

Abstract
Electrocatalytic methods for organic synthesis could offer sustainable alternatives to traditional redox reactions, but strategies are needed to enhance the performance of molecular catalysts designed for this purpose. Herein, we describe the synthesis of a pyrene-tethered TEMPO derivative (TEMPO = 2,2,6,6-tetra-methylpiperidinyl-N-oxyl) that undergoes facile in situ non-covalent immobilization onto a carbon-cloth electrode. Cyclic voltammetry and controlled potential electrolysis studies demonstrate that the immobilized catalyst exhibits much higher activity relative to, 4-acetamido-TEMPO, an electronically similar homogeneous catalyst. Turnover numbers and frequencies approach 2000 and 4000 h−1, respectively, in preparative electrolysis experiments with a series of alcohol substrates. The synthetic utility of the method is further demonstrated in the oxidation of a sterically hindered hydroxymethylpyrimidine precursor to the blockbuster drug, rosuvastatin.
Keywords: Alcohol oxidation, immobilization, TEMPO, electrocatalysis, pyrene
COMMUNICATION
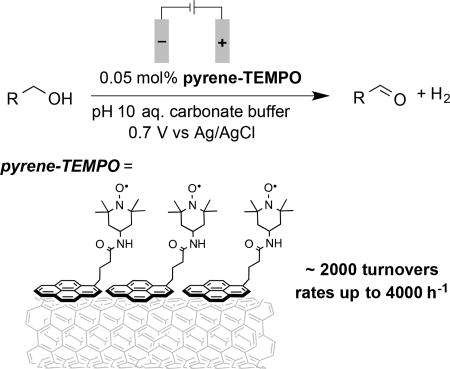
Performance link: Non-covalent immobilization of a TEMPO-based catalyst tethered to pyrene on a carbon-cloth electrode results in very efficient catalytic alcohol oxidation. The facile in situ catalyst immobilization protocol demonstrates a strategy to achieve improved catalytic performance that could find widespread utility in electroorganic synthesis.
Molecular electrocatalysis has been the focus of substantial recent attention in the area of energy storage and conversion,[1] with applications ranging from water oxidation and hydrogen production to the conversion of CO2 to liquid fuels. Advances in this area have important implications for the complementary field of electroorganic synthesis.[2,3] In the present study, we show how a non-covalent catalyst immobilization method widely used in energy-based applications greatly enhances the performance and practicality of TEMPO-based electrocatalysts for electrochemical alcohol oxidation (TEMPO = 2,2,6,6-tetra-methylpiperidinyl-N-oxyl).
Molecular electrocatalysis resembles traditional homo-geneous catalysis, but the reactions use an electrode rather than a chemical reagent as the electron donor or acceptor to promote redox transformations. This approach has the potential to improve the sustainability of chemical reactions, but key challenges must be overcome to achieve such benefits. For example, electrochemical reactions with homogeneous catalysts can experience lower rates because catalyst turnover only occurs when the catalyst is in diffusive contact with the electrode.[4] This limitation underlies the considerable efforts directed toward immobilizing molecular catalysts on electrode surfaces, whereby the catalyst can retain electronic communication with the electrode.[5] The performance benefits associated with catalyst immobilization can be offset, however, by the added cost or complexity of the immobilization process and by the limited stability of many molecular catalysts.
TEMPO and related organic nitroxyl derivatives have been used extensively as catalysts for alcohol oxidation in combination with a chemical oxidants.[6, 7] Electrochemical methods are appealing because anodic nitroxyl-mediated alcohol oxidation may be coupled to cathodic proton reduction, resulting in generation of H2 as the sole by-product of the reaction (Scheme 1). A number of examples of this chemistry have been reported, including those with TEMPO as a dissolved catalytic mediator[8] and immobilized on electrode surfaces.[9,10] High catalyst loadings (up to 1 equiv TEMPO) are common when the nitroxyl is used as a dissolved mediator in order to offset the low rates observed at lower catalyst loading.[8a,c–e] Various methods have been investigated for TEMPO immobilization, the majority of which use polymer-immobilization methods.[9a–e] Common examples include coupling of 4-amino- TEMPO with carboxylic acids on a polyacrylic acid-coated electrode,[9b,c,e] as well as electropolymerization of pyrrole tethered to TEMPO.[9a] While these methods lead to significantly better catalytic rates, TEMPO instability (especially in its oxoammonium form) limits the catalyst lifetime, eventually degrading the electrode performance, and mass transport through polymeric networks can reduce performance.[11]
Scheme 1.

Electrochemical TEMPO-mediated alcohol oxidation with the formation of H2 as the sole by-product.
In a recent analysis of a series of organic nitroxyl derivatives for electrocatalytic alcohol oxidation, we identified 4-acetamido- TEMPO (ACT) as an especially effective catalyst.[10b] The high redox potential of this derivative contributes to much higher turnover rates than those observed with TEMPO or other nitroxyls. Because the electrode potential may be tuned to match the nitroxyl redox potential, the slow catalyst reoxidation observed with chemical oxidants (e.g., NaOCl) is avoided. The present study was motivated by an interest in developing a practical synthetic protocol for ACT-mediated electrochemical alcohol oxidation. Previous strategies for TEMPO immobilization were considered, but a preferable method would achieve the rate benefits of catalyst immobilization without causing irreversible fouling of the electrode due to catalyst decay. Our attention was drawn to the widespread use of pyrene as a non-covalent anchor for molecular electrocatalysts in energy-related applications.[12] This catalyst immobilization method has not yet been applied to synthetic applications. The results reported herein describe the development, testing, and application of pyrene-TEMPO conjugates for electrochemical alcohol oxidation, and highlight the potential utility of pyrene-tethered electrocatalysts for synthetic organic chemistry.
A pyrene-TEMPO derivative was readily synthesized in excellent yield via amide coupling between the commercially available pyrene-linked carboxylic acid derivative and 4-amino TEMPO, as shown in Eq. (1). The identity of the pyrene-TEMPO conjugate was confirmed by 1H and 13C NMR spectroscopy, following reduction of the nitroxyl to the corresponding hydroxylamine with ascorbic acid (see Supporting Information for details). A cyclic voltammogram of the pyrene-TEMPO species dissolved in 1:1 acetonitrile/water reveals reversible electrochemical oxidation to the corresponding oxoammonium species at a potential nearly identical to that of 4-acetamido-TEMPO (ACT) (E1/2 = 0.69 V and 0.70 V vs Ag/AgCl for pyrene-TEMPO and ACT, respectively; Figure 1).[13]
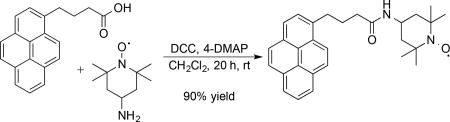 |
(1) |
Figure 1.

Cyclic voltammograms of the pyrene-TEMPO conjugate (solid line) and 4-acetamido-TEMPO (dashed line) in 1:1 acetonitrile/water containing 0.1 M NaHCO3/Na2CO3 (1:1). Conditions: TEMPO derivative (1 mM), glassy carbon working electrode, Pt-wire counter electrode, Ag/AgCl reference electrode; 25 mV·s−1 scan rate.
Pyrene has been shown to adsorb especially well to multi-walled carbon nanotubes (MWCNTs) via π-π stacking interactions.[14] Consequently, MWCNTs were drop-casted onto a glassy carbon (GC) electrode. A surface immobilized pyrene-TEMPO electrode was then prepared by dipping the GC-MWCNT electrode into an acetonitrile solution containing the pyrene-TEMPO derivative, followed by rinsing with acetonitrile to remove loosely bound pyrene-TEMPO (Figure 2A; see Supporting Information for details). The electrochemical behavior of pyrene-TEMPO-functionalized electrode was then analyzed by cyclic voltammetry (CV) (Figure 2B). The voltammograms reveal a reversible oxidation feature at E1/2 = 0.66 V vs Ag/AgCl in aqueous solution (pH 10). At slow scan rate (5 mV·s−1), the peak-to-peak separation between the anodic and cathodic waves is 26 mV, considerably less than the 59 mV separation that would be expected for a diffusion-controlled redox couple. A linear relationship between the peak current and scan rate (Figure 2C) provides further evidence for effective immobilization of the pyrene-TEMPO derivative on the surface of the electrode.[15]
Figure 2.

(A) Schematic representation of pyrene-TEMPO immobilized on the surface of carbon nanotubes. (B) Cyclic voltammograms of pyrene-TEMPO immobilized on the surface of the GC-MWCNTs electrode at various scan rates. [Conditions: pyrene-TEMPO/GC-MWCNT as working electrode, Pt-wire counter electrode, Ag/AgCl reference electrode, 0.2 M NaHCO3/Na2CO3 (1:1)] (C) Scan rate vs peak current plot. See Supporting Information for detailed experimental procedures for B and C.
Electrocatalytic alcohol oxidation by surface-immobilized pyrene-TEMPO was then analyzed by CV, with benzyl alcohol as the substrate (Figure 3). A catalytic feature is clearly evident in the anodic CV scan at a potential that corresponds to formation of the oxoammonium species, and the catalytic current increases at higher pH. The pH dependence observed with the pyrene-immobilized TEMPO resembles the trend observed previously with dissolved TEMPO derivatives.[10a,b] The enhanced rate at higher pH reflects the established mechanism of oxoammonium-mediated alcohol oxidation,[6] whereby deprotonation of the alcohol leads to formation of an oxoammonium-alkoxide adduct en route to the aldehyde product. A CV study was then carried out to compare the electrocatalytic performance of pyrene-immobilized TEMPO with the electronically similar dissolved electrocatalyst, ACT. The concentration of dissolved ACT was adjusted until the recorded CV showed a similar peak current to that of the immobilized pyrene-TEMPO (see insets of Figures 4A and 4B). CVs recorded after addition of benzyl alcohol (100 mM) revealed a relatively small catalytic feature from the ACT solution, while very high catalytic current was observed with pyrene-TEMPO (Figures 4A and 4B), reflecting dramatically improved electrocatalytic performance of the immobilized catalyst.
Figure 3.

The pH dependence of electrocatalytic oxidation of benzyl alcohol with surface-immobilized pyrene-TEMPO on GC-MWCNT as the working electrode in presence of 25 mM benzyl alcohol.
Figure 4.

Comparison of the electrochemical activity of 4-acetamido-TEMPO and pyrene-TEMPO for benzyl alcohol oxidation by CV. (scan rate: 20 mV/s). (A) CV of 2.5 mM 4-acetamido-TEMPO in the absence of benzyl alcohol (solid line) and the presence of benzyl alcohol (dashed line), (B) CV of surface immobilized pyrene-TEMPO (2.1 nmol electrochemically active pyrene-TEMPO based on CV) in the absence of benzyl alcohol (solid line) and the presence of benzyl alcohol (dashed line). See Supporting Information for details.
Efforts then turned to evaluating the effectiveness of the immobilized pyrene-TEMPO catalyst in bulk electrolysis experiments. The GC electrode used for CV experiments was replaced with carbon-cloth (CC), which has a higher surface area. MWCNTs were integrated into the CC electrode by soaking the CC in an ethanolic suspension of MWCNTs and 0.02 wt% Nafion, followed by drying (see Supporting Information for details).[16] An image of the resulting CC-MWCNT electrode obtained by scanning electron microscopy confirmed the presence of MWCNTs on the surface of carbon fibers (see Supporting Information, Figure S3).
Controlled potential electrolysis experiments were performed in an undivided cell containing aqueous carbonate electrolyte buffered to pH 10, using a CC-MWCNT working electrode (6 cm2), a Pt-wire counter electrode, and a Ag/AgCl reference electrode. Benzyl alcohol and an acetonitrile solution of pyrene- TEMPO were then added to the cell to achieve concentrations of 0.1 M and 5 × 10−5 M (0.05 mol%), respectively. Upon applying a potential of 0.7 V vs. Ag/AgCl, benzyl alcohol underwent full conversion to benzaldehyde within 50 min (Figure 5). When the electrolysis was repeated using ACT (5 × 10−5 M) rather than pyrene-TEMPO as the catalyst, negligible benzyl alcohol oxidation was observed (Figure 5). These results demonstrate the vastly superior performance of the pyrene-TEMPO catalyst relative to a non-immobilized homogeneous analog. The simple in situ adsorption of the pyrene-TEMPO catalyst to the electrode surface allows this enhanced performance to be achieved with no added operational complexity. Moreover, the electrode may be restored to its original form by washing with acetone/water (1:1), followed by neat acetone.
Figure 5.

Comparison of the bulk electrochemical oxidation of 0.1 M benzyl alcohol by 5 × 10−5 M pyrene-TEMPO (black) and 5 × 10−5 M of 4-acetamido-TEMPO (gray) at 0.7 V vs. Ag/AgCl. Total reaction volume = 10 mL.
The immobilized pyrene-TEMPO was then tested to assess its utility for preparative-scale oxidation of a series of benzylic alcohols (Scheme 2). Excellent results were observed for oxidation of various alcohols to the corresponding aldehydes with 0.05 mol% pyrene-TEMPO. In most cases, ≥90% yield of the aldehyde was obtained in 80 min or less, including examples with electron donating, electron withdrawing, and ortho substituents. These results reflect >1800 turnovers of the immobilized TEMPO catalyst. With the activated substrate 4-methoxybenzyl alcohol, the turnover frequency was nearly 4000 h−1. The somewhat reduced yield observed for oxidation of 4-NO2-benzyl alcohol (74%) could arise from reduction of the nitro group at the counter electrode.[17] In addition, oxidation of 2-aminobenzyl alcohol was unsuccessful (not shown). This substrate and other anilines are not compatible with oxoammonium reagents,[6] and they undergo nonselective direct oxidation at the electrode at potentials needed to promote catalytic alcohol oxidation. Small amounts of carboxylic acids were observed, arising from overoxidation of the aldehydes, but these side products were readily removed via extraction of the aldehydes from the basic reaction media (see Supporting Information for details).
Scheme 2.

Preparative electrolysis reactions with representative benzylic alcohols.a
In a more stringent test of the methodology, we evaluated the oxidation of a hydroxymethylpyrimidine precursor to rosuvastatin (Crestor™), a blockbuster HMG-CoA reductase inhibitor (i.e., a statin) for which the patent recently expired (Scheme 3).[18] This alcohol oxidation step is featured in one of the important routes to this compound.[19, 20] To improve the solubility of the substrate, a 1:1 mixture of acetonitrile and water was used as a solvent mixture. The pyrene-TEMPO catalyst has lower affinity for the electrode in the presence of organic co-solvent. Nevertheless, this sterically hindered benzylic alcohol substrate underwent successful oxidation to the aldehyde in 91% yield with 0.1 mol% catalyst loading in 6 h (Scheme 3). This result and those in Scheme 2 highlight prospects for the use of non-covalent immobilization of molecular catalysts in preparative-scale electroorganic synthesis. Moreover, future process optimization (e.g., development of a continuous-flow electrolysis) could provide the basis for use of this methodology in large-scale applications. Efforts directed toward the latter goal have been initiated.
Scheme 3.

Oxidation of a hydroxymethylpyrimidine precursor to rosuvastatin (Crestor™).
In conclusion, we have prepared and demonstrated the efficacy of a pyrene-tethered TEMPO catalyst for electrochemical alcohol oxidation, using both CV and bulk electrolysis methods. The use of a non-covalent immobilization method, arising from π-π stacking interactions between the pyrene fragment and MWCNTs on the electrode surface, allows for facile preparation of the functionalized electrode, which shows excellent electrocatalytic performance. The approach described herein holds promise for widespread implementation in electroorganic synthesis and enhancement of the practical utility of methods that use molecular electrocatalysts.
Supplementary Material
Acknowledgments
Financial support for this project was provided by an NSF CCI grant (CHE-1305124; reversible tethering strategy) and AbbVie (organic chemistry applications). We are grateful to Dr. Paul Alsters (DSM) for supplying a sample of the rosuvastatin precursor. We thank Dr. Mohammad Rafiee for helpful discussions and suggestions during the course of this project, and Maaz Ahmed for acquisition of the SEM image of the CC and CC-MWCNT electrodes. NMR spectroscopy facilities were partially supported by the NSF (CHE-9208463) and NIH (S10 RR08389).
References
- 1.a) DuBois MR, DuBois DL. Catalysis without Precious Metals. Wiley-VCH Verlag GmbH & Co. KGaA; 2010. pp. 165–180. [Google Scholar]; b) Lavacchi A, Miller H, Vizza F. Nanotechnology in Electrocatalysis for Energy. Springer New York; New York, NY: 2013. pp. 273–315. [Google Scholar]; c) Rakowski Dubois M, Dubois DL. Acc. Chem. Res. 2009;42:1974–1982. doi: 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]; d) Artero V, Chavarot-Kerlidou M, Fontecave M. Angew. Chem. Int. Ed. 2011;50:7238–7266. doi: 10.1002/anie.201007987. [DOI] [PubMed] [Google Scholar]; e) Bullock RM, Helm ML. Acc. Chem. Res. 2015;48:2017–2026. doi: 10.1021/acs.accounts.5b00069. [DOI] [PubMed] [Google Scholar]; f) Queyriaux N, Jane RT, Massin J, Artero V, Chavarot-Kerlidou M. Coord. Chem. Rev. 2015;304–305:3–19. doi: 10.1016/j.ccr.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Coutard N, Kaeffer N, Artero V. Chem. Commun. 2016;52:13728–13748. doi: 10.1039/c6cc06311j. [DOI] [PubMed] [Google Scholar]; h) Elgrishi N, Chambers MB, Wang X, Fontecave M. Chem. Soc. Rev. 2017;46:761–796. doi: 10.1039/c5cs00391a. [DOI] [PubMed] [Google Scholar]
- 2.a) Fleischmann M, Pletcher D. R. Inst. Chem., Rev. 1969;2:87–116. [Google Scholar]; b) Pletcher D. J. Appl. Electrochem. 1984;14:403–415. [Google Scholar]; c) Moeller KD. Tetrahedron. 2000;56:9527–9554. [Google Scholar]; d) Bard AJ, Stratmann M, Schäfer HJ. Encyclopedia of Electrochemistry: Organic Electrochemistry. Vol. 8. Wiley-VCh; 2002. [Google Scholar]; e) Pletcher D. Developments in Electrochemistry. John Wiley & Sons, Ltd; 2014. pp. 77–94. [Google Scholar]; f) Hammerich O, Speiser B, editors. Organic Electrochemistry. 5. CRC Press; Boca Raton, FL: 2015. [Google Scholar]
- 3.For recent reviews, see: Yoshida J-i, Kataoka K, Horcajada R, Nagaki A. Chem. Rev. 2008;108:2265–2299. doi: 10.1021/cr0680843.Frontana-Uribe BA, Little RD, Ibanez JG, Palma A, Vasquez-Medrano R. Green Chem. 2010;12:2099–2119.Waldvogel SR, Elsler B. Electrochim. Acta. 2012;82:434–443.Francke R, Little RD. Chem. Soc. Rev. 2014;43:2492–2521. doi: 10.1039/c3cs60464k.Ogawa KA, Boydston AJ. Chem. Lett. 2015;44:10–16.Turygin VV, Tomilov AP. Rus. J. Electrochem. 2015;51:999–1020.Horn EJ, Rosen BR, Baran PS. ACS Cent. Sci. 2016;2:302–308. doi: 10.1021/acscentsci.6b00091.
- 4.Savéant J-M. Chem. Rev. 2008;108:2348–2378. doi: 10.1021/cr068079z. [DOI] [PubMed] [Google Scholar]
- 5.a) Bullock RM, Das AK, Appel AM. Chem. Eur. J. 2017 doi: 10.1002/chem.201605066. [DOI] [PubMed] [Google Scholar]; b) Axet MR, Dechy-Cabaret O, Durand J, Gouygou M, Serp P. Coord. Chem. Rev. 2016;308(Part 2):236–345. [Google Scholar]
- 6.For a comprehensive survey, see: Bobbitt JM, Brückner C, Merbouh N. Organic Reactions. John Wiley & Sons, Inc.; 2009.
- 7.For more-focused reviews, see: Sheldon RA, Arends IWCE. Adv. Syn. Cat. 2004;346:1051–1071.Tebben L, Studer A. Angew. Chem. Int. Ed. 2011;50:5034–5068. doi: 10.1002/anie.201002547.Ryland BL, Stahl SS. Angew. Chem. Int. Ed. 2014;53:8824–8838. doi: 10.1002/anie.201403110.Miles KC, Stahl SS. Aldrichimica Acta. 2015;48:8–10.
- 8.For examples of synthetic applications, see: Semmelhack MF, Chou CS, Cortes DA. J. Am. Chem. Soc. 1983;105:4492–4494.Inokuchi T, Matsumoto S, Torii S. J. Org. Chem. 1991;56:2416–2421.Kashiwagi Y, Kurashima F, Kikuchi C, Anzai J-i, Osa T, Bobbitt JM. Tetrahedron Lett. 1999;40:6469–6472.Mitsudo K, Kumagai H, Takabatake F, Kubota J, Tanaka H. Tetrahedron Lett. 2007;48:8994–8997.Cha HG, Choi K-S. Nat. Chem. 2015;7:328–333. doi: 10.1038/nchem.2194.
- 9.For examples of synthetic applications, see: Deronzier A, Lìmosin D, Moutet J-C. Electrochim. Acta. 1987;32:1643–1647.Osa T, Kashiwagi Y, Mukai K, Ohsawa A, Bobbitt JM. Chem. Lett. 1990;19:75–78.Kashiwagi Y, Yanagisawa Y, Kurashima F, Anzai J-i, Osa T, Bobbitt JM. Chem. Commun. 1996:2745–2746. doi: 10.1039/b209871g.Belgsir EM, Schäfer HJ. Electrochem. Commun. 2001;3:32–35.Kashiwagi Y, Kurashima F, Chiba S, Anzai J-i, Osa T, Bobbitt JM. Chem. Commun. 2003:114–115. doi: 10.1039/b209871g.Palmisano G, Ciriminna R, Pagliaro M. Adv. Syn. Cat. 2006;348:2033–2037.Karimi B, Rafiee M, Alizadeh S, Vali H. Green Chem. 2015;17:991–1000.
- 10.Many other studies have focused on fundamental studies of TEMPO electrochemistry and catalysis, without focusing on synthetic utility (e.g., yields, substrate scope, etc.). For leading references, see: Rafiee M, Karimi B, Alizadeh S. ChemElectroChem. 2014;1:455–462.Rafiee M, Miles KC, Stahl SS. J. Am. Chem. Soc. 2015;137:14751–14757. doi: 10.1021/jacs.5b09672.Hickey DP, Schiedler DA, Matanovic I, Doan PV, Atanassov P, Minteer SD, Sigman MS. J. Am. Chem. Soc. 2015;137:16179–16186. doi: 10.1021/jacs.5b11252.Hickey DP, Milton RD, Chen D, Sigman MS, Minteer SD. ACS Catal. 2015;5:5519–5524.Badalyan A, Stahl SS. Nature. 2016;535:406–410. doi: 10.1038/nature18008.
- 11.For context, see: Andrieux CP, Saveant JM. J. Electroanal. Chem.y. 1978;93:163–168.
- 12.For examples of pyrene-tethered molecular electrocatalysts focusing on energy-related applications see: Tran PD, Le Goff A, Heidkamp J, Jousselme B, Guillet N, Palacin S, Dau H, Fontecave M, Artero V. Angew. Chem. Int. Ed. 2011;50:1371–1374. doi: 10.1002/anie.201005427.Li F, Zhang B, Li X, Jiang Y, Chen L, Li Y, Sun L. Angew. Chem. Int. Ed. 2011;50:12276–12279. doi: 10.1002/anie.201105044.Toma FM, Sartorel A, Iurlo M, Carraro M, Rapino S, Hoober-Burkhardt L, Da Ros T, Marcaccio M, Scorrano G, Paolucci F, Bonchio M, Prato M. ChemSusChem. 2011;4:1447–1451. doi: 10.1002/cssc.201100089.Blakemore JD, Gupta A, Warren JJ, Brunschwig BS, Gray HB. J. Am. Chem. Soc. 2013;135:18288–18291. doi: 10.1021/ja4099609.Kang P, Zhang S, Meyer TJ, Brookhart M. Angew. Chem. Int. Ed. 2014;53:8709–8713. doi: 10.1002/anie.201310722.Tran PD, Morozan A, Archambault S, Heidkamp J, Chenevier P, Dau H, Fontecave M, Martinent A, Jousselme B, Artero V. Chem. Sci. 2015;6:2050–2053. doi: 10.1039/c4sc03774j.Gentil S, Serre D, Philouze C, Holzinger M, Thomas F, Le Goff A. Angew. Chem. Int. Ed. 2016;55:2517–2520. doi: 10.1002/anie.201509593.Creus J, Matheu R, Peñafiel I, Moonshiram D, Blondeau P, Benet-Buchholz J, García-Antón J, Sala X, Godard C, Llobet A. Angew. Chem. Int. Ed. 2016;55:15382–15386. doi: 10.1002/anie.201609167.Reuillard B, Warnan J, Leung JJ, Wakerley DW, Reisner E. Angew. Chem. Int. Ed. 2016;55:3952–3957. doi: 10.1002/anie.201511378.Lalaoui N, Means N, Walgama C, Le Goff A, Holzinger M, Krishnan S, Cosnier S. ChemElectroChem. 2016;3:2058–2062.Koelewijn JM, Lutz M, Detz RJ, Reek JNH. ChemPlusChem. 2016;81:1098–1106. doi: 10.1002/cplu.201600235.Maurin A, Robert M. J. Am. Chem. Soc. 2016;138:2492–2495. doi: 10.1021/jacs.5b12652.Elouarzaki K, Holzinger M, Le Goff A, Thery J, Marks RS, Cosnier S. J. Mater. Chem. A. 2016;4:10635–10640.Lalaoui N, Reuillard B, Philouze C, Holzinger M, Cosnier S, Le Goff A. Organometallics. 2016;35:2987–2992.
- 13.For redox potentials of diverse nitroxyl species, see: Lauber MB, Stahl SS. ACS Catal. 2013;3:2612–2616.Shibuya M, Pichierri F, Tomizawa M, Nagasawa S, Suzuki I, Iwabuchi Y. Tetrahedron Lett. 2012;53:2070–2073.
- 14.a) Zhao Y-L, Stoddart JF. Acc. Chem. Res. 2009;42:1161–1171. doi: 10.1021/ar900056z. [DOI] [PubMed] [Google Scholar]; b) Rodriguez-Perez L, Herranz MaA, Martin N. Chem. Commun. 2013;49:3721–3735. doi: 10.1039/c3cc38950b. [DOI] [PubMed] [Google Scholar]
- 15.The current exhibits a square-root dependence on the scan rate for dissolved redox species, but a linear dependence for immobilized (non-diffusive) redox species. See: Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. 2. John Wiley& Sons; New York: 2001.
- 16.Nigović B, Sadiković M, Sertić M. Talanta. 2014;122:187–194. doi: 10.1016/j.talanta.2014.01.026. [DOI] [PubMed] [Google Scholar]
- 17.The side products arising from reduction of the nitroaromatic will reduce the yield and are susceptible to further reaction and/or decomposition. Similar observations have been noted previously; see ref. 9g.
- 18.a) Olsson AG, McTaggart F, Raza A. Cardiovasc. Drug Rev. 2002;20:303–328. doi: 10.1111/j.1527-3466.2002.tb00099.x. [DOI] [PubMed] [Google Scholar]; b) McTaggart F, Jones P. Cardiovasc. Drugs Ther. 2008;22:321. doi: 10.1007/s10557-008-6113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Watanabe M, Koike H, Ishiba T, Okada T, Seo S, Hirai K. Bioorg. Med. Chem. 1997;5:437–444. doi: 10.1016/s0968-0896(96)00248-9. [DOI] [PubMed] [Google Scholar]; b) Andrushko N, Andrushko V, König G, Spannenberg A, Börner A. Eur. J. Org. Chem. 2008;2008:847–853. [Google Scholar]; c) Tartaggia S, Ferrari C, Pontini M, De Lucchi O. Eur. J. Org. Chem. 2015;2015:4102–4107. [Google Scholar]; d) Tartaggia S, Fogal S, Motterle R, Ferrari C, Pontini M, Aureli R, De Lucchi O. Eur. J. Org. Chem. 2016;2016:3162–3165. [Google Scholar]
- 20.For representative oxidation methods used for this step, see: Šterk D, Časar Z, Jukič M, Košmrlj J. Tetrahedron. 2012;68:2155–2160.Guan Y, Zhou G, Yang W. Heterocycl. Commun. 2014;20:11–13.Steves JE, Preger Y, Martinelli JR, Welch CJ, Root TW, Hawkins JM, Stahl SS. Org. Process Res. Dev. 2015;19:1548–1553. doi: 10.1021/op500328f.Steves JE, Stahl SS. J. Org. Chem. 2015;80:11184–11188. doi: 10.1021/acs.joc.5b01950.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.