

Abstract
IMPORTANCE
Several kindreds having germline BAP1 mutations with a propensity for uveal and cutaneous melanomas and other internal malignancies have been described in an autosomal dominant tumor predisposition syndrome. However, clinically atypical moles have not been previously recognized as a component of this syndrome, to our knowledge. We describe the first kindred to date with a germline mutation in BAP1 associated with multiple cutaneous melanomas and classic dysplastic nevus syndrome.
OBSERVATIONS
We describe a 53-year-old man who was initially seen in 2003 with dysplastic nevus syndrome, multiple atypical melanocytic proliferations showing loss of immunostaining for BAP1, and 7 cutaneous melanomas. Germline testing was performed in the proband, his 16-year-old son, and his 13-year-old daughter, revealing a germline mutation in the BAP1 gene (c.592G>T, p.Glu198X) in the proband and in his 16-year-old son. CDKN2A and CDK4 genes were wild type. No members of this kindred reported a history of uveal melanoma.
CONCLUSIONS AND RELEVANCE
To our knowledge, this is the first report of a patient with multiple melanomas, dysplastic nevus syndrome, and an inactivating germline BAP1 mutation. The coexistence of dysplastic nevus syndrome and a BAP1 germline mutation extends the spectrum of the BAP1 tumor predisposition syndrome and may confer a greater risk for cutaneous melanomas.
Several kindreds with germline mutations in the BRCA1-associated protein 1 gene (BAP1 [OMIM 603089]) and a propensity for uveal and cutaneous melanomas have been described.1–4 BAP1 mutation carriers often develop crops of small (<10 mm) orange-red translucent papules, plaques, or nodules, typically located on the torso or the scalp.1 Histologically, these BAP1-deficient tumors (BDTs) consist of conventional nevus cells and a second population of epithelioid cells with spitzoid cytomorphology occupying the dermis.3 These spitzoid cells show a large pale eosinophilic cytoplasm and may be highly atypical. However, these lesions lack other features of Spitz nevi such as epidermal hyperplasia, Kamino bodies, clefting, or spindle-shaped melanocytes.3,5 Cytogenetically, BDTs in these patients often exhibit allelic loss on chromosome 3p21 at the BAP1 locus.3 These same combined melanocytic tumors may also occur sporadically.5
Patients with germline mutations in BAP1 have an increased risk for internal cancers such as mesothelioma, lung adenocarcinoma, meningiomas, and renal cell carcinoma, indicating that BAP1 inactivation leads to a mixed cancer phenotype.2,4,6 Some researchers have suggested that the constellation of cutaneous or ocular melanomas, atypical melanocytic proliferations, and other internal neoplasms should be termed COMMON syndrome to distinguish it from familial atypical multiple mole melanoma or from dysplastic nevus syndrome.1 In the latter, patients exhibit a high cutaneous melanoma risk but lack an apparent ocular melanoma risk. Moreover, individuals with familial atypical multiple mole melanoma have a large number of dysplastic nevi, which are histologically distinct from BDTs. Familial atypical multiple mole melanoma kindreds have also been shown to harbor inactivating germline events in CDKN2A (OMIM 600160) or activating mutations in CDK4 (OMIM 123829) and are at an increased risk for pancreatic cancer,7 which is seen less often in COMMON syndrome. We describe a new kindred with a novel germline mutation in BAP1 in which the proband phenotypically has dysplastic nevi, BDTs, and multiple conventional superficial spreading melanomas (SSMs).
Report of Cases
During the last 10 years, the pigmented lesion clinic in the Department of Dermatology at Northwestern University followed a man who was initially seen in 2003 at age 43 years with numerous clinically atypical nevi (Figure 1). His personal history included 7 cutaneous melanomas. One was a nodular melanoma, and 6 were SSMs with typical intraepidermal changes of melanoma, including extensive pagetoid and lentiginous growth (Figure 2). All 6 SSMs were reviewed by our histopathologist (P.G.), while the nodular melanoma was part of his medical history and was not available for our review. In all 6 SSMs, the cells were epithelioid but lacked spitzoid cytomorphology. The patient also had several biopsy specimens of dysplastic nevi (Figure 1) and 13 biopsy specimens of classic BDTs (Figure 3). BAP1 (C-4, 1:400; Santa Cruz Biotechnology) and BRAF V600E (VE1, 1:100; Spring Bioscience) immunostains were performed in the SSMs and in 6 BDTs on an autostainer (Leica BOND-MAX; Leica Biosystems) using a kit (DS9390, Polymer Refine Red Detection; Leica Biosystems). All 6 SSMs were negative for immunostaining for BRAF V600E. Two SSMs were also evaluated for BRAF (OMIM 164757) mutation using a test (cobas 4800 BRAF V600; Roche), and were both negative. All 6 cases also showed loss of nuclear immunostaining with anti-BAP1 antibodies (Figure 2). Six of the BDT cases were also evaluated by immunohistochemistry for BRAFV600E mutation, with 3 positive and 3 negative results. One of these negative cases was tested using the mutation test (cobas 4800 BRAF V600), confirming the negative result. All the BDTs showed loss of nuclear BAP1 immunostaining.
Figure 1. Clinical and Histologic Findings of Dysplastic Nevi in the Proband.
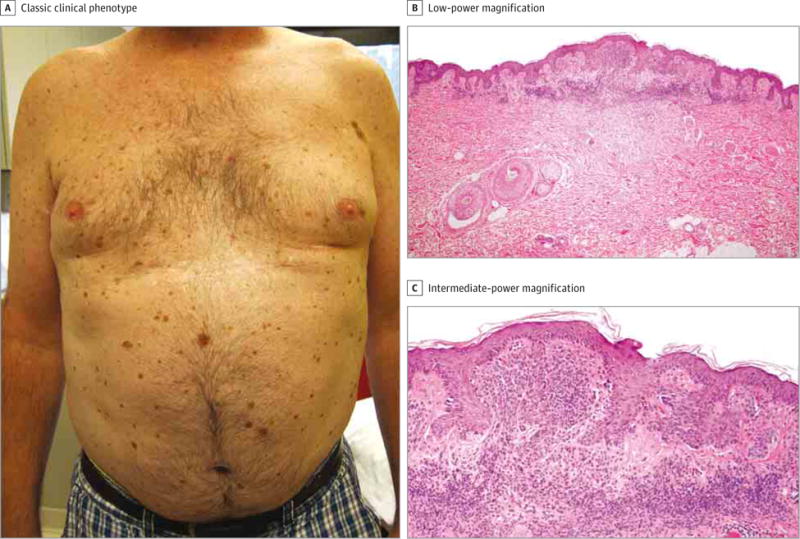
A, Shown is a patient with dysplastic nevus syndrome. B, The dysplastic compound nevus shows size variation of the junctional nests, elongation of the rete ridges, and shoulder phenomenon (hematoxylin-eosin, original magnification ×20). C, Bridging of nests and perireteal fibroplasia are shown (hematoxylin-eosin, original magnification ×100).
Figure 2. Histologic Findings in a Superficial Spreading Melanoma.
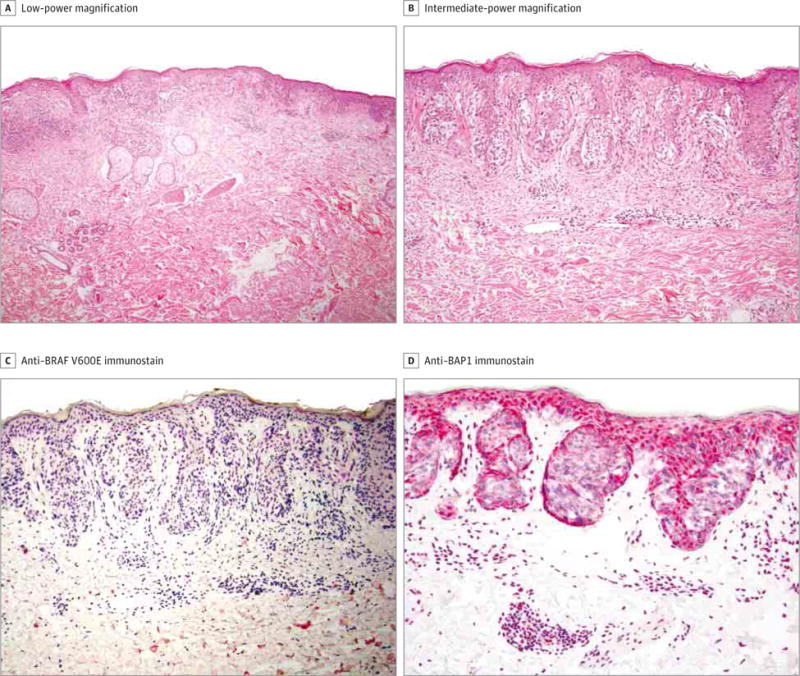
A, Shown is a junctional proliferation of atypical melanocytes (hematoxylin-eosin, original magnification ×40). B, An irregular junctional proliferation of atypical melanocytes with suprabasal movement is shown (hematoxylin-eosin, original magnification ×100). C and D, Shown at intermediate-power magnification (red chromogen, ×100), the tumor cells do not immunostain with anti–BRAF V600E (C) or anti-BAP1 (D).
Figure 3. Histologic Findings in a BAP1-Deficient Tumor.
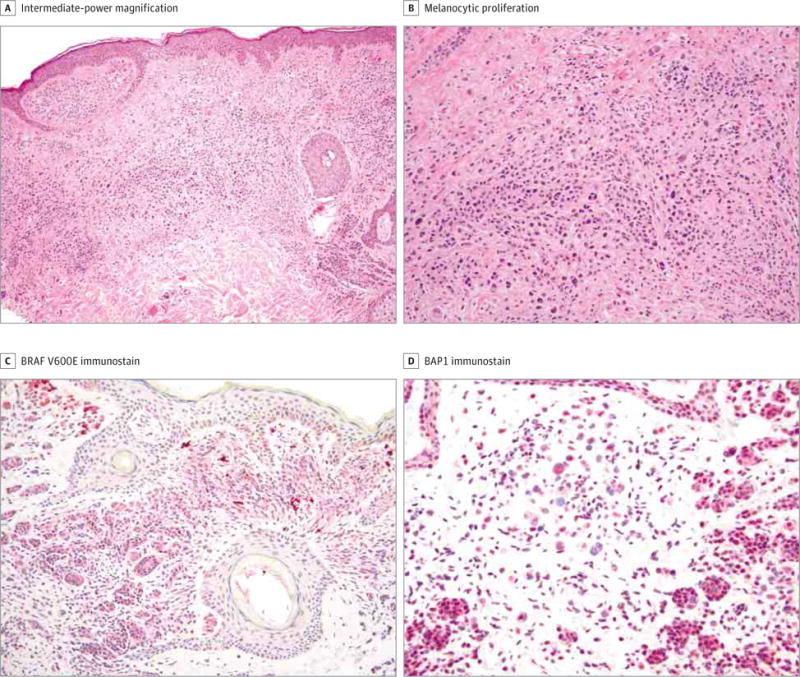
A, Shown is a dermal proliferation of epithelioid melanocytes (hematoxylin-eosin, original magnification ×20). B, This proliferation comprises one component of normal-appearing nevoid cells and a second component of atypical epithelioid spitzoid cells (hematoxylin-eosin, original magnification ×100). C and D, The tumor is positive for BRAF V600E immunostaining (shown at intermediate-power magnification, red chromogen, ×100) (C), and the larger epithelioid cells show dotlike perinuclear BAP1 immunostaining but loss of nuclear expression (shown at high-power magnification, red chromogen, ×200) (D).
One of the melanomas involved a sentinel lymph node, but the complete lymph node dissection was negative. Computed tomographic scans and blood test results suggested no evidence of further metastatic disease. The patient was given interferon alfa but tolerated the therapy for only 1½ months because of dysgeusia and palmoplantar paresthesias. He has had ophthalmologic examinations every 6 months for the last 2 years, which have revealed no evidence of uveal melanoma.
The proband’s family history was significant in that the patient’s mother died of lung adenocarcinoma. He denied any personal or family history of pancreatic cancer or additional cutaneous or uveal melanomas. The patient has a 16-year-old son who also had a BDT.
After obtaining approval from the Northwestern University Cancer Center and the Northwestern University Institutional Review Board and following receipt of written informed consent from the patient and his son and daughter, DNA was extracted from the paraffin-embedded blocks of one of the proband’s BDTs and from one SSM, as well as from his saliva and the saliva of his 16-year-old son and 13-year-old daughter. Sanger sequencing was performed to analyze the germline DNA for BAP1, CDKN2A, and CDK4 mutations. The proband and his son had identical germline nonsense mutations in BAP1 (c.592G>T, p.Glu198X) (Figure 4C and D). One of the patient’s BDTs also showed loss of heterozygosity at BAP1 (Figure 4A), with loss of the wild-type allele. Analysis of a melanoma from the same patient demonstrated the presence of the germline mutation, while the other allele was wild type (Figure 4B). There were no germline CDKN2A or CDK4 mutations.
Figure 4. Electropherogram Showing a Nonsense Mutation in Exon 8 in the BAP1 Gene.
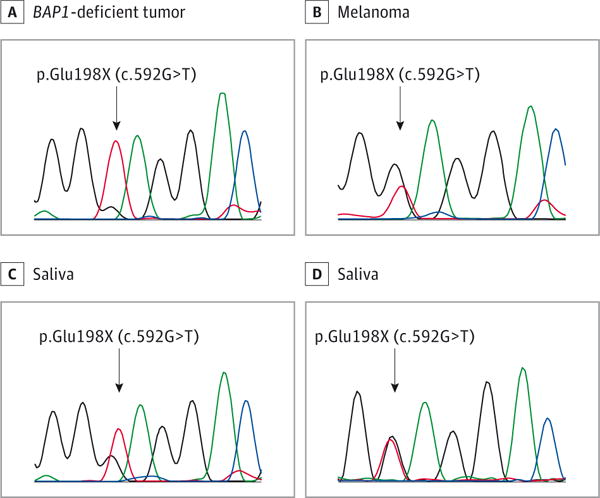
A–C, In the proband, the mutation was found in the DNA from a BAP1-deficient tumor (A), a melanoma (B), and the saliva (C), thereby demonstrating germline origination. D, The same mutation was identified in the saliva of the patient’s 16-year-old son.
Tissue Preparation and Analysis
The tissues were mounted on positively charged slides and allowed to dry in an oven. The slides were dewaxed on the autostainer. BAP1 immunohistochemistry was performed on the autostainer with a high-pH retrieval for 20 minutes. For detection of the antibody, an alkaline phosphatase–based chromagen was used with the kit (Polymer Refine Red Detection). Tumors were scored as positive or negative depending on whether or not their nuclei immunostained with BAP1. The BRAF V600E immunostain was also performed on the autostainer using a low-pH retrieval for 20 minutes, followed by detection using the kit (Polymer Refine Red Detection). The BRAF mutation was detected using DNA extracted from formalin-fixed paraffin-embedded tissue with the mutation test (cobas 4800 BRAF V600), performed according to the manufacturer’s protocol.
Discussion
Multiple cutaneous melanomas have been associated with mutations in different genes (eg, CDKN2A or CDK4).7 Germline variants in BAP1 have an increased susceptibility for different malignancies, with cancers appearing earlier than sporadic cases.8 These malignancies include uveal melanoma and (less frequently) cutaneous melanoma, as well as other cancers.8 To our knowledge, this is the first report of a patient with dysplastic nevus syndrome and an inactivating germline BAP1 mutation.
While all the SSMs from our patient demonstrated loss of BAP1 expression, none of them were found to have a BRAF mutation by polymerase chain reaction or by immunostaining. Conversely, 3 of the 6 BDTs that were evaluated for a BRAFV600E mutation were positive. Hence, there may be distinct tumorigenic pathways in patients with constitutive BAP1 inactivation. If the cooperating driver mutation is BRAFV600E, the melanocytic proliferation may more likely evolve into a characteristic BDT. This is supported by another recent study9 in which a high concordance between nuclear BAP1 loss and BRAFV600E expression was reported. On the other hand, if a cooperating driver mutation is an oncogenic hit other than BRAFV600E, there may be a higher likelihood of melanoma considering that none of our patient’s SSMs had a BRAF mutation, although this model is speculative. Nevertheless, 3 of our 6 BDTs tested did not have a BRAF mutation. Therefore, BDTs can develop in the context of other potential oncogenic stimuli.
To our knowledge, the p.Glu198X is a novel germline BAP1 mutation, although this mutation has been observed in an unrelated tumor specimen.10 The BAP1 protein is a tumor suppressor that acts as a ubiquitin carboxyl-terminal hydrolase functioning as a deubiquitinizing enzyme. It is a binding partner to BRCA1 (OMIM 113705),11 as well as to other transcription factors such as HCF1,1,4 and is implicated in DNA damage repair and regulation of apoptosis, senescence, and cell cycle regulation.8 BAP1 also interacts with ataxia telangiectasia mutated and ataxia telangiectasia and Rad3-related proteins, which regulate the DNA damage induced by UV radiation.8 Therefore, mutations in BAP1 could increase the susceptibility to UV radiation and the risk for melanomas.8 BRAF mutations have been identified in melanomas resulting from intense intermittent bouts of UV exposure.12
This case also illustrates the importance of histopathology in guiding patient management. Patients with BDTs should undergo a thorough review of their family history as it pertains to all cancers. Lesional skin biopsy specimens may be screened for BAP1 loss by immunohistochemistry. It is thought that loss of nuclear expression correlates with absence of BAP1 function.
Conclusions
Identification of this germline BAP1 mutation in a patient with a constellation of multiple cancers and multiple atypical melanocytic tumors expands the cutaneous phenotype of the BAP1 tumor predisposition syndrome. To date, most reports suggest a favorable prognosis for BDTs and BAP1-deficient Spitz tumors,13 including the patient described herein. We speculate that the presence of a BAP1 germline mutation in a patient who phenotypically has dysplastic nevus syndrome may result in a considerable increased risk for cutaneous melanomas. However, further research is needed to determine if the BAP1 germline mutation increases the risk of cutaneous melanoma beyond the dysplastic nevus phenotype alone. Although no formal guidelines exist for patients with germline BAP1 alterations, strict sun protection and regular dermatologic and ophthalmologic evaluations are advisable.
Acknowledgments
Funding/Support: This study was supported in part by the IDP Foundation (Dr Gerami) and by grant K24 CA149202 from the National Institutes of Health (Dr Tsao).
Role of the Funder/Sponsor: The sponsors had no role in the design and conduct of the study; in the collection, analysis, and interpretation of data; in the preparation, review, or approval of the manuscript; and in the decision to submit the manuscript for publication. The sponsors provided no funding or materials for this study.
Footnotes
Author Contributions: Drs Gerami and Tsao had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Gerami, Tsao.
Acquisition, analysis, or interpretation of data: Yélamos, Lee, Obregon, Yazdan, Sholl, Guitart, Njauw.
Drafting of the manuscript: Gerami, Yélamos, Lee, Obregon, Yazdan, Tsao.
Critical revision of the manuscript for importance intellectual content: Gerami, Yélamos, Tsao.
Administrative, technical, or material support: Yélamos, Lee, Sholl, Guitart, Njauw.
Study supervision: Gerami, Tsao.
Conflict of Interest Disclosures: Dr Gerami reported serving as a consultant to Castle Biosciences Inc, Myriad Genetics, and DermTech Inc and receiving honoraria. Dr Guitart reported serving as a consultant to Castle Biosciences Inc and receiving honoraria. No other disclosures were reported.
Contributor Information
Pedram Gerami, Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois; Robert H. Lurie Cancer Center, Feinberg School of Medicine, Northwestern University, Chicago, Illinois.
Oriol Yélamos, Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois.
Christina Y. Lee, Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois.
Roxana Obregon, Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois.
Pedram Yazdan, Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois.
Lauren M. Sholl, Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois.
Gerta E. Guitart, Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois
Ching-Ni Njauw, Wellman Center for Photomedicine and Department of Dermatology, Massachusetts General Hospital, Harvard University, Boston; Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, Boston.
Hensin Tsao, Wellman Center for Photomedicine and Department of Dermatology, Massachusetts General Hospital, Harvard University, Boston; Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, Boston.
References
- 1.Njauw CN, Kim I, Piris A, et al. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One. 2012;7(4):e35295. doi: 10.1371/journal.pone.0035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43(10):1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43(10):1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdel-Rahman MH, Pilarski R, Cebulla CM, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48(12):856–859. doi: 10.1136/jmedgenet-2011-100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiesner T, Murali R, Fried I, et al. A distinct subset of atypical Spitz tumors is characterized by BRAF mutation and loss of BAP1 expression. Am J Surg Pathol. 2012;36(6):818–830. doi: 10.1097/PAS.0b013e3182498be5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, et al. BAP1 loss defines a new class of renal cell carcinoma [published correction appears in Nat Genet. 2012;44(9):1072] Nat Genet. 2012;44(7):751–759. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marzuka-Alcalá A, Gabree MJ, Tsao H. Melanoma susceptibility genes and risk assessment. Methods Mol Biol. 2014;1102:381–393. doi: 10.1007/978-1-62703-727-3_20. [DOI] [PubMed] [Google Scholar]
- 8.Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. 2013;13(3):153–159. doi: 10.1038/nrc3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Busam KJ, Sung J, Wiesner T, von Deimling A, Jungbluth A. Combined BRAFV600E-positive melanocytic lesions with large epithelioid cells lacking BAP1 expression and conventional nevomelanocytes. Am J Surg Pathol. 2013;37(2):193–199. doi: 10.1097/PAS.0b013e318263648c. [DOI] [PubMed] [Google Scholar]
- 10.Yeh I, Mully TW, Wiesner T, et al. Ambiguous melanocytic tumors with loss of 3p21. Am J Surg Pathol. 2014;38(8):1088–1095. doi: 10.1097/PAS.0000000000000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen DE, Proctor M, Marquis ST, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16(9):1097–1112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- 12.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 13.Gammon B, Traczyk TN, Gerami P. Clumped perinuclear BAP1 expression is a frequent finding in sporadic epithelioid Spitz tumors. J Cutan Pathol. 2013;40(6):538–542. doi: 10.1111/cup.12133. [DOI] [PubMed] [Google Scholar]