

Abstract
Modular polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs) comprise giant multidomain enzymes responsible for the “assembly line” biosynthesis of many genetically encoded small molecules. Site-directed mutagenesis, protein biochemical, and structural studies have focused on elucidating the catalytic mechanisms of individual multidomain proteins and protein domains within these megasynthases. However, probing their functions at the cellular level typically has invoked the complete deletion (or overexpression) of multidomain-encoding genes or combinations of genes and comparing those mutants with a control pathway. Here we describe a “domain-targeted” metabolomic strategy that combines genome editing with pathway analysis to probe the functions of individual PKS and NRPS catalytic domains at the cellular metabolic level. We apply the approach to the bacterial colibactin pathway, a genotoxic NRPS–PKS hybrid pathway found in certain Escherichia coli. The pathway produces precolibactins, which are converted to colibactins by a dedicated peptidase, ClbP. Domain-targeted metabolomics enabled the characterization of “multidomain signatures”, or functional readouts of NRPS–PKS domain contributions to the pathway-dependent metabolome. These multidomain signatures provided experimental support for individual domain contributions to colibactin biosynthesis and delineated the assembly line timing events of colibactin heterocycle formation. The analysis also led to the structural characterization of two reactive precolibactin metabolites. We demonstrate the fate of these reactive intermediates in the presence and absence of ClbP, which dictates the formation of distinct product groups resulting from alternative cyclization cascades. In the presence of the peptidase, the reactive intermediates are converted to a known genotoxic scaffold, providing metabolic support of our mechanistic model for colibactin-induced genotoxicity. Domain-targeted metabolomics could be more widely used to characterize NRPS–PKS pathways with unprecedented genetic and metabolic precision.
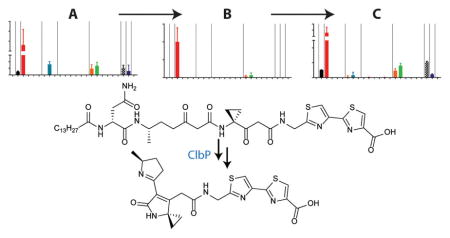
Graphical Abstract
INTRODUCTION
Polyketides, nonribosomal peptides, and hybrid polyketide–nonribosomal peptides constitute a large portion of our naturally produced small molecule drugs.1 Many of these small molecules are produced by giant multidomain (type-I) polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) enzyme systems. PKSs primarily convert a variety of simple acyl-CoA substrates into a structural panoply of molecules through Claisen condensation reactions.2 In a parallel strategy, NRPSs sample from about 500 available amino acid substrates, activate their carboxylic acids, and condense them through amide and ester bond-forming events.2b,3 Further structural complexity, such as epimerizations, N- and C-alkylations, cyclizations, and redox modifications, can be installed during assembly by accessory domains or proteins. Thiolation (T) domains, components of both PKS and NRPS systems, represent acyl- and peptidyl-carrier proteins, which are post-translationally modified with phosphopantetheine at an active-site serine. The phosphopantetheine arm tethers the transforming intermediates to the biosynthetic enzymes via a labile thioester bond (the “thiotemplate mechanism”). This commonality enables “hybrid” NRPS–PKS enzyme architectures to dramatically enhance the structural repertoire of their products. Importantly, however, because individual genes within these systems often encode multidomain proteins, gene deletions often destroy multiple catalytic domains within a given pathway. Thus, discrete domain functions are typically probed through site-directed mutagenesis, protein biochemical, and structural studies. Here we employ a genome editing strategy (multiplex automated genome engineering, MAGE)4 to systematically and site-selectively inactivate individual protein domains within a representative hybrid NRPS–PKS pathway and determine their relative effects on pathway-dependent metabolite production. This allows each metabolite to be assigned a “multidomain signature”, a functional metabolic readout of NRPS–PKS domain contributions. We show how multidomain signatures can be used to illuminate the metabolic contributions and general enzymatic timing events of individual catalytic domains in the colibactin biosynthetic pathway.5
The colibactin pathway consists of an ~55 kb hybrid NRPS–PKS biosynthetic gene cluster (clb or pks).6 The clb gene cluster is found in certain strains of Escherichia coli among other bacteria and is genotoxic to eukaryotic cells.6,7 The pathway was first reported in 2006,6 and its domain architecture was revised in 2014 to include additional domains, an embedded dehydratase (DH) and a series of “deteriorated” acyltransferase (AT*) domains.8 The clb locus has been shown to initiate tumor formation in colitis mouse models and is epidemiologically correlated to colorectal cancer patients.9 Paradoxically, a gene in the locus (clbA) has also been shown to be required for the efficacy of probiotic E. coli Nissle 1917 used to treat ulcerative colitis.10 Despite the importance of this pathway, natural colibactins capable of recapitulating cellular activities have not been isolated. Current efforts are focused on elucidating the functions and resulting metabolite structures of PKS ClbO and peptidase ClbL, the two remaining biosynthetic enzymes in the gene cluster yet to be experimentally included in the colibactin biosynthetic model. Many studies have elucidated the structures of clb-pathway-dependent metabolites,8a,11 which have aided our understanding of colibactin structure and reactivity. These structures have led to the synthesis of colibactin-type analogues with potent DNA alkylation activities in vitro.12 Colibactins are synthesized as prodrug scaffolds called “precolibactins”, which are cleaved in the bacterial periplasm to liberate an N-acyl-D-Asn side chain and genotoxic colibactins.8a,11a,13 With the available structural information, coarse full gene deletion studies, and select protein biochemical studies, a steadily growing colibactin biosynthetic model has been supported for these observed metabolites. A current biosynthetic model, including the interpretation of new results from this study, is shown in Figure 1.
Figure 1.

Proposed precolibactin biosynthesis. Domains: C, condensation; A, adenylation; T, thiolation; E, epimerase; KS, ketosynthase; AT, acyltransferase; KR, ketoreductase; DH, dehydratase; ER, enoyl reductase; AT*, deteriorated AT; Cy, dual condensation/cyclization; Ox, oxidase/dehydrogenase; TE, thioesterase.
To delineate the noncanonical assembly steps of colibactin biosynthesis, we employed the MAGE genome editing approach to individually and site-selectively inactivate bio-synthetic domains and proteins beginning with those required for 1-aminocyclopropane-1-carboxylate (ACC) incorporation (Figure 1, ClbHIJKLOQ; Tables S1 and S2). All of the upstream assembly line steps represent largely canonical transformations in PKS and NRPS biosynthesis and hence were excluded in this study. We carried out our studies in a clbP point mutant background (serine peptidase active-site mutant S78A) to monitor the metabolic effects on precolibactin production. We also analyzed ClbHI domain contributions in a wild-type background, as the only metabolites reported from the wild-type pathway are dependent on enzymes up through ClbI. Conserved active-site point mutants for individual domains were constructed on the basis of biochemical and structural knowledge about PKS and NRPS enzymes (see Figures S1 and S2). These mutations were created in a site-selective, scarless manner through the use of MAGE, a genome engineering technique that mediates allelic replacement on the E. coli chromosome or on bacterial artificial chromosomes (BACs) using single-stranded synthetic oligonucleotides. The advantage of MAGE is that it enables targeted mutations with single-base-pair resolution. We measured the production of previously reported clb-dependent ions,8a,11b known (pre)-colibactin metabolites, and some additional biosynthetic products. We report the domain dependencies of each of these metabolites (multidomain signatures, Tables S3–S5). We show that multidomain signatures can be generally utilized to support or exclude the involvement of individual domains in a biosynthetic transformation, determine the order of biosynthetic events even within a multidomain protein, and experimentally support points of biosynthetic divergence, such as module skipping. We also show that multidomain signatures are conserved in metabolites that have undergone post-assembly-line processing. To illustrate the power of multidomain signatures, we describe their use to delineate the unexpected timing events in the heterocycle assembly steps of colibactin biosynthesis.
RESULTS AND DISCUSSION
Domain-Targeted Metabolomics Decouples the Activities of ClbH Adenylation Domains
Recent structure–activity relationship experiments have demonstrated that colibactin-type compounds alkylate DNA by cyclopropane ring opening.12 ClbH is hypothesized to incorporate this functional group using a 1-aminocyclopropane-1-carboxylate (ACC) extender unit, although its biosynthesis remains unknown. In plants, ACC is an intermediate in ethylene biosynthesis and is derived from methionine via a pyridoxal phosphate (PLP)-dependent mechanism.14 ACC is not known to be produced by E. coli, and while isotopic labeling studies have shown that the ACC moiety in precolibactins is derived from methionine,11b,d the colibactin gene cluster does not encode apparent PLP-dependent enzymes. ClbH, the bio-synthetic enzyme that incorporates this moiety, has an unusual domain architecture for an NRPS. In addition to the canonical C–A–T module (containing condensation, adenylation, and thiolation domains), ClbH contains a second N-terminal A domain, A1–C–A2–T (Figure 2a). Bioinformatic analysis11b and biochemical studies11f,15 indicate that the noncanonical A1 domain activates serine and loads the carrier protein ClbE in trans. Dehydrogenases ClbD and ClbF then oxidize the enzyme-bound serine to produce the polyketide extender unit α-aminomalonyl-ClbE.11f,15 ClbG transfers the α-aminomalonyl unit onto the T domain of ClbK.11f It is currently unknown whether additional α-aminomalonyl units are incorporated or compete with malonyl building blocks, a phenomenon that has been observed in vivo for polyketide structural diversification16 and in in vitro protein biochemical studies.11f It has also been speculated that ClbH-A1 may participate in ACC biosynthesis on the basis of the traditional deletion of the multidomain gene clbH.11d To determine the functional contributions of these A domains, we individually inactivated all of the ClbH domains and measured the relative production of clb-dependent metabolites. As shown in Figure 2b, metabolite 1 (and downstream products) containing a cyclopropyl moiety are completely abolished when any of the canonical C–A2–T domains are inactivated, but the production of 1 is unaffected when ClbH-A1 is inactivated. The ClbP-cleaved products, whose structures were confirmed by ClbP-dependent cleavage of lactam 2 (Figure S3), also showed similar multidomain signatures in a wild-type background (Table S4). Thus, ClbH-A1 is dispensable for cyclopropane biosynthesis in vivo. Its metabolic effects are only observed downstream (see below). Notably, these individual domain contributions to metabolite production could not have been observed in a typical clbH knockout strain.
Figure 2.

ClbH-A1 is dispensable whereas ClbH-C–A2–T is required for cyclopropane incorporation. (a) ClbH-A1 loads serine onto carrier protein ClbE, dehydrogenases ClbD and ClbF convert it to α-aminomalonyl-ClbE, and AT ClbG transfers this extender unit downstream. (b) Multidomain signatures of precolibactins 1 and 2. Cellular trans-AT complementation for the ClbI-cis-AT mutant is noted. Domains are color-coded by function, and domains of individual multimodular proteins are grouped together by vertical lines.
The colibactin biosynthetic pathway unusually contains a mixture of cis- and trans-AT PKSs. ClbI contains an embedded functional AT domain (a cis-AT PKS). Production of metabolite 2,11b–d the decarboxylated and cyclized product of ClbI, is abolished when its ketosynthase (KS) and T domains are inactivated, as expected (Figure 2). However, inactivation of the ClbI-AT domain (GXSXG active-site motif → GXAXG) only partially reduces production. For this active-site mutant, malonyl extender units would be complemented in trans in vivo to generate products such as 2. Consistent with this observation, fatty acid biosynthetic proteins (FabD) have been shown to complement malonyl-transfer activity in vitro.11f Through structural modeling and protein homology studies, we previously reported that the other trans-AT PKSs in this pathway showed signs of accelerated evolution in “deteriorated” AT (AT*) domains.8b Their active-site GXSXG motifs were naturally mutated to GXGXG motifs. Similar types of inactivated AT domains have been reported in other select trans-AT (a.k.a., AT-less) PKS biosynthetic pathways.17 These observations are reminiscent of inactivated starter unit AT (SAT) domains in fungal iterative PKSs (SAT* domains, GXCXG → GXGXG),18 which are thought to have evolved predominantly through gene duplication and functional divergence.19 The functional in vivo redundancy observed here for our inactivated ClbI-AT domain is consistent with a reduction in evolutionary pressure to maintain functional AT domains in the clb gene cluster.
Domain-Targeted Metabolomics Establishes General Timing Events in (Bi)thiazole Biosynthesis
In the next enzyme, NRPS ClbJ, there is a bioinformatic disconnect between its domain architecture and the observed products. Specifically, the timing of oxidation to establish the first precolibactin thiazole ring remains unknown, as ClbJ lacks a dedicated oxidase (Ox) domain (Figure 3). It has been presumed that the oxidation required to establish the first thiazole ring is either spontaneous, processed by generic cellular oxidases, and/or catalyzed by the Ox domain in downstream ClbK. Only a single Ox domain exists in ClbK. By analogy to the bleomycin bithiazole biosynthetic proposals,20 it is possible that this domain is responsible for oxidizing both the first and second thiazoline rings to establish the bithiazole observed in metabolites like precolibactin C (5). To delineate these general timing events, we individually inactivated every domain in ClbJK. In our Ox mutant, we observed a reduction in monothiazole-containing precolibactin B (3) (Figure 3). Its residual detection was consistent with the proposed spontaneous (or cellular) oxidation of hydrolyzed monothiazoline-containing precolibactin 4. However, in our Ox domain mutant, we observed a dramatic upregulation of a metabolite consistent with this thiazoline 4, as supported by tandem MS comparisons to its oxidation product precolibactin B and isotopic labeling studies (Figures S4 and S5). Importantly, in the Ox mutant, we also did not observe any advanced products beyond precolibactin B. These data suggest that the assembly line oxidation of the first thiazoline is catalyzed by the Ox domain in ClbK and that the thiazole product is a preferred substrate for further assembly line processing events (i.e., oxidation to 3 occurs prior to amide bond formation and subsequent tailoring events to generate 5).
Figure 3.

Multidomain signatures reveal unexpected biosynthetic events. (A) The thiazoline introduced by ClbJ is oxidized on the assembly line by the downstream Ox domain in ClbK. Module skipping of ClbK-PKS results in diversified products. (B) Accumulation of thiazoline 4 in the Ox mutant supports that ClbK Ox is responsible for oxidizing the thiazoline installed by ClbJ to the thiazole. (C) Module skipping is observed in the multidomain signature of 5. Bithiazole 5 does not require the ClbK-PKS module or the ClbH-A1 domain. Macrocycle 6 requires the ClbK-PKS module for α-aminomalonyl extender unit incorporation.
Multidomain Signatures Experimentally Support Complete Module Skipping as a Strategy for Product Diversification
Recent structural elucidation efforts suggest that the α-aminomalonyl unit can be incorporated into ClbK products, leading to macrocyclic product 6 with an α-aminomalonyl polyketide extension bisecting the bithiazole.11h Thus, it was proposed on the basis of precolibactin structural analysis that precolibactin C (5) is derived from module skipping in ClbK. To experimentally test these predictions, we analyzed the multidomain signatures for precolibactins 5 and 6. As expected, the first committed step in aminomalonyl biosynthesis (ClbH-A1) and all of the NRPS–PKS domains in ClbK were required for the production of precolibactin 6, which incorporates an aminomalonyl-derived unit (Figure 3). Interestingly, we showed that the PKS domains in ClbK, as well as ClbH-A1, are completely dispensable for bithiazole biosynthesis in precolibactin C, and in fact, these mutations lead to exclusive and enhanced bithiazole formation. These studies demonstrate that multidomain signatures can be used to experimentally support points of divergence in a complex biosynthetic pathway and that module skipping in the clb pathway does not require non-elongating transfer through the PKS domains.
Multidomain Signatures Illuminate Post-Assembly-Line Processing Events and Reactive (Pre)colibactin Metabolites
To date, three different structural classes of precolibactins have been characterized: the lactams, such as 2; the aromatic pyridones, such as 3–5; and macrocycle 6. The lactams and pyridones can currently be accessed synthetically through common linear intermediates via selective cyclization reactions.11g,12 When the linear products are synthesized as their deacylated (ClbP-cleaved) forms, they readily cyclize to lactams, which exhibit potent DNA alkylation activities in vitro.12 N-acylated linear precursors, however, readily form pyridones through a double cyclodehydration sequence under mildly acidic or basic conditions.11g Control experiments established that pyridones do not alkylate DNA. These data suggested that the linear precolibactins, which were based on a theoretical biomimetic synthesis, should also exist in the cell. To address this question, we analyzed our metabolomic data for these precolibactin metabolites, and indeed, we could assign multidomain signatures for two new precolibactin metabolites, 7 and 8 (Figure 4a). The multidomain signatures of metabolites 7 and 8 closely resemble that of pyridone 5. This similarity was expected and indicates that multidomain signatures can be used to predict post-assembly-line processing events and that the cyclization from 7 to 8 to 5 occurs after hydrolytic release from the assembly line. These reactive precolibactin metabolites were confirmed by comparisons to synthetic standards (coinjections, Figure 4b; tandem MS, Figures S6 and S7). Linear metabolite 7 was robustly detected, particularly in mutants that exclusively generate bithiazole products (Figure 4b). Precolibactin lactam 8 was detected in trace amounts, an observation consistent with its facile in vitro cyclization to the corresponding pyridone.11g These metabolic data lend further support to a model in which the pyridones are irreversible, off-path products deriving from the linear precursor 7 in the absence of functional peptidase ClbP. Analogous linear species of macrocycle 6 were not detected under the conditions of our experiments. It is currently unclear whether the cyclizations leading to macro-cycle 6, which have only weak cytotoxic activity, are similarly off-path.
Figure 4.

Similarities in multidomain signatures support post-assembly-line transformations. (A) The multidomain signatures of reactive precolibactins, linear (7) and lactam (8), en route to precolibactin C (5) (top, bottom, and Figure 3, bottom left, respectively) show similar multidomain dependencies, indicating that the chemical transformations occur off the assembly line. (B) New precolibactin metabolites were confirmed via comparisons with synthetic standards. (C) ClbP cleaves supplemented 7 to form lactam 10. Pyridone 9 was also detected. Coinjections with synthetic standards via the intermediacy of 12 confirmed the production of these two colibactin metabolites. (D) Proposed biosynthetic fate of reactive precolibactin 7 metabolite in the presence and absence of peptidase ClbP. ClbP is required in order to access lactam 10, a known genotoxic scaffold.
To assess the structural fate of the newly observed linear precolibactin 7 metabolite in the presence and absence of peptidase ClbP, we fed 7 to E. coli DH10B either carrying an empty control plasmid (pBAD18) or expressing ClbP (pClbP) (Figure 4c). In the absence of ClbP, no cleavage was observed. However, the linear reactive metabolite was converted to the stable aromatic precolibactin pyridone 5. In the presence of ClbP, several cleavage products were observed, including N-myristoyl-D-asparagine and colibactin pyridone 9, as we previously reported for cleavage of precolibactin pyridone 5,11g and new colibactin lactam scaffold 10. The unsaturated imine 10 had not been previously detected in E. coli extracts and was identical to a synthetic standard produced by cyclizing precursor 12 in saturated sodium bicarbonate. The presence of 9 in this sample likely arises from spontaneous cyclization of 7 to 8 to 5 prior to cleavage. The colibactin metabolites were confirmed by comparisons to synthetic standards (coinjections, pClbP + 9 or + 10; Figures 4c, S8, and S9). Promiscuous ClbP thus cleaves precolibactins independent of their cyclization states. These data support the proposed model, in which reactive linear precolibactin intermediates (e.g., 7) are synthesized by the assembly line. In the absence of ClbP, these linear intermediates form inactive products (e.g., pyridones), whereas they are converted to known genotoxic lactam scaffolds (e.g., 10) in the presence of ClbP (Figure 4d).
CONCLUSIONS
In the assembly line biosynthesis of polyketides and non-ribosomal peptides, the intermediates are attached to carrier proteins (T domains) through labile thioester bonds, which are subject to hydrolysis. Detection of these hydrolyzed intermediates provides a “Sanger sequencing-like” view of the pathway. Previous studies have inactivated the terminal off-loading thioesterase (TE) domains of NRPS–PKS pathways to build up these intermediates and promote their hydrolysis and detection.21 However, in the colibactin pathway, an atypical editing-type TE, ClbQ, promotes promiscuous off-loading of most of the precolibactin assembly intermediates, as observed in the multidomain signatures here and in other recent studies.11h We speculate that these assembly line derailment products unusually enhanced by ClbQ could have functional cellular roles, as has been suggested from our previous in vitro assays.8a Nevertheless, the enzyme-bound thioester intermediates are labile and hydrolyze even in the absence of a functional ClbQ. Domain-targeted metabolomic analyses of these detectable hydrolysis products enabled the systematic assessment of the functions of individual domains in the colibactin pathway, provided unexpected insights for the general timing events of colibactin heterocycle formation, and led to the characterization of reactive metabolites that metabolically supported our mechanistic model for colibactin-induced genotoxicity. Because MAGE can be used to site-selectively mutate bacterial artificial chromosomes (BACs), domain-targeted metabolomics could be generally applied to many heterologous expression systems. With the emergence of genome editing tools, such as MAGE used here, and the use of sensitive modern-day mass spectrometers, we expect that domain-targeted metabolomics could be widely utilized to study the functions of multidomain pathways at the cellular level with high genetic and metabolic precision.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (1DP2-CA186575 to J.M.C. and R01GM110506 to S.B.H.), the Damon Runyon Cancer Research Foundation (DRR-39-16 to J.M.C.), the Searle Scholars Program (13-SSP-210 to J.M.C.), and Yale University is gratefully acknowledged. E.E.S. was supported by the National Science Foundation Graduate Research Fellowships Program. We thank Ryan R. Gallagher, Jaymin R. Patel, and Farren J. Isaacs (Yale University) for providing the EcNR1 variant used in this study and guidance in initiating MAGE technologies in the lab.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b00659.
Experimental procedures and supplementary figures and tables (PDF)
References
- 1.(a) Newman DJ, Cragg GM. J Nat Prod. 2016;79:629. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]; (b) Shen B. Cell. 2015;163:1297. doi: 10.1016/j.cell.2015.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]; (b) Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; (c) Khosla C, Herschlag D, Cane DE, Walsh CT. Biochemistry. 2014;53:2875. doi: 10.1021/bi500290t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Finking R, Marahiel MA. Annu Rev Microbiol. 2004;58:453. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]; (b) Walsh CT, O’Brien RV, Khosla C. Angew Chem, Int Ed. 2013;52:7098. doi: 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Nature. 2009;460:894. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gallagher RR, Li Z, Lewis AO, Isaacs FJ. Nat Protoc. 2014;9:2301. doi: 10.1038/nprot.2014.082. [DOI] [PubMed] [Google Scholar]
- 5.(a) Trautman EP, Crawford JM. Curr Top Med Chem. 2016;16:1705. doi: 10.2174/1568026616666151012111046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Balskus EP. Nat Prod Rep. 2015;32:1534. doi: 10.1039/c5np00091b. [DOI] [PubMed] [Google Scholar]; (c) Taieb F, Petit C, Nougayrede JP, Oswald E. EcoSal Plus. 2016 doi: 10.1128/ecosalplus.ESP-0008-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Science. 2006;313:848. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 7.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP. Proc Natl Acad Sci U S A. 2010;107:11537. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Vizcaino MI, Engel P, Trautman E, Crawford JM. J Am Chem Soc. 2014;136:9244. doi: 10.1021/ja503450q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engel P, Vizcaino MI, Crawford JM. Appl Environ Microbiol. 2015;81:1502. doi: 10.1128/AEM.03283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. Science. 2012;338:120. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. Gut Microbes. 2014;5:675. doi: 10.4161/19490976.2014.969989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olier M, Marcq I, Salvador-Cartier C, Secher T, Dobrindt U, Boury M, Bacquie V, Penary M, Gaultier E, Nougayrede JP, Fioramonti J, Oswald E. Gut Microbes. 2012;3:501. doi: 10.4161/gmic.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Bian X, Fu J, Plaza A, Herrmann J, Pistorius D, Stewart AF, Zhang Y, Muller R. ChemBioChem. 2013;14:1194. doi: 10.1002/cbic.201300208. [DOI] [PubMed] [Google Scholar]; (b) Vizcaino MI, Crawford JM. Nat Chem. 2015;7:411. doi: 10.1038/nchem.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brotherton CA, Wilson M, Byrd G, Balskus EP. Org Lett. 2015;17:1545. doi: 10.1021/acs.orglett.5b00432. [DOI] [PubMed] [Google Scholar]; (d) Bian XY, Plaza A, Zhang YM, Muller R. Chem Sci. 2015;6:3154. doi: 10.1039/c5sc00101c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li ZR, Li Y, Lai JY, Tang J, Wang B, Lu L, Zhu G, Wu X, Xu Y, Qian PY. ChemBioChem. 2015;16:1715. doi: 10.1002/cbic.201500239. [DOI] [PubMed] [Google Scholar]; (f) Zha L, Wilson MR, Brotherton CA, Balskus EP. ACS Chem Biol. 2016;11:1287. doi: 10.1021/acschembio.6b00014. [DOI] [PubMed] [Google Scholar]; (g) Healy AR, Vizcaino MI, Crawford JM, Herzon SB. J Am Chem Soc. 2016;138:5426. doi: 10.1021/jacs.6b02276. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Li ZR, Li J, Gu JP, Lai JYH, Duggan BM, Zhang WP, Li ZL, Li YX, Tong RB, Xu Y, Lin DH, Moore BS, Qian PY. Nat Chem Biol. 2016;12:773. doi: 10.1038/nchembio.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Healy AR, Nikolayevskiy H, Patel JR, Crawford JM, Herzon SB. J Am Chem Soc. 2016;138:15563. doi: 10.1021/jacs.6b10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Dubois D, Baron O, Cougnoux A, Delmas J, Pradel N, Boury M, Bouchon B, Bringer MA, Nougayrede JP, Oswald E, Bonnet R. J Biol Chem. 2011;286:35562. doi: 10.1074/jbc.M111.221960. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cougnoux A, Gibold L, Robin F, Dubois D, Pradel N, Darfeuille-Michaud A, Dalmasso G, Delmas J, Bonnet R. J Mol Biol. 2012;424:203. doi: 10.1016/j.jmb.2012.09.017. [DOI] [PubMed] [Google Scholar]; (c) Brotherton CA, Balskus EP. J Am Chem Soc. 2013;135:3359. doi: 10.1021/ja312154m. [DOI] [PubMed] [Google Scholar]
- 14.Adams DO, Yang SF. Proc Natl Acad Sci U S A. 1979;76:170. doi: 10.1073/pnas.76.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brachmann AO, Garcie C, Wu V, Martin P, Ueoka R, Oswald E, Piel J. Chem Commun. 2015;51:13138. doi: 10.1039/c5cc02718g. [DOI] [PubMed] [Google Scholar]
- 16.Park HB, Crawford JM. J Nat Prod. 2015;78:1437. doi: 10.1021/np500974f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohman JR, Ma M, Osipiuk J, Nocek B, Kim YC, Chang CS, Cuff M, Mack J, Bigelow L, Li H, Endres M, Babnigg G, Joachimiak A, Phillips GN, Shen B. Proc Natl Acad Sci U S A. 2015;112:12693. doi: 10.1073/pnas.1515460112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Crawford JM, Dancy BC, Hill EA, Udwary DW, Townsend CA. Proc Natl Acad Sci U S A. 2006;103:16728. doi: 10.1073/pnas.0604112103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crawford JM, Vagstad AL, Ehrlich KC, Townsend CA. Bioorg Chem. 2008;36:16. doi: 10.1016/j.bioorg.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kroken S, Glass NL, Taylor JW, Yoder OC, Turgeon BG. Proc Natl Acad Sci U S A. 2003;100:15670. doi: 10.1073/pnas.2532165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galm U, Wendt-Pienkowski E, Wang L, Huang SX, Unsin C, Tao M, Coughlin JM, Shen B. J Nat Prod. 2011;74:526. doi: 10.1021/np1008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helfrich EJ, Piel J. Nat Prod Rep. 2016;33:231. doi: 10.1039/c5np00125k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.