

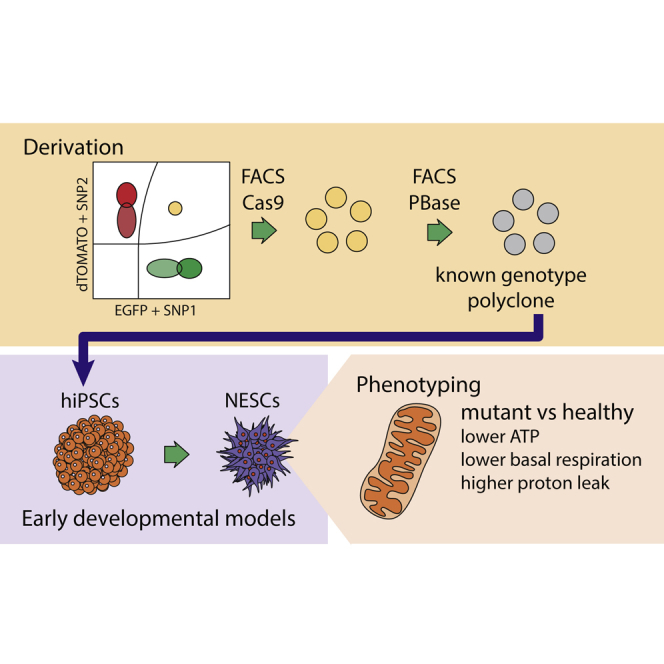
Summary
Genome editing and human induced pluripotent stem cells hold great promise for the development of isogenic disease models and the correction of disease-associated mutations for isogenic tissue therapy. CRISPR-Cas9 has emerged as a versatile and simple tool for engineering human cells for such purposes. However, the current protocols to derive genome-edited lines require the screening of a great number of clones to obtain one free of random integration or on-locus non-homologous end joining (NHEJ)-containing alleles. Here, we describe an efficient method to derive biallelic genome-edited populations by the use of fluorescent markers. We call this technique FACS-assisted CRISPR-Cas9 editing (FACE). FACE allows the derivation of correctly edited polyclones carrying a positive selection fluorescent module and the exclusion of non-edited, random integrations and on-target allele NHEJ-containing cells. We derived a set of isogenic lines containing Parkinson's-disease-associated mutations in α-synuclein and present their comparative phenotypes.
Keywords: stem cells, neuro stem cell, genome editing, repetitive elements, random integration, FACS, α-synuclein, mitochondria
Graphical Abstract
Highlights
-
•
Fluorescent protein-SNP pairs enable deterministic genotypes for genome editing
-
•
Repetitive elements modulate off-targeting random integration
-
•
Parkinson's disease NESCs present impaired mitochondrial energy performance
In this article, Arias-Fuenzalida and colleagues show a platform to achieve deterministic genotypes for genome editing. The combinatorial use of fluorescent proteins and defined SNPs facilitates the genome editing endeavor. Using neuroepithelial stem cells, they demonstrate early mitochondrial phenotypes in SNCA mutants.
Introduction
Parkinson's disease (PD) is a multifactorial neurodegenerative disorder characterized by motor and non-motor symptoms (Caligiore et al., 2016). Some cases of PD cases result from autosomal dominant mutations in the SNCA gene, which encodes α-synuclein. Physiologically, α-synuclein is implicated in synaptic transmission and vesicle transport, while pathologically it is part of the protein aggregates known as Lewy bodies and Lewy neurites (Goedert et al., 2013). Patients carrying mutations in the SNCA gene suffer from early onset of PD. Mutations in SNCA include increase in gene dosage (Devine et al., 2011) and heterozygous missense mutations such as p.A30P and p.A53T (Bendor et al., 2013, Soldner et al., 2011). Mutations in SNCA can account for up to 15% of cases of familial early-onset PD (Bozi et al., 2014).
Importantly, genome editing tools can assist in parsing PD phenotypes. The reliability of CRISPR-Cas9 as an editing tool has been extensively validated by whole-genome sequencing (Veres et al., 2014). Furthermore, Cas9 specificity has been improved with high-fidelity variants (Kleinstiver et al., 2016). However, eliminating uncertainties in genotype outcomes of edited lines has remained challenging. Screening of correctly edited clones is a labor-consuming process that entails the selection of on-target knockin clones and the exclusion of random integrations, on-target indels, and second-allele indel events. To leverage the power of genome editing tools in the evaluation of polygenic diseases such as PD, it is necessary to overcome such labor- and time-consuming limitations. Hence, the fast generation of genome-edited populations carrying a known genotype outcome is highly necessary.
Results
Deterministic Genotype Outcomes for the Generation of Isogenic Lines
The use of donors containing fluorescent protein (FP) reporters associated with defined SNP variants enables editing outcomes of known genotype (Figures 1A and 1E): heterozygous, homozygous healthy, and homozygous pathogenic (Figure 1E). One-step biallelic targeting occurs with a mean frequency of 37.5% using double-stranded DNA (dsDNA) templates (Table S1). Donor vectors for SNCA exon 2 and exon 3 were cloned with an internal positive selection module (PSM) coding EGFP or dTOMATO, and an external negative selection module (NSM) containing tagBFP (Figure 1A). SNCA mutations are dominant, and missense SNCA PD patients are heterozygote. Hence, donor pairs for the SNCA mutations rs104893878 (p.A30P) and rs104893877 (p.A53T) were designed to match heterozygous genotype outcomes (Figure 1E). In the case of SNCAe2, an EGFP donor carried the transversion c.88g>c. For SNCAe3, an EGFP donor carried the transition c.209g>a. For each locus, a corresponding dTOMATO donor carried the healthy variant, as shown for population type 1A (Figure 1E). A similar expression level of the FP reporters was observed from each allele in SNCA chromosome 4, as demonstrated by a symmetric fluorescence-activated cell sorting (FACS) analysis (Figure 1D). In order to test whether similar PSM expression levels are observed in other loci, the gene PINK1 exon 5 of chromosome 1 was targeted. In contrast to SNCA mutant PD patients, PINK1 PD patients are homozygote or compound heterozygote (Ishihara-Paul et al., 2008). Hence, for PINK1e5, both donors, EGFP and dTOMATO, carried the pathogenic transversion c.1197t>a, matching population type 1C (Figure 1E). FACS analysis showed that biallelic targeted populations separated clearly from other genotype outcomes for PINK1 chromosome 1 (Figure S1C) and SNCA chromosome 4 (Figure 1D). These results validate the approach to target both alleles of a gene of interest, independent of the locus.
Figure 1.

Biallelic Integration of FP-SNP Pairs Enable Deterministic Genotype Outcomes
(A) Donor vectors contain a PSM expressing EGFP or dTOMATO, and an NSM expressing tagBFP. PSMs contain puromycin resistance gene (Puro).
(B) Representative example of SNCAe3 polyclone 636. Random integration tagBFPpos cells are excluded from the correctly edited on-target cells tagBFPneg.
(C) Theoretical distribution of populations for non-random outcomes.
(D) Representative example of SNCAe3. On-target cells include homozygous populations, EGFPpos/EGFPpos or dTOMATOpos/dTOMATOpos (type 2), and heterozygous populations of undefined second-allele state EGFPpos/WT-NHEJ or dTOMATOpos/WT-NHEJ (type U). WT, wild-type.
(E) Outcomes of the derived population are defined according to the donor vector design.
(F) The tagBFP NSM allows removal of random integration events, assisting in the derivation of defined outcomes.
Repetitive Elements Reduce On-Target Genome Editing Efficiency by Increasing Random Integration
Silent point mutations were introduced in protospacer adjacent motif (PAM) sequences of the donors (Table S2). The PAM edited template is resistant to Cas9-induced linearization, avoiding linear DNA-induced random integration. Thus, properly targeted alleles are shielded from Cas9-induced secondary incisions, eliminating the risk of on-target indels (Merkle et al., 2015) (Table S2). Two weeks after electroporation, each edited population was expanded up to 15 × 106 puromycin-resistant and FP-positive cells. The inclusion of tagBFP in the NSM allowed random integration events to be quantified, visualized, and excluded (Figures 1A, 1B, and 1F). The tagBFP NSM avoids bystander toxicity or incomplete negative selection from systems such as thymidine kinase (Ruby and Zheng, 2009). The percentage of tagBFPpos random integration ranged from 5.8% to 14.6% for SNCAe2, from 42.6% to 64.2% for SNCAe3, and from 27.2% to 30.4% for PINK1e5 (Figures S2A–S2C). The extent of random integration correlated with the type and proportion of repetitive elements present in the homology arms of the donors. We assessed random integration using donors for six loci with known repetitive element composition and tested 12 single-guide RNAs (sgRNAs). The loci evaluated included chromosome 1 (PINK1 exon 5), chromosome 4 (SNCA exon 2 and exon 3), and chromosome 16 (ceroid lupofuscinosis 3, CLN3 exon 5–8, exon 10–13, and exon 14–15) (Figure S2G). For the analysis, we performed a linear optimization model of the form Ax = b (Figure S2H). The resulting matrix A corresponds to the frequency of repetitive elements in the homology arms (Figure S2G). The vector x corresponds to the type of repetitive elements present in the analyzed dataset (Figure S2G) and a variable of all non-included repetitive elements (upsilon). The vector b corresponds to the experimentally measured random integration level, given by the percentage of tagBFPpos cells (Figures S2A–S2F). Based on this, we derived a model to predict random integration frequency intrinsic to the composition of repetitive elements in the homology arms (Figures S2H and S2I). The solution allows assigning weight coefficients to each repetitive element. Their value indicates which repetitive element contributes the most to the random integration frequency observed. The solution space is constrained for a maximum of 100% random integration and sequence length boundaries of each repetitive element. The optimization solution indicates that the most relevant repetitive elements correspond to the Short Interspersed Nuclear Elements (SINE) family, specifically Alu and Mir (Figures S2H and S2I).
FACS Purification Increases the Speed and Yield of Isogenic Derivation
For the on-target tagBFPneg cells, the ratio of EGFP to dTOMATO was ∼50% in all cases analyzed, which is consistent with a comparable efficiency for both donors (Figures 2A and S1C). The initial percentage of double-positive EGFPpos/dTOMATOpos cells ranged from a mean 2.15% for SNCAe2, 3.4% for SNCAe3, to 3.75% for PINK1e5 (Figures 2A and S1C). Quantifications were conducted independently using different sgRNAs (Figures 2A and S1C). One sorting step yielded a population of up to 3 × 105 EGFPpos/dTOMATOpos cells (Figures 2C and 2D). The gating position of the double-positive population afforded nearly complete purity with either purity-purity or yield-purity sorting masks (Figures 2B and S1D). Although it is possible to isolate single-channel double-positive EGFPpos/pos or dTOMATOpos/pos populations (type 2) (Figure 2A) using the FSC-A dimension, there is an extensive overlap with the indel-bearing single-positive population (type U) (Figures S1G–S1J). A high frequency of non-homologous end joining (NHEJ) events was detected in the non-targeted allele of the single-positive population (type U) (Figures S1J and S1K). Hence, purification of the double-positive EGFPpos/pos or dTOMATOpos/pos populations presents the risk of co-purifying overlapping indel-bearing cells (Figures S1J and S1K). In this combination of events, only the biallelic EGFPpos/dTOMATOpos group offers a deterministic genotype outcome. Sanger sequencing of biallelic targeted SNCA mutations demonstrated the heterozygous integration of the pathogenic SNP rs104893878 (p.A30P) and rs104893877 (p.A53T) in each polyclone (Figures 2E and 2F), the homozygous integration of the edited PAM, and the transition from genome to PSM (Figures 2E and 2F). Sequencing-isolated single clones from the polyclonal populations permitted composition analysis (Figures 2G, S3F, and S3G and Table S3).
Figure 2.

FACS Purification Increases the Speed and Yield of Isogenic Derivation
(A) Post-selection sorting of double-positive biallelic edited cells for SNCAe2 and SNCAe3 using independent sgRNAs. FACS plots are represented with 2% contour lines. For SNCAe3 sgRNA-636, a dotplot is included to show the distribution of 1.2%. Diagram of knockin population types is shown (right).
(B) Yield-purity and purity-purity sorting strategies permit the generation of a homogeneous biallelic knockin population. Diagram of purification population types is shown (right).
(C) Representative post-sorting of single cells for SNCA polyclone. Single-cell gating structures yields high-purity biallelic edited cells. Scale bar, 50 μm.
(D) Representative post-sorting culture for biallelic EGFPpos/dTOMATOposSNCA polyclone. Scale bar, 200 μm.
(E) Sanger sequencing chromatogram of SNCAe2 p.A30P polyclone 632 knockin (KI).
(F) Sanger sequencing chromatogram of SNCAe3 p.A53T polyclone 636 knockin.
(G) Analysis of the polyclone composition as in Figure S3.
Transposase-Mediated Generation of Footprint-free Isogenic Lines
The PSMs in each double-positive polyclone were excised using a codon-optimized hyperactive and excision-only variant of the piggybac transposase (Li et al., 2013b, Yusa et al., 2011) (Figures 3A and 3B). Even though the excision-only variant presents an activity of 0.85 times that of wild-type (Figures S1M and S1N), it is preferred as it lacks the reintegration cycle of wild-type variants (Li et al., 2013a). The heterozygous SNCAe2 and SNCAe3 EGFPpos/dTOMATOpos polyclonal populations were transfected with in vitro transcribed mRNA encoding excision-only transposase. Subsequently, the excised EGFPneg/dTOMATOneg population was sorted (Figures 3A and 3B). Using the excision-only variant and two transfection steps, we observed average excision efficiencies of 3.65% for SNCAe2, 2.15% for SNCAe3, and 6.5% for PINK1e5 (Figures 3A and S1E). A second sorting step to purify cells that underwent selection module removal yielded up to 2.5 × 106 EGFPneg/dTOMATOneg SNP knocked in cells. In the FACS analysis, it is possible to observe transition states for single-copy excision and complete removal of both selection modules (Figures 3A, 3B, and S1E). We observed a curved population shifting from the double-positive EGFPpos/dTOMATOpos quadrant to the double-negative EGFPneg/dTOMATOneg quadrant in all cases. Sanger sequencing of the SNCA targeted and transposed genomic region demonstrated the heterozygous integration of the pathogenic SNP rs104893878 (p.A30P) and rs104893877 (p.A53T) in each polyclone (Figures 3C and 3D). Isolation of single-cell-derived clones from the polyclones and sequencing permitted quantification of their composition (Figures 3E, S3H, and S3I and Table S3). The polyclone composition analysis demonstrated that PSMs were excised and the edited SNPs and edited PAM sites remained in the non-coding sequence (Figures S3H and S3I). Karyotype assessment was conducted for each polyclone and parental control (Figures S4A–S4C). Pluripotency of lines was assessed by immunostaining for OCT4, SOX2, TRA1-81, and SSEA4 (Figures S4D–S4F).
Figure 3.

Transposase-Mediated Excision of PSMs
(A) FACS analysis for PSM removal. Two transfection steps of excision-only transposase result in removal of the PSMs for SNCAe2 and SNCAe3. Purification of EGFPneg/dTOMATOneg cells yields footprint-free edited lines. Diagram of removal population types is shown (right).
(B) Cultures after transposase transfection for SNCAe2 present single and double PSM removal events (in arrowheads) as shown in (A). Scale bar, 200 μm.
(C) Sanger sequencing chromatogram of transposed SNCAe2 p.A30P polyclone 632 and parental control.
(D) Sanger sequencing chromatogram of transposed SNCAe3 p.A53T polyclone 636 and parental control.
(E) Analysis of the respective polyclone composition as in Figure S3.
SNCA Mutants Present Early Mitochondrial Impairment
In order to validate the edited SNCA lines, a phenotypic characterization was conducted (Figure 4). Isogenic induced pluripotent stem cells (iPSCs) were differentiated into neuroepithelial stem cells (NESCs) (Reinhardt et al., 2013) (Figures 4A, S4G, and S4H). NESCs typically express the SNCA transcript at 0.86 and 0.7 times the level of GAPDH and TUBG1, respectively (Figures 4B and S4I). Western blot analysis indicated a similar protein level of monomeric α-synuclein for all genotypes (Figure 4C). Extracellular energy flux analyses were conducted for parental healthy NESCs, and mutant isogenic α-synuclein p.A30P and p.A53T NESCs (Figure 4D). Cells expressing the α-synuclein mutation p.A53T showed a significantly reduced maximal respiration capacity compared with the parental isogenic control (Figures 4D–4F). Moreover, both the p.A30P and p.A53T α-synuclein mutant NESCs showed comparatively reduced energy performance, manifested by a lower basal respiration, ATP production, and non-mitochondrial respiration (Figures 4D–4F).
Figure 4.

Edited SNCA Isogenic Lines Present PD-Associated Phenotypes
(A) NESC differentiation protocol.
(B) Microarray expression level for SNCA, TUBG1, and GAPDH in healthy control NESCs. Data represent three replicates.
(C) Western blot subsequent to denaturing SDS-PAGE for α-synuclein and GAPDH for NESCs.
(D) Wave plot of oxygen consumption rates for the α-synuclein isogenic set. Each wave corresponds to three biological replicates. SD of the sample is included.
(E) Maximal respiration, proton leak, basal respiration, ATP production, and non-mitochondrial respiration for the extracellular energy flux analysis in (D).
(F) Radar plot of fold changes for the parameters in (E). Significance levels correspond to the higher p value assigned to a mutant per category. Significance determined by unpaired Student's t-test.
Significance levels are ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001; n.s., not significant.
Discussion
Overall, FACE constitutes a robust method to achieve deterministic genotype outcomes for the generation of isogenic cell lines. The selection of biallelic editing events ensures a defined genotype. It should be noted that, due to transient disruption of the coding sequence, this approach is restricted to genes with non-essential function in the target cell type. The use of fluorescent NSM excludes random integration events, enabling clearer sorting gates and isolated biallelic populations. This constitutes an advancement over similar approaches (Eggenschwiler et al., 2016). However, potential limitations are that PSMs could be subjected to position-effect variegation or promotor silencing. Nevertheless, usage of the FP markers expedites the selection, reducing the timescale in comparison with potential position-effect variegation (Norrman et al., 2010). It should be noted that editing approaches that use single-stranded DNA (ssDNA) or dsDNA could be subjected to cleavage within non-functional or functional sequences. Hence, donor break points within PSM cannot be fully excluded. The advantage of dsDNA approaches, in comparison with ssDNA, are their flexibility to carry larger cargos in order to deposit designer insertions, designer deletions, or PSMs. In addition, larger sequences of donors are easier to detect by conventional methods in comparison with short ssDNA. Similarly, potential imperfect integration of dsDNA donor templates can be readily detected by simple methods such as PCR, in comparison with ssDNA-based methods.
Conventional derivation of single nucleotide mutations, not associated with a direct selection phenotype or selection marker, can require screening an average of 911 ± 375 clones and using 8.8 ± 5.9 sgRNAs. Conversely, early elimination of undesirable outcomes obviates the need to perform extensive colony screening and results in a faster, more efficient derivation process. Thus, FACE constitutes an attractive alternative to conventional methods. The efficiency of homology-directed repair is influenced by the length of the homology arms used (Hasty et al., 1991). We and others have used homology arms of ∼1 kbp, which provides a balance between efficiency and specificity (Soldner et al., 2011). The sequence conversion from an endogenous sequence to that carried in donor templates extends from ∼400 bp in dsDNA (Elliott and Jasin, 2001) to ∼30 bp in ssDNA donors (Paquet et al., 2016). Hence, it is of critical importance to include the edited bases close to the dsDNA break point and close to the PSM unit, independently of the length of the homology arms or the type of template used. Post-knockin and post-transposition clonal composition analysis confirmed that FACE enables the derivation of polyclones and significantly reduces the screening efforts if individual clones are needed. On the other hand, the derivation of edited polyclones presents the advantage of avoiding the risk inherent with clone-specific biases. Extensive expansion, required for clonal derivation, is reported to subject cells to culture aberrations (Martins-Taylor and Xu, 2012). It is widely accepted that single-cell passaging for any type of cell-culture application, including the process of FACS-based enrichment described here, imposes an unavoidable risk of genome instability (Chan et al., 2008). The derivation of polyclones reduces the culture time needed for each step, since sufficient material is available earlier. Karyotype analysis of the edited lines demonstrated that the process did not induce chromosomal abnormalities when compared with the parental line. Previous reports also support the possibility of achieving a low incidence of modification with genome editing tools (Tsai and Joung, 2014, Veres et al., 2014).
In order to protect the dsDNA donor template from Cas9-induced linearization and to avoid post-integration cleavage of targeted sequences, we introduced silent mutations in the PAM sequences. This requires special attention to the design in order to introduce the edited PAM in a non-coding sequence or as a synonymous mutation. We and others have successfully used this mechanistic insight to protect post-integration targeted sequences from secondary cleavage events (Inui et al., 2014, Paquet et al., 2016). Similarly, design considerations are needed to identify adjacent transposase excision sequences, or to generate a de novo TTAA sequence in non-coding regions or by silent editions. Protocol optimization for the use of an excision-only transposase variant (Li et al., 2013b) allowed the derivation of footprint-free isogenic sets for disease modeling. We were able to observe transition states that represent the removal of one or both PSMs. The transition populations presented a curve pattern that accounts for the dissimilar stability of the FPs (Snapp, 2009) and transcripts after the CDS module was removed.
The influence of repetitive elements on the efficiency of genome editing has been reported previously (Ishii et al., 2014). Recognizable repetitive elements constitute up to 45% of the human genome (Lander et al., 2001). Repetitive elements in humans can be classified in four families: SINE, LINE, LTR retrotransposons, and DNA transposons. Each category presents multiple sub-families. Using linear optimization modeling, we determined that, in our dataset, the repetitive elements of the SINE family, Alu and Mir, contribute the most to random integration events. These repetitive elements have 1.5 million copies and constitute ∼13% of the human genome (Lander et al., 2001). Although this discrete dataset does not include all existing human repetitive elements, it demonstrates their direct contribution to random integration. Other aspects, such as the composition of repetitive elements and distance to the dsDNA break point, might modulate the frequency of random integration. Our data confirm previous reports that repetitive elements act as templates for off-target homologous recombination (Ishii et al., 2014). These sequences should be avoided when designing homology arms in order to enhance on-target recombination and edition.
In summary, we generated an isogenic set of human SNCA mutants for PD-specific cellular modeling. The set carries disease-associated mutations p.A30P or p.A53T in the SNCA gene. We observed energy metabolism phenotypes in human NESCs, an early neurodevelopment disease model. Such traits have been previously described in SNCA p.A30P mutant differentiated neurons (Ryan et al., 2013). This validates the applicability of the approach described here for the generation of disease-relevant models. We envision that FACE could be efficiently implemented for automated high-throughput genome editing, enabling fast phenotype assessment in the future.
Experimental Procedures
Stem cells A13777 were cultured in Essential 8 medium on Geltrex. Cells were passed with accutase and plated with Y27632 (10 μM) for 24 hr after dissociation. Cells were electroporated using 4D-Nucleofector. Selection was conducted with puromycin (0.5 μg/mL). FACS was conducted on an ARIA III sorter. Cells were purified with single-cell exclusive gating. Post-knockin cells were transfected with in vitro transcribed mRNA coding transposase. Human iPSCs were characterized for OCT4, SOX2, TRA1-81, and TRA1-61. Microarray karyotype was conducted using Illumina iScan technology. NESCs were differentiated as represented in Figure 4A. NESCs were characterized for NESTIN and SOX2. Transcription levels for NESCs were evaluated using Affymetrix human gene arrays (GEO: GSE101534). Extracellular energy flux analysis was conducted on NESCs using a Seahorse XFe96 assay as indicated in Figure 4D. Comprehensive information on the experimental procedures is described in the Supplemental Information.
Author Contributions
J.A.-F., J.J., X.Q., G.G.-G., J.W., and S.L.N. designed the study and conducted the experiments. J.C.S., H.Z., and H.R.S. supervised. J.A.-F., J.J., and J.C.S. wrote the manuscript and organized the display items. All the authors read and agreed to the final version of the manuscript.
Acknowledgments
We would like to thank Prof. F. Zhang from the McGovern Institute for Brain Research for providing the Cas9 vector. We acknowledge G. Preciat for valuable feedback on optimization. We acknowledge Prof. J. Hejna, E. Berger, and S. Bolognin for their valuable comments on the manuscript. This project was supported by the LCSB Pluripotent Stem Cell Core Facility. J.J., J.W., and X.Q. were supported by fellowships from the FNR (AFR, Aides à la Formation-Recherche). G.G.-G. was supported by NCL-Stiftung. J.J. is supported by a Pelican award from the Fondation du Pelican de Mie et Pierre Hippert-Faber. This is an EU Joint Programme-Neurodegenerative Disease Research (JPND) project (INTER/JPND/14/02; INTER/JPND/15/11092422). Further support comes from the SysMedPD project, which has received funding from the European Union's Horizon 2020 Research and Innovation Program under grant agreement no. 668738. Jens Schwamborn is founder, CSO, and shareholder of Braingineering Technologies Sarl. J.C.S., J.A.F., J.J., and X.Q. are inventors on the patent (LU92964).
Published: October 5, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.08.026.
Accession Numbers
The accession number for the gene expression data (Figure 4B) reported in this paper is GEO: GSE101534.
Supplemental Information
References
- Bendor J.T., Logan T.P., Edwards R.H. The function of alpha-synuclein. Neuron. 2013;79:1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozi M., Papadimitriou D., Antonellou R., Moraitou M., Maniati M., Vassilatis D.K., Papageorgiou S.G., Leonardos A., Tagaris G., Malamis G. Genetic assessment of familial and early-onset Parkinson's disease in a Greek population. Eur. J. Neurol. 2014;21:963–968. doi: 10.1111/ene.12315. [DOI] [PubMed] [Google Scholar]
- Caligiore D., Helmich R.C., Hallett M., Moustafa A.A., Timmermann L., Toni I., Baldassarre G. Parkinson's disease as a system-level disorder. NPJ Parkinsons Dis. 2016;2:16025. doi: 10.1038/npjparkd.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.M., Yates F., Boyer L.F., Schlaeger T.M., Daley G.Q. Enhanced plating efficiency of trypsin-adapted human embryonic stem cells is reversible and independent of trisomy 12/17. Cloning Stem Cells. 2008;10:107–118. doi: 10.1089/clo.2007.0064. [DOI] [PubMed] [Google Scholar]
- Devine M.J., Ryten M., Vodicka P., Thomson A.J., Burdon T., Houlden H., Cavaleri F., Nagano M., Drummond N.J., Taanman J.W. Parkinson's disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat. Commun. 2011;2:440. doi: 10.1038/ncomms1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler R., Moslem M., Fraguas M.S., Galla M., Papp O., Naujock M., Fonfara I., Gensch I., Wahner A., Beh-Pajooh A. Improved bi-allelic modification of a transcriptionally silent locus in patient-derived iPSC by Cas9 nickase. Sci. Rep. 2016;6:38198. doi: 10.1038/srep38198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B., Jasin M. Repair of double-strand breaks by homologous recombination in mismatch repair-defective mammalian cells. Mol. Cell. Biol. 2001;21:2671–2682. doi: 10.1128/MCB.21.8.2671-2682.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Spillantini M.G., Del Tredici K., Braak H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- Hasty P., Rivera-Perez J., Bradley A. The length of homology required for gene targeting in embryonic stem cells. Mol. Cell. Biol. 1991;11:5586–5591. doi: 10.1128/mcb.11.11.5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui M., Miyado M., Igarashi M., Tamano M., Kubo A., Yamashita S., Asahara H., Fukami M., Takada S. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci. Rep. 2014;4:5396. doi: 10.1038/srep05396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara-Paul L., Hulihan M.M., Kachergus J., Upmanyu R., Warren L., Amouri R., Elango R., Prinjha R.K., Soto A., Kefi M. PINK1 mutations and parkinsonism. Neurology. 2008;71:896–902. doi: 10.1212/01.wnl.0000323812.40708.1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A., Kurosawa A., Saito S., Adachi N. Analysis of the role of homology arms in gene-targeting vectors in human cells. PLoS One. 2014;9:e108236. doi: 10.1371/journal.pone.0108236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Li M.A., Pettitt S.J., Eckert S., Ning Z., Rice S., Cadinanos J., Yusa K., Conte N., Bradley A. The piggyBac transposon displays local and distant reintegration preferences and can cause mutations at noncanonical integration sites. Mol. Cell. Biol. 2013;33:1317–1330. doi: 10.1128/MCB.00670-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Burnight E.R., Cooney A.L., Malani N., Brady T., Sander J.D., Staber J., Wheelan S.J., Joung J.K., McCray P.B., Jr. piggyBac transposase tools for genome engineering. Proc. Natl. Acad. Sci. USA. 2013;110:E2279–E2287. doi: 10.1073/pnas.1305987110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Taylor K., Xu R.H. Concise review: genomic stability of human induced pluripotent stem cells. Stem Cells. 2012;30:22–27. doi: 10.1002/stem.705. [DOI] [PubMed] [Google Scholar]
- Merkle F.T., Neuhausser W.M., Santos D., Valen E., Gagnon J.A., Maas K., Sandoe J., Schier A.F., Eggan K. Efficient CRISPR-Cas9-mediated generation of knockin human pluripotent stem cells lacking undesired mutations at the targeted locus. Cell Rep. 2015;11:875–883. doi: 10.1016/j.celrep.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrman K., Fischer Y., Bonnamy B., Wolfhagen Sand F., Ravassard P., Semb H. Quantitative comparison of constitutive promoters in human ES cells. PLoS One. 2010;5:e12413. doi: 10.1371/journal.pone.0012413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D., Kwart D., Chen A., Sproul A., Jacob S., Teo S., Olsen K.M., Gregg A., Noggle S., Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- Reinhardt P., Glatza M., Hemmer K., Tsytsyura Y., Thiel C.S., Hoing S., Moritz S., Parga J.A., Wagner L., Bruder J.M. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One. 2013;8:e59252. doi: 10.1371/journal.pone.0059252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby K.M., Zheng B. Gene targeting in a HUES line of human embryonic stem cells via electroporation. Stem Cells. 2009;27:1496–1506. doi: 10.1002/stem.73. [DOI] [PubMed] [Google Scholar]
- Ryan S.D., Dolatabadi N., Chan S.F., Zhang X., Akhtar M.W., Parker J., Soldner F., Sunico C.R., Nagar S., Talantova M. Isogenic human iPSC Parkinson's model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell. 2013;155:1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp E.L. Fluorescent proteins: a cell biologist's user guide. Trends Cell Biol. 2009;19:649–655. doi: 10.1016/j.tcb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F., Laganiere J., Cheng A.W., Hockemeyer D., Gao Q., Alagappan R., Khurana V., Golbe L.I., Myers R.H., Lindquist S. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S.Q., Joung J.K. What's changed with genome editing? Cell Stem Cell. 2014;15:3–4. doi: 10.1016/j.stem.2014.06.017. [DOI] [PubMed] [Google Scholar]
- Veres A., Gosis B.S., Ding Q., Collins R., Ragavendran A., Brand H., Erdin S., Cowan C.A., Talkowski M.E., Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K., Zhou L., Li M.A., Bradley A., Craig N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.