

Abstract
An electrochemical method has been developed for selective benzylic iodination of methylarenes. The reactions feature the first use of N-hydroxyphthalimide (NHPI) as an electrochemical mediator for C–H oxidation to non-oxygenated products. The method provides the basis for direct (in situ) or sequential benzylation of diverse nucleophiles using methylarenes as the alkylating agent. The hydrogenatom transfer mechanism for C–H iodination allows C–H oxidation to proceed with minimal dependence on the substrate electronic properties and at electrode potentials 0.5 – 1.2 V lower than that of direct electrochemical C–H oxidation.
Graphical Abstract
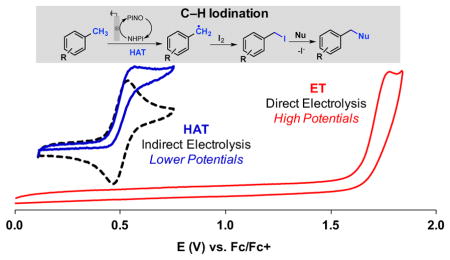
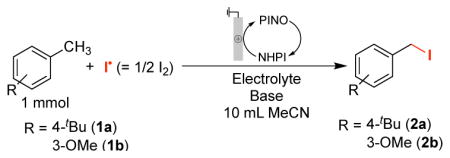
Organic electrochemistry represents an appealing strategy to perform redox reactions;1, 2 however, many organic molecules do not readily undergo direct electron transfer (ET) at an electrode. Redox mediators provide a means to overcome this limitation by expanding the scope of accessible mechanisms that may be coupled to an electrochemical driving force.3 The utility of N-hydroxyphthalimide (NHPI) as a hydrogen-atom transfer (HAT) mediator was introduced by Masui and coworkers in the 1980s. 4 NHPI undergoes proton-coupled oxidation at an electrode to afford phthalimido-N-oxyl (PINO), which then mediates HAT from weak C–H bonds (allylic, benzylic, adjacent to heteroatoms) (Scheme 1). Trapping of the organic radical by O2 leads to site-selective oxygenation products. Subsequent work by Ishii5 and others6 introduced numerous cobalt/NHPI (non-electrochemical) catalytic methods for related aerobic C–H oxygenation. Recent studies highlight pharmaceutically relevant applications of these methods. For example, Baran and coworkers developed improved electrochemical conditions for allylic C–H oxygenation, including scalable oxidation of terpenes (cf. Scheme 1),7 and we implemented cobalt and electrochemical NHPI/O2-based methods for benzylic oxygenation of heterocyclic compounds. 8 Each of the aforementioned methods affords oxygenated products. This outcome is inevitable for the Co/NHPI-catalyzed methods because O2, which is needed for NHPI oxidation to PINO, reacts at near-diffusion-controlled rates with organic radicals. Electrochemical oxidation of NHPI to PINO, however, presents the opportunity to trap organic radicals with reagents other than O-atom sources. We report herein the first example of this concept, showing that iodine (I2) is an effective radical trap for electrochemical NHPI-mediated oxidation of methylarenes to benzyl iodides (Scheme 2).9,10 The broader implications of HAT-mediated electrochemical C–H oxidation are demonstrated by comparison of this approach to related direct-ET electrochemical benzylic oxidation methods. Building on previous electrochemical studies of NHPI, 11 cyclic voltammetry was used to assess the reactivity of NHPI under relevant reaction conditions.
Scheme 1.
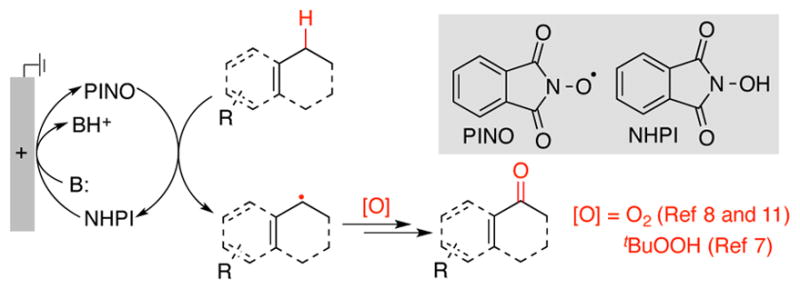
Electrochemical NHPI/PINO-mediated strategy for oxygenation of benzylic and allylic C–H bonds.
Scheme 2.
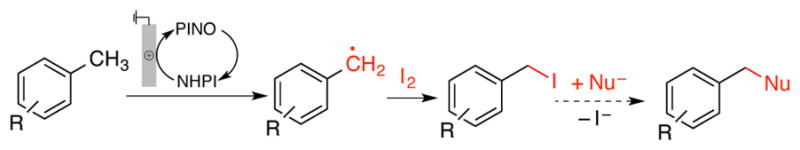
Electrochemical NHPI/PINO-mediated iodination/functionalization of methyl arenes.
The cyclic voltammogram (CV) of NHPI in acetonitrile is quasi-reversible with a mid-point potential (Emid) of 0.90 V versus Fc/Fc+ (Fc = ferrocene; Figure 1a). The proton-coupled nature of this redox process is evident upon inclusion of pyridine as a weak base in the solution, which shifts the Emid to a significantly lower potential (0.41 V; Figure 1c). The lower cathodic-to-anodic peak current ratio (Ic/Ia= 0.77), however, reflects the instability of PINO under these conditions. More basic conditions, with [NBu4]OAc as the base, result in near-complete disappearance of the cathodic peak (not shown),12 whereas use of a buffered pyridine/pyridinium solution led to improved PINO stability (Ic/Ia= 0.96) while still lowering the redox potential for PINO generation (0.49 V; Figure 1b). Upon addition of 4-tBu-toluene (1a, 20 mM) to the solution, the CV exhibits an increase in the anodic current, reflecting regeneration of NHPI on the CV timescale via PINO-mediated HAT from 1a (Figure 1d). No cathodic current is present in the reverse scan, as is expected if PINO is consumed by reaction with the substrate.
Figure 1.
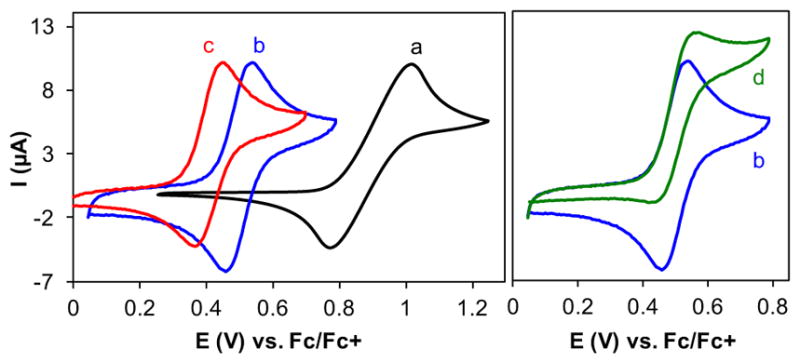
CVs of NHPI (1 mM) in acetonitrile (a); in the presence of pyridine/pyridinium perchlorate (0.1 M each) (b); pyridine (0.01 M) and solid KHCO3 (100 equiv) (c); and in the presence of pyridine/pyridinium perchlorate (0.1 M each), and 4-tBu-toluene (20 mM) (d). Other conditions: glassy carbon working electrode, scan rate = 10 mV/s, and 0.1 M KPF6 electrolyte for (a) and (c).
In order to extend these observations from voltammetry to synthetic bulk-electrolysis, we sought a suitable trap to functionalize the in situ-generated benzylic radical. Preliminary tests probed the reactivity of 4-tBu-toluene in the presence of NHPI (2.5 mol% with respect to 1a) and different trapping agents, including PhS–SPh, TsCN, TsC≡CPh, CuCl2 and I2 (10:1 ratio of 1a:trap).12,13 A divided cell configuration was used to avoid reduction of the trap (e.g., I2, CuCl2) at the cathode. No productive reactivity was observed, however, except with iodine, which led to the benzyl iodide product in quantitative yield with respect to I• (both iodine atoms of I2 are used) (Table 1, entry 1). Successful benzylic C–H iodination presumably reflects the rapid reaction of I2 with radicals relative to other traps.14 The reaction of PINO with benzylic C–H bonds is nearly thermoneutral,15 and inefficient trapping of the organic radical could prevent product formation due to the inevitable degradation of PINO during the electrolysis.16
Table 1.
Optimization of preparative electrolysis reactions.
| ||||||
|---|---|---|---|---|---|---|
| entry | NHPI (mol%) | R | Base (equiv) | Electrolyte | I• (equiv) | % yield vs ArCH3 (vs I•) |
| 1 | 2.5% | 4-tBu | Lut, KHCO3 (0.1, 1.1) | KPF6 | 0.1 | 10 (100) |
| 2 | 15% | 4-tBu | Lut, KHCO3 (0.1, 3.3) | KPF6 | 1.0 | 38 (38) |
| 3 | 20% | 4-tBu | Lut (1.5) | [LutH]ClO4a | 1.0 | 57 (57) |
| 4 | 20% | 4-tBu | Lut (1.5) | [LutH]ClO4 | 1.5 | 52 (35) |
| 5 | 20% | 3-OMe | Lut (1.5) | [LutH]ClO4 | 1.0 | 83 (83) |
| 6 | 20% Cl4NHPI | 3-OMe | Lut (1.5) | [LutH]ClO4 | 1.0 | 71 (71) |
| 7 | 20% NHSI | 3-OMe | Lut (1.5) | [LutH]ClO4 | 1.0 | 54 (54) |
| 8 | 20% Ph4NHPI | 3-OMe | Lut (1.5) | [LutH]ClO4 | 1.0 | 78 (78) |
|
| ||||||
|
| ||||||
The synthetic utility of electrochemical methods of this type will ultimately require the methylarene to be used as the limiting reagent. Various reaction conditions were tested in an effort to optimize the product yield with respect to the methylarene.12 Increasing the NHPI loading and optimization of the base and electrolyte composition led to a 57% yield of 4-tBu-benzyl iodide (2a) (Table 1, entry 3) (see Supporting Information for full optimization data). The combination of lutidine/lutidinium supplies the base and the supporting electrolyte for the reaction, while lutidinium provides a proton source to facilitate production of H2 at the counter electrode. Overoxidation and side reactions of the product 2a limited the accessible yield, and use of excess equivalents of I• did not improve the outcome (entry 4). An improved yield of benzyl iodide was obtained with 3-methoxytoluene (1b) (83%; entry 5) owing to the increased stability of the benzyl iodide 2b under the reaction conditions.
Testing of other imidoxyl mediators did not improve the results. Ph4NHPI has been reported to be more stable than NHPI,17 but no improvement in mediator stability or product yield was observed with this mediator (entry 6). Cl4NHPI and NHSI have stronger O–H bonds and, therefore, the corresponding imidoxyl species should undergo more rapid reaction with the methylarene. However, these imidoxyl species undergo more rapid degradation and result in lower product yields (entries 7 and 8).
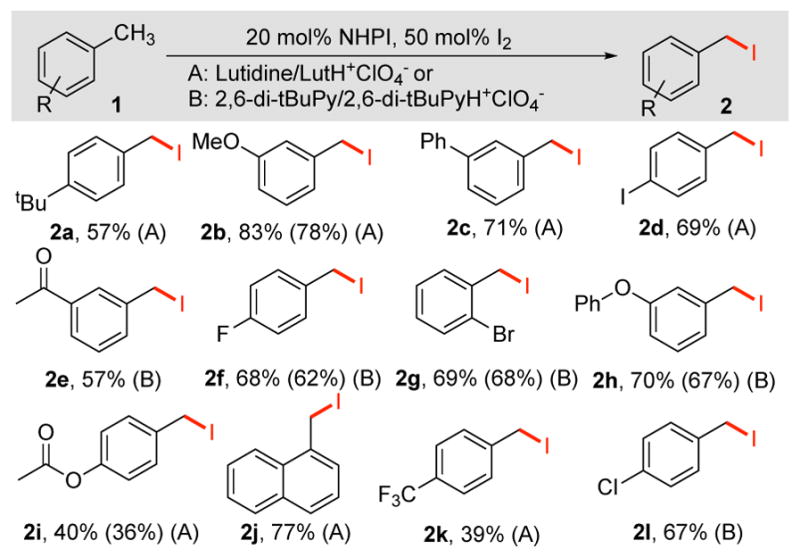
Good reactivity was observed with a range of methylarenes under these electrochemical iodination conditions (Table 2). The method tolerates various functional groups, including halide, methoxy, phenoxy, acetyl and acetoxy groups. Reactions with some electron-deficient substrates (e.g., 1e, 1k) revealed significant amounts of competing iodination of the benzylic methyl groups of lutidine, but improved yields could be obtained by using 2,6-di-tert-butylpyridine/pyridinium (DTBP)/DTBP-H+ as the buffering electrolyte. In contrast, substrates with electron-donating groups (e.g., 1a) are susceptible to facile displacement of the iodide. In these cases, replacement of lutidine by DTBP did not improve the yield.
Table 2.
Electrochemical iodination of methylarenes mediated by NHPI.a
|
A: 0.1 M 1, 20 mol% NHPI, 50 mol% I2, 0.2 M Lut/0.1 M LutH+ClO4−, 10 mL MeCN, 5 mA. B: 0.1 M substrate 1, 20 mol% NHPI, 50 mol% I2, 0.2 M 2,6-di-tBuPy, 0.1 M 2,6-di-tBuPyH+ClO4−, 10 mL MeCN, 5 mA. 1H NMR yields (isolated yields in parentheses).12
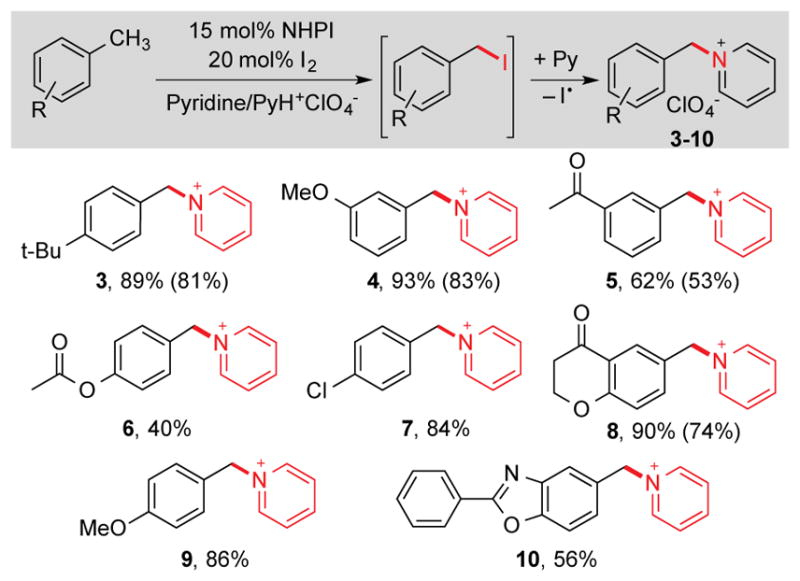
The in situ nucleophilic substitution of iodide has potential synthetic utility. Use of pyridine/pyridinium as the electrolyte led to efficient displacement of iodide by pyridine, leading to diverse benzyl pyridinium products under the iodination electrolysis conditions (Table 3). Generation of iodide in the nucleophilic substitution step has the benefit of allowing reoxidation of iodide to iodine at the anode, and permitting these reactions to be performed with catalytic quantities of iodine (20 mol%). Benzyl pyridinium derivatives of this type have been used in a number of synthetic methods, such as [3+2] cycloadditions, to access valuable heterocyclic compounds. 18 It is worth noting that the yields of these reactions are generally significantly higher than those from the iodination reactions (e.g., 89% yield of 3 vs. 57% yield of 2a from the reaction of 1a) owing to the enhanced product stability relative to benzyl iodides. In addition to in situ functionalization, the electrochemical iodination protocol may be employed in sequential iodination/alkylation. This concept is illustrated by the facile preparation of the three key pharmaceutically important target molecules 11–13 (Scheme 3).19 Together, these in situ and sequential protocols illustrate an appealing strategy for the use of methylarene derivatives as alkylating agents.
Table 3.
In situ methylarene iodination/alkylation of pyridine under NHPI-mediated electrolysis conditions.a
|
Conditions: 0.3 M substrate 1, 15 mol% NHPI, 20 mol% I2, 0.6 M Pyridine, 0.6 M PyH+ClO4−, 10 mL MeCN, 15 mA. 1H NMR yield (ext. std. = mesitylene). 1H NMR yields (isolated yields in parentheses).12
Scheme 3.
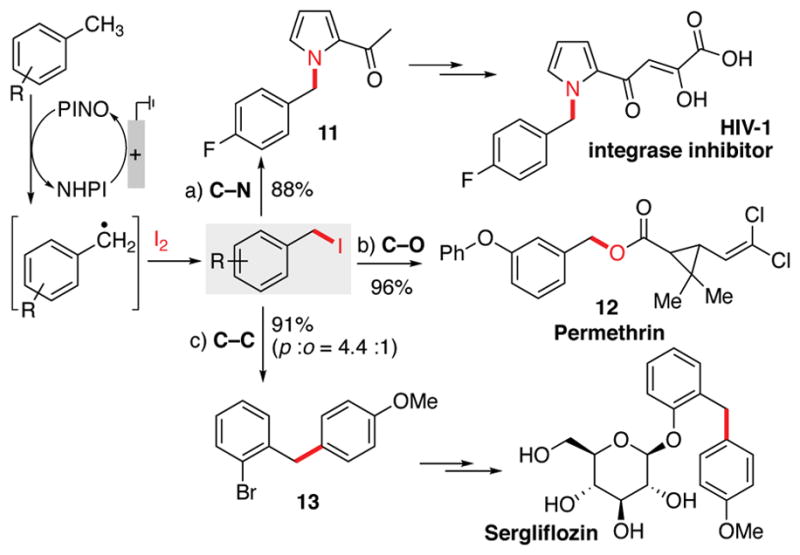
Sequential methylarene iodination/alkylation to access pharmaceutical intermediates and compounds.
a) 2f (0.12 mmol), acetylpyrrole (2.0 equiv.), NaH (2.0 equiv.), DMF (1 mL), RT; b) 2h (0.2 mmol), permethric acid (1.1 equiv.), Na2CO3 (4.0 equiv.), DMF (2.0 mL), 50 °C; c) 2g (0.1 mmol), anisole (4.0 equiv.), AgOTf (1.5 equiv.), lutidine (2.0 equiv.), CDCl3 (1.0 mL), RT.
These observations have important implications within the broader context of electrochemical C–H oxidation, beyond the synthetic considerations defined herein. The present HAT-mediated benzylic oxidation reactions may be compared to related electrochemical methods initiated by electron transfer.20a The CVs of different methylarene derivatives in Figure 2B depict the strong electronic dependence of redox potentials on arene substituents, and the trends are in agreement with gas-phase ionization potentials of these compounds (Figure 2B-2).15a On the other hand, C–H bond strengths exhibit negligible electronic dependence (Figure 2A-2).15b The latter trend reflects the offsetting electronic effects of substituents on the redox potential and the pKa of the methylarene and accounts for the similar catalytic reactivity of PINO with electron-rich, -neutral, and -deficient substrates (Figure 2A-1 and 2A-3). Thus, the use of the HAT mediator negates substrate electronic effects and allows for methylarene oxidation at electrode potentials more than 0.5–1.2 V lower than the single-electron redox potential of these substrates. Electrochemical ET-initiated reactions of methylarenes, such as the “cation pool” and related reactions reported by Yoshida20b–d and others,20a,e,f predominantly feature electron-rich substrates. A recent study of electrochemical ET-initiated oxidation of methylarenes to benzaldehydes featured electronically diverse substrates, and showed that electron-deficient derivatives exhibited significantly lower yields (Figure 2B-3).20a This outcome may be contrasted to HAT-mediated conversion of methylarenes to benzylpyridinium derivatives, which show uniform yields for the same set of electronically varied substrates (Figure 2A-3).
Figure 2.

Comparison of PINO-mediated-HAT- and direct-ET-initiated C–H oxidation of methylarenes, including the influence of electronic effects (1 and 2) and product yields from representative methods (3). Data for the Figure 2B-3 adapted from Ref. 20a.
This analysis and the other results described herein draw attention to unique opportunities associated with the use of HAT mediators for electrochemical C–H oxidation. Stoichiometric chemical oxidants that are used to generate reactive radicals (e.g., O2, peroxides, and related reagents) are also efficient radical traps. Therefore, radical generation and functionalization are intimately coupled with these oxidants. By using an electrode to generate the PINO HAT mediator, functionalization of the organic radical may be achieved with a reagent, such as I2, that is not capable of serving as the stoichiometric oxidant. This work further highlights the advantages of mediated electrolysis methods that access mechanisms distinct from direct electron transfer, specifically by allowing reactions to proceed at much lower applied potentials and with less sensitivity to substrate electronic properties. These features suggest that development of new mediators, such those with improved stability or other desirable properties, could provide the basis for important new electrochemical oxidation methods that access products not readily accessed by more-traditional chemical approaches.
Supplementary Material
Acknowledgments
Financial support for this project was provided by Merck Research Laboratories. Additional support was provided by an SIOC fellowship (for FW) and the NIH (R01 GM100143 for SSS, F32 GM113399 for DPH). Spectroscopic instrumentation was partially supported by the NIH (S10 OD020022) and the NSF (CHE-1048642).
Footnotes
Additional electrochemical data, experimental details and product characterization data; this material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Shono T, editor. Electroorganic Synthesis. Academic Press; London: 1991. [Google Scholar]; (b) Little RD, Weinberg NL, editors. Electroorganic Synthesis. Marcel Dekker; New York: 1991. [Google Scholar]; (c) Grimshaw J. Electrochemical Reactions and Mechanisms in Organic Chemistry. Elsevier; Amsterdam: 2000. [Google Scholar]; (d) Hammerich O, Speiser B, editors. Organic Electrochemistry. 5. CRC Press; Boca Raton: 2015. [Google Scholar]
- 2.For recent reviews, see: Moeller KD. Tetrahedron. 2000;56:9527.Sperry JB, Wright DL. Chem Soc Rev. 2006;35:605. doi: 10.1039/b512308a.Yoshida JI, Kataoka K, Horcajada R, Nagaki A. Chem Rev. 2008;108:2265. doi: 10.1021/cr0680843.Horn EJ, Rosen BR, Baran PS. ACS Cent Sci. 2016;2:302. doi: 10.1021/acscentsci.6b00091.
- 3.(a) Steckhan E. Angew Chem, Int Ed. 1986;28:683. [Google Scholar]; (b) Ogibin YN, Elinson MN, Nikishin GI. Russ Chem Rev. 2009;78:89. [Google Scholar]; (c) Francke R, Little RD. Chem Soc Rev. 2014;43:2492. doi: 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- 4.For leading references, see: Masui M, Hara S, Ueshima T, Kawaguchi T, Ozaki S. Chem Pharm Bull. 1983;31:4209.Masui M, Kawaguchi T, Ozaki S. J Chem Soc, Chem Commun. 1985:1484.
- 5.For reviews, see: Ishii Y, Sakaguchi S, Iwahama T. Adv Synth Catal. 2001;343:393.Ishii Y, Sakaguchi S. J Synth Org Chem, Jpn. 2003;61:1056.
- 6.Recupero F, Punta C. Chem Rev. 2007;107:3800. doi: 10.1021/cr040170k. [DOI] [PubMed] [Google Scholar]
- 7.Horn EJ, Rosen BR, Chen Y, Tang J, Chen K, Eastgate MD, Baran PS. Nature. 2016;533:77. doi: 10.1038/nature17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hruszkewycz DP, Miles KC, Thiel OR, Stahl SS. Chem Sci. 2017;8:1282. doi: 10.1039/c6sc03831j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For representative examples of other approaches to electrochemical C–H functionalization, including iodination methods, see: Shono T, Matsumura Y, Katoh S, Ikeda K, Kamada T. Tetrahedron Lett. 1989;30:1649.Yoshida JI, Suga S. Chem Eur J. 2002;8:2651. doi: 10.1002/1521-3765(20020617)8:12<2650::aid-chem2650>3.0.co;2-s.Campbell JM, Xu HC, Moeller KD. J Am Chem Soc. 2012;134:18338. doi: 10.1021/ja307046j.Ashikari Y, Shimizu A, Nokami T, Yoshida JI. J Am Chem Soc. 2013;135:16070. doi: 10.1021/ja4092648.Francke R. Beilstein J Org Chem. 2014;10:2858. doi: 10.3762/bjoc.10.303.Arai T, Tateno H, Nakabayashi K, Kashiwagi T, Atobe M. Chem Commun. 2015;51:4891. doi: 10.1039/c5cc01253h.Hou ZW, Mao ZY, Zhao HB, Melcamu YY, Lu X, Song J, Xu HC. Angew Chem Int Ed. 2016;55:9168. doi: 10.1002/anie.201602616.Schulz L, Enders M, Elsler B, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Angew Chem Int Ed. 2017;17:4877. doi: 10.1002/anie.201612613.Kawabata Y, Yan M, Liu Z, Bao D-H, Chen J, Starr JT, Baran PS. J Am Chem Soc. 2017;139:7448. doi: 10.1021/jacs.7b03539.
- 10.Some NHPI-mediated methods with chemical oxidants afford products not derived from O-atom trapping: Sakaguchi S, Hirabayashi T, Ishii Y. Chem Commun. 2002:516. doi: 10.1039/b110638d.Minisci F, Recupero F, Gambarotti C, Punta C, Paganelli R. Tetrahedron Lett. 2003;44:6919.Minisci F, Porta O, Recupero F, Gambarotti C, Paganelli R, Pedulli GF, Fontana F. Tetrahedron Lett. 2004;5:1607.Kiyokawa K, Takemoto K, Minakata S. Chem Commun. 2016;52:13082. doi: 10.1039/c6cc07164c.
- 11.(a) Ueda C, Noyama M, Ohmori H, Masui M. Chem Pharm Bull. 1987;35:1372. [Google Scholar]; (b) Gorgy K, Lepretre JC, Saint-Aman E, Einhorn C, Einhorn J, Marcadal C, Pierre JL. Electrochim Acta. 1998;44:385. [Google Scholar]; (c) Kishioka S, Yamada A. J Electroanal Chem. 2005;578:71. [Google Scholar]
- 12.See Supporting Information for details.
- 13.For other applications of these reagents as radical traps, see: Crossley SWM, Obradors C, Martinez RM, Shenvi RA. Chem Rev. 2016;116:8912. doi: 10.1021/acs.chemrev.6b00334.Yi H, Zhang G, Wang H, Huang Z, Wang J, Singh AK, Lei A. Chem Rev. 2017;117:9016. doi: 10.1021/acs.chemrev.6b00620.
- 14.Iodine has even been shown to be a more efficient trap than O2. See ref. 10b and the following: Hook SCW, Saville B. J Chem Soc Perkin Trans II. 1975:589.
- 15.The BDE of toluene derivatives typically ranges from 88 to 90 kcal/mol, and the BDE of NHPI is 88 kcal/mol. Fox T, Kollman PA. J Phys Chem. 1996;100:2950.Bean GP. Tetrahedron. 2002;58:9941.Pratt DA, DiLabio GA, Mulder P, Ingold KU. Acc Chem Res. 2004;37:334–340. doi: 10.1021/ar010010k.Xue X-S, Ji P, Zhou B, Cheng J-P. Chem Rev. 2017;117:8622. doi: 10.1021/acs.chemrev.6b00664.Hirai N, Sawatari N, Nakamura N, Sakaguchi S, Ishii Y. J Org Chem. 2003;68:6587. doi: 10.1021/jo034313z.
- 16.Both bimolecular and unimolecular pathways have been reported for decomposition of PINO. See ref. 11a, c.
- 17.Nechab M, Einhorn C, Einhorn J. Chem Commun. 2004:1500. doi: 10.1039/b403004d. [DOI] [PubMed] [Google Scholar]
- 18.(a) Tsuge O, Kanemasa S, Takenaka S. Chem Lett. 1985;14:355. [Google Scholar]; (b) Kajigaeshi S, Mori S, Fujisaki S, Kanemasa S. Bull Chem Soc Jpn. 1985;58:3547. [Google Scholar]; (c) Liu Y, Hu H, Zhou J, Wang W, He Y, Wang C. Org Biomol Chem. 2017;15:5016. doi: 10.1039/c7ob00980a. [DOI] [PubMed] [Google Scholar]
- 19.Sechi M, Bacchi A, Carcelli M, Compari C, Duce E, Fisicaro E, Rogolino D, Gates P, Derudas M, Al-Mawsawi LQ, Neamati N. J Med Chem. 2006;49:4248. doi: 10.1021/jm060193m. [DOI] [PubMed] [Google Scholar]; (b) Bicker W, Kacprzak K, Kwit M, Lammerhofer M, Gawronski J, Lindner W. Tetrahedron: Asymmetry. 2009;20:1027. [Google Scholar]; (c) Lee J, Lee S-H, Seo HJ, Son EJ, Lee SH, Jung ME, Lee MW, Han HK, Kim J, Kang J, Lee J. Bioorg Med Chem. 2010;18:2178. doi: 10.1016/j.bmc.2010.01.073. [DOI] [PubMed] [Google Scholar]
- 20.For leading references, see: Zhu Y, Zhu Y, Zeng H, Chen Z, Little RD, Ma C. J Electroanal Chem. 2015;751:105.Hayashi R, Shimizu A, Yoshida JI. J Am Chem Soc. 2016;138:8400. doi: 10.1021/jacs.6b05273.Morofuji T, Shimizu A, Yoshida JI. J Am Chem Soc. 2013;135:5000. doi: 10.1021/ja402083e.Hayashi R, Shimizu A, Song Y, Ashikari Y, Nokami T, Yoshida JI. Chem Eur J. 2017;23:61. doi: 10.1002/chem.201604484.Sarma BB, Efremenko I, Neumann R. J Am Chem Soc. 2015;137:5916. doi: 10.1021/jacs.5b01745.Kramer K, Robertson PM, Ibl N. J Appl Electrochem. 1980;10:29.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.