 7A). High mRNA expression of these genes was shown by qRT–PCR as well as single-cell RNA-seq analysis in early B-cell progenitor populations in BM (Fig. 7B,C). To determine whether these factors are essential for normal B-cell differentiation, iLS cells were retrovirally transduced with shRNA antagonists of these genes, and the targeted cells were then induced to differentiate into the B-cell lineage. We found that B-cell numbers were significantly decreased in Cbx2, Uhrf1, and Mysm1 knockdown cells, similar to Pax5 knockdown cells (Fig. 7D–F). These results suggested that epigenetic regulation by these factors acts in concert with B-cell-specific TFs during B-cell lineage commitment.
Figure 7.

Essential roles of epigenetic modifiers for the generation of B cells. (A) Gene expression profiles of TFs among clusters VIII and IX in the iLS system shown in Figure 1F. (B) qRT–PCR analysis of the indicated genes in HSPC and B-cell progenitor fractions in BM. The gating strategy for each fraction is shown in Supplemental Figure S9. Values represent mean ± SD in a representative of two independent experiments. (C) t-SNE projection of the indicated genes in LMPP, CLP, and pro-B cells in BM revealed by single-cell RNA-seq analysis. The numbers indicate the percentage of expressing cells in each cluster. (D–F) shRNA-mediated knockdown of the indicated genes using iLS cells. Knockdown efficiency assessed by qRT–PCR (D), FACS profiles (E), and B-cell numbers (F) are shown. Values represent mean ± SD in three independent experiments. (*) P < 0.05; (**) P < 0.01 compared with control.
Discussion
By taking advantage of a uniquely efficient B-cell programming system, we characterized transitions of transcriptional landscape and dynamic TF behavior during the B-cell fate determination (Fig. 3; Supplemental Fig. S5). Single-cell transcriptomic analysis strongly supported the transcriptional priming that regulates the B-lineage commitment in vivo. We also identified the “transitional” state between multipotent and B-lineage-committed cells (Figs. 1F, 3B–D, 4B,C), consistent with the previous report that described a similar transition state during embryonic stem (ES)-to-epiblast differentiation process by single-cell transcriptome analysis (Klein et al. 2015).
Among the components of the transcriptional waves, the functional importance of some of the genes in early B-cell development was already reported. In particular, major B-cell-associated TFs such as Pax5, Irf4, Bach2, Ebf1, and Foxo1 were induced at the late stage of B-cell commitment (Figs. 1F, 3C, 4C,D). The consistency of the in vitro and in vivo data clearly demonstrates that the iLS system used in this study is highly suitable to determine the mechanisms underlying B-cell fate decisions in vivo. Strikingly, several epigenetic factors in this cluster, such as Dnmt1, Uhrf1, and Cbx2, have been characterized as novel regulators for normal B-cell differentiation (Fig. 7). Moreover, the master TFs and epigenetic factors were closely associated with each other (Figs. 3, 7). The histone H2A deubiquitinase Mysm1 (Jiang et al. 2011) and the chromatin remodeler Smarca4 (Brg1) (Bossen et al. 2015) have been shown to be essential for early B-cell development. Ctcf is also shown to be essential for normal VDJ recombination by binding the intergenic control region between VH and D gene clusters (Guo et al. 2011). These results suggest that there still remain unknown essential players that mediate epigenetic regulation in this cluster. In fact, we found that perturbation of the PRC1 component Cbx2 and the cofactor of DNA methyltransferase Uhrf1 causes defects in B-cell generation (Fig. 7D–F). Consistent with this, it was reported that deficiency of another PRC1 component, Bmi1, or the PRC2 component Ezh2 leads to the arrest of B-cell development at the pro-B-cell stage (Van Der Lugt et al. 1994; Su et al. 2003). Therefore, these epigenetic factors will be strong candidates for novel essential regulators of B-cell development.
The clustering analysis identified 10 representative expression patterns (Fig. 1F). Among the gene clusters, we found the rapid (0.5–4 h) and dramatic mRNA induction of a series of TFs that may correspond to IEGs, which were originally implicated in virus infection (Watson and Clements 1980). The importance of IEGs has also been suggested for the establishment and maintenance of long-lasting memory in neural cells (Okuno 2011). It has become more evident recently that various stimuli, such as growth factors, hormones, or cytokines, can induce rapid and transient mRNA synthesis in mammalian cells even without any protein synthesis (Okuno 2011). TSSs of IEGs are occupied with poised RNA polymerase II (Pol II), but productive elongation by Pol II is stalled by the negative elongation factor (NELF) in the neuronal system (Saha et al. 2011). Strikingly, Pol II stalling, which allows for rapid induction, is widespread throughout the genome (Kwak et al. 2013). Moreover, the function and transcriptional program of IEGs are totally distinct from secondary response genes in many aspects (Tullai et al. 2007), suggesting that such a regulatory mechanism may play a fundamental role in development. In fact, c-Fos knockout mice showed a marked reduction of B cells in addition to bone formation defects (Wang et al. 1992). Overexpression of Fos and Jun in hematopoietic cells also perturbs B-cell development at the pro-B-cell or pre-B-cell stage (Fujita et al. 1993). The number of B-cell precursors is increased in Egr1 knockout mice (Gururajan et al. 2008), whereas sustained expression of Egr2 in CD2+ lymphocytes causes a severe developmental block in pro-B cells (Li et al. 2011). We also found multiple effects of the first-wave TFs (Egr1, Nr4a1/2, Klf4, and Nfil3) on B-cell generation (Fig. 6; Supplemental Figs. S8,10). Moreover, most of the genes in this cluster, such as Jun, Junb, Fos, Egr1, and Nr4a1, were indicated as the targets of E2A (Lin et al. 2010). Given that IEGs are sensitive to induction by environmental changes, it is possible that the first-wave factors, including IEGs, could be the “first trigger,” setting off B-cell fate determination. However, it still remains to be determined which type of extracellular or intracellular signaling pathway induces IEGs in B-cell progenitors in vivo. Furthermore, the functional relevance among E2A, IEGs, and secondary responding TFs is still obscure. Detailed analysis of gene regulatory regions of IEGs and the downstream genes in early B-cell progenitors in BM will facilitate our understanding of IEGs in normal B-cell development.
Several models of gene regulatory networks have been described in the immune system (Singh et al. 2014). In particular, TF networks controlling TH17 cell differentiation have been examined extensively by multiple groups (Ciofani et al. 2012; Yosef et al. 2013). Yosef et al. (2013) described dynamic molecular circuits by combining transcriptome profiling of multiple time-course samples, computational modeling, and careful validation of the model. Consistent with our data, they divided transcriptional regulators into three clusters based on their functional activities. Of note, they identified Myc, Egr1, and Smarca4 (which also contribute to promote B-cell commitment) as positive modules in the differentiation of TH17 cells. These data suggest that the transcriptional waves that we observed here might be the general architecture of the genetic circuits that determine cell fate during differentiation of many cell types. The data also suggest that the TFs essential for TH17 cell differentiation may share many features (e.g., timing of expression, correlation with other factors, and function) with TFs seen in B-cell progenitors. Further network modeling, including the relationship between enhancer and promoter regions of the crucial gene loci, should provide a comprehensive genome-wide resource for a better understanding of transcriptional regulation in immune cell development.
Recently, a novel concept of “superenhancers” has been proposed; the definition is genomic regions with clusters of putative enhancers that are densely occupied by unusually high levels of master regulators and the Mediator coactivator complex. Whyte et al. (2013) found that the superenhancers contain binding motifs for cell type-specific master TFs (OCT4, SOX2, and NANOG in ES cells and PU.1, EBF1, and FOXO1 in pro-B cells), demonstrating that these superenhancers play essential roles in establishing cellular identity. Intriguingly, they also showed that the E2A-binding motif was considerably more enriched at superenhancer regions than at typical enhancer regions in pro-B cells, suggesting that E2A is a master regulator for B-cell commitment. Superenhancers are also suggested to underlie the lineage commitment and plasticity of stem cells (Adam et al. 2015). Consistent with this, our data indicated that a significantly high percentage (∼10%–30%) of the differentially expressed genes were the targets of E2A, EBF1, and PAX5 (Fig. 2). Thus, the iLS system should be ideal for in-depth investigation of enhancer activities in immune cell development.
In summary, our work demonstrates the transcriptional dynamics displaying complex but highly coordinated sequential events during B-cell fate determination at the single-cell resolution. More than 4000 genes are differentially expressed in multipotent progenitors differentiating into B-lineage cells. The differentially expressed genes are divided into three distinct steps according to the expression pattern. It still remains to be determined why and how the transcriptional priming occurs in three steps. It could be that the commitment may occur generally in the three steps, primed by nonspecific TFs setting up the lineage decision process (suppression of inappropriate genes involved in the maintenance of multipotency or differentiation into alternative lineages) followed by the change of cell cycle status and metabolism (Cdk1, Ccna2, Wnt, Insulin, and BMPs) prior to the onset of the master regulators (E2A, EBF1, and PAX5 in the case of B-cell commitment). Unknown regulatory factors (enhancer RNA, long noncoding RNA, and genomic interaction) might have profound functions connecting TFs and epigenetic factors during the stepwise programming. Further investigation will be required for deciphering the precise nature of transcriptional maps to determine the cell fate.
Materials and methods
Mice
C57BL/6J (B6), B6.Rag1 knockout, and B6.CD45.1 congenic mice were purchased from CLEA Japan, Inc. NOD/Shi-scid, IL2Rγnull (NOG) mice were purchased from the Central Institute for Experimental Animals. Nfil3−/− and Eμ-Nfil3 Tg mice were maintained in our animal facility. Embryos at 14 d post-coitum (dpc) were obtained from timed pregnancies. The day that a vaginal plug was observed was designated as 0 dpc. All experiments were conducted according to guidelines approved by the Institutional Animal Care and Use Committee of RIKEN's Yokohama Branch.
Establishment and B-cell induction of iLS cells
LSK cells from fetal livers or BM were transduced with hId3-ERT2-IG virus supernatants by spin infection as described previously (Ikawa et al. 2015). The transduced cells were maintained on TSt-4 stromal cells in the presence of 20 ng/mL IL-7 (R&D), 10 ng/mL SCF (R&D), 10 ng/mL Flt-3L (R&D), and 40 nM 4-OHT (Sigma). To induce B-cell differentiation of iLS cells, the cells were cultured on TSt-4 cells with 5 ng/mL IL-7 in the absence of 4-OHT.
Organelle fractionation and Western blotting
The iLS cells (2 × 106) were fractionated using NE-PER nuclear and cytoplasmic extraction reagents (Pierce). Each lysate was subjected to SDS-PAGE using Mini-Protean TGX precast gel (Bio-Rad) and then transferred to PVDF membrane by Transblot Turbo transfer system (Bio-Rad). Membranes were blocked by PVDF blocking reagent for Can Get Signal (TOYOBO) for 1 h at room temperature and then incubated with the following primary antibodies diluted in Can Get Signal solution 1 (TOYOBO) for 1 h at room temperature: anti-human ID3 (Cell Signaling, 9837), anti-α-tubulin (Santa Cruz Biotechnology, sc-8035), and anti-Lamin A/C (Santa Cruz Biotechnology, sc-7292). The membrane was washed three times by TBS-T and then incubated with secondary antibody (HRP-conjugated anti-mouse or rabbit IgG [Life Technologies] diluted in Can Get Signal solution 2 [TOYOBO]) for 1 h at room temperature. After extensive washing of the membrane (four washes) by TBS-T, immunoreactive proteins were detected by Western Lightning Plus-ECL reagent (Perkin Elmer).
shRNA-mediated knockdown experiments
The iLS cells were transduced with respective shRNA retrovirus by spin infection, and the cells were cultured on TSt-4 cells in the presence of 4-OHT. At 2 d after infection, hCD25+ cells were collected by magnetic-activated cell sorting (MACS) (Myltenyi Biotec), and the 1 × 105 purified cells were cultured in B-cell induction culture conditions (described above). LSK cells in BM were transduced with shRNA in the same manner as iLS cells except in the absence of 4-OHT.
RNA extraction and qRT–PCR
Total RNA was extracted by Maxwell 16 LEV simplyRNA purification kit (Promega) following the manufacturer's protocol. cDNA was synthesized with VILO cDNA synthesis kit (Invitrogen). qPCR was performed with SYBR Premix Ex Taq (TaKaRa). Primer sequences are listed in Supplemental Table 2.
RNA-seq
Poly(A)+ mRNA was isolated from total RNA by NEBNext poly(A) mRNA magnetic isolation module (New England Biolabs), and then the library was prepared by NEBNext Ultra RNA library preparation kit for Illumina (New England Biolabs). The average size and quantity of each library were measured by Agilent 2100 Bioanalyzer (Agilent Technologies) and KAPA library quantification kit (KAPA Biosystems), respectively. Sequences were read by a HiSeq 1500 system (Illumina).
Single-cell RNA-seq
Single-cell RNA-seq was performed according to the manufacturer's protocol as described previously (Zheng et al. 2017). Briefly, iLS cells at day 0 and day 8 of B-cell induction or LMPP, CLP, and pro-B cells in BM were sorted, and ∼3000 cells were loaded onto a GemCode single-cell instrument (10x Genomics) to generate single-cell gel beads in emulsion (GEMs). Single-cell RNA-seq libraries were prepared using GemCode single-cell 3′ gel bead and library kit (10x Genomics). Sequencing libraries were loaded onto an Illumina HiSeq 2500. Cell Ranger single-cell software was used to perform sample demultiplexing, barcode processing, and single-cell 3′ gene counting and to combine libraries. For secondary analysis, including t-SNE projection and clustering, Cell Ranger R kit was used.
Network construction based on time-course gene expression profiles
DNA-binding factors were picked up by their gene ontology term. Read counts for the genes annotated in University of California at Santa Cruz mm9 were assembled using featureCounts (Liao et al. 2014), and differential gene expression was determined using DESeq2 version 1.8.1. We integrated additional data obtained from GEO with the ChEA2 database (Kou et al. 2013) to construct a ChIP database containing a total of 25,468,840 binding interactions for 486 transcription regulators. The network was shown in an organic layout using Cytoscape (Shannon et al. 2003). The node colors indicate the enrichment scores of TFs calculated with weighted parametric enrichment analysis, indicating the weighted average expression changes of their target genes in each stage (early, mid, and late) of B-cell commitment. The width of the line represents the experimental score of the protein interaction obtained from STRING database version 10.
Additional materials and methods are described in the Supplemental Material.
Data availability
The data set for the RNA-seq and ChIP-seq analyses in this study was deposited in GEO. The accession number for the time course series of RNA-seq (Fig. 1) and ChIP-seq (Supplemental Fig. 5) is GSE106795, and the accession number for the single-cell RNA-seq data (Fig. 4; Supplemental Figs. 6, 7) is GSE107527.
Supplementary Material
Acknowledgments
We are grateful to J. Sharif, K. Moro, H. Koseki, and S. Koyasu for sharing the protocols, supporting this project, and helpful comments; P. Burrows for critical reading of the manuscript; A. Shibano-Satoh, M. Ohno, and M. Nakano-Ikegaya for technical assistance; and C. Murre for the shRNA retrovirus construct. This work was supported in part by grants from the Japan Society for the Promotion of Science (25118732 to T.I.), the RIKEN IMS Young Chief Investigator program (to T.I.), the RIKEN Junior Research Associate Program (to T.M.), and the Takeda Science Foundation (to T.I.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.309575.117.
References
- Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, Polak L, Kadaja M, Asare A, Zheng D, et al. 2015. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 521: 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. 2005. Reversing histone methylation. Nature 436: 1103–1106. [DOI] [PubMed] [Google Scholar]
- Boller S, Grosschedl R. 2014. The regulatory network of B-cell differentiation: a focused view of early B-cell factor 1 function. Immunol Rev 261: 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossen C, Murre CS, Chang AN, Mansson R, Rodewald H-R, Murre C. 2015. The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat Immunol 16: 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, et al. 2012. A validated regulatory network for Th17 cell specification. Cell 151: 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobaleda C, Schebesta A, Delogu A, Busslinger M. 2007. Pax5: the guardian of B cell identity and function. Nat Immunol 8: 463–470. [DOI] [PubMed] [Google Scholar]
- Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. 2014. A committed precursor to innate lymphoid cells. Nature 508: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel I, Murre C. 2001. The function of E- and Id proteins in lymphocyte development. Nat Rev Immunol 1: 193–199. [DOI] [PubMed] [Google Scholar]
- Fujita K, Miki N, Mojica MP, Takao S, Phuchareon J, Nishikawa S, Sudo T, Tokuhisa T. 1993. B cell development is perturbed in bone marrow from c-fos/v-jun doubly transgenic mice. Int Immunol 5: 227–230. [DOI] [PubMed] [Google Scholar]
- Geiger TL, Abt MC, Gasteiger G, Firth MA, O'Connor MH, Geary CD, O'Sullivan TE, van den Brink MR, Pamer EG, Hanash AM, et al. 2014. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med 211: 1723–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Yoon HS, Franklin A, Jain S, Ebert A, Cheng H-L, Hansen E, Despo O, Bossen C, Vettermann C, et al. 2011. CTCF-binding elements mediate control of V(D)J recombination. Nature 477: 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gururajan M, Simmons A, Dasu T, Spear BT, Calulot C, Robertson DA, Wiest DL, Monroe JG, Bondada S. 2008. Early growth response genes regulate B cell development, proliferation, and immune response. J Immunol 181: 4590–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Kawamoto H, Wright LYT, Murre C. 2004. Long-term cultured E2A-deficient hematopoietic progenitor cells are pluripotent. Immunity 20: 349–360. [DOI] [PubMed] [Google Scholar]
- Ikawa T, Masuda K, Huijskens MJAJ, Satoh R, Kakugawa K, Agata Y, Miyai T, Germeraad WTV, Katsura Y, Kawamoto H. 2015. Induced developmental arrest of early hematopoietic progenitors leads to the generation of leukocyte stem cells. Stem Cell Rep 5: 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X-X, Nguyen Q, Chou Y, Wang T, Nandakumar V, Yates P, Jones L, Wang L, Won H, Lee H-R, et al. 2011. Control of B cell development by the histone H2A deubiquitinase MYSM1. Immunity 35: 883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwada M, Pham NL, Pewe LL, Harty JT, Rothman PB. 2011. NFIL3/E4BP4 is a key transcription factor for CD8α+ dendritic cell development. Blood 117: 6193–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami E, Nakaoka S, Ohta T, Kitano H. 2016. Weighted enrichment method for prediction of transcription regulators from transcriptome and global chromatin immunoprecipitation data. Nucleic Acids Res 44: 5010–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. 2015. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161: 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou Y, Chen EY, Clark NR, Duan Q, Tan CM, Ma'ayan A. 2013. ChEA2: gene-set libraries from ChIP-X experiments to decode the transcription regulome. In Availability, reliability, and security in information systems and HCI. CD-ARES 2013. Lecture Notes in Computer Science, Vol. 8127 (ed. Cuzzocrea A, et al. ), pp. 416–430. Springer, Berlin. [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, Lis JT. 2013. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339: 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Symonds ALJ, Zhu B, Liu M, Raymond MV, Miao T, Wang P. 2011. Early growth response gene-2 (egr-2) regulates the development of B and T cells. PLoS One 6: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. 2010. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11: 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer EM, Lin YC, Benner C, Jhunjhunwala S, Dutkowski J, Flores M, Sigvardsson M, Ideker T, Glass CK, Murre C. 2011. Multilineage priming of enhancer repertoires precedes commitment to the B and myeloid cell lineages in hematopoietic progenitors. Immunity 35: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Chen S, Chandra V, Wagatsuma K, Agata Y, Rodewald HR, Saito R, Chang AN, Varki N, et al. 2015. The E-Id protein axis modulates the activities of the PI3K–AKT–mTORC1–Hif1a and c-myc/p19Arf pathways to suppress innate variant TFH cell development, thymocyte expansion, and lymphomagenesis. Genes Dev 29: 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motomura Y, Kitamura H, Hijikata A, Matsunaga Y, Matsumoto K, Inoue H, Atarashi K, Hori S, Watarai H, Zhu J, et al. 2011. The transcription factor E4BP4 regulates the production of IL-10 and IL-13 in CD4+ T cells. Nat Immunol 12: 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Urbánek P, Relink A, Busslinger M. 1997. Essential functions of Pax5 (BSAP) in pro-B cell development: difference between fetal and adult B lymphopoiesis and reduced V-to-DJ recombination at the IgH locus. Genes Dev 11: 476–491. [DOI] [PubMed] [Google Scholar]
- Okuno H. 2011. Regulation and function of immediate-early genes in the brain: beyond neuronal activity markers. Neurosci Res 69: 175–186. [DOI] [PubMed] [Google Scholar]
- Pongubala JMR, Northrup DL, Lancki DW, Medina KL, Treiber T, Bertolino E, Thomas M, Grosschedl R, Allman D, Singh H. 2008. Transcription factor EBF restricts alternative lineage options and promotes B cell fate commitment independently of Pax5. Nat Immunol 9: 203–215. [DOI] [PubMed] [Google Scholar]
- Revilla-i-Domingo R, Bilic I, Vilagos B, Tagoh H, Ebert A, Tamir IM, Smeenk L, Trupke J, Sommer A, Jaritz M, et al. 2012. The B-cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. EMBO J 31: 3130–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV. 2014. Transcriptional control of early T and B cell developmental choices. Annu Rev Immunol 32: 283–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Wissink EM, Bailey ER, Zhao M, Fargo DC, Hwang J-Y, Daigle KR, Fenn JD, Adelman K, Dudek SM. 2011. Rapid activity-induced transcription of Arc and other IEGs relies on poised RNA polymerase II. Nat Neurosci 14: 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, Huntington ND, Belz GT, Carotta S. 2014. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med 211: 1733–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semerad CL, Mercer EM, Inlay MA, Weissman IL, Murre C. 2009. E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc Natl Acad Sci 106: 1930–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Khan AA, Dinner AR. 2014. Gene regulatory networks in the immune system. Trends Immunol 35: 211–218. [DOI] [PubMed] [Google Scholar]
- Su I-H, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, Tarakhovsky A. 2003. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 4: 124–131. [DOI] [PubMed] [Google Scholar]
- Treiber T, Mandel EM, Pott S, Györy I, Firner S, Liu ET, Grosschedl R. 2010. Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription- independent poising of chromatin. Immunity 32: 714–725. [DOI] [PubMed] [Google Scholar]
- Tullai JW, Schaffer ME, Mullenbrock S, Sholder G, Kasif S, Cooper GM. 2007. Immediate-early and delayed primary response genes are distinct in function and genomic architecture. J Biol Chem 282: 23981–23995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Lugt NMT, Domen J, Linders K, Van Roon M, Robanus-Maandag E, Te Riele H, Van Der Valk M, Deschamps J, Sofroniew M, Van Lohuizen M, et al. 1994. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev 8: 757–769. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. 1992. Bone and haematopoietic defects in mice lacking c-fos. Nature 360: 741–745. [DOI] [PubMed] [Google Scholar]
- Watson RJ, Clements JB. 1980. A herpes simplex virus type 1 function continuously required for early and late virus RNA synthesis. Nature 285: 329–330. [DOI] [PubMed] [Google Scholar]
- Welinder E, Mansson R, Mercer EM, Bryder D, Sigvardsson M, Murre C. 2011. The transcription factors E2A and HEB act in concert to induce the expression of FOXO1 in the common lymphoid progenitor. Proc Natl Acad Sci 108: 17402–17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153: 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, et al. 2013. Dynamic regulatory network controlling TH17 cell differentiation. Nature 496: 461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, Mcdermott GP, Zhu J, et al. 2017. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data set for the RNA-seq and ChIP-seq analyses in this study was deposited in GEO. The accession number for the time course series of RNA-seq (Fig. 1) and ChIP-seq (Supplemental Fig. 5) is GSE106795, and the accession number for the single-cell RNA-seq data (Fig. 4; Supplemental Figs. 6, 7) is GSE107527.