

Abstract
A goal of synthetic biology is to develop new nucleobases that retain the desirable properties of natural nucleobases at the same time as expanding the genetic alphabet. The non-standard Watson-Crick pair between imidazo[1,2-a]-1,3,5-triazine-2(8H)-4(3H)-dione (X) and 2,4-diaminopyrimidine (K) does exactly this, pairing via complementary arrangements of hydrogen bonding in these two nucleobases, which do not complement any natural nucleobase. Here, we report the crystal structure of a duplex DNA oligonucleotide in B-form including two consecutive X:K pairs in GATCXKDNA determined as a host-guest complex at 1.75 Å resolution. X:K pairs have significant propeller twist angles, similar to those observed for A:T pairs, and a calculated hydrogen bonding pairing energy that is weaker than that of A:T. Thus, although inclusion of X:K pairs results in a duplex DNA structure that is globally similar to that of an analogous G:C structure, the X:K pairs locally and energetically more closely resemble A:T pairs.
TOC graphic
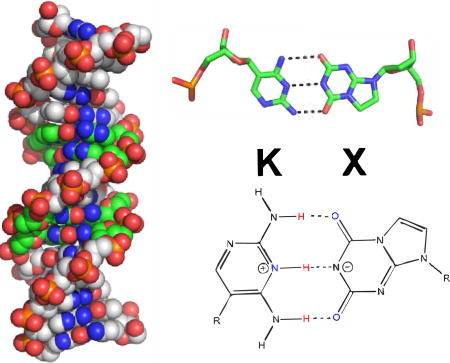
INTRODUCTION
A quarter-century has passed since the first experiments showed that standard DNA does not exploit all hydrogen bonding patterns possible within the Watson-Crick nucleobase pairing scheme1. In standard pairs, the nucleobase heterocycles attached to a deoxyribose sugar through N-glycosidic bonds on antiparallel phosphodiester strands interact on a Watson-Crick edge via hydrogen bonding interactions. However, this Watson-Crick geometry can accommodate as many as twelve nucleobases forming up to as many as six orthogonal nucleobase pairs (Figure 1) 2–6. These form an artificially expanded genetic information system (AEGIS). Explorations of AEGIS have created new technology and medicine as they have opened new frontiers for the study of nucleic acids in general 7.
Figure 1.

Chemical structures with atom numbers for A:T, G:C, P:Z, and X:P.
In particular, no standard pyrimidine nucleotide presents a hydrogen bond donor-acceptor-donor pattern to a purine that presents the complementary acceptor-donor-acceptor hydrogen bonding pattern. In AEGIS, this non-standard pattern can be implemented by the nucleotide analogs 2,4-diamino-5-(1'-beta-D-2'-deoxyribofuranosyl)-pyrimidine (implementing the pyrimidine donor-acceptor-donor hydrogen bonding pattern, K), and 8-(β−D-2'-deoxyribofuranosyl)imidazo[1,2-a]-1,3,5-triazine-2(8H)-4(3H)-dione (implementing the purine acceptor-donor-acceptor hydrogen bonding pattern, X). The first nucleobase K has a pKa of ~6.7 (as a free species, in its protonated form8. The second nucleobase X has a pKa of ~ 8.5, reported here for the first time based on a trace of UV as a function of pH (SI Figure S1). The X:K pair differs from natural pairs in two ways. First, a pyrimidine analog implementing a donor-acceptor-donor hydrogen bonding pattern (like K) must be a C-glycoside, joining the heterocycle to the sugar via a carbon-carbon bond. Second, that analog presents a hydrogen bond donor, an NH2 moiety, to the minor groove. These features of K differ in both respects from natural T and C. These are both N-glycosides, and both present a C=O unit (the oxygen being O2) to the minor groove. Further, K differs in the second respect from the AEGIS 5-nitro-1H-pyridin-2-one heterocycle (presenting the pyrimidine donor-donor-acceptor pattern, trivially Z); Z also presents a C=O unit to the minor groove 9–12. The fact that a nearly complete molecular biology has been developed for the P:Z pair suggests that a pair with a C-glycosidic component is compatible with a synthetic biology (Figure 1).
As noted in previously published work, this particular X:K AEGIS pair is of interest because it has no tautomeric forms, protonated states, or deprotonated states that allow either the purine or the pyrimidine to mispair with any natural nucleobase to form a pair with a Watson-Crick geometry, i.e. edge-on hydrogen bonded pair 13,14. Thus, the fidelity of X:K replication depends only on interactions made directly by the DNA polymerase involved. Native E. coli DNA polymerase I makes little full-length product for a template containing a single K in the absence of dXTP, but incorporates dTTP opposite X in the absence of dKTP in primer extension assays14. This mismatch arises through the formation of a type 1 X:T wobble structure13. Further, when challenged with a substrate containing two consecutive X:K pairs, DNA polymerase I has difficulty extending the primer; this is true whether the template includes KK or a mixture of K and X14.
In earlier work, xanthosine implemented the X hydrogen bonding pattern, and five cycles of replications were carried out using an HIV-1 reverse transcriptase variant12. The fidelity per cycle was calculated by taking the fifth root of the overall extent of loss, which was determined through the generation of a restriction site as a consequence of the loss. The wobble pair that created the loss was studied using standing start primer extension, followed by analysis of the products using gel electrophoresis and band quantitation. Here, fidelity was measured in the absence of the complement 13.
Fidelity has also been assessed by reference to the fidelity of replication of two other AEGIS pairs, the S:B pair and the Z:P pair. With these two AEGIS pairs, in contrast to the X:K pair, mismatching without geometric distortion is possible from a minor tautomer form or a minor deprotonated form. Of course, the exact fidelity depends on the exact polymerase used, the exact molecule implementing the X hydrogen bonding pattern, and the exact conditions where the fidelity is examined.
The ability of E. coli to rescue selectable markers by mismatching was examined in living cells, which ensures fidelity of DNA replication14. Here, the S:B pair and the Z:P pairs, absent their complementary triphosphates, behave in large part as expected based on their tautomeric forms (for the first) and deprotonated forms (for the second)15. The X:K pair is "seen" by the mismatch repair enzyme MutS to be a legitimate match, meaning that this DNA repair system will not excise X:K pairs in living bacterial cells. This makes the X:K pair a prime candidate to enter living systems as a fifth and sixth DNA pair, one that does not suffer from geometric distortion displayed by purely size-complementary pairs16,17.
To advance in a corresponding way the synthetic biology of GACTKX DNA, we began by recognizing that nucleobases play an important role in dictating the overall structure and properties of a DNA duplex 18. In general, duplex structures and their overall local and helical parameters play an important role in recognition of the DNA in processes such as protein binding, replication, gene regulation and subsequent transcriptional events. Exploration of the structure of duplex DNA containing unnatural nucleotides can expand our understanding of this relation. Studies of DNA duplexes having multiple and consecutive non-natural nucleobase pairs are likely to be more informative than studies with duplexes containing only one. This was the case for Z and its partner P (7-amino-9-(1'-beta-D-2'-deoxyribofuranosyl)-imidazo[1,2-c]pyrimidin-5(1H)-one), where both structural and dynamic studies of duplexes containing multiple and consecutive Z:P pairs have advanced our understanding of GACTZP DNA, as well as DNA in general 11,19.
With the goal of understanding how multiple X:K pairs might affect the structure of duplex DNA, which is of particular interest given that E. coli DNA polymerase I has difficulty incorporating two consecutive X:K pairs, we first examined the circular dichroism (CD) spectra of this system in aqueous solution. Then, we used a host-guest system to crystallize 5’- CTTATXXTAKKATAAG −3’, referred to as 2X2K. In this system, the N-terminal fragment (residues 24-278) of Moloney murine leukemia virus reverse transcriptase (MMLV-RT) serves as the host and a 16-mer duplex DNA oligonucleotide as the guest 20,21. The N-terminal fragment of MMLV-RT consists of the fingers and palm domains, and the DNA binds to a site within the fingers domain involving minor groove and backbone hydrogen bonding22.
The use of the host guest system enables us to compare the structures of DNA containing unnatural nucleobases to those of natural DNA duplexes. Since the central ten base pairs out of the sixteen base pairs are free of interaction with the protein, the structures of DNA are dictated by the sequence 20,22–24. Moreover, different DNA sequences crystallize in the same crystal lattice; therefore, the structural comparisons are subject to the same lattice constraints. Our structural analysis of X:K pairs is supported by computational analysis of the electrostatic potential surface, dipole, and hydrogen bonding energy.
RESULTS AND DISCUSSION
Solution properties of GATCKX
The CD spectra of all four sequences (AT, GC, 2X2K and 3K3X) exhibit B-like properties (Figure 2), with subtle differences in the peak position and heights pertaining to differences in the primary sequence of the DNA. The spectra for the AT-rich sequence exhibits a negative peak around 248 nm and a positive, longer wavelength peak at about 279 nm, typical of right-handed B-DNA. The GC- rich sequence on the other hand has a broad negative peak at around 245 nm and a positive peak for the GC-rich sequence shifted to 270 nm instead of 279 observed for the AT-rich sequence, still indicative of right-handed B-form DNA. Both 2X2K and 3K3X sequences exhibit spectra that are more similar to the GC-rich than AT-rich sequence with their broad negative peak at 250 nm and the positive peak at 273 nm. This finding suggests that the overall helical forms and the X:K chromophores more closely resemble B-form GC-rich DNA than AT-rich DNA.
Figure 2.

Characterization of duplex DNA including X:K pairs by circular dichroism (CD). The ellipticity is plotted versus the wavelength for 2X2K (long dash line), 3X3K (dash dot dot line) along with control sequences for GC (dotted line), and AT (solid line). All of the duplexes have CD spectra indicative of right-handed B-form DNA.
Structure of the host-guest complex
To obtain a detailed understanding of the XK-containing DNA, the self-complementary 2X2K 16-mer oligonucleotide was crystallized as a host-guest complex. The host in this system is the N-terminal fragment of Moloney murine leukemia virus reverse transcriptase. Guest DNA molecules including 2X2K that crystallize in this system are B-form in the host-guest complex. 3X3K, like the corresponding oligonucleotide containing P:Z pairs (3/6ZP), did not crystallize as a host-guest complex. DNA-only crystals of 3X3K did not diffract to high resolution in contrast to those obtained for 3/6ZP11. The asymmetric unit of the 2X2K crystal includes one protein molecule and half of the self-complementary 16-mer DNA oligonucleotide or an 8-mer duplex. The host-guest complex is generated by crystallographic symmetry comprising two molecules of the host protein and a 16mer DNA duplex (Figure 3). Both dK and dX yielded well-defined electron density (Figure 3) in the structure and exhibited the expected hydrogen-bonding interactions, namely three hydrogen bonds between the edge-on nucleobases. The hydrogen bonding distances between the two X:K nucleobase pairs vary from 2.64 to 3.29 Å in the 2X2K structure (Figure 3) and will be discussed in more detail below. An advantage of using this system is the ability to directly compare the structures of sequences containing G:C, A:T, or P:Z base pairs in the equivalent positions, crystallized in the same host-guest complex and thus subject to the same lattice constraints.
Figure 3.

Crystal structure of host-guest complex including self-complementary 16 base pair oligonucleotide. (A) The N-terminal fragment of Moloney murine leukemia virus reverse transcriptase serves as the host in the complex including two protein molecules, shown as cartoon renderings in cyan and green, and a 16-mer duplex, each strand shown as a stick rendering C, cyan or green, O, red, N, blue, and phosphorous in orange; X:K pairs are shown with C in magenta. The complex depicted is that of the host-guest complex for the oligonucleotide shown with two consecutive X:K nucleobase pairs (2X2K). Within our crystals, the asymmetric unit includes only half of the complex depicted and thus the equivalent of 8 nucleobase pairs and one protein molecule, indicated by the dashed line. (B) The sequence of the 2X2K sequence and position numbering for the nucleobases within the duplex. (C) The final 2Fo-Fc electron density map is shown as gray mesh renderings contoured at 1.0 σ for the X:K pair at position 6.
The X:K pair presents a total of three electronegative atoms in the major groove as found in the natural counterparts, with potential hydrogen bond acceptors, N5 of K and O6 of X and a potential hydrogen bond donor, the N4 amino group amino group of K (Figure 4). G:C, A:T and P:Z present potential hydrogen bond acceptors including O6 of G or P and N7 of G or A and O4 of T, while the N6 amino group of A and N4 amino group of C or Z can serve as hydrogen bond donors. Z also presents the zwitterionic nitro group in the major groove, which can serve as a hydrogen bond acceptor.
Figure 4.

Van der Waals sphere renderings are shown with O in red, N, blue, P, orange, and C in green for X:K, yellow for P:Z, cyan for G:C, and pink for A:T for major and minor groove presentation faces of the nucleobase pairs.
The presence of a hydrogen bond acceptor, either O2 or N3, associated with the pyrimidine or purine nucleobase, respectively, within the minor groove is thought to be an essential feature in natural pairs, G:C and A:T, that is “read” by DNA polymerases to ensure proper pairing during replication 25,26. The term “read” refers to specific hydrogen bonding interactions formed in the active site between the DNA polymerase and the template/primer substrate. The unnatural X:K pair differs from natural pairs in lacking a hydrogen bond acceptor in the minor groove of K, which instead has a hydrogen bond donor, the N2 amino group, while the unnatural P:Z pair retains the N3 and O2 atoms, respectively, in the minor groove. Specifically, in the minor groove, X presents N3 and O2 hydrogen bond acceptors, while P and G presents N3 and a hydrogen bond donor, the N2 amino group. Thus, the hydrogen bonding capabilities of X:K are unique from those of the other nucleobase pairs. Despite this, X:K is replicated faithfully 13,14.
Helical properties of duplex DNA containing X:K pairs
The helical parameters of the 2X2K structure were analyzed using 3DNA, which uses El Hassan and Calladine’s method 27 to calculate the major and minor groove widths as well as other base pair parameters 28–30, and compared to other host-guest structures of B-form DNA in which X:K is replaced with G:C, A:T and P:Z at the same positions, PDB IDs 4XPE, 4XPC, and 4XO0, respectively 11. The sequences used for comparison are listed in Table 2. Overall, the 2X2K structure maintains B-helical form with an average helical twist of 34.9° +/− 4.18°, corresponding to 10.3 base pairs per turn. In comparison to 2X2K structure, GC, AT and PZ structures have an average helical twist of 34.7° corresponding to 10.4 base pairs per turn. Thus, incorporation of two consecutive X:K pairs in the DNA sequence does not perturb the overall helical form of the DNA duplex significantly. Variations in helical twist of the X:K base pair steps are similar to those observed in all of the structures for the same base pair positions.
Table 2.
Oligonucleotides used for analysis
| Name | Sequence (5’ – 3’) | PDB ID |
|---|---|---|
| 2X2K | 5’-CTTATXXTAKKATAAG | 5VBS |
| 2P | 5’-CTTATPPTAZZATAAG | 4XO0 |
| AT | 5’-CTTATAAATTTATAAG | 4XPC |
| GC | 5’-CTTATGGGCCCATAAG | 4XPE |
All four structures including 2X2K, PZ, GC and AT are B-form throughout, but exhibit some differences in minor and major groove widths. Overall, the major and minor groove widths for 2X2K resemble those of GC and PZ (Figure 5 and Table 3). The average minor and major groove widths of 12.3 Å and 18.3 Å, respectively, associated with the dinucleotide steps containing X:K pairs are similar to those of the corresponding dinucleotide steps in the GC structure of 12.4 Å and 18.0 Å and PZ structure of 12.5 Å and 18.7 Å, respectively (Table 3). AT-rich sequences have a narrower minor groove, as observed in our AT structure with an average minor groove width of 9.7 Å. The minor groove of 2X2K is wider by 2.6 Å than that of the AT structure. On the other hand, the major groove of 2X2K is narrower by 0.8 Å than the AT structure with major groove width of 19.1 Å.
Figure 5.

Comparison of helical properties for host-guest complexes including X:K, G:C, A:T, and P:Z. The unique 8-mer DNA structure including X:K pairs (green) is shown superimposed in (A) with A:T (gray), (B) with G:C (blue), and (C) with P:Z (yellow). (D) The associated minor groove widths for the 16-mer DNA structures are shown in the same colors as designated in (A-C).
Table 3.
Helical parameters and local base pair parameters: AT, GC, 2X2K and PZ
| Local base pair parameters at position 6 | ||||
|---|---|---|---|---|
|
| ||||
| X-K | P-Z | G-C | A-T | |
| Shear (Å) | − 0.44 | − 0.87 | − 1.40 | − 0.22 |
| Stretch (Å) | 0.08 | − 0.42 | − 0.47 | − 0.34 |
| Stagger (Å) | 0.23 | − 0.41 | − 0.02 | 0.18 |
| Buckle (°) | 6.19 | −11.91 | 5.38 | − 1.40 |
| Propeller (°) | −10.61 | − 6.69 | − 5.60 | −15.89 |
| Opening (°) | 6.64 | 2.96 | 12.60 | 8.06 |
|
| ||||
| Local base pair parameters at position 7 | ||||
|
| ||||
| X-K | P-Z | G-C | A-T | |
| Shear (Å) | 0.08 | 0.84 | − 0.68 | 0.39 |
| Stretch (Å) | − 0.07 | − 0.25 | − 0.66 | − 0.21 |
| Stagger (Å) | 0.23 | 0.17 | 0.32 | 0.33 |
| Buckle (°) | −3.09 | 0.05 | − 0.45 | 3.23 |
| Propeller (°) | −19.41 | − 10.05 | − 12.64 | −17.58 |
| Opening (°) | − 8.10 | 0.30 | − 4.85 | 5.93 |
|
| ||||
| Local base pair step parameters | ||||
|
| ||||
| XX/KK | PP/ZZ | GG/CC | AA/TT | |
| Shift (Å) | − 2.03 | − 0.93 | − 1.62 | − 0.29 |
| Slide (Å) | 0.84 | − 0.02 | 0.99 | − 0.22 |
| Rise (Å) | 3.43 | 3.03 | 3.49 | 3.13 |
| Tilt (°) | − 5.12 | − 6.26 | − 5.74 | − 1.17 |
| Roll (°) | − 4.82 | 4.31 | − 5.58 | − 3.15 |
| Twist (°) | 39.29 | 39.03 | 43.67 | 39.74 |
|
| ||||
| Helical parameters | ||||
|
| ||||
| XX/KK | PP/ZZ | GG/CC | AA/TT | |
| X-displacement (Å) | 1.83 | − 0.51 | 1.87 | 0.02 |
| Y-displacement (Å) | 2.35 | 0.68 | 1.58 | 0.30 |
| Helical – rise (Å) | 3.54 | 3.12 | 3.52 | 3.14 |
| Inclination (°) | − 7.10 | 6.38 | − 7.43 | − 4.63 |
| Tip (°) | 7.53 | 9.27 | 7.64 | 1.71 |
| Helical-twist (°) | 39.89 | 39.74 | 44.36 | 39.88 |
|
| ||||
| Other parameters | ||||
|
| ||||
| X:K | P:Z | G:C | A:T | |
| Overall Helical twist (°) | 34.93 (4.18 SD) | 34.69 (8.05 SD) | 34.70 (7.19 SD) | 34.70 (3.49 SD) |
| # Average Minor Groove width (Å) | 12.3 | 12.5 | 12.4 | 9.7 |
| # Average Major Groove width (Å) | 18.3 | 18.7 | 18.0 | 19.1 |
Average values obtained for dinucleotide steps 5–7 containing X:K, Z:P, G:C, or A:T, respectively.
Other helical parameters including x-displacement, y-displacement, helical rise, inclination and tip are similar to the GC structure as shown in Table 3. The helical twist angle for the X:K base pairs also lies in the range usually reported for B form DNA, with X:K having a helical twist of 39.89° (B-form DNA has helical twist values ranging between 27.9° to 40°) 31–34. X:K pairs resemble more closely G:C pairs than P:Z pairs in terms of local helical conformations. Minor variations in the values of helical parameters between GC and 2X2K are a result of sequence dependent effects on the structure.
Local properties of the X:K nucleobase pair
Local base pair parameters, local base pair step parameters, and groove widths for the X:K pair were analyzed using 3DNA (Table 3). In comparing the local base pair parameters for X:K with P:Z, G:C, and A:T, the buckle values for positions 6 and 7 (refer to Figure 1 for numbering scheme), 6.19° and −3.09°, respectively, are similar to G:C values, 5.38° and −0.45°, but less similar to those for P:Z, −11.91° and 0.05° (Table 3). Shear, stagger, and opening values are within the range observed for the other base pairs, while stretch values of 0.08 and −0.07 Å for X:K pairs in positions 6 and 7 are smaller in magnitude than those observed for other base pairs.
The X:K base pair is not planar, as evidenced by propeller twist angles of −10.61° and −19.41° for positions 6 and 7, respectively, closer to those observed for A:T (−15.98° and −17.58°) than G:C (−5.6° and −12.64°) or P:Z (−6.69° and −10.05°) (Figure 6A and Table 3). Significant propeller angles in A:T pairs are often attributed to the fact that there are only two hydrogen bonds in the pair. However, this feature in the X:K pair, which has three hydrogen bonds, suggests that it is perhaps an inherent property of the base pair itself and not dependent upon the number of hydrogen bonds. Stacking interactions of X:K and A:T pairs are superficially similar as shown in Figure 6B.
Figure 6.

Comparison of local base pair properties of X:K and A:T. (A) Cartoon/stick renderings for X:K and A:T pairs (7th nucleotide step, X7:K10 as shown in Fig.3) with a semi-transparent gray molecular surface superimposed show significant propeller twist angles for the base plane of X or A vs. K or T. A similar rendering is shown for a relatively planar G:C base pair (6th nucleotide step G6:C11). (B) Stacking of XX/KK and AA/TT shown as stick renderings are overall similar in accommodating the nucleobase pairs with significant propeller twist angles.
It was therefore of interest to consider the electrostatic potential surface (ESP), dipole, and hydrogen bonding energies for X:K, A:T, G:C, and P:Z pairs. The ESP approximates the short-range electronic environment, while the dipole describes a longer-range electronic effect and the hydrogen bond energies, the tightness of the Watson-Crick pair. The ESP visually displays that there are different hydrogen bonding opportunities in X:K vs. A:T (Figure 7). Within the ESP map, we have also displayed the electrical dipole moments. X:K has a very different electric dipole moment than A:T, both in magnitude and direction. The electrical dipole moment for the X:K pair is rotated ~90 degrees relative to A:T. Moreover, the A:T dipole is 2.1 Debye whereas the dipole in X:K is only 1.3 Debye. The Watson-Crick hydrogen bonding energy of A:T is greater than X:K by 3.6 kcal/mol (Table 4). Thus, the contributions from three hydrogen bonds in X:K energetically are weaker than for two hydrogen bonds in A:T. The estimate of the A:T electronic binding energy matches previous calculations done35. The Gibbs energy of forming the X:K pair is just barely negative at −1.9 kcal/mol. Note this only tells us what the hydrogen bonding contribution to the energy is, and previous studies have shown that base-pair stacking dispersion interactions are more numerically significant to the total Gibbs energy 36. The Watson-Crick hydrogen bonding free energy utilized CCSD(T)37–39 /aug’’-cc-pVTZ for the electronic energy, which has been shown to be accurate to ~1 kcal/mol of the true gas-phase energy of single-reference wave functions; entropic contribution was based on the M06-2X/aug’’-cc-pVTZ partition function. The overall point is clear that X:K hydrogen bonding energy per hydrogen bond is significantly weaker than A:T, allowing the net contribution of the X:K pair towards duplex stability that is slightly less than the net contribution by A:T pair, notwithstanding it having three hydrogen bonds. These calculations are consistent with lower experimental melting temperatures determined for oligonucleotides including X:K pairs (SI, Table S1).
Figure 7.

Dipole moments and electrostatic potential maps (ESPs) for (A) X:K, (B) A:T, (C) G:C, (D) P:Z nucleobase pairs. Methyl substituents are used in place of the C1’ carbon in the deoxyribose ring. The ESP color gradation is such that red is ~-40 kcal/mol and blue is ~+40 kcal/mol; strict energetic interpretation is limited to indications of broad differences in reactivity. Dipole moments are shown as a blue arrow, following the convention that a positive vector points toward a positive charge density. The magnitude of the vectors is not proportional to length (length modified for ease of view).
Table 4.
Watson-Crick Pair Hydrogen Bonding Energy Contributions (kcal/mol) for X:K and A:T Nucleobase Pairs.
| X:K | A:T | Δ | ||
|---|---|---|---|---|
| ΔU (Electronic) | −15.2 | −16.1 | +0.9 | |
| ΔH | −13.8 | −13.1 | −0.7 | |
|
| ||||
| −TΔS | 11.9 | 7.6 | +4.3 | |
|
| ||||
| ΔG | −1.9 | −5.5 | +3.6 | |
Of note, in the calculated structures of the X:K pair, the proton on N1 of X is apparently transferred to N3 of K. This transfer appears to produce the most favorable arrangement of secondary interactions proposed by Jorgensen and Pranata 40. In their analysis, a symmetric pattern of partial positive and negative charges in forming three hydrogen bonds is the least favorable arrangement. This is the hydrogen bonding arrangement depicted for X:K in Figure 1. Alternatively, deprotonation of N1 in X and protonation of N3 in K produces the most favorable secondary arrangement with three partial positive charges associated with K and three partial negative charges associated with X as depicted in Figure 8 along with the calculated electron density supporting this arrangement for the protons. Alternatively, one could say that the energy of the endergonic acid-base transfer (given the pKa values of ~6.7 for K8 and pKa of 8.5 for X, SI Figure S1) comes at the expense of the exergonic hydrogen bonds formed. This helps us to understand why three hydrogen bonds are not better than two: we only achieved this at the expense of a proton transfer.
Figure 8.

The calculated X:K pair has a proton transfer. (A) The chemical structure is shown for the calculated X:K pair with bond distances in the vicinity of the hydrogen bonding pairs. Bond lengths (Å) of Key Species for Watson-Crick Binding of X:K. All calculations are based on M06-2X/aug''-cc-pVDZ geometries, expected to be accurate to within 0.03Å. Note that the proton transfer from X to K, as shown by the bond lengths. (B) The calculated electron density is shown for the X:K pair and clearly shows that the proton involved in the central hydrogen bonding pair is associated with N3 of K and not N1 of X. Electron density contours given at 0.04 electron charge/Bohr3 from the KS-DFT density matrix.
One might be skeptical of the computational claim that an acid-base transfer is necessary to form the Watson-Crick pair. This finding is independent of the gas-phase DFT calculation, however. If one uses an implicit solvent model (Cramer and Truhlar’s SMD model SMD) to represent an aqueous solvent with the same DFT protocol, the proton transfer is also observed (1.05 Å hydrogen bond distance to the K nitrogen, 1.79 Å to the X nitrogen). Similarly, if one uses an ab initio wave function calculation with no empirical parameters such as MP2 (otherwise known as MBPT(2)), the same features are observed (1.09 Å to the K nitrogen, 1.62 Å to the X nitrogen).
The fact that the hydrogen bonding is weaker in X:K than in A:T, likely contributes to the observed propensity to propeller. As noted above, the X:K pairs each include one hydrogen bond longer than 3.0 Å. For position 6, O2-N2, N1-N3, and O6-N4 hydrogen bonding distances are 2.64, 2.98, and 3.18 Å, respectively; for position 7, O2-N2, N1-N3, and O6-N4 are 3.28, 2.98, and 2.69Å, respectively (See Figure 1 for atom numbers, Figure 3 for position numbers). Thus, for position 6, the long hydrogen bond is the O6-N4 bond, while for position 7, it is the O2-N2 bond. This finding is consistent with weaker theoretical hydrogen bonding in general, larger propeller angles, and potentially a contribution from buckling as well. The X:K pair at position 6 has a buckle angle of 6.2 ° and propeller angle of −10.6 °, while position 7 buckles in the opposite direction, buckle angle of −3.1 °, and has a propeller angle of −19.4 °. Similarly, the A:T pairs have one long hydrogen bond for positions 6 and 7 (numbering shown in Figure 3), in this case the same bond, N6-O4, 3.08 and 3.13 Å, respectively. Both of the A:T pairs have large propeller angles, −15.9 and −17.6 ° with small buckle angles, −1.4 ° and 3.2 °, respectively.
On the other hand, P:Z hydrogen bonding distances at position 6 for N2-O2, N1-N3, and O6-N4 are 2.57, 2.79, and 2.94 Å, respectively, and the values for position 7P:Z are 2.63, 2.80, and 2.90 Å, respectively 11. This finding is consistent with stronger theoretical hydrogen bonding reported for P:Z41 than G:C or A:T. P:Z pairs have also been shown to be more stable than possible mispairs experimentally42. Finally, the G:C pair in position 6 exhibits one long hydrogen bond for position 6, O6-N4 distance of 3.37 Å, potentially due to a combination of shearing by −1.4 Å, buckling of 5.4 °, and opening of 12.6 °. For position 7, all three hydrogen bonds are 2.71 Å. Thus, in the absence of a large shearing effect, G:C hydrogen bonding interactions are all less than 3.0 Å. The two P:Z pairs are also sheared by −0.87 and 0.84 Å, respectively for position 6 and 7, but retain normal hydrogen bonding distances.
On comparing other local base pair step parameters of the 2X2K structure with other B-form DNA structures (G:C, A:T and P:Z), including axial rise per nucleotide (B-DNA values range between 3.03 Å to 3.37 Å) and base pair tilt (B-DNA values range between −5.9° to −16.4°) 31–34, both rise and tilt values for 2X2K lie in the same range. The XX/KK dinucleotide step base has a rise and tilt of 3.43 Å and – 5.12°. The phosphate-phosphate (P-P) distances also show little deviation from that of standard B-form DNA. The P-P distance for B-DNA is around 7 Å while for XX and KK, the P-P distances are 6.5 Å and 6.3 Å, respectively 34. The chi angles of all the residues fall in anti- conformation as found in B-form DNA. Our results suggest that the 2X2K structure resembles B-form DNA, specifically B-form DNA sequences rich in G:C base pairs.
The bigger picture. Why terran DNA uses the standard nucleobase pairs?
As experimental work associated with AEGIS pairs has developed, we have learned that pairs with non-standard hydrogen bonding arrangements are able to robustly support duplex structures. This includes duplexes between strands where multiple consecutive nonstandard pairs are present. This is not the case for pairs whose pairing principle is based solely on hydrophobic interactions or size complementarity, lacking interbase hydrogen bonding. One example is Romesberg’s nonstandard bases d5SICS and dNaM that pair by hydrophobic and geometric complementarity and stack via an intercalative mode rather than edge on in duplex DNA16,43. Another is Hirao’s hydrophobic unnatural base pair system between 7-(2-thienyl)imidazo[4,5-b]pyridine (Ds) and 2-nitro-4-propynylpyrrole (Px)44,45. Hirao’s hydrophobic nucleobases Ds:Px are successfully amplified in a PCR reaction; and the crystal structure of the ternary complex of Klentaq incorporating dPxTP opposite Ds indicates that the hydrophobic pairs can also act as promising candidates for incorporation by DNA polymerases46. However, these studies do not describe the effect of multiple hydrophobic base pairs on the overall structure of DNA duplex. A clash of templating dDs with side chain oxygen atom of Thr664, more flexible thumb domain in the ternary structure and lack of a binary structure incorporating these hydrophobic nucleotides suggest that more studies would be needed for hydrophobic base pairs before their use for the expansion of the genetic alphabet46.
To date, the vast majority of functional and structural data existing for nonstandard nucleobases mainly focuses on the incorporation of a single non-natural base pair in a DNA duplex. It is important to understand the effect of incorporation of multiple nonstandard base pairs in a duplex DNA that will not distort the overall structure of DNA significantly and also provide a basis for their retention in duplex DNA after multiple rounds of replication. Thus, the X:K pair meets a goal of synthetic biology, to develop expanded genetic systems that retain the desirable properties of natural nucleobases, including full evolvability.
These observations, however, also raise the question as to why natural DNA uses the nucleobases that it does. For example, we struggle to understand why adenine presents only two hydrogen bonding units to its thymine complement, while advanced biotechnologists must struggle to obtain uniform hybridization and priming in a system that contains a "weak" nucleobase pair and a "strong" nucleobase pair. Why not use 2-aminoadenine (diaminopurine) instead of adenine and get a pair joined by three canonical hydrogen bonds?
Under a Darwinian model, one cannot speak of "optimization" of a genetic system without presuming that alternative systems were accessible through random variation. In fact, some viruses are known to use 2-aminoadenine in their DNA, suggesting that this alternative was in fact available to terran life during its natural history47. Given this, one might interpret the surprisingly weak pair between 2-aminoadenine and thymidine, especially in DNA as compared to RNA 48, as a second example of the disadvantage of symmetry in Watson-Crick pairing. This disadvantage is also present in the similarly symmetrical X:K pair.
Conclusion
In summary, properties of interest associated with the X:K pair include a unique pattern of hydrogen bond donor and acceptors presented in the major and minor grooves that differs from those of A:T, G:C, or P:Z. As we observed for P:Z, inclusion of two consecutive X:K base pairs and four X:K pairs total in a self-complementary 16-mer oligonucleotide is readily accommodated in B-form DNA within our host-guest system. It is essential that any artificial components be accommodated in B-form DNA, as that is the form in which genomic DNA is most often found in cells. Sequence-specific properties associated with dinucleotide steps independent of their position in the oligonucleotide (excluding the three terminal base pairs involved in interactions with the protein) were previously demonstrated for a host-guest study of the CA dinucleotide integrase processing site 24. Thus, the host-guest system has been vetted for analysis of sequence specific effects free of differing lattice constraints that might impact the structural properties of the oligonucleotides.
In this study, we analyzed the structural properties of the dinucleotide base pair steps for positions 6 and 7 including X:K, P:Z, G:C, or A:T. Structurally, the X:K pairs exhibit propeller angles similar to those in A:T pairs, while buckle values and other base pair parameter values were similar to those of G:C in the same position. The propensity for X:K pairs to exhibit significant propeller twist angles is supported by melting temperature data and calculation of hydrogen bonding energies for X:K, which are in fact weaker than A:T. These calculations support a hydrogen bonding pair of X:K comprising a deprotonated X and protonated K. Overall, the helical properties of 2X2K are B-form and most similar to G:C and P:Z with major and minor groove widths similar to those observed for G:C.
Thus, the inability of E. coli DNA polymerase I to incorporate two consecutive X:K pairs does not result from significant distortions in duplex DNA as a result of consecutive X:K pairs. Rather, it more likely results from the chemical properties of the nucleobases, the lack of a hydrogen bond acceptor in K, for example; DNA polymerases are known to “read” the minor groove through specific hydrogen bonding interactions25,26. This is not a significant limitation for the use of X:K in expanding the genome as its use as a single pair is supported. In summary, we conclude that inclusion of X:K pairs provides unique properties while still maintaining compatibility with biologically relevant forms of DNA and therefore has the potential to expand the genetic alphabet.
METHODS
Synthesis and purification of KX containing oligonucleotides
AEGIS-containing oligonucleotides were prepared by solid phase synthesis using phosphoramidites of dX and dK synthesized using procedures described elsewhere. These phosphoramidites are now available via Firebird Biomolecular Sciences, LLC (www.firebirdbio.com, Alachua, FL). The GACTKX "six letter" DNA molecules 5’- CTTATXXTAKKATAAG −3’ (2X2K) and 5’- CTTATXXXKKKATAAG −3’ (3K3X) were prepared in house, as described below. The remaining sequences used here were purchased from IDT (Coralville IA).
Experiments showed that deprotection of GACTKX DNA oligonucleotides by treatment under standard condition (ammonium hydroxide, 55 °C, overnight) led to substantial decomposition of the dX heterocycle. Therefore, the A, G, C, and K exocyclic amines were protected as the phenoxyacetyl, phenoxyacetyl, acetyl, and isobutyroyl groups, respectively. Then, all GACTKX oligonucleotides used in this study were deprotected using 50 mM K2CO3 in MeOH at 55 °C, overnight. They were then purified by HPLC.
Circular dichroism analyses
Circular dichroism (CD) studies were used to assess the helical form of the oligonucleotide duplexes in aqueous solution that was buffered at neutral pH with low salt concentrations. The self-pairing DNA sequences analyzed included 2X2K (5’- CTTATXXTAKKATAAG −3’), 3X3K (5’- CTTATXXXKKKATAAG −3’), the corresponding sequence with A:T pairs (5’ CTTATAAATTTATAAG 3’) and the corresponding sequence with G:C pairs (5’ CTTATGGGCCCATAAG 3’).
For CD analysis, stock solutions (2.5 mM) of these DNA sequences were diluted to 5 µM with buffer containing 10 mM HEPES pH 7.0 and 10 mM MgCl2. The CD spectra for DNA sequences were collected on a Jasco J-810 CD instrument at a temperature of 25 °C, at a rate of 50 nm/min and a wavelength increment of 0.1 nm. Ellipticity, Ø (mdegrees) was recorded for the DNA sequences from a wavelength of 320 to 220 nm. The final spectrum is the average of five scans corrected for ellipticity readings obtained for buffer (10 mM HEPES pH 7.0, 10 mM MgCl2) by itself. Spectra were initially measured for GC and AT control sequences and subsequently for 2X2K and 3K3X sequences.
Crystallization and data collection
The self-complementary 16-mer DNA oligonucleotides containing either two or three X:K pairs (2X2K or 3X3K) were resuspended in buffer containing 10 mM HEPES (pH 7.0) and 10 mM MgCl2 and then annealed by heating to 70 °C followed by slow cooling to room temperature to give a final concentration of 2.5 mM duplex DNA. The protein (the N-terminal fragment including residues 24-278 of Moloney murine leukemia virus reverse transcriptase, MMLV RT) was diluted to a concentration of 0.65 mM in two steps. A 2.9 mM stock solution of the protein was diluted to 1.4 mM using 50 mM MES (pH 6.0) and 0.3 M NaCl. This 1.4 mM sub-stock was then further diluted to 0.65 mM in 100 mM HEPES (pH 7.5) and 0.3 M NaCl. The host-2X2K or host-3X3K (protein-DNA) complex was set at a ratio of 1:2 respectively (0.43 mM Protein: 0.86 mM DNA) in buffer containing 100 mM HEPES (pH 7.5), 0.3 M NaCl and incubated at 4° C for 1 h.
The self-nucleated protein-DNA crystals of host-GC were used as seeds for crystallizing host protein with oligonucleotides containing KX. Crystals of host protein complexed with GC grew in hanging drops containing 1 μL protein-DNA complex and 1 μL of solution containing 10 % PEG 4000, 5 mM magnesium acetate and 50 mM ADA (pH 6.5). The reservoir solution consisted of 500 μL 10 % PEG 4000, 5 mM magnesium acetate, and 50 mM ADA (pH 6.5). Host −2X2K microseeded crystals grew at 8 % PEG 4000, 5 mM magnesium acetate, and 50 mM ADA (pH 6.5). No host-guest crystals were obtained for the 3X3K complex. DNA-only 3X3K crystals did not diffract to high resolution and were not further pursued. The host-2X2K crystals were cryoprotected in 9 % PEG 4000, 5 mM magnesium acetate, 100 mM HEPES (pH 7.5) and 20 % ethylene glycol before flash freezing in liquid nitrogen for data collection.
The 2X2K host-guest complex data were collected to a resolution of 1.75 Å at SBC-19-BM beamline at the Advanced Photon Source, Argonne National Laboratory, Darien, IL (Table). Data reduction was carried out in space group P21212, and data were indexed, integrated and scaled using HKL3000 49. The host-guest crystal structure was determined by molecular replacement using the CCP4 program MOLREP 50 using the protein model from PDB ID 4XO0 11. Use of the protein model alone for phasing provided unbiased electron density for the DNA in the host guest complex. Adjustments to the protein model and addition of water molecules were done in COOT 51 and initially refined in REFMAC 52.
Table 1.
Data and refinement statistics for 2X2K
| a (Å) | 55 |
| b (Å) | 145.6 |
| c (Å) | 46.9 |
| Space group | P21212 |
| Resolution | 33.72 – 1.75 |
| Total observations | 484673 |
| Unique reflections | 38305 |
| Completeness | 98.4 (92.3) |
| Rmeas (%) | 4.2 (33.8) |
| Rpim (%) | 1.7 (15.4) |
| I/σ | 37.4 (4.65) |
| Refinement statistics | |
| R value (%) | 20.9 |
| R free (%) | 23.9 |
| RMSD bonds (Å) | 0.007 |
| RMSD angles (degree) | 1.114 |
| Number of Atoms | |
| Protein/DNA | 1968/324 |
| Water | 188 |
| Average B-factors | |
| Protein/DNA | 24.13/48.36 |
| Water | 26.06 |
For building the DNA, initially 3 base pairs were built, followed by refinement in REFMAC to improve the electron density for the next consecutive base pairs. The protein-DNA refinement was followed by the addition of two more base pairs and refinement in REFMAC. The parameter and linking files for KX were created in PHENIX 53. Finally, the last three base pairs were added including the K s and X s, and refinement was done using PHENIX. Multiple rounds of model adjustment in COOT and refinement in PHENIX yielded an R-work and R-free values of 20.86 % and 23.85 %, respectively. Coordinates have been deposited for 2X2K with PDB (Table 1) with PDB identifier 5VBS.
Quantum Mechanical Calculations
Structures of the X:K, P:Z, G:C, and A:T pairs were optimized using M06-2X54/aug’’-cc-pVTZ55 in the Gaussian09 software 56. In each structure, the 2'-deoxyribose was modeled by a methyl group to represent the surrounding electronic environment. The single prime notation refers to the use of aug’-cc-pVTZ on all heavy atoms (not hydrogen); the use of double primes indicates that no diffuse functions were used on either hydrogen or carbon atoms. This approach was chosen due to the linear dependencies present by having so many diffuse functions over aromatic rings, indicating that their absence helps SCF convergence with no loss in the basis representation.
A structure was deemed to be “optimized” when the RMS force was no greater than 1.0x10−4 Hartree/Bohr over all geometric parameters and no single geometric parameter having a force greater than 3.3x10−4 Hartree/Bohr. The KS-DFT 57,58 Lebedev integration grid used 99 radial points and 590 solid angle points. The SCF was deemed to be “converged” when the change in SCF matrix elements was less than 10−6. Spherical d functions were used throughout for all basis sets in this paper. The construction of all electrostatic potential maps used the KS determinant density with isocontours 0.001 elementary charge per cubic Bohr. The M06-2X functional was chosen for its strong performance in calculating organic molecule energies, geometries, and dispersion phenomena, despite being parameterized empirically. All dipole moments calculated were based on this SCF reference.
The Watson-Crick hydrogen bonding free energy utilized the composite method of CCSD(T) 37–39 /aug’-cc-pVDZ describing the electronic energy, which has been shown to be accurate to ~1 kcal/mol of the true gas-phase energy of single-reference wave functions59. The basis set extrapolation used the Helgaker 36 scheme for aug’’-cc-pVTZ and aug’’-cc-pVQZ extrapolation. The coupled cluster equations were considered “converged” when the coupled cluster energy equation tensor amplitudes changed less than 10−6. All coupled cluster and MP2 calculations utilized the ACES3 software 60 for its ability to parallelize over thousands of processors. All calculations were performed on the Big Red II supercomputer of Indiana University. The entropic contributions to the free energy used the vibrational partition function based on M06-2X/aug’’-cc-pVTZ optimization. The Watson-Crick hydrogen bond energy is here defined to be the difference between the interacting pair (say PZ) vs. the lone fragments, each individually optimized (P and Z, separately, in this case).
Supplementary Material
Figure S1. UV absorbance as a function of pH for the dX nucleoside gives pKa of 8.5 ±0.1.
Table S1. Melting point temperatures for oligonucleotides containing X:K pairs Coordinates of M06-2X/aug’’-cc-pVDZ optimized structures, Å.
Acknowledgments
Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. Calculations were performed on the Indiana University Big Red 2 supercomputer. The authors thank the Lilly Endowment, Inc. for support of the Indiana University Pervasive Technology Institute and the Indiana METACyt Initiative. The material is based in part upon work supported by NASA under awards NNX14AK37G and NNX15AF46G. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Aeronautics and Space Administration. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM111386. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This material is also based in part upon work supported by the National Science Foundation under Grant No. CHE 1507816.
Footnotes
Author contributions
M.M.G., I.S., and S.A.B. designed the study. I.S. prepared the protein and M.J.K. synthesized the oligonucleotides containing X:K pairs, originally designed and characterized by S.A.B. and coworkers. M.J.K. measured the pKa of X, and H.S. performed melting temperature measurements. I.S. performed the crystallizations and measured C.D. data; I.S. measured X-ray diffraction data, phased the structure by molecular replacement, and completed the crystallographic refinement. I.S. and M.M.G. performed the structural analysis of the host-guest complex. R.W.M. performed the quantum mechanical calculations. I.S., M.M.G., R.W.M., and S.A.B. wrote and edited the manuscript.
References
- 1.Ghosh A, Bansal M. A glossary of DNA structures from A to Z. Acta Crystallogr D Biol Crystallogr. 2003;59:620. doi: 10.1107/s0907444903003251. [DOI] [PubMed] [Google Scholar]
- 2.Switzer C, Moroney SE, Benner SA. Enzymatic incorporation of a new base pair into DNA and RNA. J Am Chem Soc. 1989;111:8322. [Google Scholar]
- 3.Piccirilli JA, Krauch T, Moroney SE, Benner SA. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature. 1990;343:33. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 4.Benner SA. Understanding nucleic acids using synthetic chemistry. Acc Chem Res. 2004;37:784. doi: 10.1021/ar040004z. [DOI] [PubMed] [Google Scholar]
- 5.Geyer CR, Battersby TR, Benner SA. Nucleobase pairing in expanded Watson-Crick-like genetic information systems. Structure. 2003;11:1485. doi: 10.1016/j.str.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Yakovchuk P, Protozanova E, Frank-Kamenetskii MD. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006;34:564. doi: 10.1093/nar/gkj454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benner SA, Karalkar NB, Hoshika S, Laos R, Shaw RW, Matsuura M, Fajardo D, Moussatche P. Alternative Watson-Crick Synthetic Genetic Systems. Cold Spring Harbor perspectives in biology. 2016;8 doi: 10.1101/cshperspect.a023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lutz MJ, Horlacher J, Benner SA. Recognition of a non-standard base pair by thermostable DNA polymerases. Bioorg Med Chem Lett. 1998;8:1149. doi: 10.1016/s0960-894x(98)00177-2. [DOI] [PubMed] [Google Scholar]
- 9.Yang Z, Chen F, Chamberlin SG, Benner SA. Expanded genetic alphabets in the polymerase chain reaction. Angew Chem Int Ed Engl. 2010;49:177. doi: 10.1002/anie.200905173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z, Chen F, Alvarado JB, Benner SA. Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J Am Chem Soc. 2011;133:15105. doi: 10.1021/ja204910n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Georgiadis MM, Singh I, Kellett WF, Hoshika S, Benner SA, Richards NG. Structural basis for a six nucleotide genetic alphabet. J Am Chem Soc. 2015;137:6947. doi: 10.1021/jacs.5b03482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sismour AM, Lutz S, Park JH, Lutz MJ, Boyer PL, Hughes SH, Benner SA. PCR amplification of DNA containing non-standard base pairs by variants of reverse transcriptase from Human Immunodeficiency Virus-1. Nucleic Acids Res. 2004;32:728. doi: 10.1093/nar/gkh241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winiger CB, Kim MJ, Hoshika S, Shaw RW, Moses JD, Matsuura MF, Gerloff DL, Benner SA. Polymerase Interactions with Wobble Mismatches in Synthetic Genetic Systems and Their Evolutionary Implications. Biochemistry. 2016;55:3847. doi: 10.1021/acs.biochem.6b00533. [DOI] [PubMed] [Google Scholar]
- 14.Winiger CB, Shaw RW, Kim MJ, Moses JD, Matsuura MF, Benner SA. Expanded Genetic Alphabets: Managing Nucleotides That Lack Tautomeric, Protonated, or Deprotonated Versions Complementary to Natural Nucleotides. ACS Synth Biol. 2017;6:194. doi: 10.1021/acssynbio.6b00193. [DOI] [PubMed] [Google Scholar]
- 15.Benner SA, Shaw RW. 2015 [Google Scholar]
- 16.Betz K, Malyshev DA, Lavergne T, Welte W, Diederichs K, Dwyer TJ, Ordoukhanian P, Romesberg FE, Marx A. KlenTaq polymerase replicates unnatural base pairs by inducing a Watson-Crick geometry. Nat Chem Biol. 2012;8:612. doi: 10.1038/nchembio.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malyshev DA, Dhami K, Lavergne T, Chen T, Dai N, Foster JM, Correa IR, Jr, Romesberg FE. A semi-synthetic organism with an expanded genetic alphabet. Nature. 2014;509:385. doi: 10.1038/nature13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neidle S. Nucleic Acid Structure. Oxford University Press; New York: 1999. [Google Scholar]
- 19.Molt RW, Jr, Georgiadis MM, Richards NGJ. Consecutive non-natural PZ nucleobase pairs in DNA impact helical structure as seen in 50 mus molecular dynamics simulations. Nucleic Acids Res. 2017;45:3643. doi: 10.1093/nar/gkx144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun D, Jessen S, Liu C, Liu X, Najmudin S, Georgiadis MM. Cloning, expression, and purification of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase: crystallization of nucleic acid complexes. Protein Sci. 1998;7:1575. doi: 10.1002/pro.5560070711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cote ML, Yohannan SJ, Georgiadis MM. Use of an N-terminal fragment from moloney murine leukemia virus reverse transcriptase to facilitate crystallization and analysis of a pseudo-16-mer DNA molecule containing G-A mispairs. Acta Crystallogr D Biol Crystallogr. 2000;56:1120. doi: 10.1107/s0907444900008246. [DOI] [PubMed] [Google Scholar]
- 22.Najmudin S, Cote ML, Sun D, Yohannan S, Montano SP, Gu J, Georgiadis MM. Crystal structures of an N-terminal fragment from Moloney murine leukemia virus reverse transcriptase complexed with nucleic acid: functional implications for template-primer binding to the fingers domain. J Mol Biol. 2000;296:613. doi: 10.1006/jmbi.1999.3477. [DOI] [PubMed] [Google Scholar]
- 23.Cote ML, Georgiadis MM. Structure of a pseudo-16-mer DNA with stacked guanines and two G-A mispairs complexed with the N-terminal fragment of Moloney murine leukemia virus reverse transcriptase. Acta Crystallogr D Biol Crystallogr. 2001;57:1238. doi: 10.1107/s090744490100943x. [DOI] [PubMed] [Google Scholar]
- 24.Montano SP, Cote ML, Roth MJ, Georgiadis MM. Crystal structures of oligonucleotides including the integrase processing site of the Moloney murine leukemia virus. Nucleic Acids Res. 2006;34:5353. doi: 10.1093/nar/gkl693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joyce CM, Steitz TA. Function and structure relationships in DNA polymerases. Annu Rev Biochem. 1994;63:777. doi: 10.1146/annurev.bi.63.070194.004021. [DOI] [PubMed] [Google Scholar]
- 26.Hendrickson CL, Devine KG, Benner SA. Probing minor groove recognition contacts by DNA polymerases and reverse transcriptases using 3-deaza-2'-deoxyadenosine. Nucleic Acids Res. 2004;32:2241. doi: 10.1093/nar/gkh542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El Hassan Ma, Calladine CR. Two distinct modes of protein-induced bending in DNA. J Mol Biol. 1998;282:331. doi: 10.1006/jmbi.1998.1994. [DOI] [PubMed] [Google Scholar]
- 28.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu XJ, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nature protocols. 2008;3:1213. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colasanti AV, Lu XJ, Olson WK. Analyzing and building nucleic acid structures with 3DNA. Journal of visualized experiments : JoVE. 2013:e4401. doi: 10.3791/4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prive GG, Heinemann U, Chandrasegaran S, Kan LS, Kopka ML, Dickerson RE. Helix geometry, hydration, and G.A mismatch in a B-DNA decamer. Science. 1987;238:498. doi: 10.1126/science.3310237. [DOI] [PubMed] [Google Scholar]
- 32.Grzeskowiak K. Sequence-dependent structural variation in B-DNA. Chemistry & biology. 1996;3:785. doi: 10.1016/s1074-5521(96)90062-9. [DOI] [PubMed] [Google Scholar]
- 33.Berman HM. Crystal studies of B-DNA: the answers and the questions. Biopolymers. 1997;44:23. doi: 10.1002/(SICI)1097-0282(1997)44:1<23::AID-BIP3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 34.Drew HR, Wing RM, Takano T, Broka C, Tanaka S, Itakura K, Dickerson RE. Structure of a B-DNA dodecamer: conformation and dynamics. Proc Natl Acad Sci U S A. 1981;78:2179. doi: 10.1073/pnas.78.4.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sponer J, Jurecka P, Hobza P. Accurate interaction energies of hydrogen-bonded nucleic acid base pairs. J Am Chem Soc. 2004;126:10142. doi: 10.1021/ja048436s. [DOI] [PubMed] [Google Scholar]
- 36.Bak KL, Jørgensen P, Olsen J, Helgaker T, Klopper W. Accuracy of atomization energies and reaction enthalpies in standard and extrapolated electronic wave function/basis set calculations. J Chem Phys. 2000;112:9229. [Google Scholar]
- 37.Watts JD, Gauss J, Bartlett RJ. Coupled-cluster methods with noniterative triple excitations for restricted open-shell Hartree–Fock and other general single determinant reference functions. Energies and analytical gradients. J Chem Phys. 1993;98:8718. [Google Scholar]
- 38.Coester F, Kümmel H. Short-Range Correlations in Nuclear Wave Functions. Nuclear Physics. 1960;17:477. [Google Scholar]
- 39.Purvis GD, Bartlett RJ. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J Chem Phys. 1982;76:1910. [Google Scholar]
- 40.Jorgensen WL, Pranata J. Importance of secondary interactions in triply hydrogen bonded complexes: guanine-cytosine vs uracil-2,6-diaminopyridine. J Am Chem Soc. 1990;112:2008. [Google Scholar]
- 41.Wang W, Sheng X, Zhang S, Huang F, Sun C, Liu J, Chen D. Theoretical characterization of the conformational features of unnatural oligonucleotides containing a six nucleotide genetic alphabet. Phys Chem Phys. 2016;18:28492. doi: 10.1039/c6cp05594j. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Hoshika S, Peterson RJ, Kim MJ, Benner SA, Kahn JD. Biophysics of Artificially Expanded Genetic Information Systems. Thermodynamics of DNA Duplexes Containing Matches and Mismatches Involving 2-Amino-3-nitropyridin-6-one (Z) and Imidazo[1,2-a]-1,3,5-triazin-4(8H)one (P) ACS Synth Biol. 2017;6:782. doi: 10.1021/acssynbio.6b00224. [DOI] [PubMed] [Google Scholar]
- 43.Malyshev DA, Pfaff DA, Ippoliti SI, Hwang GT, Dwyer TJ, Romesberg FE. Solution structure, mechanism of replication, and optimization of an unnatural base pair. Chemistry. 2010;16:12650. doi: 10.1002/chem.201000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimoto M, Kawai R, Mitsui T, Yokoyama S, Hirao I. An unnatural base pair system for efficient PCR amplification and functionalization of DNA molecules. Nucleic Acids Res. 2009;37:e14. doi: 10.1093/nar/gkn956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashige R, Kimoto M, Takezawa Y, Sato A, Mitsui T, Yokoyama S, Hirao I. Highly specific unnatural base pair systems as a third base pair for PCR amplification. Nucleic Acids Res. 2012;40:2793. doi: 10.1093/nar/gkr1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Betz K, Kimoto M, Diederichs K, Hirao I, Marx A. Structural Basis for Expansion of the Genetic Alphabet with an Artificial Nucleobase Pair. Angew Chem Int Ed Engl. 2017 doi: 10.1002/anie.201704190. [DOI] [PubMed] [Google Scholar]
- 47.Kirnos MD, Khudyakov IY, Alexandrushkina NI, Vanyushin BF. 2-aminoadenine is an adenine substituting for a base in S-2L cyanophage DNA. Nature. 1977;270:369. doi: 10.1038/270369a0. [DOI] [PubMed] [Google Scholar]
- 48.Howard FB, Miles HT. 2NH2A X T helices in the ribo- and deoxypolynucleotide series. Structural and energetic consequences of 2NH2A substitution. Biochemistry. 1984;23:6723. doi: 10.1021/bi00321a068. [DOI] [PubMed] [Google Scholar]
- 49.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 50.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 53.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2008;120:215. [Google Scholar]
- 55.Kendall RA, Dunning TH, Jr, Harrison RJ. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys. 1992;96:6796. [Google Scholar]
- 56.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JAJ, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision E.01 [Google Scholar]
- 57.Hohenberg P, Kohn W. Inhomogeneous Electron Gas. Physical Review. 1964;136:B864. [Google Scholar]
- 58.Kohn W, Sham LJ. Self-Consistent Equations Including Exchange and Correlation Effects. Physical Review. 1965;140:A1133. [Google Scholar]
- 59.Řezáč J, Hobza P. Benchmark Calculations of Interaction Energies in Noncovalent Complexes and Their Applications. Chem Rev. 2016;116:5038. doi: 10.1021/acs.chemrev.5b00526. [DOI] [PubMed] [Google Scholar]
- 60.Lotrich VF, Flocke N, Ponton M, Yau AD, Perera AS, Deumens E, Bartlett RJ. Parallel implementation of electronic structure energy, gradient, and Hessian calculations. J Chem Phys. 2008;128:194104. doi: 10.1063/1.2920482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. UV absorbance as a function of pH for the dX nucleoside gives pKa of 8.5 ±0.1.
Table S1. Melting point temperatures for oligonucleotides containing X:K pairs Coordinates of M06-2X/aug’’-cc-pVDZ optimized structures, Å.