

Abstract
Microbes are the oldest and most widespread, phylogenetically and metabolically diverse life forms on Earth. However, they have been discovered only 334 years ago, and their diversity started to become seriously investigated even later. For these reasons, microbial studies that unveil novel microbial lineages and processes affecting or involving microbes deeply (and repeatedly) transform knowledge in biology. Considering the quantitative prevalence of taxonomically and functionally unassigned sequences in environmental genomics data sets, and that of uncultured microbes on the planet, we propose that unraveling the microbial dark matter should be identified as a central priority for biologists. Based on former empirical findings of microbial studies, we sketch a logic of discovery with the potential to further highlight the microbial unknowns.
Keywords: metagenomics, eukaryogenesis, microbial evolution, tree of life, web of life, CPR bacteria
Introduction
Microbial studies are fascinating. Not only their findings can deeply transform knowledge in a broad range of scientific fields (from evolutionary biology to zoology and medical and environmental sciences) but also, whereas philosophers of sciences debate whether there is such thing as a logic of scientific discovery (Schickore 2014), microbial studies provide biologists with a set of empirical rules to enhance one’s chances to discover novel and unexpected life forms. This unique potential of microbial studies to reshape knowledge has been recognized relatively recently, even though there is a long standing history of studies of microbial pathogens, involving famous early researchers such as Robert Koch, Louis Pasteur, or Martinus Beijerinck. If the laymen nowadays appreciate that microbes impact our everyday life (i.e., via their fermentative roles in food production), and know that microbes also impacted our recent human histories (i.e., via their contribution to major pandemics; Diamond 1997), from a scientific perspective, microbes are nonetheless rather novel objects of studies. There are both technical and conceptual reasons for this late yet broad recognition of microbes, as we will highlight below, whereas providing an empirical recipe for further insights into the microbial dark matter.
In 1619, the famous astronomer Galileo, whose observations of the moons of Jupiter had threatened the geocentric theory, modified a telescope to magnify nearby terrestrial objects. Although he clearly was a revolutionary thinker, he found these observations of the minute world of limited interest, and, only 6 years later, did his friends name microscopio the strange inverted telescope Galileo had invented (Falkowski 2015). By contrast, Robert Hooke, an English polymath scientist, and, later, Anton van Leeuwenhoek, who did not belong to the academic world, were much more excited by describing their microscopic observations. In 1671, van Leeuwenhoek, who had substantially changed the design of the microscope to enhance its magnifying power, initiated a series of striking findings: microscopic lifeforms are abundant and everywhere to be seen. Microbes, who had populated Earth for over 3.5 billion years, were for the first time exposed to the human eye (Falkowski 2015). Both a technical progress and an uncommon ability to delve into an unseen world were critical components of that progress. However, since biological theory at the time considered the living world was distributed into two major groups: plants and animals, van Leeuwenhoek naturally assumed he was observing populations of minute animals (with tiny organs), when microbes were mobile, rather a new kind of living beings. In that sense, the unveiled microbiological world was first rationalized in ways that fit within preexisting theoretical categories derived from the known living world. Importantly, neither Hooke nor van Leeuwenhoek had immediate scientific successors. Arguably, it took another 200 years (Falkowski 2015), and several novel conceptual and technological developments to formulate an issue, currently at the forefront of microbial studies: « is it possible that unknown microorganisms, with different properties than those currently associated with the known living world, are thriving in nature? ».
The potential theoretical importance of such “known unknowns” and even “unknown unknowns” of the microbial world (e.g., unknown genes, genomes, functions, organisms, processes, and communities associated with uncultured microbes and viruses), that were often popularized under the catch-phrase “microbial dark matter,” should not be underestimated. Interestingly, the relevance of this sentence is debatted in microbiology. Many scientists find the metaphor misleading or inaccurate, because the “microbial dark matter” does not correspond to the dark matter studied by astronomers and physicists. This latter represents a hypothetical, still unobserved, although widely accepted, kind of matter, which does not interact with light but interacts through gravity. Taking the mass of this unseen astronomic dark matter into account would explain the uncorrect predictions of the movement of galaxies by classic astronomy theories. This astronomic dark matter is thus unquestionably different from the microbial dark matter. However, other microbiologists have endorsed the analogy (Rinke et al. 2013; Lobb et al. 2015; Lok 2015; Saw et al. 2015; Bruno et al. 2017; Krishnamurthy and Wang 2017; Lewis 2017), since the sentence nonetheless conveniently stresses that, to some extent, newly discovered microbes can harbor a different biology from those that had been cultured. Although we agree that microbial and astronomic dark matter are very different notions, we also find the sentence “microbial dark matter,” popularized by (Rinke et al. 2013) to be more useful than detrimental. First, it is a convenient short hand for the idea that unknown microbial life may be playing important and even dominant role in ecosystem processes. Second, it has some editorial and educational virtues, as it effectively helps raising the interest for microbiology studies beyond the field of microbiology (in which none would really conflate astronomic and microbial dark matter), surely enhancing the general interest for the unexplored diversity of microbes and their genes. We recommend however a more careful rather than sensationialistic use of the term, to describe the (overwhelming) amount of microbes, microbial genes, and microbial contributions to processes that were unknown at the time at which scientists performed their analyses.
Precisely, much of the extant knowledge in biology, that is, about biological entities and biological processes, heavily relies on analyses conducted on macro-organisms and on cultured microbes. Yet, 60–99% of the microbial diversity are not easily culturable, or are not culturable using standard techniques (Staley and Konopka 1985; Barer and Harwood 1999). Unraveling the microbial dark matter could thus led to two (nonexclusive) types of observations. Either the discovery of hidden microbes will show that microbes unveiled from the microbial dark matter are comparable in terms of genetic diversity, ecological roles, abundance, evolutionary history, and affected by processes similar to those affecting cultured microbes, in which case our current knowledge of microbes is representative of what’s really going on in nature (we will simply find more of what we already knew by mining the microbial world); or the microbial dark matter will prove to host entities and processes that differ from those already described, with the major consequence that scientific knowledge will not only need to be completed but also corrected as microbiologists gain access to this still hidden microbial world in order to consider new phenomena, poorly explained in extant theories. Such significant theoretical transformations have arguably occurred when 1) microbiologists looked for life in extreme environments, 2) detected life under unexpected (i.e., very diverged) forms, and 3) unveiled new processes involving microbes, which allows us to stress some key features for the success of a scientific research oriented toward the discovery of microbiological novelty.
Searching Life in Extreme Environment: A Few Lessons
The developments of molecular markers and sequencing techniques were instrumental for the discovery of extremophiles. By unveiling the archaea, a novel early branching Domain of life, possibly sister-group to eukaryotes, Carl Woese’s phylogenetic studies of the 16 S RNA revolutionized the views on the entire biological world (Woese and Fox 1977; Woese et al. 1990). Woese argued that, rather than being partitioned into two major groups, the eukaryotes and the prokaryotes, the living world encompassed a much broader microbial diversity, justifying its classification into three Domains of life. Subsequently, Woese and his colleagues (referred to as “the Woese army” by Lynn Margulis; Doolittle 2013) actively promoted this position, bringing the newly termed “archaea” into full light, while intending to ban the use of the “older” term “prokaryotes” (Pace 2006).
Importantly, this comparative approach of molecular phylogenetics was later coupled to a phase of exploratory science (Waters 2007). Exploratory science is in essence a strategy of data mining. It goes from the data to the hypotheses (Burian 2013), seeking (robust) patterns in the data or unraveling new phenomena. Although microbiology has a long history of exploratory research (O’Malley 2014), this mode of science appears in strong contrast with the more classic hypothetico-deductive strategy, heralded by Karl Popper. This deductive approach has inspired much of microbiology and biochemistry studies, since these studies largely operated from the hypotheses to the data, that is, using data to reject preexisting hypotheses, or eventually to corroborate them. Since exploratory science is not first aimed at rejecting (or confirming) preestablished hypotheses (thus deepening current knowledge), it can potentially produce novel, unexpected knowledge, or simply fail, making the financial and scientific investment in exploratory studies especially risky.
Fortunately, the pioneering approach, first largely based on the development of 16 S rRNA gene sequencing (Schmidt et al. 1991; Barns et al. 1996; Hugenholtz et al. 1998), then on the sequencing of other makers (Beja et al. 2000), and latter on the development of metagenomics (Breitbart et al. 2002; Tyson et al. 2004; Tringe et al. 2005) and single-cell genomics, bypassed the need for culture studies, thereby lifting a blind spot imposed by culture-based investigation to comparative analyses. These studies returned a diversity of exciting findings. By the beginning of the 2000s, microbial ecologists had started characterizing the gene content, diversity, and relative abundance of environmental microbes (Venter et al. 2004). They had identified new functions of major importance in the ocean (e.g., ammonia oxidation by archaea; Francis et al. 2005), possibly affecting the global nitrogen cycle, as well as unexpected photosynthesis (and other) genes in viruses (Sullivan et al. 2005). They had also gained unprecedented insights into the survival strategies of microbes (Tyson et al. 2004), into their community structures (Tyson et al. 2004; DeLong et al. 2006), and into their niche-specific adaptations (Tringe et al. 2005), for example, by unraveling unknown iron-oxidizing and free-living diazotroph in acid mine drainage biofilms (Ram et al. 2005; Tyson et al. 2005).
Environmental genomics in particular produced remarkable results when microbiologists turned their eyes to extreme regions (in terms of temperature, pH, pressure, mineralization, radiations) that many considered a priori devoid of life (Pikuta et al. 2007). The seemingly counter-intuitive idea to sample lifeforms in environments hostile to life unveiled a broad diversity of extremophiles in the three Domains. Granted, finding DNA in extreme environments does not in itself constitute an ultimate proof that the life forms bearing this DNA existed there, but analyses of environmental DNA (be they nonassembly based, assembly based or even of genome resolved metagenomics) are nonetheless an important step in the discovery of new microbes in extreme environment. Cultivation of microbes from these extreme locations offers a much stronger evidence, that is, Karl-Otto Stetter, by this cultivation approach discovered life at the extreme temperature limits, pushing the boundaries of life as it was then known (Stetter 2013).
Using these strategies, microbiologists realized that life was possible at temperature 122 °C, at negative pH (!), and at pH> 11, at pressures exceeding 1,200 atmospheres; that microbes could be resurrected after 20–40 millions of years of dormancy, survive 2.5 years of travel in space, and thrive within rocks as well as in the terrestrial stratosphere (at > 44 km of altitude) (de los Rios et al. 2003; Pikuta et al. 2007) (see, e.g., https://www.slideshare.net/AnjaliMalik3/extremophiles-imp-1). Some of these statistics were so unexpected that Pikuta et al. (Pikuta et al. 2007), summarizing the ongoing knowledge on extremophiles drew too short axes for temperature, pH, and salinity on plots showing the physico-chemical conditions compatible with life. Some environmental microbes were definitely outliers with respect to the majority of known creatures. This counter-intuitive search for extremophiles likely reaches his summit in astrobiological studies, which search for life beyond Earth, seeking to define biomarkers in exoplanetary analogs and to train to detect these biomarkers in regions of the universe that currently fit the minimal requirements for life in C, H, N, O, P, S, liquid water, and energy (Olsson-Francis and Cockell 2010). No one knows whether extraterrestrial microbes will ultimately be discovered this way, but, at least, ironically terrestrial microbes, which can grow in the International Space Station and Spacecraft Assembly Facilities (Checinska et al. 2015) have potentially increased chances to spread in space, a problem known as the issue of planetary protection (McKay and Davis 1989).
Searching for Very Divergent Homologs: A Few Lessons
In as much as environmental genomics enhance microbial dark matter studies, for example, by unraveling extremophiles, it also raises issues, since environmental genomics has its own blind spots. The selection of samples, of genes of interests (e.g., in metabarcoding projects, or more generally in targeted environmental genomics) and the many filtering decisions and heuristics in the subsequent bioinformatic treatments imposed by the wealth of environmental sequences (i.e., reads and contigs), as well as the increased standardization of the methods and questions of environmental genomics studies (a logical scientific development for a comparative science; Vigliotti et al. 2017) raise the risk that the most unexpected of life forms, even if already sequenced, remain drowned under this deluge of data. This risk has notorious roots: our observations are strongly constrained by what our theory makes us prone to expect, and therefore by former perspectives informing various criteria in the sampling process.
This limit is obvious in the process of size-fractioning associated with metagenomics analyses, such as the one conducted in the Tara expedition, which a priori optimized the net sizes of its filter to capture different taxa of marine microbes (Karsenti et al. 2011). This procedure entails the inherent risk that important players of the microbial world may be overlooked if their sizes do not satisfy these filtering conditions. For example, 10 years ago, few (or even no) microbiologists nor virologists would have assumed that bacteria in the range of 0.2 microns and viruses >0.2 microns existed (Council 1999). This view radically changed with the discovery of ultrasmall bacteria, aka nanoorganisms, such as the CPR in 2015 (Brown et al. 2015; Luef et al. 2015) or some DPANN in 2010 (Baker et al. 2010), and with the discovery of giant viruses, such as Mimiviridae, in 2003 (La Scola et al. 2003). These taxa are now found in diverse environments, albeit at low abundance(Brown et al. 2015). CPR are remarkably phylogenetically diverse (Hug et al. 2016), representing up to 50% of the bacterial domain (Anantharaman et al. 2016), and present an unusual biology (i.e., 16 S RNA with insertion, lack of metabolic genes usually considered as essential), which suggests that CPR depend on other life forms (Kantor et al. 2013; Gong et al. 2014; Brown et al. 2015; Nelson and Stegen 2015; Danczak et al. 2017). CPR cells occupy an extremely tiny average volume of 0.009 ± 0.002 μm3, for a spherical diameter of 253 ± 25 nm (Luef et al. 2015). Mimivirus biology is not less striking. In particular, they are hosts to yet another new kind of viruses: virophages, that is, viruses of giant viruses (Boyer et al. 2011). The phylogenetic position of these relatively newcomers, especially regarding how deep CPR and giant viruses branch (if they do) with respect to the other Domains of life, is heavily debated (Colson et al. 2012; Moreira and Lopez 2015; Hug et al. 2016), even though, regarding the phylogenetic position of CPR, Hug et al. did not committ themselves strongly, stressing instead that their method did not result in a well resolved phylogeny (Hug et al. 2016). Such debates illustrates that attempts to establish novel groups inevitably (and logically) arise resistances, but no one questions that an accurate picture of the microbial world and its evolution can any longer satisfactorily be achieved without including nanoorganisms and viruses, be they giant or not.
Environmental genomics has not merely unraveled new microbial lineages, it has also reported new gene families (Riesenfeld et al. 2004; Lok 2015), new CRISPR-Cas systems (Burstein et al. 2017), and unusual gene forms (i.e., very divergent homologs from known genes). In principle, newly sequenced environmental genes could fall into one of 4 groups (fig. 1). The in silico functional and taxonomical annotations of environmental genes using existing ontologies (here, applied to 339 metagenomes; Fondi et al. 2016, sampling a diversity of environments, that is, soil, seawater, inland-water, wastewater, host, air, bioremediation, biotransformation, and sludge waste) indicates that most environmental genes have unknown functions, and belong to uncharacterized microbial lineages (fig. 2). In fact, at the minimum %ID threshold of 95%, >50% of these genes are neither functionally nor taxonomically annotated, and at the minimum %ID threshold of 50%, >30% of these genes are neither functionally nor taxonomically annotated, which stresses the genuine abundance of microbial dark matter in metagenomic data.
Fig. 1.—
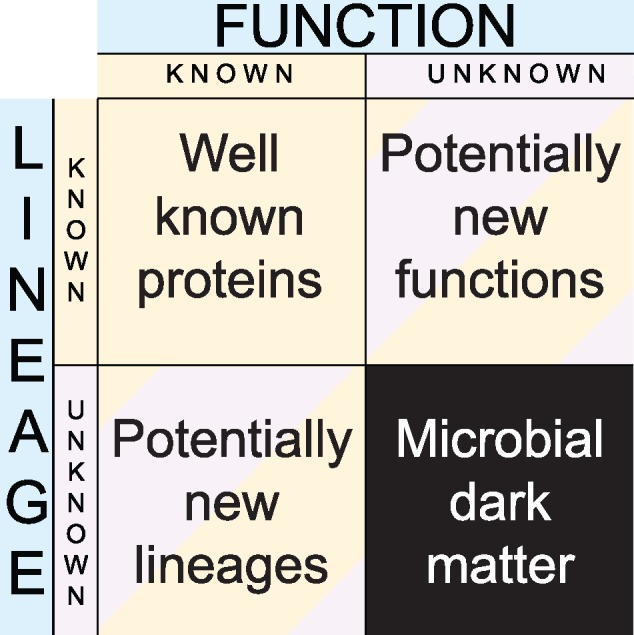
Four types of environmental sequences. Environmental sequences can be classified based on their taxonomical annotation (horizontal line) and their functional annotation (vertical column), which defines four categories. The cells in purple and black correspond to categories that are not readily explained based on current biological knowledge.
Fig. 2.—

Microbial dark matter across a diversity of environmental samples. Proteins inferred (with FragGeneScan; Rho et al. 2010) based on Metagenomic sequences from (Fondi et al. 2016), clustered based on their taxonomy (using MEGAN 6; Huson et al. 2016) and functional (using EggNOG-mapper; Huerta-Cepas et al. 2017) annotation. The pie charts represent the proportion of proteins from each type of environment. The taxonomy annotation was performed using three minimum percentage of identity: 50% (panels A and B), 85% (panels C and D), and 95% (panels E and F). In panels A, C, and E, the proteins were clustered based on their functional annotation including the category S (“Function unknown”). Panels B, D, and F were clustered with the exclusion of the category S.
Bioinformatic developments are currently designed to associate these unknown genes to reference gene families. For example, the search for highly divergent homologs using sequence similarity networks (Lopez et al. 2015) highlighted that a large majority of the ancient gene families that are well-conserved in cultured microbes have extremely divergent homologs in nature. Lopez et al. (2015) proposed that at least some of these very divergent homologs might sign the existence of deep branching yet unseen major divisions of life. Discovering environmental deeper lineages, branching below the currently recognized prokaryotic domains, could reopen the debate on the number of Domains of life, questioning our fundamental knowledge in terms of biological classifications and regarding early life evolution. Bioinformatic studies of random environmental sequences however need to be complemented by another type of experimental evidence, that is, individual sequences of genomes from putative very early branching microbes or even isolations of these organisms. The former type of evidence typically obtains by genome resolved metagenomics, that is, genome binning from metagenomics data sets. Genome binning consists in assembling metagenomic contigs using relative abundance and/or tetra nucleotide abundance (Sedlar et al. 2017). This protocol allows to recover synteny and to identify conserved or unusual/unexpected genes for related microorganisms. This approach is invaluable to recover genomes for uncultured organisms and to study their metabolic capabilities. Moreover, within the field of environmental genomics, single cell genomics offers an additional alternative approach to produce environmental data sets, identifying genes from the same genomes. Even though these approaches are gaining popularity and data start accumulating, so far, despite the actual high number of environmental “known unknowns” no scientists (i.e., peer-reviewers) working with major scientific journals have yet been convinced that enough evidence for new candidate Domains of life is available. For example, the remarkable work by (Parks et al. 2017) did not use universally shared ribosomal proteins to build a tree of life, including simultaneously novel environmental lineages, as well as known archaeal and bacterial lineages, whereas this strategy could have identified deep branching environmental groups.
Microbial Processes as a Yet Unexhausted Source of Knowledge
At the same time that new microbes were discovered, our knowledge on processes involving or affecting microbes evolved substantially. The focus on interactions and the use of networks rather than trees to frame microbial studies is emerging as a major trend. It is becoming obvious that simple tree-based models, aiming at reconstructing the divergence of lineages from a last common ancestor, are not fully doing justice to the diversity and complexity of the processes explaining microbial evolution. For example, in nature, diversity generating retroelements contribute to rapid, targeted sequence diversification in Archaea and their viruses (Paul et al. 2015), and in CPR (Paul et al. 2017). Introgressive processes such as lateral gene transfer stress the collective dimension of microbial evolution (Doolittle 1999; Ochman et al. 2000; Bapteste et al. 2012). Likewise, the discovery of environmental microbes with genuinely incomplete genomes (i.e., lacking genes considered as essential) and of syntrophic consortia insists on the importance of metabolic, ecological, and evolutionary scaffolding in the microbial world (DeLong 2007; Morris et al. 2012; Sachs and Hollowell 2012; Caporael et al. 2013; Brown et al. 2015; Ereshefsky and Pedroso 2015). The claim that in nature microbes depend on other microbes to survive, contrasts strongly with the notion that natural selection ultimately favors individual optimized lineages via the success of the fittest cells among large and phylogenetically homogeneous microbial populations. It matches however well with the empirical observation that pure culture fails for most microbes (Staley and Konopka 1985), and in fact provides an explanation for this great plate anomaly. Microbes belong to collectives rather than they live alone. Other striking interactions are also unveiled as scientists dig further into the microbial world. For example, unheard forms of communication impact microbial and viral population dynamics (Erez et al. 2017). Microbiomes and their hosts coconstruct a broad range of animal and plant phenotypes (Gill et al. 2006; Gilbert et al. 2015), to the point that some propose to introduce holobionts (the emergent associations of hosts and microbes) as a novel kind of central evolutionary player (Bordenstein and Theis 2015; Moran and Sloan 2015; Theis et al. 2016). At an even broader scale, in the environment, microbes, most of which are unknown, are now assumed to affect the geochemical processes that shape our planet (Guidi et al. 2016) and, by a process called niche construction (Laland et al. 2016), these microbes are considered likely to impact ecosystems and the future of life. All these processes (lateral gene transfer, scaffolding, communication, microbial coconstruction, and niche construction), while widespread in the microbial world, are still rather peripheral in biological explanations. Introducing the processes to which microbial dark matter contribute within biological theory thus requires revising the relative priority currently attributed to concepts in scientific explanations, which is likely to be a slow and tedious epistemic process. For example, prokaryotic biology, especially when considering microbiomes, appears in fact so different from the biology of model eukaryotic organisms that several evolutionary biologists and theoreticians have independently suggested that key aspects of the classic Darwinian theory and of the Modern Synthesis would have been very different had microbial studies been more central during the early development of the evolutionary theory. Others however disagree that the structure and content of the evolutionary theory requires to be reshaped, even in the light of this new knowledge in microbiology (Wray 2014). Yet, debates around the gene content, nature, and phylogenetic position of Asgard archaea (Saw et al. 2015; Da Cunha et al. 2017; Zaremba-Niedzwiedzka et al. 2017) powerfully illustrates that an enhanced knowledge of the microbial dark matter has unquestionably the potential to transform central elements in the evolutionary theory. If Asgard archaea, currently only known via assemblies of environmental reads, prove to be sister-groups of eukaryotes, this should (at least) impact the very notion of a tree of life, bring further evidence regarding the number of Domains of life (since a convincing argument that the 2 domains tree is better supported than the 3 domains tree predates the discovery of Asgard; Williams et al. 2013), and, depending on the intimate structural biology and metabolisms of these Asgard, it will also help testing among competing hypotheses for the origin of eukaryotes (Koonin 2015; Sousa et al. 2016).
On a different level, newly discovered microbial genes have also impacted, and could further impact, critical societal needs. Discovering enzymes, such as lipases (Rogalska et al. 1997) or organo-phosphorus degrading enzymes (Singh 2009), with greater activity, specificity, or stability, or new antibiotics in the environment (Lok 2015), such as Teixobactin (Ling et al. 2015), is central to the development of the industrial enzymes market, which is expected to represent up to 6.20 billion of dollars in 2020. Scientific research, as acknowledged by several Nobel Prizes, has also greatly benefited from the discovery of microbial enzymes, including restrictions enzymes, such as HindII (Smith and Wilcox 1970), or the DNA polymerases (Brock and Freeze 1969), which allowed the development of the Polymerase Chain Reaction (Saiki et al. 1988). More recently, the discovery of Crispr-cas9 systems (Jinek et al. 2012), now used for genome editing, also highlights the significant potential of microbial genes discovery to enhance the evolution of drugs, biotechnologies, and research tools.
Conclusion
The discovery of an increasing number of types of microbes has consistently shown that our planet hosts microbes with properties that were not simply identical to the ones formerly described. Studies of the microbial dark matter have brought forward the existence of novel entities (e.g., nanoorganisms, giant viruses, and virophages) and novel relationships within the microbial world (e.g., viral languages, high divergence, and scaffolding). This formerly dark microbial matter has not been unraveled randomly. To sum up its logic of discovery, it has required: to think outside the box (e.g., Woese’s definition of a novel Domain), to take scientifically and financially risky decisions (e.g., sampling sites where life was unlikely), to develop novel methods pushing back the limits of detection (e.g., better microscopes, inclusive networks), to prepare one’s mind to detect unknowns and unexpected forms (e.g., biomarkers), to identify and to seek to explain anomaly (e.g., the great plate count anomaly), to change perspectives (e.g., embracing the notion of nanoorganisms, or of multiple prokaryotic domains), to use analogies to uncover new microbial systems (e.g., for the study of extremophiles in space), to purposely depart from normal scientific practices and background knowledge (e.g., network studies of divergent gene forms, exploration of increasingly extreme environments), to be willing to create novel groups (e.g., Archea, CPR, Mimiviridae,…), and finally to convince (e.g., by banning competing notions, or by establishing new attractive fields, such as environmental genomics). Indeed, many of these discoveries presented in this work generated resistances. These resistances are perfectly explainable. Unraveling the unknown is especially difficult, because although we could empirically sketch a logic of scientific discovery, at the time each novel finding was made, their inventors could not yet rely on a standard method but essentially they had to convince the rest of the community that both their unusual approaches and finding were relevant. Convincing its own peers is finally essential, and possibly one of the largest and commonest challenge for microbial dark matter studies, and this seems especially difficult even for creative outsiders. Van Leeuwenhoek’s pioneering example offers indeed a great reminder that extraordinary results can easily be forgotten.
Acknowledgments
R.L., G.B., J.S.P., and E.B. are funded by the European Research Council (FP7/2017-2013 Grant Agreement #615274). We thank Dr Karen Olsson-Francis, Dr Yan Boucher, and Dr Lucie Bittner for stimulating discussion.
Literature Cited
- Anantharaman K, et al. 2016. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 7:13219.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BJ, et al. 2010. Enigmatic, ultrasmall, uncultivated Archaea. Proc Natl Acad Sci U S A. 10719:8806–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E, et al. 2012. Evolutionary analyses of non-genealogical bonds produced by introgressive descent. Proc Natl Acad Sci U S A. 10945:18266–18272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barer MR, Harwood CR.. 1999. Bacterial viability and culturability. Adv Microb Physiol. 41:93–137. [DOI] [PubMed] [Google Scholar]
- Barns SM, Delwiche CF, Palmer JD, Pace NR.. 1996. Perspectives on archaeal diversity, thermophily and monophyly from environmental rRNA sequences. Proc Natl Acad Sci U S A. 9317:9188–9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beja O, et al. 2000. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 2895486:1902–1906. [DOI] [PubMed] [Google Scholar]
- Bordenstein SR, Theis KR.. 2015. Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol. 138:e1002226.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer M, et al. 2011. Mimivirus shows dramatic genome reduction after intraamoebal culture. Proc Natl Acad Sci U S A. 10825:10296–10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, et al. 2002. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci U S A. 9922:14250–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock TD, Freeze H.. 1969. Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile. J Bacteriol. 981:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CT, et al. 2015. Unusual biology across a group comprising more than 15% of domain bacteria. Nature 5237559:208–211. [DOI] [PubMed] [Google Scholar]
- Bruno A, et al. 2017. Exploring the under-investigated “microbial dark matter” of drinking water treatment plants. Sci Rep. 7:44350.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burian RM. 2013. Exploratory experimentation. New York: Springer; p. 720–723. [Google Scholar]
- Burstein D, et al. 2017. New CRISPR-Cas systems from uncultivated microbes. Nature 5427640:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporael L, Griesemer J, Wimsatt W.. 2013. Scaffolding in evolution, culture, and cognition. Massachusetts: MIT Press. [Google Scholar]
- Checinska A, et al. 2015. Microbiomes of the dust particles collected from the International Space Station and Spacecraft Assembly Facilities. Microbiome 3:50.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson P, de Lamballerie X, Fournous G, Raoult D.. 2012. Reclassification of giant viruses composing a fourth domain of life in the new order Megavirales. Intervirology 555:321–332. [DOI] [PubMed] [Google Scholar]
- Council NR. editor. 1999. Report from the National Research Council. Washington (DC).
- Da Cunha V, Gaia M, Gadelle D, Nasir A, Forterre P.. 2017. Lokiarchaea are close relatives of Euryarchaeota, not bridging the gap between prokaryotes and eukaryotes. PLoS Genet. 13:e1006810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danczak RE, et al. 2017. Members of the Candidate Phyla Radiation are functionally differentiated by carbon- and nitrogen-cycling capabilities. Microbiome 51:112.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Rios A, Wierzchos J, Sancho LG, Ascaso C.. 2003. Acid microenvironments in microbial biofilms of antarctic endolithic microecosystems. Environ Microbiol. 54:231–237. [DOI] [PubMed] [Google Scholar]
- DeLong EF. 2007. Microbiology. Life on the thermodynamic edge. Science 3175836:327–328. [DOI] [PubMed] [Google Scholar]
- DeLong EF, et al. 2006. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 3115760:496–503. [DOI] [PubMed] [Google Scholar]
- Diamond J. 1997. Guns, germs, and steel: the fates of human societies. New York city: W. W. Norton. [Google Scholar]
- Doolittle WF. 1999. Phylogenetic classification and the universal tree. Science 2845423:2124–2129. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. 2013. Carl R. Woese (1928–2012). Curr Biol. 235:R183–R185. [DOI] [PubMed] [Google Scholar]
- Ereshefsky M, Pedroso M.. 2015. Rethinking evolutionary individuality. Proc Natl Acad Sci U S A. 11233:10126–10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez Z, et al. 2017. Communication between viruses guides lysis-lysogeny decisions. Nature 5417638:488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkowski P. 2015. Leeuwenhoek’s lucky break. Discover 1–5. [Google Scholar]
- Fondi M, et al. 2016. “Every Gene Is Everywhere but the Environment Selects”: global geolocalization of gene sharing in environmental samples through network analysis. Genome Biol Evol. 85:1388–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB.. 2005. Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proc Natl Acad Sci U S A. 10241:14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SF, Bosch TC, Ledon-Rettig C.. 2015. Eco-Evo-Devo: developmental symbiosis and developmental plasticity as evolutionary agents. Nat Rev Genet. 1610:611–622. [DOI] [PubMed] [Google Scholar]
- Gill SR, et al. 2006. Metagenomic analysis of the human distal gut microbiome. Science 3125778:1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J, Qing Y, Guo X, Warren A.. 2014. “Candidatus Sonnebornia yantaiensis”, a member of candidate division OD1, as intracellular bacteria of the ciliated protist Paramecium bursaria (Ciliophora, Oligohymenophorea). Syst Appl Microbiol. 371:35–41. [DOI] [PubMed] [Google Scholar]
- Guidi L, et al. 2016. Plankton networks driving carbon export in the oligotrophic ocean. Nature 5327600:465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Forslund K, Pedro Coelho L, Szklarczyk D, Juhl Jensen L, von Mering C, Bork P.. 2017. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. 348:2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug LA, et al. 2016. A new view of the tree of life. Nat Microbiol. 1:16048.. [DOI] [PubMed] [Google Scholar]
- Hugenholtz P, Goebel BM, Pace NR.. 1998. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 18018:4765–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, et al. 2016. MEGAN Community Edition – interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol. 126:e1004957.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, et al. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 3376096:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor RS, et al. 2013. Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. MBio 45:e00708–e00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenti E, et al. 2011. A holistic approach to marine eco-systems biology. PLoS Biol. 910:e1001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. 2015. Archaeal ancestors of eukaryotes: not so elusive any more. BMC Biol. 13:84.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy SR, Wang D.. 2017. Origins and challenges of viral dark matter. Virus Res. 239:136–142. [DOI] [PubMed] [Google Scholar]
- La Scola B, et al. 2003. A giant virus in amoebae. Science 2995615:2033.. [DOI] [PubMed] [Google Scholar]
- Laland K, Matthews B, Feldman MW.. 2016. An introduction to niche construction theory. Evol Ecol. 30:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. 2017. Antibiotics from the microbial dark matter. FASEB J. 31(Suppl 257):252. [Google Scholar]
- Ling LL, et al. 2015. A new antibiotic kills pathogens without detectable resistance. Nature 5177535:455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb B, Kurtz DA, Moreno-Hagelsieb G, Doxey AC.. 2015. Remote homology and the functions of metagenomic dark matter. Front Genet. 6:234.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok C. 2015. Mining the microbial dark matter. Nature 5227556:270–273. [DOI] [PubMed] [Google Scholar]
- Lopez P, Halary S, Bapteste E.. 2015. Highly divergent ancient gene families in metagenomic samples are compatible with additional divisions of life. Biol Direct. 10:64.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luef B, et al. 2015. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat Commun. 6:6372.. [DOI] [PubMed] [Google Scholar]
- McKay CP, Davis WL.. 1989. Planetary protection issues in advance of human exploration of Mars. Adv Space Res. 96:197–202. [DOI] [PubMed] [Google Scholar]
- Moran NA, Sloan DB.. 2015. The hologenome concept: helpful or hollow? PLoS Biol. 1312:e1002311.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira D, Lopez GP.. 2015. Evolution of viruses and cells: do we need a fourth domain of life to explain the origin of eukaryotes? Philos Trans R Soc Lond B Biol Sci. 3701678:20140327.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JJ, Lenski RE, Zinser ER.. 2012. The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. MBio 32:e00036-12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WC, Stegen JC.. 2015. The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front Microbiol. 6:713.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA.. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 4056784:299–304. [DOI] [PubMed] [Google Scholar]
- Olsson-Francis K, Cockell CS.. 2010. Experimental methods for studying microbial survival in extraterrestrial environments. J Microbiol Methods 801:1–13. [DOI] [PubMed] [Google Scholar]
- O’Malley MA. 2014. Philosophy of microbiology. Cambridge: Cambridge University Press. [Google Scholar]
- Pace NR. 2006. Time for a change. Nature 4417091:289.. [DOI] [PubMed] [Google Scholar]
- Parks DH, Rinke C, Chuvochina M, Chaumeil PA, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW.. 2017. Recovery of nearly 8, 000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2:1533–1542. [DOI] [PubMed] [Google Scholar]
- Paul BG, et al. 2015. Targeted diversity generation by intraterrestrial archaea and archaeal viruses. Nat Commun. 6:6585.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BG, et al. 2017. Retroelement-guided protein diversification abounds in vast lineages of Bacteria and Archaea. Nat Microbiol. 2:17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikuta EV, Hoover RB, Tang J.. 2007. Microbial extremophiles at the limits of life. Crit Rev Microbiol. 333:183–209. [DOI] [PubMed] [Google Scholar]
- Ram RJ, et al. 2005. Community proteomics of a natural microbial biofilm. Science 3085730:1915–1920. [PubMed] [Google Scholar]
- Rho M, Tang H, Ye Y.. 2010. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 3820:e191.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesenfeld CS, Goodman RM, Handelsman J.. 2004. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ Microbiol. 69:981–989. [DOI] [PubMed] [Google Scholar]
- Rinke C, et al. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 4997459:431–437. [DOI] [PubMed] [Google Scholar]
- Rogalska E, Douchet I, Verger R.. 1997. Microbial lipases: structures, function and industrial applications. Biochem Soc Trans. 251:161–164. [DOI] [PubMed] [Google Scholar]
- Sachs JL, Hollowell AC.. 2012. The origins of cooperative bacterial communities. MBio 33:e00099-12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki RK, et al. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 2394839:487–491. [DOI] [PubMed] [Google Scholar]
- Saw JH, et al. 2015. Exploring microbial dark matter to resolve the deep archaeal ancestry of eukaryotes. Philos Trans R Soc Lond B Biol Sci. 3701678:20140328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schickore J. 2014. Scientific discovery Stanford: The Stanford Encyclopedia of Philosophy. [Google Scholar]
- Schmidt TM, DeLong EF, Pace NR.. 1991. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol. 17314:4371–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlar K, Kupkova K, Provaznik I.. 2017. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Comput Struct Biotechnol J. 15:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh BK. 2009. Organophosphorus-degrading bacteria: ecology and industrial applications. Nat Rev Microbiol. 72:156–164. [DOI] [PubMed] [Google Scholar]
- Smith HO, Wilcox KW.. 1970. A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J Mol Biol. 512:379–391. [DOI] [PubMed] [Google Scholar]
- Sousa FL, Neukirchen S, Allen JF, Lane N, Martin WF.. 2016. Lokiarchaeon is hydrogen dependent. Nat Microbiol. 15:1–3. [DOI] [PubMed] [Google Scholar]
- Staley JT, Konopka A.. 1985. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 39:321–346. [DOI] [PubMed] [Google Scholar]
- Stetter KO. 2013. A brief history of the discovery of hyperthermophilic life. Biochem Soc Trans. 411:416–420. [DOI] [PubMed] [Google Scholar]
- Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW.. 2005. Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 35:e144.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis KR, Dheilly NM, Klassen JL, Brucker RM, Baines JF, Bosch TC, Cryan JF, Gilbert SF, Goodnight CJ, Lloyd EA, et al. 2016. Getting the hologenome concept right: an eco-evolutionary framework for hosts and their microbiomes. mSystems 1 (2): DOI: 10.1128/mSystems.00028-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe SG, et al. 2005. Comparative metagenomics of microbial communities. Science 3085721:554–557. [DOI] [PubMed] [Google Scholar]
- Tyson GW, et al. 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 4286978:37–43. [DOI] [PubMed] [Google Scholar]
- Tyson GW, et al. 2005. Genome-directed isolation of the key nitrogen fixer Leptospirillum ferrodiazotrophum sp. nov. from an acidophilic microbial community. Appl Environ Microbiol. 7110:6319–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, et al. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 3045667:66–74. [DOI] [PubMed] [Google Scholar]
- Vigliotti C, Lopez P, Bapteste E.. 2017. Microbial diversity studies: the (paradoxical) challenge to have a broad view with metagenomics In: Maurel PGMC, editor. Evolution and biodiversity. ISTE Editions. Amsterdam: Elsevier. [Google Scholar]
- Waters CK. 2007. The nature and context of exploratory experimentation: an introduction to three case studies of exploratory research. Hist Philos Life Sci. 293:275–284. [PubMed] [Google Scholar]
- Williams TA, Foster PG, Cox CJ, Embley TM.. 2013. An archaeal origin of eukaryotes supports only two primary domains of life. Nature 5047479:231–236. [DOI] [PubMed] [Google Scholar]
- Woese CR, Fox GE.. 1977. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A. 7411:5088–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Kandler O, Wheelis ML.. 1990. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 8712:4576–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba-Niedzwiedzka K, et al. 2017. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 5417637:353–358. [DOI] [PubMed] [Google Scholar]